

The role of calyceal KCNQ channels and associated M-current in normal mammalian vestibular function is unknown. Our results show that calyceal KCNQ channels are critical for normal vestibular function in the intact mammal. The findings provide evidence that efferent modulation of M-currents may act normally to differentially adjust the sensitivity of vestibular neurons to transient and tonic stimulation and that such mechanisms may be targeted to achieve effective clinical management of vestibular disorders.
Keywords: KCNQ channels, vestibular afferent, calyx, M-current, mammal, VsEP, XE991, retigabine
Abstract
The precise role and mechanisms underlying efferent modulation of peripheral vestibular afferent function are not well understood in mammals. Clarifying the details of efferent action may lead to new strategies for clinical management of debilitating disturbances in vestibular and balance function. Recent evidence in turtle indicates that efferent modulation of M-currents is likely one mechanism for modifying afferent discharge. M-currents depend in part on KCNQ potassium conductances (Kv7), which can be adjusted through efferent activation of M1, M3, and/or M5 muscarinic acetylcholine receptors (mAChRs). How KCNQ channels and altered M-currents affect vestibular afferent function in vivo is unclear, and whether such a mechanism operates in mammals is unknown. In this study we used the KCNQ antagonist XE991 and the KCNQ activator retigabine in anesthetized mice to evaluate the effects of M-current modulation on peripheral vestibular responses to transient head motion. At low doses of XE991, responses were modestly enhanced, becoming larger in amplitude and shorter in latency. Higher doses of XE991 produced transient response enhancement, followed by steady-state suppression where latencies and thresholds increased and amplitudes decreased. Retigabine produced opposite effects. Auditory function was also impacted, based on results of companion auditory brain stem response testing. We propose that closure of KCNQ channels transforms vestibular afferent behavior by suppressing responses to transient high-frequency stimuli while simultaneously enhancing responses to sustained low-frequency stimulation. Our results clearly demonstrate that KCNQ channels are critical for normal mammalian vestibular function and suggest that efferent action may utilize these mechanisms to modulate the dynamic characteristics and gain of vestibular afferent responses.
NEW & NOTEWORTHY The role of calyceal KCNQ channels and associated M-current in normal mammalian vestibular function is unknown. Our results show that calyceal KCNQ channels are critical for normal vestibular function in the intact mammal. The findings provide evidence that efferent modulation of M-currents may act normally to differentially adjust the sensitivity of vestibular neurons to transient and tonic stimulation and that such mechanisms may be targeted to achieve effective clinical management of vestibular disorders.
there is growing interest and progress in identifying the mechanisms underlying vestibular efferent neural action on primary vestibular afferents and the role that efferent vestibular system (EVS) action plays in modulating afferent discharge behavior. The interest in part stems from the idea that EVS synaptic mechanisms and downstream signal pathways may be amenable to pharmacological manipulation. Understanding these mechanisms may reveal new strategies and clinical targets for treatment of debilitating balance disorders. A role for KCNQ channels and M-currents in vestibular efferent action has been recently demonstrated in the turtle (Holt et al. 2017). A similar role for KCNQ in the mammalian periphery is possible but has not been definitively demonstrated (Holt et al. 2015a).
Four of the five KCNQ channel subunits (2, 3, 4, and 5, here designated KCNQ*, Kv7 gene family) mediate an M-current consisting of a low-voltage-activated outward K+ current (IKL) that is found widely in neurons throughout the nervous system (Brown and Passmore 2009). The current is modulated by G protein-coupled receptors and acts to influence the general excitability and dynamic response characteristics of neurons.
In mammalian embryos and neonates, KCNQ* channels are upregulated and present in both type I hair cells and calyceal afferent endings (Hurley et al. 2006; Kharkovets et al. 2000; Rocha-Sanchez et al. 2007), where they have been shown to mediate an M, or M-like, K+ current in isolated vestibular ganglia and hair cells (Holt et al. 2007; Hurley et al. 2006; Pérez et al. 2009, 2010). In contrast, in the mature mammalian vestibular sensory epithelium, KCNQ* channels are primarily found at relatively high densities in the membranes of calyx-bearing terminals with little evidence for their presence in vestibular type I hair cells (Hurley et al. 2006; Lysakowski et al. 2011; Spitzmaul et al. 2013). The importance and role of these channels in normal adult mammalian vestibular function, however, is largely unknown. Null mutations of KCNQ4 reportedly do not produce behaviors suggestive of vestibular deficits (Kharkovets et al. 2006; Spitzmaul et al. 2013), drawing into question a putative critical role for KCNQ4 subunits in vestibular function despite being a dominant channel subunit in calyx surface microdomains (Lysakowski et al. 2011; Spitzmaul et al. 2013). However, in many cases behavior is not a sensitive indicator of peripheral vestibular deficits (e.g., Goodyear et al. 2012; Jones et al. 2005; Jones and Jones 2014; Lee et al. 2013; Mathur et al. 2015), and thus there could be significant vestibular dysfunction in the KCNQ4 null mouse. Indeed, Spitzmaul et al. (2013) reported reduced vestibulo-ocular reflex (VOR) gain in mice across all frequencies (1/16 to 4 Hz) in channel knockouts (KCNQ4−/−) and knockdowns (KCNQ5dn/dn). These findings provided the first evidence that KCNQ4 and KCNQ5 channels may influence central or peripheral VOR circuitry in mammals. Moderate changes in the VOR and lack of morphological changes in the vestibular epithelium, however, are a striking contrast to the well-known autosomal dominant progressive deafness (DFNA2) and loss of cochlear outer hair cells produced by KCNQ4 mutation (e.g., human: Coucke et al. 1999; De Leenheer et al. 2002; mouse: Kharkovets et al. 2000, 2006).
The central question addressed in this study is whether or not KCNQ* channels play a critical role specifically in peripheral vestibular function in the mammal. Understanding and clarifying the role of KCNQ channels expressed in the inner ear may lead to better therapeutic approaches to the prevention and/or treatment of auditory and vestibular disorders. To examine this question, we measured vestibular compound action potentials in the intact mouse using vestibular sensory evoked potentials (VsEPs) before and after systemic administration of the potent KCNQ channel antagonist XE991 (M-current blocker; Wang et al. 1998) and the KCNQ activator retigabine (Tatulian et al. 2001). Our findings show that KCNQ* channels are critical for normal mammalian vestibular function and provide strong evidence in support of the hypothesis that KCNQ* channels modulate vestibular afferent sensitivity and response dynamics normally in the intact mammal.
METHODS
All experimental procedures were approved by the Institutional Animal Care and use committee at University of Nebraska-Lincoln and undertaken in accordance with National Institutes of Health guidelines. Fifty-three C57BL/6J mice (age 8–11 wk; weight 18.1 ± 1.2 g; female) were obtained from The Jackson Laboratory. The animals were anesthetized with urethane-xylazine (1.2 g/kg urethane:20 mg/kg xylazine), administered intraperitoneally (volume 0.1 ml). No animals required maintenance doses following initial anesthesia. A thermocouple microwire (Cole Parmer, Vernon Hills, IL) was implanted in the brain 2–3 mm below the skull surface for the measurement and control of brain temperature. A rectal temperature probe was placed to monitor and control core rectal temperature. Brain and rectal temperatures were maintained at 36.0 ± 0.2°C and 37.0 ± 0.2°C, respectively, by using a laboratory heating lamp and homeothermic heating pad (FHC, Bowdoin, ME). During data collection, heart rate (beats/min), respiration rate (inspirations/min), and blood oxygen saturation (, %) were monitored and recorded every 5 min. To prevent hypoxemia under anesthesia, oxygen gas supplement (50% N2 and 50% O2) was presented to anesthetized animals as a mixture with room air. levels remained stable over the course of studies.
Drug preparation and doses.
XE991 (dihydrochloride) was obtained from Tocris Bioscience (Bristol, UK) and was dissolved in sterile 0.9% sodium chloride saline (Nova-Tech, Grand Island, NE). Mice received a single dose of XE991 (0.05, 0.5, 1, 2.5, 3.5, 5, 12.5, 25, or 50 mg/kg; maximum volume of 0.7 ml) or saline (volume 0.1 ml) intraperitoneally to characterize drug effects over a period of at least 80 min. Retigabine was obtained from Alomone Laboratories (Jerusalem, Israel) and was dissolved in 100% dimethyl sulfoxide (DMSO; Fisher Science, Pittsburgh, PA). Mice received a single dose of 20 mg/kg retigabine or 100% DMSO in a 0.1-ml volume intraperitoneally.
Vestibular sensory evoked potentials.
The VsEP recording method has been described in detail elsewhere (e.g., Jones et al. 2015) and is briefly described here. The linear VsEP is the compound neural response to a transient linear acceleration of the head and is composed of several positive and negative electrical response peaks occurring within ~4–5 ms following the onset of the stimulus. The first three peaks (P1, N1, and P2) were scored and analyzed. P1 and N1 response peaks represent the compound action potential of the peripheral vestibular nerve innervating macular sensors (Jones 1992; Jones et al. 1999, 2004; Nazareth and Jones 1998) and are referred to as “peripheral” response peaks. Response peak P2 is a “central peak” reflecting the compound activity of central vestibular relays in brain stem vestibular nuclei. VsEP threshold is a measure of the sensitivity of peripheral macular sensors to transient linear acceleration of the head (Jones and Jones 1999). Threshold was defined as the stimulus level midway between the highest stimulus failing to produce a response and the lowest stimulus level producing a response. Response latencies were scored as the time of occurrence for each peak (P1, N1, P2) relative to the onset of the stimulus in micro- or milliseconds. Amplitudes were calculated as the voltage differences between positive and negative peaks: P1-N1 and P2-N1 in microvolts. VsEP response parameters (thresholds, latencies, and amplitudes) were quantified and used for evaluating functional changes.
To produce an adequate head motion stimulus (Jones et al. 2011), a linear voltage ramp of 2-ms duration was applied to an electromechanical shaker (model ET-132-203; LabWorks, Costa Mesa, CA). The animal’s head was securely coupled to the shaker using a noninvasive clip (Jones et al. 2015; Zhao et al. 2008). The direction of motion was parallel to the naso-occipital axis and in a vertical direction with the animal oriented nose up. Motion was measured using a calibrated accelerometer mounted to the shaker platform. The output of the accelerometer (100 mV/g, where g = 9.81 m/s2) was electronically differentiated to provide a monitor of stimulus jerk levels (Jones et al. 2011). The differentiated output (jerk magnitude) was expressed in units of decibels relative to 1.0 g/ms (dB re: 1.0 g/ms). The resulting head translation stimulus was a rectangular (step) jerk having a duration of 2 ms. A stimulus repetition rate used during all studies was 17 stimuli/s.
Three subcutaneous needle electrodes were placed over the nuchal crest (G1), behind the right pinna (G2), and on the hip (ground). Electrophysiological signals were amplified (200,000 times; Grass P511, model K), filtered (0.3–3 kHz, −6-dB amplitude points) for VsEP recordings. Analog-to-digital conversion of the electrophysiological signals was triggered at the onset of each stimulus (1,024 points at 10 µs/point). Initial upward or downward stimulus motions defined opposite stimulus polarities. Signal averaging was used to resolve responses out of background noise and generate a VsEP response trace. To produce each VsEP trace, separate averaged responses to each polarity (128 sweeps each) were collected and then averaged for a total of 256 sweeps. Two trace pairs were recorded for each stimulus level used. A binaural forward masker (90 dB SPL, bandwidth: 50 Hz to 50 kHz; Jones and Jones 1999) was presented via sound field to confirm the absence of auditory responses during VsEP recording. The masker was presented with the use of a free-field speaker (model FF1; Tucker Davis Technologies, Alachua, FL).
VsEPs were recorded at a stimulus level of +6 dB re: 1.0 g/ms at 5-min intervals during the 20-min stable baseline period before drug or vehicle injection, then at 2-min intervals for the first 20 min following drug or vehicle injection, and thereafter at 5-min intervals as needed. VsEP thresholds were determined at ~20-min intervals before and after intraperitoneal XE991 or retigabine administration.
Auditory brain stem responses.
In a portion of the animals, we sought to characterize the time course and extent of effects on auditory function. To do this, we measured auditory brain stem responses (ABRs). The short-latency evoked ABR occurs within ~6–7 ms following the onset of the click stimulus. P1 and N1 response peaks (peripheral origins) and P4 and N4 peaks (central origins) were scored and quantified in the current study (e.g., Buchwald and Huang 1975; Henry 1979; Jewett 1970). ABR latencies (P1, N1, and P4) and amplitudes (P1-N1 and P4-N4) were analyzed to characterize drug effects on auditory function. ABR thresholds were not determined.
ABR was recorded at 15-min intervals between VsEP measurements. ABR was evoked by a single rectangular voltage pulse of 0.03-ms duration, which generated a click at a stimulus level of ~114 dB peSPL (peak equivalent sound pressure level). ABR stimuli were calibrated using a Brüel & Kjær ⅛-in. microphone (SN 4138) and analyzed by fast-Fourier transform (bandwidth: 0–25.2 kHz, −3 dB point). Click stimuli were presented via a free-field acoustic driver, which was also used for acoustic masking during VsEP recording (noted above). The standard signal averaging technique described above for VsEP was also used to record ABRs.
Data analysis and statistics.
The collected response data were normalized by subtracting the mean baseline value obtained before drug or vehicle administration from the value measured after drug or vehicle administration [e.g., a normalized data point = value (at time t) minus mean baseline value]. The normalized data were statistically analyzed using either multivariate (rmMANOVA) or univariate repeated-measures analysis of variance (rmANOVA) for response parameters at multiple time points. Post hoc comparisons for determining within-group differences over time were made using Bonferroni procedures as noted. Linear regression was also used to evaluate effects over time. For the VsEPs recorded at 5-min intervals, 4 to 5 consecutive 5-min measurements were averaged to characterize each 20-min period where desired. Descriptive statistics are also expressed as means ± SD (n = sample size) unless otherwise stated. A P value <0.05 was considered significant. Statistical analyses were performed using SPSS (version 22.0; SPSS, Chicago, IL). Sigmoid curve fits (“dose-response curve,” Hill equation) were generated using SigmaPlot (version 12.5; Systat Software, San Jose, CA). The equation used is given by
where Y is the expected response at dose x, max is the maximum drug effect, min is the minimum drug effect, ED50 is the drug dose that gives half-maximal drug effect, and Hill slope is the steepest slope of the curve.
RESULTS
The effects of XE991 administration on vestibular function were evaluated at 10 dose levels in 53 animals using VsEPs. KCNQ channel blockade by XE991 significantly altered vestibular function. Figure 1A illustrates representative vestibular response waveforms obtained before and after administration of three different XE991 dose levels (3.5, 12.5, and 50 mg/kg) as well as treatment with vehicle alone (saline). All response waveforms in the saline group (Fig. 1A, top) represent typical normal vestibular (VsEP) responses.
Fig. 1.

A: VsEP response waveforms before (Bas, baseline) and 20 and 40 min after XE991 (doses as shown) or saline injection. Stimulus level used for all responses was +6 dB re: 1.0 g/ms. Response traces begin at the onset of the transient head movement stimulus (arrow). Response peaks P1, N1, and P2 are labeled for the first trace pair of the saline group (top) and P1 and N1 (marked with lines) for other trace pairs. Note the absence of changes in vestibular responses following saline injection (top 3 trace pairs). Significant delays in latency and reduction in amplitudes can be seen at 20 and 40 min following XE991 injection, where the magnitude of change increases with increasing dose (3.5, 12.5, and 50 mg/kg). Maximum drug effects were reached before 40 min after drug. Calibration bars indicate 1.0 ms and 1.0 μV for graphic. Dashed vertical lines identify latencies of P1, N1, and P2 before drug injection. B: suppression of vestibular responses following higher doses of XE991 (2.5 to 50 mg/kg). Normalized mean changes (difference from baseline) in VsEP latencies (P1 and N1; in µs) and amplitudes (P1-N1; in µV) are plotted over time for different dose groups of XE991 and saline. Data at left of vertical dashed line represent the baseline period before drug administration, and data at right indicate drug effects following injection. XE991 injection was made at time 0. Different doses are color coded the same for all latency and amplitude graphs and are marked to aid comparisons. Sample sizes were as follows: 50 mg/kg, n = 5; 25 mg/kg, n = 6; 12.5 mg/kg, n = 5; 5 mg/kg, n = 6; 3.5 mg/kg, n = 6; 2.5 mg/kg, n = 5; 1 mg/kg, n = 5; 0.5 mg/kg, n = 5; 0.05 mg/kg, n = 5; and saline, n = 5. Stimulus level was +6 dB re: 1.0 g/ms.
Several features of the responses are worth noting. First, the initial three positive and negative peaks are marked and labeled (P1, N1, and P2, top trace pair, Fig. 1A). P1 signals the onset of the response. The latency of P1 reflects the amount of time required to activate gravity receptor neurons in response to the onset of the transient head motion stimulus. The stimulus onset is indicated with an arrow in Fig. 1A, occurring at the beginning of each response trace. The latencies of N1 and P2 reflect the time required for the conduction of the resulting vestibular compound action potential along the eighth nerve and transmission across the first brain stem relay synapse in vestibular nuclei. VsEP response amplitudes (P1-N1, P2-N1) reflect the number of synchronously activated neurons contributing to the response. Second, normal vestibular responses are highly replicable and stable over time as can be seen from the pairs of vestibular responses recorded before (Fig. 1A, baseline) and at 20 and 40 min after saline injection (Fig. 1A, saline). In this case, the timing of activation and response amplitudes remained the same. These normal features of responses in the “saline group” should be contrasted with responses recorded before and after XE991. All mice had robust VsEP responses before XE991 treatment (baseline). However, a prominent dose-dependent suppression of vestibular responses occurred within 20 min following XE991, and this can be seen in the waveforms of Fig. 1A for doses of 3.5, 12.5, and 50 mg/kg. Figure 1B summarizes quantitative changes in latencies and amplitudes over the course of 80 min following XE991 administration for 8 of the 10 doses used in the present study. Suppression was signaled by a substantial delay of response onset latency and reduction of response amplitudes at 20 and 40 min following injection. There was also an early period of modest response enhancement, including decreases in the latency of response onset and increased amplitudes. An example of enhancement is seen in Fig. 2A at 20 and 40 min following a low-dose XE991 injection. Figure 2B summarizes response changes across animals during enhancement, where changes in latency and amplitude are plotted vs. time relative to XE991 injection for several lower doses (0.05 to 2.5 mg/kg). A period of enhancement was present at doses as low as 0.5 mg/kg (Fig. 2). At doses of 2.5 mg/kg and higher, responses showed a biphasic pattern over time. This pattern consisted of an initial period of response enhancement as noted followed by a second phase of sustained response suppression (i.e., delayed and diminished responses, Fig. 1). At lower doses between 0.05 and 2.5 mg/kg, only enhancement of vestibular responses was seen (Fig. 2). In contrast, such changes were absent in animals receiving vehicle alone (saline; Figs. 1 and 2). The detailed characteristics of VsEP response enhancement and suppression are considered below.
Fig. 2.

Vestibular response enhancement at low doses of XE991. A: VsEP response waveforms shown were recorded before (B, baseline) and 20 and 40 min after a dose of 0.5 mg/kg XE991. Notice the shortening of latencies for all peaks (P1, N1, P2) at 20 and 40 min after injection. A modest increase in amplitudes for P1-N1 is present as well. Calibration bars indicate 1.0 ms and 2.0 μV for graphic. Dashed vertical lines identify latencies of P1, N1, and P2 before drug injection. Stimulus level was 6 dB re:1 g/ms. B: mean values during enhancement of vestibular responses following low doses of XE991 (0.5 and 1.0 mg/kg). Values are normalized to baseline mean value for responses before injection. Values are mean changes (difference) from baseline (0) for response latencies (P1, N1; in μs) and amplitudes (P1-N1; in μV) over time for each dose group indicated. Significant reduction in latencies and increased amplitudes were found for 0.5 and 1.0 mg/kg only. Dose groups are represented by different colors as follows: 0.05 (dark green; n = 5), 0.5 (dark red; n = 5), and 1 mg/kg (dark blue; n = 6) and saline (black; n = 5). These low doses of XE991 led to decreased VsEP peripheral latencies (P1, N1) and increased amplitudes (P1-N1) systematically. Time 0 is time of XE991 injection. Data at left of vertical dashed lines represent the baseline period before drug administration, and data at right indicate drug effects after drug injection. Stimulus level was 6 dB re: 1.0 g/ms.
VsEP response enhancement.
The initial evidence of drug effects in all cases was signaled by a relatively modest response enhancement (Fig. 2). For doses of 0.5 and 1.0 mg/kg, the decrease in mean latencies approached a maximum of 0.1 ms and mean amplitudes increased on the order of 0.1 to 0.2 μV (rmMANOVA, 0.5 mg/kg: amplitudes, P = 0.019, latencies P = 0.015; 1 mg/kg: amplitudes, P = 0.030, latencies, P = 0.007). Although enhancement at these doses began within 10 min of drug administration, the maximum magnitude of enhancement was achieved slowly within ~40 min, depending on dose, and remained through the rest of the recording period (Fig. 2). At doses of 0.05 mg/kg or following saline injection, VsEP responses showed no change in latencies and amplitudes over time. There also were no significant changes in vestibular threshold for doses less than 3.5 mg/kg (Fig. 3A). At higher doses, enhancement was interrupted by a dose-dependent response suppression beginning within 10–20 min of drug administration, depending on dose, as described below.
Fig. 3.

Changes in mean VsEP response parameters following various doses of XE991. Different doses are color coded the same for all panels as marked and identified in A to aid comparisons. A: changes in vestibular response threshold over time following XE991 injection. Mean VsEP thresholds are represented as the difference from mean baseline threshold in dB re: 1 g/ms. Threshold was determined every 20 min. Each line represents the mean difference from baseline across individual animals in each dose group as a function of time. For low doses from 0.05 to 0.5 mg/kg, threshold changes were not significant during the period of enhanced latencies and amplitudes. For higher doses (3.5 to 50 mg/kg), significant threshold increases above baseline over time occurred during response suppression. B: high-resolution sampling in time. VsEPs were recorded every 2 min for the first 20 min following XE991 administration. Peripheral response components of the VsEP (P1, N1, P1-N1) are represented. Enhancement and suppression of responses developed systematically in a dose-dependent manner. Onset timing of suppression was generally between 5 and 10 min after drug. Sample sizes are as follows: 50 mg/kg, n = 5; 25 mg/kg, n = 2; 12.5 mg/kg, n = 5; 5 mg/kg, n = 6; 3.5 mg/kg, n = 6; 2.5 mg/kg, n = 5; 1 mg/kg, n = 5; 0.5 mg/kg, n = 5; 0.05 mg/kg, n = 5; and saline, n = 4. Stimulus level was +6 dB re: 1.0 g/ms.
VsEP response suppression.
The effects of suppression emerged at a dose of 2.5 mg/kg, where enhancement began within minutes of injection as usual but was reversed over the remaining period of 80 min (Fig. 1B). At doses above 2.5 mg/kg, XE991 dramatically suppressed VsEP responses (Figs. 1 and 3, Table 1) following a brief period of modest enhancement. Changes in responses during suppression included increased mean latencies (Fig. 4A; dose-response regression: P1, P < 0.0001, R2 = 0.91; N1, P < 0.0001, R2 = 0.95), decreased mean amplitudes (dose-response regression: P1-N1, P < 0.0001, R2 = 0.81), and elevated mean thresholds (dose-response regression: P < 0.0001, R2 = 0.71), all of which changed in a dose-dependent manner (e.g., Figs. 1, 3, and 4A: data for P1 latency shown only).
Table 1.
VsEP latency, amplitude, and threshold before and after different doses of XE991 and saline
| XE991 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| VsEPs | 50 mg/kg (5, 3) | 25 mg/kg (6) | 12.5 mg/kg (5) | 5 mg/kg (6) | 3.5 mg/kg (6) | 2.5 mg/kg (5) | 1 mg/kg (5) | 0.5 mg/kg (5) | 0.05 mg/kg (5) | Saline (5) |
| Latency, µs | ||||||||||
| P1 | ||||||||||
| Baseline | 1,375 ± 49 | 1,336 ± 55 | 1,354 ± 24 | 1,327 ± 58 | 1,347 ± 41 | 1,330 ± 68 | 1,356 ± 34 | 1,325 ± 54 | 1,264 ± 56 | 1,337 ± 68 |
| 0–20 min | 1,531 ± 101 | 1,412 ± 41 | 1,459 ± 43 | 1,371 ± 66 | 1,353 ± 59 | 1,306 ± 65 | 1,313 ± 40 | 1,315 ± 55 | 1,258 ± 56 | 1,338 ± 78 |
| 25–40 min | 1,655 ± 51 | 1,580 ± 36 | 1,627 ± 24 | 1,512 ± 77 | 1,449 ± 73 | 1,318 ± 58 | 1,275 ± 38 | 1,276 ± 50 | 1,245 ± 52 | 1,338 ± 83 |
| 45–60 min | 1,640 ± 59 | 1,585 ± 38 | 1,632 ± 26 | 1,519 ± 85 | 1,462 ± 45 | 1,351 ± 60 | 1,268 ± 30 | 1,265 ± 40 | 1,238 ± 61 | 1,326 ± 84 |
| N1 | ||||||||||
| Baseline | 1,701 ± 82 | 1,629 ± 59 | 1,662 ± 18 | 1,610 ± 82 | 1,651 ± 48 | 1,624 ± 55 | 1,639 ± 27 | 1,626 ± 47 | 1,572 ± 30 | 1,639 ± 791 |
| 0–20 min | 1,856 ± 144 | 1,706 ± 39 | 1,768 ± 53 | 1,662 ± 89 | 1,643 ± 62 | 1,590 ± 61 | 1,599 ± 19 | 1,604 ± 50 | 1,567 ± 30 | 1,643 ± 90 |
| 25–40 min | 1,957 ± 23 | 1,939 ± 65 | 1,959 ± 28 | 1,844 ± 88 | 1,773 ± 51 | 1,585 ± 70 | 1,569 ± 35 | 1,562 ± 41 | 1,536 ± 45 | 1,637 ± 97 |
| 45–60 min | 1,943 ± 46 | 1,935 ± 60 | 1,963 ± 23 | 1,848 ± 84 | 1,780 ± 45 | 1,631 ± 66 | 1,566 ± 47 | 1,550 ± 36 | 1,524 ± 44 | 1,625 ± 95 |
| P2ab | ||||||||||
| Baseline | 2,069 ± 41 | 1,973 ± 81 | 2,090 ± 41 | 2,072 ± 86 | 2,023 ± 72 | 2,032 ± 67 | 2,046 ± 64 | 1,988 ± 24 | 1,915 ± 38 | 2,015 ± 80 |
| 0–20 min | 2,358 ± 110 | 2,164 ± 94 | 2,265 ± 84 | 2,188 ± 104 | 2,133 ± 71 | 2,047 ± 83 | 2,060 ± 62 | 1,966 ± 44 | 1,919 ± 31 | 2,008 ± 84 |
| 25–40 min | 2,572 ± 33 | 2,526 ± 204 | 2,511 ± 164 | 2,465 ± 101 | 2,357 ± 75 | 2,161 ± 62 | 2,164 ± 48 | 2,006 ± 56 | 1,926 ± 35 | 2,003 ± 92 |
| 45–60 min | 2,572 ± 24 | 2,536 ± 183 | 2,523 ± 167 | 2,457 ± 78 | 2,381 ± 94 | 2,257 ± 89 | 2,207 ± 39 | 2,042 ± 34 | 1,935 ± 36 | 1,999 ± 96 |
| Amplitude, µV | ||||||||||
| P1N1 | ||||||||||
| Baseline | 0.93 ± 0.22 | 0.81 ± 0.15 | 1.01 ± 0.29 | 0.97 ± 0.15 | 0.97 ± 0.18 | 1.01 ± 0.21 | 0.89 ± 0.24 | 1.02 ± 0.17 | 0.89 ± 0.16 | 0.84 ± 0.16 |
| 0–20 min | 0.63 ± 0.17 | 0.69 ± 0.14 | 0.79 ± 0.14 | 0.86 ± 0.14 | 0.96 ± 0.12 | 1.08 ± 0.18 | 0.94 ± 0.26 | 1.05 ± 0.14 | 0.91 ± 0.22 | 0.86 ± 0.17 |
| 25–40 min | 0.33 ± 0.06 | 0.38 ± 0.13 | 0.50 ± 0.13 | 0.61 ± 0.17 | 0.80 ± 0.14 | 1.18 ± 0.23 | 1.06 ± 0.32 | 1.12 ± 0.14 | 0.96 ± 0.19 | 0.87 ± 0.15 |
| 45–60 min | 0.31 ± 0.06 | 0.45 ± 0.15 | 0.56 ± 0.14 | 0.31 ± 0.06 | 0.87 ± 0.11 | 1.15 ± 0.23 | 1.11 ± 0.26 | 1.16 ± 0.21 | 1.03 ± 0.22 | 0.86 ± 0.20 |
| P2abN1 | ||||||||||
| Baseline | 0.68 ± 0.23 | 0.65 ± 0.15 | 0.94 ± 0.21 | 0.91 ± 0.33 | 0.91 ± 0.20 | 0.74 ± 0.17 | 0.79 ± 0.12 | 0.83 ± 0.18 | 0.78 ± 0.23 | 1.10 ± 0.46 |
| 0–20 min | 0.52 ± 0.22 | 0.66 ± 0.12 | 0.75 ± 0.33 | 0.99 ± 0.42 | 1.07 ± 0.16 | 0.89 ± 0.14 | 1.05 ± 0.28 | 0.96 ± 0.12 | 0.85 ± 0.28 | 1.06 ± 0.45 |
| 25–40 min | 0.52 ± 0.24 | 0.46 ± 0.24 | 0.31 ± 0.21 | 0.62 ± 0.27 | 0.97 ± 0.27 | 1.03 ± 0.17 | 1.27 ± 0.32 | 1.03 ± 0.16 | 0.85 ± 0.27 | 1.05 ± 0.51 |
| 45–60 min | 0.52 ± 0.27 | 0.43 ± 0.21 | 0.31 ± 0.16 | 0.58 ± 0.27 | 0.95 ± 0.29 | 1.14 ± 0.18 | 1.27 ± 0.34 | 1.04 ± 0.16 | 0.95 ± 0.28 | 1.08 ± 0.49 |
| Threshold, dB re:1 g/ms | ||||||||||
| Baseline | −9.9 ± 1.3 | −10.5 ± 1.9 | −11.1 ± 1.3 | −10.5 ± 0.0 | −10.0 ± 1.2 | −9.3 ± 1.6 | −11.1 ± 1.3 | −9.9 ± 2.5 | −8.7 ± 1.6 | −9.9 ± 1.3 |
| 0–20 min | −3.5 ± 1.7 | −3.0 ± 4.1 | −3.3 ± 1.6 | −5.0 ± 2.3 | −8.5 ± 3.6 | −11.1 ± 1.3 | −12.3 ± 1.6 | −10.5 ± 2.1 | −9.3 ± 1.6 | −9.3 ± 1.6 |
| 25–40 min | −1.5 ± 0.0 | −4.0 ± 2.3 | −3.3 ± 2.7 | −5.0 ± 2.3 | −7.0 ± 2.3 | −9.9 ± 1.3 | −11.7 ± 2.78 | −11.1 ± 2.5 | −10.5 ± 0.0 | −9.9 ± 1.3 |
| 45–60 min | −1.5 ± 3.0 | −4.0 ± 3.0 | −3.9 ± 3.3 | −6.0 ± 1.6 | −7.5 ± 1.9 | −9.3 ± 1.6 | −12.3 ± 1.6 | −12.3 ± 1.6 | −8.7 ± 1.6 | −9.3 ± 1.6 |
Values are means ± SD of vestibular response (VsEP) latency, amplitude, and threshold over a period of 60 min following a 20-min stable baseline period for each of 9 different XE991 doses and saline; sample size (n) is indicated in parentheses for each group. Each 20-min period was an average of 4–5 consecutive measurements ~5 min apart. In the 50 mg/kg dose group, responses were essentially eliminated following XE991 administration in 2 animals. All mice were anesthetized with urethane-xylazine and monitored with rectal and brain temperature probes throughout the experiments. Stimulus level was +6 dB re: 1.0 g/ms for latency and amplitude measurements. The latency for P2 is represented as P2ab = (P2a + P2b)/2, which defined a composite value for P2 when in bifed form of P2a and P2b.
Fig. 4.

A: XE991 dose-response curves for VsEP latencies (P1) during vestibular response suppression. Normalized P1 latency delays (mean maximum delay for each dose minus the mean baseline value for each dose) are plotted as the percentage of the maximum delay across all doses as a function of dose for 2.5 mg/kg and higher. The XE991 50% effective dose (ED50) for VsEP was 3.9 mg/kg (Hill slope = 3.9% per mg/kg). The largest drug effect occurred in the highest dose group, and the mean latency delay value for this group was defined as the maximum delay for the drug. This was used to calculate the %maximum delay for each of the respective dose groups. Values for each dose are based on mean latency delay observed during the steady-state period of maximal effect following injection; 0% represents the %maximum delay observed for the saline group. Mean %maximum delay (±SE) is represented for each dose. B: XE991 dose-response curves for ABR latencies (P1) during response suppression. Normalized ABR P1 latencies were significantly delayed by XE991 administration at doses of 2.5 mg/kg and higher. The largest drug effect occurred in the highest dose group. For the highest ABR dose group, the average dose of 50 and 12.5 mg/kg [(50 + 12.5)/2 = 31.25 mg/kg] was adopted and used for graphic display, and the mean latency delay for this group was used for calculation of %maximum delay. Calculations were made using the same strategy outlined in A. Mean %maximum delay(±SE) is represented for each dose. XE991 ED50 for ABR was 3.1 mg/kg (Hill slope = 3.2% per mg/kg).
The onset time of suppression following drug injection was also dose dependent (Figs. 1B and 3B). At a dose of 2.5 mg/kg, suppression began to interrupt enhancement after ~20 min (Fig. 1B). At successively higher doses, this onset time became shorter (Figs. 1B and 3B). This is best illustrated in Fig. 3B, where the time axis has been expanded to evaluate in detail the first 20 min following drug injection. Data shown have been truncated so all dose levels can be represented. VsEP recordings were made at 2-min intervals during this period. The steep increases in VsEP onset latencies (P1, N1) and dramatic fall in amplitudes at doses greater that 2.5 mg/kg can be appreciated from Figs. 1B and 3B. The shortest onset latency for suppression was between 5 and 8 min following IP injection, and this occurred with the highest doses (Fig. 3B). The rate of change in latencies and thresholds during the onset of suppression also increased with increasing dose and ranged from 1.2 to 41.3 µs/min for P1 and N1 latencies and from 0.075 to 0.45 dB/min for threshold (Figs. 1 and 3). The rate of decrease in P1-N1 amplitudes at the onset of suppression also increased with dose, producing rates ranging from −0.005 to −0.063 µV/min. Following the rapid suppression onset, the responses stabilized such that amplitudes, latencies, and thresholds remained suppressed but stable at levels that were dose dependent over the remaining time of the experiment. Figures 1B and 3A illustrate the rapid shift in VsEP parameters and final steady state for different doses of XE991. The time required to reach a stable steady-state suppression was also a function of dose, but generally was less than 40 min. The VsEP was virtually eliminated in two of the five animals studied at the highest dose (50 mg/kg).
Dynamic characteristics of suppressed vestibular responses.
To more clearly understand the suppression state, we examined whether the suppressed vestibular system encoded different stimulus levels in a normal manner. We evaluated responses at stimulus levels between threshold and the maximum stimulus level of +6 dB re: 1g/ms for XE991 doses of 2.5 to 25 mg/kg. As noted, the changes in responses reached a steady state by 40 min postdrug for doses above 2.5 mg/kg. At steady state, VsEP response parameters showed the normal relationships to stimulus level, that is, increasing amplitudes and decreasing latencies with increasing stimulus level [Fig. 5; input/output (I/O) functions]. Stimulus level in Fig. 5 was expressed relative to each animal’s threshold in decibels above threshold, which is normally referred to as “sensation level” (dB SL). Thus threshold shifts due to drug treatment were taken into consideration in Fig. 5. The I/O function for the baseline period before drug (filled circles) was contrasted with that for each treatment group measured 40 min after treatment (open circles). Evaluation was made over the three stimulus levels providing the largest sample sizes (levels 4.5, 7.5, and 10.5 dB SL). Note that the two I/O functions (before and after drug) are generally parallel. Indeed, there were no significant differences in amplitude and latency regression slopes on stimulus level for baseline compared with drug treatments (2.5 to 50 mg/kg). At doses of 2.5 mg/kg, both latency and amplitude I/O functions virtually superposed with predrug values. For dose levels of 3.5 to 25 mg/kg, postdrug I/O functions of both latencies and amplitudes followed predrug functions closely. However, amplitudes tended to be slightly reduced and latencies slightly longer than predrug values at dose levels from 3.5 to 12.5 mg/kg (rmMANOVA: P1-N1, P = 0.02; P1, P = 0.014; N1, P = 0.011). This was particularly true at the highest stimulus levels and doses. At doses of 12.5 and 25 mg/kg, sample size decreased at higher stimulus levels, which contributed to the amplitude reductions (Fig. 5, arrows). The fact that these I/O functions otherwise align so well indicates that the effects of XE991 were, to a large extent, related to the reduced response sensitivity to transient stimuli signaled by VsEP threshold increases at these doses (Fig. 3A).
Fig. 5.

VsEP response input-output (I/O) functions. Plots represent mean response latency and amplitude as a function of stimulus level in dB SL. I/O plots for 5 XE991 dose groups (2.5 to 25 mg/kg) are represented. Stimulus level is expressed relative to each animal’s threshold in dB above threshold (referred to as “sensation level” in dB SL). The I/O functions for the baseline condition before drug are also shown (filled circles). Postdrug data were collected at 40 min after injection (open circles). Amplitudes tended to be slightly reduced and latencies slightly longer than predrug values at dose levels from 3.5 to 12.5 mg/kg (rmMANOVA: P1-N1, P = 0.02; P1, P = 0.014; N1, P = 0.011). The highest dose group (50 mg/kg) was not included because of the reduced sample sizes associated with very high thresholds. At doses of 12.5 and 25 mg.kg, sample sizes changed with increasing stimulus level due to reduced dynamic ranges accompanying elevated thresholds (arrows), thus introducing the appearance of reduced amplitudes in I/O functions.
Actions on central relays.
Changes in central peaks were observed at low doses of 1 mg/kg and higher. The changes in central response peaks involved latency prolongation that was independent of peripheral response latencies (data not shown). The central VsEP response component P2, often separated into two peaks, P2a and P2b, where P2b shifted to longer latencies (post hoc paired t-test, P = 0.006). These central changes occurred during peripheral enhancement, thus contrasting with the shortening of latencies in the periphery (P1 and N1). A composite representation of the two peaks was used for quantification: P2ab = (P2a + P2b)/2 (see Table 1). Independent action of XE991 on central vestibular relays is of interest and contrasts with effects on central ABR peaks (see below).
Effects of KCNQ blockade on auditory function.
Sensory neural hearing loss is known to occur in mutations in the KCNQ4/KCNQ5 genes, and such mutations are understood to be the causative agent in human DFNA2 (reviewed in Wang and Li 2016). Auditory deficits have also been shown in KCNQ4−/− and KCNQ5dn/dn genetic mouse mutants (Kharkovets et al. 2000, 2006). However, the effects of KCNQ blockade by XE991 on auditory function have not been reported. Therefore, we evaluated this question in 31 of the animals tested using the ABR. Seven different dose groups were investigated. ABR latencies and amplitudes were significantly altered by XE991 administration, and the effects of XE991 on auditory responses were generally similar to those on vestibular responses, showing, in a dose-dependent manner, some limited characteristics of response enhancement (Fig. 6, 0.5 mg/kg; Fig. 7, ABR P1-N1 amplitude), full suppression (Figs. 4B and 6, 12.5 mg/kg; Fig. 7, ABR), and a more gradual onset of effects (Fig. 7). At doses above 1 mg/kg, XE991 produced a dose-dependent severe-to-profound suppression of the ABR. ABRs were eliminated by the drug in two of the five animals studied at the highest doses (one at 12.5 and one at 50 mg/kg). Because of the large similar effects at these two doses, and to preserve sample size, data for 12.5 and 50 mg/kg were combined to form a single highest dose group (12.5/50 mg/kg, Fig. 7).
Fig. 6.
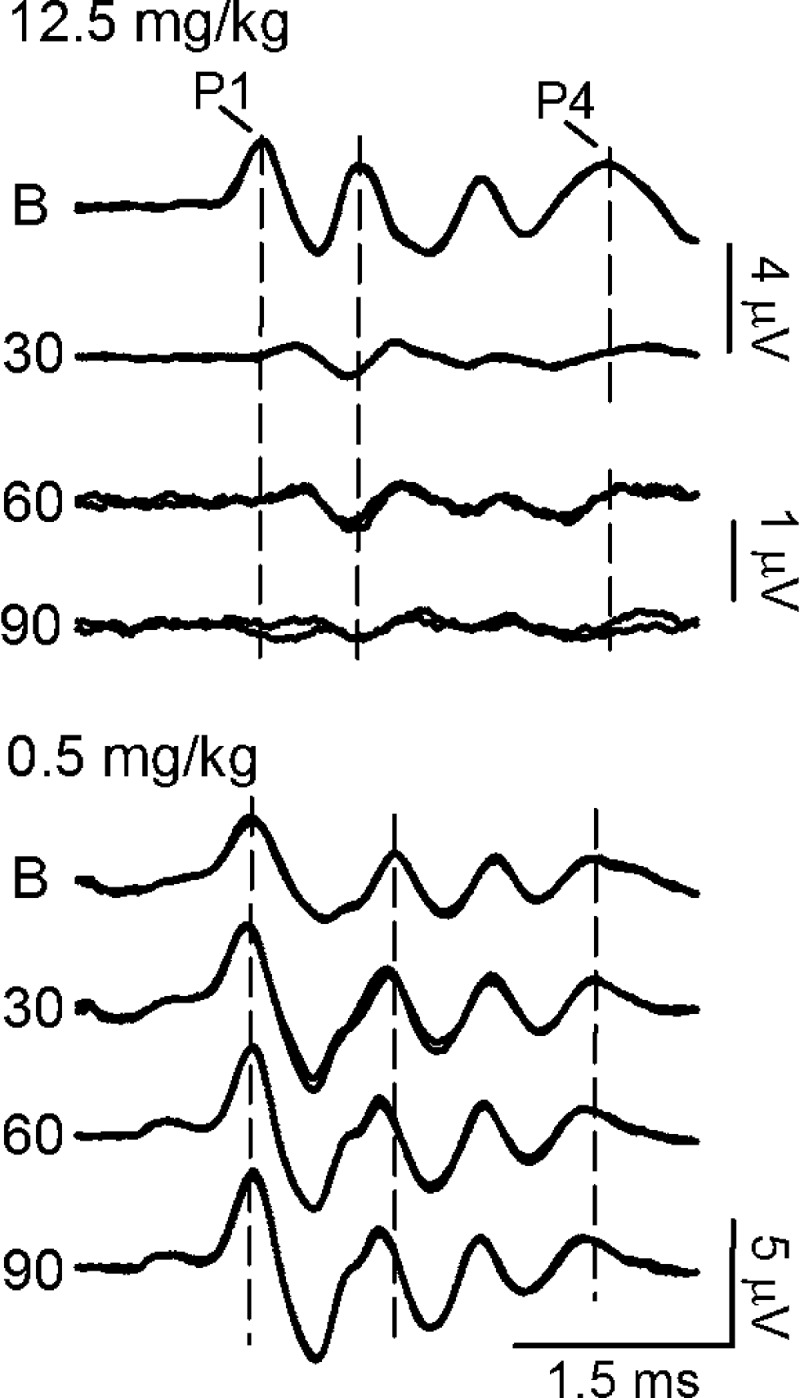
ABR waveforms following XE991 administration. Suppression of the ABR following a 12.5 mg/kg dose of XE991 is illustrated at top. Enhancement is illustrated following a 0.5 mg/kg dose of XE991 at bottom. ABR responses to 110 dB peSPL clicks were recorded at baseline (B; before drug) and 30, 60, and 90 min after drug. ABR waveforms demonstrate that XE991 gradually alters the ABRs over time. Vertical dashed lines represent the baseline latencies of P1, P2, and P4 response peaks.
Fig. 7.

Comparison of XE991 effects on VsEP (left) and ABR (right) peripheral response components. Response amplitudes (bottom) and latencies (top) are represented as a percentage of baseline; 100% indicates no drug effects on the responses, and 0% indicates elimination of responses following drug injection. For latencies, proportionate increases or decreases relative to baseline are indicated. Sampling for VsEP values were at 5-min intervals, whereas 15-min intervals were collected for ABR. Except for the highest dose, changes in ABR were more gradual than changes in VsEP. Different doses are color coded the same for all panels and marked where possible. Below 2.5 mg/kg at doses of 0.5 and 1.0 mg/kg, ABR amplitudes increased over the first 40 min following injection (rmMANOVA: 1 mg/kg: P1-N1, P = 1.7 × 10−5; P4-N4, P = 0.014; 0.5 mg/kg: P1-N1, P = 9.4 × 10−7).
Figure 7 summarizes the drug effects of XE991 on ABR responses recorded before and after systemic administration. Among those doses tested, the highest three dose groups (2.5, 3.5, and 12.5/50 mg/kg) showed clear response suppression, including significantly reduced amplitudes and prolonged latencies (Figs. 4B, 6, and 7; dose-response regression: P1, P < 0.0001, R2 = 0.72; N1, P < 0.0001, R2 = 0.79; P1-N1, P < 0.0001, R2 = 0.76). Below 2.5 mg/kg, at doses of 0.5 and 1.0 mg/kg (Fig. 7), ABR amplitudes increased over the first 40 min following injection (rmMANOVA; 1 mg/kg: P1-N1, P = 1.7 × 10−5; P4-N4, P = 0.014; 0.5 mg/kg: P1-N1, P = 9.4 × 10−7). Despite enhanced P1-N1 amplitudes, ABR latencies did not show a clear sustained decrease in P1 during enhancement at low doses (Fig. 7, 0.5 and 1.0 mg/kg). However, the interpeak latency between P1 and N1 decreased at doses of 0.5 and 1.0 mg/kg in association with a change in the shape and increase in amplitude of P1-N1 (Fig. 6, 0.5 mg/kg; rmANOVA, P = 3.2 × 10−7).
The central response component of the ABR (P4) provided little evidence of independent central XE991 action. ABR latency changes for P1, N1, and P4 following XE991 treatment occurred in parallel; i.e., the changes in latency were the same for all peaks such that interpeak latencies for P4 and P1 were unaffected by XE991 over time following injection. In addition, the regression slopes for latency vs. time (µs/min) were not significantly different for P1 and P4 latencies. These results imply that the drug effects reflect actions largely in the auditory periphery rather than compound actions on peripheral and central relays over the 90-min period of study.
Dose-response curves: comparing effects on vestibular and auditory responses.
Traditional dose-response curves for XE991 are shown in Fig. 4. Represented are dose-response curves for suppression of VsEP (Fig. 4A) and ABR (Fig. 4B) responses based on P1 latencies. The 50% effective dose (ED50) for XE991 was 3.9 mg/kg for the VsEP and 3.1 mg/kg for ABR. Similar curves and ED50 values ranging from 3.8 to 4.2 mg/kg were obtained for the VsEP by using other response parameters (i.e., N1, P1-N1, and response threshold; data not shown), and the range of ED50 values for ABR was 2.3 to 3.3 mg/kg for latencies and amplitudes. It was not possible to generate a traditional dose-response curve for VsEP enhancement periods because of the overwhelming suppression that interrupted the process. In lieu of ED50, we note that the threshold doses for XE991 effects leading to response enhancement for both VsEP and ABR were comparable and between 0.05 and 0.5 mg/kg.
Figure 7 illustrates the VsEP and ABR parameter values during enhancement (low doses, 0.5 and 1.0 mg/kg) and suppression (high doses, >1.0 mg/kg) as a function of time. Latencies and amplitudes are expressed as a percentage of mean baseline to facilitate comparison of the two sensory modalities. The largest change in peripheral response components due to suppression was comparable for VsEP and ABR. For the highest dose groups, delays in latency ranged between 115% and 125% of baseline, whereas amplitudes fell to levels between 15% and 25% of baseline values and replicable responses were absent in two cases for both VsEP and ABR. For low to moderate doses (1.0 to 3.5 mg/kg), the amplitude response changes were somewhat larger for the ABR. The time course of drug effects on vestibular vs. auditory function appeared to be different across the experiment. Changes in ABRs tended to develop more gradually over 90 min for all but the highest doses, whereas drug effects on VsEPs showed an early relatively sharp onset plateau of maximum effect for all doses above 2.5 mg/kg (Fig. 7). The early enhancement period for vestibular responses was well characterized due to use of the 5-min sampling periods. ABR amplitude enhancement at low doses was slightly more robust compared with vestibular amplitude enhancement, and it persisted through the entire postdrug period. However, there was little or no consistent prolonged enhancement shown in ABR latencies (Fig. 7).
Effects of activating KCNQ channels using retigabine.
The effects of XE991 described above resulted from the closure (blockade) of KCNQ channels in the vestibular periphery. In light of our findings, it was also of interest to evaluate the effects of a KCNQ activator (channel opener). The KCNQ opener retigabine has been shown to activate KCNQ channels including subunit family members 2, 3, 4, and 5, but not KCNQ1 (Dupuis et al. 2002; Tatulian et al. 2001). In the mature mammal, KCNQ channels found in the sensory epithelia are located in calyces of dimorphic and calyx-only afferents and include all members except KCNQ1 (Lysakowski et al. 2011; Spitzmaul et al. 2013). Hypothetically, KCNQ* channels are expected to vary normally in their open-state probability and therefore be subject to control by vestibular efferent neurons and the activation of mAChRs. Therefore, administration of retigabine would be expected to increase the conductance of calyx KCNQ* channels but not KCNQ1 channels located in nonsensory regions. If the effects we report above for XE991 are in fact due primarily to the closing of calyx KCNQ* channels and the reduction of M-current, then, hypothetically, retigabine will produce effects that are opposite to the effects produced by XE991 (i.e., retigabine will produce an initial suppression followed by enhancement of vestibular responses to transient stimuli). We tested this hypothesis by administering retigabine and evaluated function using the same testing procedures described above for the XE991 experiments (intraperitoneal injection, 20 mg/kg in 100% DMSO).
Figure 8A illustrates VsEP waveforms from a representative animal treated with retigabine (20 mg/kg). Generally, VsEP responses were initially suppressed (reduced in amplitude and delayed in onset latency; Fig. 8A, “20 min,” and Fig. 8B), followed by a long enhancement period (amplitude increased and latencies decreased; Fig. 8A, “120 min,” and Fig. 8B). These changes were reversed in sequence from the sequence of enhancement followed by suppression that was induced by XE991. In contrast, such changes in the VsEP were absent in mice receiving the DMSO vehicle alone (Fig. 8B). The response differences between retigabine treatment and DMSO treatment were significant (rmMANOVA: suppression: latencies, P = 0.001; amplitudes, P = 0.038; enhancement: latencies, P = 2.2 × 10−4; amplitudes, P = 7.6 × 10−7). The initial period of vestibular suppression was characterized by a rapid delay in latencies, reaching a maximum within 10 min of drug injection and peaking with average latency delays approaching 0.1 ms for P1 and 0.13 for N1 [Fig. 8B, P1 (P = 0.022) and N1 (P = 0.026), post hoc paired t-test]. This was accompanied by reduced amplitudes on the order of 15–20% (Fig. 8B, P1-N1; post hoc paired t-test: P = 0.043). VsEP thresholds also increased by 3 dB at 20 min following drug injection (3.0 ± 2.7 dB, rmANOVA, P = 0.042). At 15 min after retigabine administration, enhancement effects began to reverse the suppression characteristics, leading to shorter latencies and increased amplitudes of the VsEP response over the subsequent 80 min. From the point of maximum suppression (10 min postdrug), VsEP latencies decreased on average by up to −0.19 ms (linear regression: P1, −1.52 μs/min, P = 7.77 × 10−16, R2 = 0.957; N1, −1.72 μs/min, P = 7.38 × 10−13, R2 = 0.918) and amplitudes increased by over 60% (0.52 µV; linear regression: P1-N1, 0.005 μV/min, P = 2.61 × 10−15, R2 = 0.952). Over the same period, thresholds improved by −6.5 dB (linear regression: −0.057 dB/min, R2 = 0.920). On the other hand, VsEP response parameters were stable through the experiments in mice treated with the DMSO vehicle alone.
Fig. 8.

Effects of retigabine on vestibular responses. A: changes in vestibular response waveforms. Responses shown were recorded before (Bas, baseline) and 20 and 120 minutes after a retigabine dose of 20 mg/kg. After 20 min, responses were delayed (P1, N1, P2) and reduced in size (P1-N1, P2-N1). Thereafter, responses were enhanced, increasing in amplitude, decreasing in latency, and lowering of threshold (120 min). Stimulus level was 6 dB re: 1 g/ms. Calibration bars indicate 2.0 ms and 1.0 μ/V for graphic. Dashed vertical lines identify latencies of P1 and P2 before drug injection. B: effects of a single dose of retigabine (filled circles) and DMSO vehicle alone (open circles) on VsEP responses. Mean values are normalized to baseline (mean value minus mean baseline) for response latencies (P1, N1), amplitudes (P1-N1), and thresholds over a period of up to 2 h following retigabine (20 mg/kg; n = 6) and DMSO (n = 9) injections. Data are means ± SD (retigabine group: black circles ± solid red lines; DMSO group: open circles ± blue dotted lines). Data to the right of the vertical dashed line represent the effects of drug or vehicle administration on VsEP responses. Baseline means were as follows: P1, 1.306 ± 0.033 ms; N1, 1.605 ± 0.071 ms; and P1-N1, 1.045 ± 0.246 μV; threshold = −10.5 ± 0.0 dB re: 1 g/ms. Stimulus level was 6 dB re: 1 g/ms.
Retigabine-treated mice also showed some aspects of enhancement effects on the ABR waveform (data not shown). In contrast to the retigabine effects on VsEP responses, ABR latencies and amplitudes did not show early suppression characteristics. Instead, ABR amplitudes showed a gradual increase over the entire postdrug recording period following retigabine injection (up to 120 min; P1-N1: rate of 0.014 μV/min, mean increase of 1.69 ± 1.22 µV; P4-N4: rate of 0.010 μV/min, mean increase of 1.16 ± 0.58 µV at 120 min; rmMANOVA, P = 4.71 × 10−4). Latencies, on the other hand, were unchanged from baseline over the same period. Enhancement of ABR P1-N1 amplitudes became significant at 60 min postdrug (post hoc Dunnett: P = 0.018). The steady increase in ABR amplitudes showed no evidence of reaching a plateau or decreasing within the period of the experiment. DMSO administration alone did not change ABR response parameters throughout the experiments.
Changes in systemic physiological parameters during XE991 and retigabine testing.
levels (median ~90%) and respiratory rates (median ~170 inspirations/min) were stable over the entire study period (80 to 120 min) following XE991 administration at all doses. Similarly, heart rates were stable for the first 40 min after XE991 injection. Thereafter, heart rates gradually and modestly increased (on the order of 12%, from a median of ~375 to 420 beats/min) over time for all treatment groups (rmANOVA, P = 8.7 × 10−25), including animals receiving saline alone. The changes in vestibular responses were not correlated with changes in heart rate. Such heart rate changes are a normal occurrence in mice anesthetized with urethane-xylazine. Heart rate for the largest XE991 dose (50 mg/kg) showed the lowest increase in rate. It is not clear whether this reduced increase for the 50 mg/kg dose group was related to XE991 effects on KCNQ1 channels in the peripheral cardiovascular system (e.g., Wang et al. 2002). All observed heart rates were well within normal physiological ranges for the anesthetized mouse.
During retigabine administration (20 mg/kg), there was a transient reduction in mean heart and respiratory rates. Mean heart rate fell from ~350 to 290 beats/min on average in the first 20 min following injection and then returned to baseline levels or higher by 60 min. Respiratory rates fell from 180 to 160 inspirations/min over the first hour and returned to baseline by 100 min following injection. Despite these changes, levels were stable throughout the experiments. These modest changes in systemic respiratory and cardiovascular parameters following XE991 and retigabine were unlikely to have contributed to the panel of changes observed in the inner ear.
DISCUSSION
The present study provides in vivo evidence for a critical role of KCNQ channels in the peripheral vestibular system of mammals. The results clearly demonstrate that the KCNQ antagonist XE991 significantly altered vestibular function in a dose-dependent manner. Following XE991 administration of doses at or above 2.5 mg/kg, a transient modest early enhancement of the VsEP response was followed by a steady-state suppression lasting at least 2 h. XE991 suppression appeared rapidly for doses above 2.5 mg/kg, reaching a steady state within 30 min of drug injection. Suppression included sustained elevation of thresholds by up to ~10 dB. These effects of KCNQ channel blockade are remarkable and represent profound changes in the ability of the macular neuroepithelium to respond to transient head motion.
Our principal hypothesis was that the effects of XE991 were mediated primarily by acting specifically on KCNQ2–5 (KCNQ*) channels in calyx-bearing afferents in the peripheral sensory neuroepithelium. However, it may be argued that the primary site of action of XE991 blocks KCNQ1/KCNE1 channels located in the apical membranes of stria marginal cells and dark cells of cochlear and vestibular epithelia, respectively.
Transduction currents in auditory hair cells are critically dependent on the endocochlear potential (EP; Sewell 1984), which is generated by the stria vascularis and appears normally in the endolymphatic compartment of the scala media with a magnitude generally of ≥80 mV relative to perilymph. Elimination of the EP leads to profound hearing loss and substantial reductions in the ABR (Freeman et al. 1995; Hirose and Liberman 2003; Sewell 1984). The EP is critically dependent on intact KCNQ1/KCNE1 channels (Lee et al. 2000; Vetter et al. 1996), suggesting that blockade of these channels (reviewed in Robbins and Passmore 2010) very likely contributed to the profound reductions in the ABR with XE991 administration. Because KCNQ1 channels participate in K+ homeostasis for both vestibular (dark cells) and cochlear (stria vascularis) epithelia (Wangemann 1995, 2006; Zdebik et al. 2009), it is possible that XE991’s effects on the VsEP were also due to interference with these more general mechanisms. To rule this out, we recently recreated similar conditions by blocking the Na+-K+-2Cl− (NKCC1; Slc12a2) cotransporter, located in basolateral membranes of both vestibular dark cells and stria marginal cells, using the loop diuretic ethacrynic acid (Lee C and Jones TA, unpublished observations). This drug reduces or eliminates the EP (e.g., Bosher et al. 1973; Bosher 1979, 1980; Matz 1976; Sellick and Johnstone 1974) by inhibiting K+ secretion into the endolymph and interfering with K+ homeostasis in a manner analogous to KCNQ1/KCNE1 blockade, but KCNQ channels remain intact (Wangemann 1995, 2006; Zdebik et al. 2009). With the use of the same study design reported in this article, the effects of ethacrynic acid (40 mg/kg) on VsEP and ABR responses were evaluated over a period of 2 h. The ABR was reduced at 15 min and eliminated within ~70 min, whereas the VsEP in the same animals was unaffected. Elimination of the ABR confirmed the inhibition of K+ secretion and its attendant sequelae. The absence of VsEP effects demonstrated that inhibition of KCNQ1 by XE991 alone could not account for the rapid and profound reductions in the VsEP reported in this study. We conclude, therefore, that changes in the VsEP were due to the action of XE991 on the neurosensory elements of the vestibular epithelium. Further support for this conclusion is provided by the results of retigabine administration in the present study, which showed that 1) opening only KCNQ* channels (i.e., retigabine does not act on KCNQ1) produces the opposite effects on vestibular function compared with closing channels (by XE991), and 2) such an effect could only be produced by action on the vestibular sensory epithelium itself and presumably on vestibular calyces where channels are located.
The most likely neurosensory elements affected directly by XE991 are the calyx-bearing afferents in the vestibular neuroepithelium. This hypothesis is based largely on the observations that 1) the VsEP is thought to be predominantly generated by irregularly discharging calyx-bearing afferents; 2) in mature mammals, KCNQ* channels are predominantly found in membranes of primary afferent calyces, but not hair cells (Hurley et al. 2006; Lysakowski et al. 2011; Spitzmaul et al. 2013); and 3) the most intense staining for KCNQ4 channels is found in central/striolar calyces, where irregularly discharging afferents reside (Lysakowski et al. 2011; Spitzmaul et al. 2013).
These observations provide the first direct in vivo evidence of a critical functional role for KCNQ* channels in the mammalian peripheral vestibular system. Based on these results, a peripheral deficit should be anticipated with KCNQ* mutations, and such a deficit could explain the reduced VOR gains reported for KCNQ4/KCNQ5 mutant animals (Spitzmaul et al. 2013). Because the actual extent of peripheral dysfunction in KCNQ4/KCNQ5 mutants is unknown, it will be important to determine the peripheral loss in these models.
Effects of KCNQ blockade on central components of the VsEP and ABR.
Although the precise central neural generators of mammalian VsEP responses have not been localized, P2 is likely generated by vestibular brain stem relays and not the cerebellum (Gaines 2012; Jones 1992; Nazareth and Jones 1998). Several studies have reported that KCNQ4/KCNQ5 channels are present in central auditory pathways, trigeminal nuclei, and dorsal root ganglia (Heidenreich et al. 2011; Kharkovets et al. 2000; Wang and Li 2016). The separation of the VsEP response component P2 into P2a and P2b and the independent prolongation of VsEP P2ab latencies clearly indicate central effects of XE991 in vestibular relay pathways. Central brain stem effects seen with XE991 in the present study suggest that central deficits may also have contributed to VOR changes in KCNQ4/5 mutants (Spitzmaul et al. 2013). We cannot exclude the possibility that XE991 could have direct effects on efferent neurons or on upstream central circuitry that influence efferent neuronal activity. Single-unit recordings from vestibular afferents during XE991 administrations would be helpful in clarifying whether such a mechanism is at play.
There was no convincing evidence for an independent acute central action by XE991 on auditory relays and responses. XE991 effects on central (P4-N4 amplitude, %change) and peripheral (P1-N1 amplitude) ABR components were comparable. Kharkovets et al. (2006) reported that gene mutation of KCNQ4 channels in mice produced little change in interpeak latencies between central and peripheral response components of the ABR despite significant reduction of peripheral amplitude. They concluded that the loss of KCNQ channel function had little effect on auditory central relays. The possibility of central auditory circuit compensation for peripheral deficits in this case (3-wk-old mice with constitutive gene mutation), however, cannot be ruled out. Taken together, our results suggest that XE991 acts most effectively on peripheral auditory function, including actions altering K+ secretion into the endolymph and inactivation of KCNQ4 channels in primary afferents and outer hair cells.
Suppression of VsEP responses by XE991 reflected shifts in macular sensitivity.
VsEP thresholds increased systematically with increasing XE991 dose (Fig. 3A and Table 1). Indeed, threshold was directly proportional to XE991 dose at levels near ED50 (0–5 mg/kg), where it increased at a rate of ~1.2 dB·mg−1·kg−1 (regression: P = 0.0001, R2 = 0.97). The slope decreased at ≥5 mg/kg, presumably as receptor binding approached saturation. VsEP I/O functions before and after XE991 (Fig. 5) were closely matched, indicating that the primary effect of KCNQ blockade was to reduce the sensitivity of the vestibular epithelium to transient head motion. Taking this threshold shift into account in the “sensation level” (dB SL) plots of Fig. 5 compensated almost entirely for the change in sensitivity. There was no evidence of major changes in onset activation timing.
Excessive prolonged calyx terminal/hair cell depolarization.
Ruling against the notion that a compromised EP or acutely disrupted K+ secretion was the primary mechanism producing VsEP suppression, two remaining candidate mechanisms may explain XE991’s effects. In calyceal endings, transduction currents in the type I hair cell are expected to transfer K+ to the synaptic space continuously. The only readily available direct diffusion path out of the synaptic cleft exists at the extreme apical rim. Thus, if the rate of K+ removal by membrane resident transporters (e.g., Na+-K+-ATPase, K+ channels, cotransporters, etc.) is inadequate to compensate for K+ entry, then K+ accumulation in the synaptic cleft could increase sufficiently to depolarize both the hair cell and calyx ending (e.g., Eatock and Lysakowski 2006; Goldberg 1996). This suggests that a delicate balance is required to prevent chronic excessive depolarization of the type I hair cell and calyx ending. Low-voltage-activated K+ channels (KLV), including KCNQ* channels in calyx terminal membranes, likely provide a low-impedance conductance path to clear K+ from the synaptic cleft (Eatock and Lysakowski 2006). Thus the blockade of KCNQ* channels by XE991 may impair K+ removal from the synaptic cleft between type I hair cells and calyces, leading to sustained excessive depolarization of the hair cell and calyx terminal, and ultimately compromise synaptic function.
Synaptic blockade following K+ accumulation cannot be ruled out in our study and thus may contribute to VsEP suppression with XE991. However, it is unlikely to be the primary mechanism mediating suppression, because a dramatic change in response onset activation timing and stimulus level encoding would be expected. However, VsEP I/O functions before and after XE991 administration, when expressed in terms of sensation level (Fig. 5), were parallel and quite comparable with only small delays in timing and reduction in amplitudes. Because KCNQ channels represent only a part of the calyceal K+ channel ensemble (e.g., Contini et al. 2017; Kalluri et al. 2010), the remaining unaffected K+ channels may be sufficient to clear K+ from the synapse. In our view the retention of nearly normal IO functions casts doubt on a major role for excessive prolonged terminal depolarization by XE991.
KCNQ-mediated transformation of afferent discharge characteristics.
Recent work by several investigators (e.g., Iwasaki et al. 2008; Kalluri et al. 2010; Songer and Eatock 2013) has underscored the importance of KLV in determining the discharge patterns and response characteristics of calyx-bearing afferents. Blockade of low-voltage-activated current (ILV) in vitro can transform ganglion cell spike discharge from transient spike patterns to sustained spike trains during step depolarization (Iwasaki et al. 2008; Kalluri et al. 2010; Pérez et al. 2009, 2010). Similar transformations occur in rapidly adapting somatosensory mechanoreceptors following linopirdine blockade of KCNQ channels (Heidenreich et al. 2011). Using linopirdine, Kalluri et al. (2010) estimated that in neonatal vestibular ganglia (P8-P15), KCNQ channels mediated about one-third of ILV. By blocking KCNQ* channels, therefore, XE991would be expected to reduce ILV by up to 33%. By closing KCNQ* channels, XE991 would reduce ILV, increase calyceal input impedance and the membrane charging time constant (τ = RC), and reduce the synaptic transfer of high-frequency stimuli while increasing the gain to low-frequency stimuli. With reduced ILV, calyx-bearing afferents would become more responsive and discharge longer and at higher levels in response to lower frequency sustained stimuli, but would lose their ability to respond precisely in time to stimulus transients. These characteristics reduce the ability of a transient stimulus to activate afferents synchronously at stimulus onset. Indeed, the collective response of the neuroepithelium to transient stimulation would be suppressed, and, as we show in this study, VsEP thresholds for example, would increase, amplitudes decrease, and latencies become delayed. The major changes in vestibular responses reported presently thus may represent an XE991-induced transformation of irregularly discharging calyx-bearing afferents into more regularly discharging, less dynamic, and less adapting sensors. Hypothetically at the same time following XE991 injection, the neuroepithelium should acquire enhanced responsiveness to sustained low-frequency stimulation inasmuch as the response to such stimuli should increase in cells harboring KCNQ channels. Although not tested in the present study, Holt et al. (2017) reported that efferent activation of mAChRs resulted in downstream closure of KCNQ channels, which led to increased vestibular afferent gain during low-frequency stimulation. This effect was blocked by mAChR antagonists and occluded by the application of XE991, thus demonstrating that the process was mediated by KCNQ channel closure. Collectively, these findings support the notion that closure of KCNQ channels suppresses responses to transient stimuli while simultaneously enhancing tonic responses to sustained low-frequency stimulation. Moreover, they further suggest that this transformation from dynamic to enhanced tonic afferent behavior may be one normal mode of action for the vestibular efferent system in both mammals and turtles.
The potent suppression of dynamic VsEP responses to transient stimuli produced by XE991 in the present study was likely accompanied by an increased response to sustained low-frequency stimulation. Both physiological outcomes (reduced dynamic and increased tonic responsiveness) result from KCNQ closure. In such a system, the adjustment from high-precision dynamic responses to more stable enhanced integrative tonic response characteristics can be made with one common mechanism without requiring additional neural circuitry. The concomitant suppression of dynamic responses we report presently with XE991 treatment was not evident in the turtle, because comparable responses to transient stimuli were not measured. Hypothetically, a reduction in afferent dynamic responses with efferent activation also has not been reported in classic studies of efferent action in the mammal for the same reason (e.g., Holt et al. 2011).
Across the vertebrate scale, activation of the EVS can produce afferent excitation or inhibition that can decrease or increase afferent gain in response to low-frequency vestibular stimulation (Boyle and Highstein 1990; Boyle et al. 2009; Goldberg and Fernández 1980; Holt et al. 2011, 2017; McCue and Guinan 1994). On the basis of the observations noted above, we propose that for irregularly discharging calyx-bearing vestibular afferent populations in mammals and turtles, efferent activation can lead to a dynamic transformation of afferent behavior, including enhancement of the response to low-frequency sustained stimulation accompanied by a potent suppression of responsiveness to high-frequency transient stimulation. We propose further that this shift in response dynamics is mediated by the closure of calyceal KCNQ channels, which is accessed by the vestibular efferent system with the release of acetylcholine (ACh) and activation of muscarinic ACh receptors (mAChRs) known to be present on calyx membranes (turtle: Holt et al. 2011, 2017; Jordan et al. 2013; mammals: Holt et al. 2015a; Hurley et al. 2006; Ishiyama et al. 1997; Pérez et al. 2009; Wackym et al. 1996). The activation of mAChRs leads to G protein-linked activation of phospholipase Cβ, hydrolysis and depletion of phosphatidylinositol 4,5-bisphosphate (PIP2), and closure of KCNQ channels (Brown and Passmore 2009). Through this mechanism, vestibular efferent action in mammals as well as lower vertebrates may serve to adjust the dynamic response characteristics and sensitivity (e.g., gain) of the neuroepithelium to accommodate a changing environment. Our results show that this is possible in vivo and in the mammal by controlling the open probability of KCNQ* channels. We used XE991 and retigabine to change the open probability of KCNQ* channels. Normally efferent action can be seen to do the same by titrating calyceal mAChR activation. This putative mechanism would represent only one component of efferent control given that ACh can also activate other cholinergic receptors on vestibular hair cells and afferent terminals (Holt et al. 2011, 2015b, 2017; Jordan et al. 2013).
A comment on XE991-induced enhancement of responses to stimulus transients.
The enhancement of vestibular responses during the initial effects of XE991 treatment reported in this study was quite modest. Latency and amplitude changes were small, threshold trends did not reach significance, and improvements were replaced quickly by dose-dependent suppression. However, the occurrence of enhancement is interesting and raises questions about mechanisms affecting calyx function.
The structural and functional characteristics of the calyx provide an appealing hypothetical explanation for enhancement and suppression. The calyx has a terminal synaptic region that serves to receive and filter signals presented by the hair cell. These filtered signals are transferred in turn to the spike trigger zone some several micrometers (e.g., Lysakowski et al. 2011; Fig. 3, D and E) away in the region of the heminode where presumably the final action potential discharge pattern is encoded. Schematically, one may view the synaptic region as the input segment that controls the nature of the stimulus reaching the trigger zone. The trigger zone itself can be seen as determining the onset spike threshold and timing of activation. In the calyceal ending, KCNQ channels are found in large numbers in various microdomains covering both synaptic and heminodal regions (Lysakowski et al. 2011). Presentation of XE991 to the general terminal area will close KCNQ channels and depolarize both synaptic and trigger zone regions. We propose that at low XE991 concentrations, depolarization has key distinct consequences in the two regions. Even low-level depolarizing action in the spike trigger zone would be expected to increase excitability and enhance onset responses to transient stimuli, whereas the change in synaptic signal transfer at such low doses will be minor, if not negligible. Thus the input remains virtually unchanged but coupled with a facilitated trigger zone. This combination would produce a net enhancement of responses to transient stimulation as observed in the present study. There are conditions that could favor differential XE991 effects in the two regions of calyceal endings. Given the physical constraints of the neuroepithelium, we suppose that KCNQ channels in the spike trigger zone may be more accessible to XE991 than the restricted cleft KCNQ channels and therefore respond to lower concentrations. Alternatively, differences in KCNQ subunit composition between KCNQ channels in the trigger zone and synaptic cleft might contribute to differences in pharmacological sensitivity to XE991 (Robbins 2001). In any case, at increasing XE991 concentrations, the effects on synaptic signal transfer become more important. With higher local epithelial concentrations of XE991, the transfer time constant (RC) increases significantly and the signal reaching the spike trigger zone loses high-frequency components. Such an underlying process at higher doses would produce a dose-dependent suppression of responses to transient stimuli accompanied by a more effective activation by low-frequency tonic stimuli, as elaborated above. Given that this hypothesis arises directly from the single action of closing KCNQ channels, one would anticipate the effects of opening of KCNQ channels (i.e., retigabine treatment) to be the opposite of those produced by XE991, which was the case in the present study. Moreover, one would expect a similar period of enhancement in auditory primary afferent responses to transient stimuli following low doses of XE991, an effect clearly demonstrated in our ABR findings for VsEP amplitudes but not latencies. The latter observation indicates that the calyceal ending is not required for some aspects of enhancement.
Clinical relevance of the vestibular periphery.
Clinically, the muscarinic antagonist scopolamine is commonly used to treat dizziness and vertigo. The action of this drug is probably mediated by both peripheral and central pathways to achieve clinical effects (Weerts et al. 2015). Scopolamine’s blockade of tonically activated peripheral vestibular mAChRs would hypothetically result in the reopening of KCNQ* channels located in calyx-bearing afferent neurons. This would decrease response gain and discharge duration of primary afferent responses to tonic stimuli, shift the calyx membrane response to higher frequencies, and increase the neuroepithelial sensitivity to dynamic stimuli. The resulting reduction in the duration of responses to tonic stimulation (shorter sustained discharge) and the reduced gain at low frequencies may be the key contributors to the effective clinical action of scopolamine. Based on the current findings, retigabine, which is currently used for treatment of epilepsy as a potent KCNQ* channel opener (e.g., Gunthorpe et al. 2012), could activate KCNQ channels in the peripheral vestibular system to produce effects that are similar to muscarinic antagonists. Retigabine may therefore be an additional candidate agent for treating vestibular disorders. Another new candidate agent for clinical use in the treatment of vestibular disturbances is the H4 receptor antagonist JNJ-7777120 (Desmadryl et al. 2012), whose effects on the VsEP are similar to those of retigabine (Lee and Jones 2016). This suggests that JNJ-7777120 may also ultimately increase IKL and enhance responses to transient stimuli but, more importantly, act simultaneously to reduce the level and duration of response discharge to steady or slowly changing stimuli.
GRANTS
This work was supported by the American Academy of Audiology Foundation, the Nebraska Tobacco Settlement Biomedical Research Foundation, and the Department of Special Education and Communication Disorders, University of Nebraska-Lincoln.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.L., J.C.H., and T.A.J. conceived and designed research; C.L. performed experiments; C.L. and T.A.J. analyzed data; C.L., J.C.H., and T.A.J. interpreted results of experiments; C.L. and T.A.J. prepared figures; C.L. drafted manuscript; C.L., J.C.H., and T.A.J. edited and revised manuscript; T.A.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Sherri Jones for comments and suggestions regarding the manuscript.
Present affiliation of C. Lee: Department of Otolaryngology, Washington University School of Medicine, St. Louis, MO 63110 (e-mail: c.lee@wustl.edu).
REFERENCES
- Bosher SK. The nature of the negative endocochlear potentials produced by anoxia and ethacrynic acid in the rat and guinea-pig. J Physiol 293: 329–345, 1979. doi: 10.1113/jphysiol.1979.sp012892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosher SK. The nature of the ototoxic actions of ethacrynic acid upon the mammalian endolymph system. I. Functional aspects. Acta Otolaryngol 89: 407–418, 1980. doi: 10.3109/00016488009127156. [DOI] [PubMed] [Google Scholar]
- Bosher SK, Smith C, Warren RL. The effects of ethacrynic acid upon the cochlear endolymph and stria vascularis. A preliminary report. Acta Otolaryngol 75: 184–191, 1973. doi: 10.3109/00016487309139694. [DOI] [PubMed] [Google Scholar]
- Boyle R, Highstein SM. Efferent vestibular system in the toadfish: action upon horizontal semicircular canal afferents. J Neurosci 10: 1570–1582, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle R, Rabbitt RD, Highstein SM. Efferent control of hair cell and afferent responses in the semicircular canals. J Neurophysiol 102: 1513–1525, 2009. doi: 10.1152/jn.91367.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol 156: 1185–1195, 2009. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald JS, Huang C. Far-field acoustic response: origins in the cat. Science 189: 382–384, 1975. doi: 10.1126/science.1145206. [DOI] [PubMed] [Google Scholar]
- Contini D, Price SD, Art JJ. Accumulation of K+ in the synaptic cleft modulates activity by influencing both vestibular hair cell and calyx afferent in the turtle. J Physiol 595: 777–803, 2017. doi: 10.1113/JP273060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coucke PJ, Van Hauwe P, Kelley PM, Kunst H, Schatteman I, Van Velzen D, Meyers J, Ensink RJ, Verstreken M, Declau F, Marres H, Kastury K, Bhasin S, McGuirt WT, Smith RJ, Cremers CW, Van de Heyning P, Willems PJ, Smith SD, Van Camp G. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet 8: 1321–1328, 1999. doi: 10.1093/hmg/8.7.1321. [DOI] [PubMed] [Google Scholar]
- De Leenheer EM, Huygen PL, Coucke PJ, Admiraal RJ, van Camp G, Cremers CW. Longitudinal and cross-sectional phenotype analysis in a new, large Dutch DFNA2/KCNQ4 family. Ann Otol Rhinol Laryngol 111: 267–274, 2002. doi: 10.1177/000348940211100312. [DOI] [PubMed] [Google Scholar]
- Desmadryl G, Gaboyard-Niay S, Brugeaud A, Travo C, Broussy A, Saleur A, Dyhrfjeld-Johnsen J, Wersinger E, Chabbert C. Histamine H4 receptor antagonists as potent modulators of mammalian vestibular primary neuron excitability. Br J Pharmacol 167: 905–916, 2012. doi: 10.1111/j.1476-5381.2012.02049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis DS, Schrøder RL, Jespersen T, Christensen JK, Christophersen P, Jensen BS, Olesen SP. Activation of KCNQ5 channels stably expressed in HEK293 cells by BMS-204352. Eur J Pharmacol 437: 129–137, 2002. doi: 10.1016/S0014-2999(02)01287-6. [DOI] [PubMed] [Google Scholar]
- Eatock RA, Lysakowski A. Mammalian vestibular hair cells. In: Vertebrate Hair Cells, edited by Eatock RA, Fay RR, and Popper AN. New York: Springer, 2006, p. 348–442. [Google Scholar]
- Freeman S, Goitein K, Attias J, Furst M, Sohmer H. Effect of hypoxemia and ethacrynic acid on ABR and distortion product emission thresholds. J Neurol Sci 131: 21–29, 1995. doi: 10.1016/0022-510X(95)00038-4. [DOI] [PubMed] [Google Scholar]
- Gaines GC. Generators of Mammalian Vestibular Surface Responses to Head Motion (PhD dissertation). Greenville, NC: East Carolina University, 2012. [Google Scholar]
- Goldberg JM. Theoretical analysis of intercellular communication between the vestibular type I hair cell and its calyx ending. J Neurophysiol 76: 1942–1957, 1996. [DOI] [PubMed] [Google Scholar]
- Goldberg JM, Fernández C. Efferent vestibular system in the squirrel monkey: anatomical location and influence on afferent activity. J Neurophysiol 43: 986–1025, 1980. [DOI] [PubMed] [Google Scholar]
- Goodyear RJ, Jones SM, Sharifi L, Forge A, Richardson GP. Hair bundle defects and loss of function in the vestibular end organs of mice lacking the receptor-like inositol lipid phosphatase PTPRQ. J Neurosci 32: 2762–2772, 2012. doi: 10.1523/JNEUROSCI.3635-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 53: 412–424, 2012. doi: 10.1111/j.1528-1167.2011.03365.x. [DOI] [PubMed] [Google Scholar]
- Heidenreich M, Lechner SG, Vardanyan V, Wetzel C, Cremers CW, De Leenheer EM, Aránguez G, Moreno-Pelayo MÁ, Jentsch TJ, Lewin GR. KCNQ4 K+ channels tune mechanoreceptors for normal touch sensation in mouse and man. Nat Neurosci 15: 138–145, 2011. doi: 10.1038/nn.2985. [DOI] [PubMed] [Google Scholar]
- Henry KR. Auditory brainstem volume-conducted responses: origins in the laboratory mouse. J Am Aud Soc 4: 173–178, 1979. [PubMed] [Google Scholar]
- Hirose K, Liberman MC. Lateral wall histopathology and endocochlear potential in the noise-damaged mouse cochlea. J Assoc Res Otolaryngol 4: 339–352, 2003. doi: 10.1007/s10162-002-3036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JC, Jordan PM, Lysakowski A, Shah A, Barsz K, Contini D. Muscarinic acetylcholine receptors and M-currents underlie efferent-mediated slow excitation in calyx-bearing vestibular afferents. J Neurosci 37: 1873–1887, 2017. doi: 10.1523/JNEUROSCI.2322-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JC, Jordan P.M., Schneider GT. Pharmacological characterization of the synaptic mechanisms governing the responses of mammalian vestibular afferents to efferent stimulation (PS-205). Assoc Res Otolaryngol Abstr 38: 100, 2015a. [Google Scholar]
- Holt JC, Kewin K, Jordan PM, Cameron P, Klapczynski M, McIntosh JM, Crooks PA, Dwoskin LP, Lysakowski A. Pharmacologically distinct nicotinic acetylcholine receptors drive efferent-mediated excitation in calyx-bearing vestibular afferents. J Neurosci 35: 3625–3643, 2015b. doi: 10.1523/JNEUROSCI.3388-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JC, Lysakowski A, Goldberg JM. The efferent vestibular system. In: Auditory and Vestibular Efferents, edited by Ryugo DK, Fay RR, and Popper AN. New York: Springer, 2011, p. 134–186. [Google Scholar]
- Holt JR, Stauffer EA, Abraham D, Géléoc GS. Dominant-negative inhibition of M-like potassium conductances in hair cells of the mouse inner ear. J Neurosci 27: 8940–8951, 2007. doi: 10.1523/JNEUROSCI.2085-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley KM, Gaboyard S, Zhong M, Price SD, Wooltorton JR, Lysakowski A, Eatock RA. M-like K+ currents in type I hair cells and calyx afferent endings of the developing rat utricle. J Neurosci 26: 10253–10269, 2006. doi: 10.1523/JNEUROSCI.2596-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama A, López I, Wackym PA. Molecular characterization of muscarinic receptors in the human vestibular periphery. Implications for pharmacotherapy. Am J Otol 18: 648–654, 1997. [PubMed] [Google Scholar]
- Iwasaki S, Chihara Y, Komuta Y, Ito K, Sahara Y. Low-voltage-activated potassium channels underlie the regulation of intrinsic firing properties of rat vestibular ganglion cells. J Neurophysiol 100: 2192–2204, 2008. doi: 10.1152/jn.01240.2007. [DOI] [PubMed] [Google Scholar]
- Jewett DL. Volume-conducted potentials in response to auditory stimuli as detected by averaging in the cat. Electroencephalogr Clin Neurophysiol 28: 609–618, 1970. doi: 10.1016/0013-4694(70)90203-8. [DOI] [PubMed] [Google Scholar]
- Jones SM, Erway LC, Bergstrom RA, Schimenti JC, Jones TA. Vestibular responses to linear acceleration are absent in otoconia-deficient C57BL/6JEi-het mice. Hear Res 135: 56–60, 1999. doi: 10.1016/S0378-5955(99)00090-8. [DOI] [PubMed] [Google Scholar]
- Jones SM, Erway LC, Johnson KR, Yu H, Jones TA. Gravity receptor function in mice with graded otoconial deficiencies. Hear Res 191: 34–40, 2004. doi: 10.1016/j.heares.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Jones SM, Johnson KR, Yu H, Erway LC, Alagramam KN, Pollak N, Jones TA. A quantitative survey of gravity receptor function in mutant mouse strains. J Assoc Res Otolaryngol 6: 297–310, 2005. doi: 10.1007/s10162-005-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Jones TA. Genetics of peripheral vestibular dysfunction: lessons from mutant mouse strains. J Am Acad Audiol 25: 289–301, 2014. doi: 10.3766/jaaa.25.3.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA. Vestibular short latency responses to pulsed linear acceleration in unanesthetized animals. Electroencephalogr Clin Neurophysiol 82: 377–386, 1992. doi: 10.1016/0013-4694(92)90007-5. [DOI] [PubMed] [Google Scholar]
- Jones TA, Jones SM. Short latency compound action potentials from mammalian gravity receptor organs. Hear Res 136: 75–85, 1999. doi: 10.1016/S0378-5955(99)00110-0. [DOI] [PubMed] [Google Scholar]
- Jones TA, Jones SM, Vijayakumar S, Brugeaud A, Bothwell M, Chabbert C. The adequate stimulus for mammalian linear vestibular evoked potentials (VsEPs). Hear Res 280: 133–140, 2011. doi: 10.1016/j.heares.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Lee C, Gaines GC, Grant JW. On the high frequency transfer of mechanical stimuli from the surface of the head to the macular neuroepithelium of the mouse. J Assoc Res Otolaryngol 16: 189–204, 2015. doi: 10.1007/s10162-014-0501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PM, Parks XX, Contini D, Holt JC. A review of synaptic mechanisms of vestibular efferent signaling in turtles: extrapolation to efferent actions in mammals. J Vestib Res 23: 161–175, 2013. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Xue J, Eatock RA. Ion channels set spike timing regularity of mammalian vestibular afferent neurons. J Neurophysiol 104: 2034–2051, 2010. doi: 10.1152/jn.00396.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J 25: 642–652, 2006. doi: 10.1038/sj.emboj.7600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA 97: 4333–4338, 2000. doi: 10.1073/pnas.97.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Jones TA. Effects of an H4 receptor antagonist on mammalian vestibular sensory evoked potentials (PS 932). Assoc Res Otolaryngol Abstr 39: 570, 2016. [Google Scholar]
- Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, Washington K, Argani P, Goldenring JR, Coffey RJ, Feinberg AP. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest 106: 1447–1455, 2000. doi: 10.1172/JCI10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Conrad T, Jones SM, Lagziel A, Starost MF, Belyantseva IA, Friedman TB, Morell RJ. A null mutation of mouse Kcna10 causes significant vestibular and mild hearing dysfunction. Hear Res 300: 1–9, 2013. doi: 10.1016/j.heares.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysakowski A, Gaboyard-Niay S, Calin-Jageman I, Chatlani S, Price SD, Eatock RA. Molecular microdomains in a sensory terminal, the vestibular calyx ending. J Neurosci 31: 10101–10114, 2011. doi: 10.1523/JNEUROSCI.0521-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur PD, Vijayakumar S, Vashist D, Jones SM, Jones TA, Yang J. A study of whirlin isoforms in the mouse vestibular system suggests potential vestibular dysfunction in DFNB31-deficient patients. Hum Mol Genet 24: 7017–7030, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz GJ. The ototoxic effects of ethacrynic acid in man and animals. Laryngoscope 86: 1065–1086, 1976. doi: 10.1288/00005537-197608000-00001. [DOI] [PubMed] [Google Scholar]
- McCue MP, Guinan JJ Jr. Influence of efferent stimulation on acoustically responsive vestibular afferents in the cat. J Neurosci 14: 6071–6083, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazareth AM, Jones TA. Central and peripheral components of short latency vestibular responses in the chicken. J Vestib Res 8: 233–252, 1998. doi: 10.1016/S0957-4271(97)00076-1. [DOI] [PubMed] [Google Scholar]
- Pérez C, Limón A, Vega R, Soto E. The muscarinic inhibition of the potassium M-current modulates the action-potential discharge in the vestibular primary-afferent neurons of the rat. Neuroscience 158: 1662–1674, 2009. doi: 10.1016/j.neuroscience.2008.11.023. [DOI] [PubMed] [Google Scholar]
- Pérez C, Vega R, Soto E. Phospholipase C-mediated inhibition of the M-potassium current by muscarinic-receptor activation in the vestibular primary-afferent neurons of the rat. Neurosci Lett 468: 238–242, 2010. doi: 10.1016/j.neulet.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther 90: 1–19, 2001. doi: 10.1016/S0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Robbins J, Passmore G. Kv7 channels. In: Ion Channels from Structure to Function, edited by Kew JN and Davies CH. New York: Oxford University Press, 2010, p. 57–71. [Google Scholar]
- Rocha-Sanchez SM, Morris KA, Kachar B, Nichols D, Fritzsch B, Beisel KW. Developmental expression of Kcnq4 in vestibular neurons and neurosensory epithelia. Brain Res 1139: 117–125, 2007. doi: 10.1016/j.brainres.2006.12.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellick PM, Johnstone BM. Differential effects of ouabain and ethacrynic acid on the labyrinthine potentials. Pflugers Arch 352: 339–350, 1974. doi: 10.1007/BF00585686. [DOI] [PubMed] [Google Scholar]
- Sewell WF. The effects of furosemide on the endocochlear potential and auditory-nerve fiber tuning curves in cats. Hear Res 14: 305–314, 1984. doi: 10.1016/0378-5955(84)90057-1. [DOI] [PubMed] [Google Scholar]
- Songer JE, Eatock RA. Tuning and timing in mammalian type I hair cells and calyceal synapses. J Neurosci 33: 3706–3724, 2013. doi: 10.1523/JNEUROSCI.4067-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzmaul G, Tolosa L, Winkelman BH, Heidenreich M, Frens MA, Chabbert C, de Zeeuw CI, Jentsch TJ. Vestibular role of KCNQ4 and KCNQ5 K+ channels revealed by mouse models. J Biol Chem 288: 9334–9344, 2013. doi: 10.1074/jbc.M112.433383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci 21: 5535–5545, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of the isk gene. Neuron 17: 1251–1264, 1996. doi: 10.1016/S0896-6273(00)80255-X. [DOI] [PubMed] [Google Scholar]
- Wackym PA, Chen CT, Ishyyama A, Pettis RM, Lopez IA, Hoffman L. Muscarinic acetylcholine receptor subtype mRNAs in the human and rat vestibular periphery. Cell Biol Int 20: 187–192, 1996. doi: 10.1006/cbir.1996.0023. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890–1893, 1998. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wang JJ, Li Y. KCNQ potassium channels in sensory system and neural circuits. Acta Pharmacol Sin 37: 25–33, 2016. doi: 10.1038/aps.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li H, Moss AJ, Robinson J, Zareba W, Knilans T, Bowles NE, Towbin JA. Compound heterozygous mutations in KvLQT1 cause Jervell and Lange-Nielsen syndrome. Mol Genet Metab 75: 308–316, 2002. doi: 10.1016/S1096-7192(02)00007-0. [DOI] [PubMed] [Google Scholar]
- Wangemann P. Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hear Res 90: 149–157, 1995. doi: 10.1016/0378-5955(95)00157-2. [DOI] [PubMed] [Google Scholar]
- Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol 576: 11–21, 2006. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerts AP, Putcha L, Hoag SW, Hallgren E, Van Ombergen A, Van de Heyning PH, Wuyts FL. Intranasal scopolamine affects the semicircular canals centrally and peripherally. J Appl Physiol (1985) 119: 213–218, 2015. doi: 10.1152/japplphysiol.00149.2015. [DOI] [PubMed] [Google Scholar]
- Zdebik AA, Wangemann P, Jentsch TJ. Potassium ion movement in the inner ear: insights from genetic disease and mouse models. Physiology (Bethesda) 24: 307–316, 2009. doi: 10.1152/physiol.00018.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Jones SM, Yamoah EN, Lundberg YW. Otoconin-90 deletion leads to imbalance but normal hearing: a comparison with other otoconia mutants. Neuroscience 153: 289–299, 2008. doi: 10.1016/j.neuroscience.2008.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]