

Abstract
Background
Succinate dehydrogenase-deficient leukoencephalopathy is a complex II-related mitochondrial disorder for which the clinical phenotype, neuroimaging pattern and genetic findings have not been comprehensively reviewed.
Methods
19 individuals with succinate dehydrogenase deficiency-related leukoencephalopathy were reviewed for neuroradiologic, clinical and genetic findings as part of Institutional Review Board approved studies at Children's National Health System (Washington, DC) and VU University Medical Center (Amsterdam, NL).
Results
All individuals had signal abnormalities in the central corticospinal tracts and spinal cord where imaging was available. Other typical findings were involvement of the cerebral hemispheric white matter with sparing of the U fibers, the corpus callosum with sparing of the outer blades, the basis pontis, middle cerebellar peduncles and cerebellar white matter, and elevated succinate on MRS. The thalamus was involved in most studies with a predilection for the anterior nucleus, pulvinar and geniculate bodies. Clinically, infantile-onset neurological regression with partial recovery and subsequent stabilization was typical. All individuals had mutations in SDHA, SDHB or SDHAF1, or proven biochemical defect.
Interpretation
Succinate dehydrogenase deficiency is a rare leukoencephalopathy, for which improved recognition by MRI in combination with advanced sequencing technologies allows non-invasive diagnostic confirmation. The MRI pattern is characterized by cerebral hemispheric white matter abnormalities with sparing of the U fibers, corpus callosum involvement with sparing of the outer blades, and involvement of corticospinal tracts, thalami and spinal cord. In individuals with infantile regression and this pattern of MRI abnormalities, the differential diagnosis should include succinate dehydrogenase deficiency, in particular if MRS shows elevated succinate.
Keywords: Succinate Dehydrogenase, Leukoencephalopathy, MRI, Next-Generation Sequencing
Introduction
The diagnosis of individuals with presumed genetic disorders of the brain white matter remains clinically challenging. MRI pattern recognition greatly facilitates the diagnosis and shortens the diagnostic process. Indeed, expert opinion on the diagnosis of a presumed genetic leukoencephalopathy recommends focused genetic testing if an MRI pattern suggests a specific diagnosis.1-3 Diagnostic algorithms for assessment of MRIs have thus far omitted succinate dehydrogenase (SDH) deficiency-related leukoencephalopathy due to limited information about the MRI phenotype of this condition.
Here we review the MRI features and clinical and genetic findings of 19 individuals with SDH deficiency-related leukoencephalopathy to better delineate the MRI spectrum of this rare genetic condition and define a specific constellation of MRI features. We first identified a series of six individuals with similar clinical and MRI features. Clinical characteristics, including abrupt decompensation in the context of illness, were suggestive of an underlying energy metabolism disorder. MRIs were similarly suggestive of a mitochondrial disease due to significant involvement of the brainstem and cystic degeneration of the white matter.3 Two index cases underwent next generation sequencing for a panel of nuclear encoded mitochondrial genes identifying variants in a subunit of SDH. The remaining four cases were subject to whole exome sequencing and subsequently identified by focused review of next generation sequencing data.4 We subsequently identified thirteen cases, some with existing diagnoses, while in others the diagnosis was confirmed after recognition of the MRI pattern.
Study Design and Methods
MRI Review and Data Collection
Relevant clinical information and MRIs were collected under Myelin Disorders Bioregistry Project at Children's National Health System (CNHS) or the Amsterdam Database of Unclassified Leukoencephalopathies with approval from the institutional review boards at CNHS and VU University Medical Center. MRIs were reviewed and scored by GH, MSvdK and AV using an MRI scoring system previously described and validated.5 T1-weighted and T2-weighted images at a minimum were reviewed along with fluid attenuated inversion recovery (FLAIR) images, contrast enhanced images, MR spectroscopy (MRS), diffusion-weighted images (DWI), apparent diffusion coefficient (ADC) maps, and MRI of the spinal cord where available. ADC maps were used in the evaluation of restricted diffusion as previously described and a cutoff of 60×10-5 mm2/s was used, values below which were considered to be restricted.6
Results
Clinical findings
The clinical findings are reported in Table 1 and Supplemental Table S1. All individuals presented in the first two years of life (19/19), with average onset at about 12 months, most with a rapid deterioration, in some cases provoked by an illness, an injury, vaccination, or with no obvious preceding event at all. Clinical history revealed in most cases only one episode of regression, lasting weeks to months (n=12). In most cases, the clinical course was subsequently static or slowly progressive, in three cases without significant further deterioration into early adulthood. Six died from either a pneumonia or respiratory failure; mean age at death was 5 years. One of the individuals who died presented at birth with dilated cardiomyopathy and remained hypotonic and severely delayed. None of the other individuals had evidence of involvement of other organs than the central nervous system. During life, individuals suffered from significant motor dysfunction (19/19), with variable degrees of spasticity (17/19) and dystonia (4/17). Eleven of the surviving individuals had moderate or severe cognitive impairment, and in two cases cognitive function was relatively preserved.
Table 1. Clinical features in cases of SDH-related leukoencephalopathy.
| Clinical Features | Percentage of Cases |
|---|---|
| Age of Onset | Average: 11.21 mo. Range: Birth – 20 mo. |
| Consanguinity | 67% (13/19) |
| Abrupt Decompensation | 72% (14/19) |
| Microcephaly | 50% (8/17) |
| Truncal Hypotonia | 75% (13/17) |
| Limb spasticity | 89% (17/19) |
| Ataxia | 92% (12/14) |
| Dystonia | 25% (4/17) |
| Acquired Independent Ambulation | 25% (5/17) |
| Current Functional Level | 7 Individuals with a severe spastic tetraparesis 11 Individuals with moderate or severe cognitive impairment 5 Individuals died at mean age of 5 years |
Key: mo. = month, N/A=Not Available, yrs.= years
Molecular findings
Mutation analysis results are provided in Supplemental Table S2. Ten individuals were found with mutations in SDHAF1 (OMIM:612848), five individuals with mutations in SDHB (OMIM:185470), and a further two with mutations in SDHA (OMIM:600857). One individual has confirmed SDH deficiency through biochemical analysis only. One individual had a single pathogenic variant in SDHB with a second splice site variant of uncertain significance.
MRI Analysis
26 studies of 19 individuals were available for review. MRI features in individual cases are summarized in Table 2 and reported in more detail in Supplemental Table S3-S5. All but one case had involvement of the corticospinal tracts in some form including the posterior limb of the internal capsule, pons (16/19) (Figure 1A) and the pyramids of the medulla. The transverse pontine fibers were often affected (14/19) (Figure 1B), the lateral portions of the middle cerebellar peduncles (13/19) (Figures 1C and D), in some cases creating a transverse band across the pons with extension into the middle cerebellar peduncles (11/19) (Figure 1E and F). In a few cases (3/19), there was involvement of the central tegmental tracts, visible as hyperintense areas in the pontine tegmentum (Figure 1B). The dorsal portion of the cervical spinal cord was found to be affected in the majority of cases with an MRI of the cervical spine (13/14) (1G and 1H).
Table 2. MRI Features of cases of SDH-related leukoencephalopathy.
| MRI Features | All Individuals | Early stage individuals | Intermediate stage individuals | Late stage individuals |
|---|---|---|---|---|
| Involvement of cerebral white matter with U fiber sparing | 89% (17/19) | 82% (9/11) | 100% (7/7) | 100% (8/8) |
| Rarefaction or cystic degeneration in affected white matter | 53% (8/15) | 33% (3/9) | 83% (5/6) | 100% (8/8) |
| Involvement of the corpus callosum | 95% (18/19) | 100% (11/11) | 100% (7/7) | 100% (8/8) |
| Involvement of thalamic nuclei | 79% (15/19) | 90% (10/11) | 57% (4/7) | 13% (1/8) |
| Corticospinal tract involvement | 95% (18/19) | 73% (8/11) | 71% (5/7) | 100% (8/8) |
| Involvement of the cerebellar white matter | 53% (10/19) | 45% (5/11) | 43% (3/7) | 25% (2/8) |
| Involvement of the middle cerebellar peduncles | 68% (13/19) | 64% (7/11) | 43% (3/7) | 63% (5/8) |
| Involvement of basis pontis | 74% (14/19) | 73% (8/11) | 71% (5/7) | 100% (8/8) |
| Involvement of spinal cord | 93% (13/14) | 78% (7/9) | 100% (5/5) | 100% (5/5) |
| MR Spectroscopy | Succinate 92% (11/12) | Succinate 100% 7/7 | Succinate 100% (3/3) | Succinate 90.1% (2/4) |
| Diffusion | Low 9 High 3 | Low 9 High 0 | Low 1 High 2 | Low 0 High 4 |
| Contrast enhancement | Yes 4 No 1 | Yes 3 No 1 | Yes 2 No 0 | N/A |
| Number of patients / MRIs | N=19 / 26 | 11 | 7 | 8 |
Figure 1. Special Features of SDH-related Leukoencephalopathy.
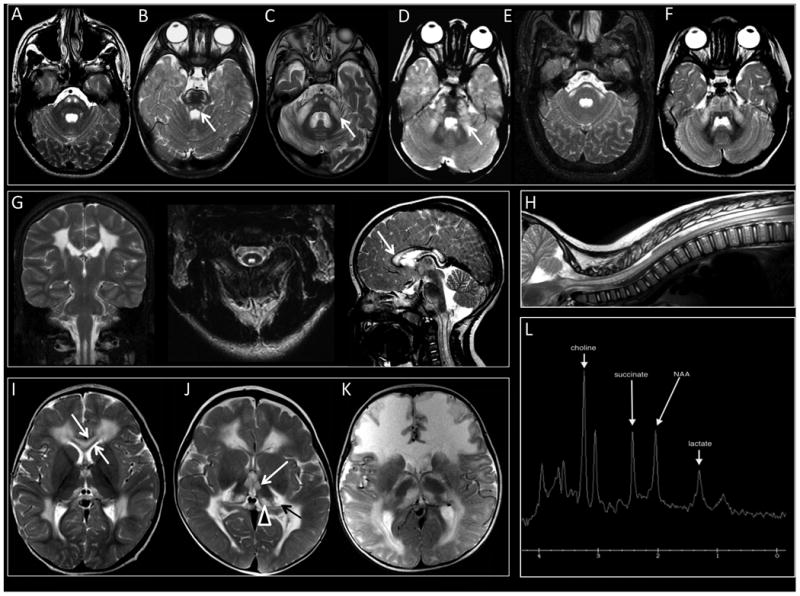
(1A) Signal abnormalities are seen in the pyramidal tracts in the pons (Image from Individual 1). (1B) The transverse pontine fibers are often affected and in a few cases, the central tegmental tracts (white arrow) (Image from Individual 13). (1C and 1D) Signal abnormalities are seen lateral portions of the middle cerebellar peduncles (Arrows, Images from Individuals 7 and 4, respectively). (1E and 1F) In some cases, a transverse band across the pons is observed with extension into the middle cerebellar peduncles (Images from Individuals 2 and 8, respectively). (1G and 1H) The dorsal portion of the cervical and sometimes also thoracic spinal cord is often affected (Images from Individual 1). The inner and outer blades are spared (white arrow). (1I and 1J) Extensive cerebral hemispheric white matter abnormalities are seen predominantly involving the frontal, parieto-occipital, and posterior temporal regions, sparing the juxtacortical fibers in all lobes (Images from Individuals 8 and 10, respectively). (1K) Diffuse abnormalities involving the juxtacortical, central and periventricular white matter may also occur (Individual 15). (1I) The inner and outer blades are spared (White Arrow), while the rest of the corpus callosum is affected (Individual 8). (1J and 1K) The thalamic nuclei anterior and medial portions of the thalamic nuclei are involved (White Arrow), as well as the medial pulvinar (Arrow Head), or medial and lateral geniculate bodies bilaterally (Black Arrow, Individual 10 (1J) and Individual 15 (1K)). (1L). Single voxel MRS (TE 35 msec) with voxel of interest placed over the mid pontine signal abnormalities. Large metabolic peak at 2.4 ppm represents marked succinate elevation. Macromolecular peak centered at 1.3 ppm is consistent with abnormal lactate.
Nearly all individuals (17/19) had extensive cerebral hemispheric white matter abnormalities predominantly involving the frontal, parieto-occipital, and posterior temporal regions, sparing the juxtacortical fibers in all lobes (Figure 1I and 1J). In a smaller number of cases, abnormalities were either more diffuse, involving the juxtacortical, central and periventricular white matter (Figure 1K) or more limited, sparing the cerebral white matter and affecting only the thalami and corticospinal tracts in the brainstem (not shown). The corpus callosum was affected and swollen in appearance (18/19), sparing the inner and outer blades (Figure 1I). The external and extreme capsules were variably involved (4/19), as was the cerebellar white matter (10/19). In many individuals the anterior thalamic nuclei (11/19), medial pulvinar (15/19) or medial and lateral geniculate bodies (7/19) were involved bilaterally (1J).
Ten of the eleven individuals who underwent proton-MRS demonstrated an abnormally elevated peak at 2.4 ppm, compatible with succinate (Figure 1L). Succinate peaks ranged from mild to markedly elevated (Supplemental Tables S3-S5) and were most often found in areas of diseased white matter. However, succinate was also observed in otherwise normal appearing occipital lobe gray matter. Mildly to markedly elevated lactate was present in some cases, mostly in regions of abnormality.
We divided the 26 MRIs of our 19 individuals in 3 groups based on age at MRI to look at the evolution of lesions over time. MRIs taken before 2 years of age were classified as early in the disease course (n=11) (Figure 2A). Of the MRIs with available FLAIR images, many (6/9) showed no evidence of white matter rarefaction. The white matter and corpus callosum was often strikingly swollen in these cases. Areas of the abnormal white matter had restricted diffusion with increased DWI signal and low ADC values (9/9) (Supplemental Table S3).
Figure 2. Evolution of lesions in SDH-related leukoencephalopathy.
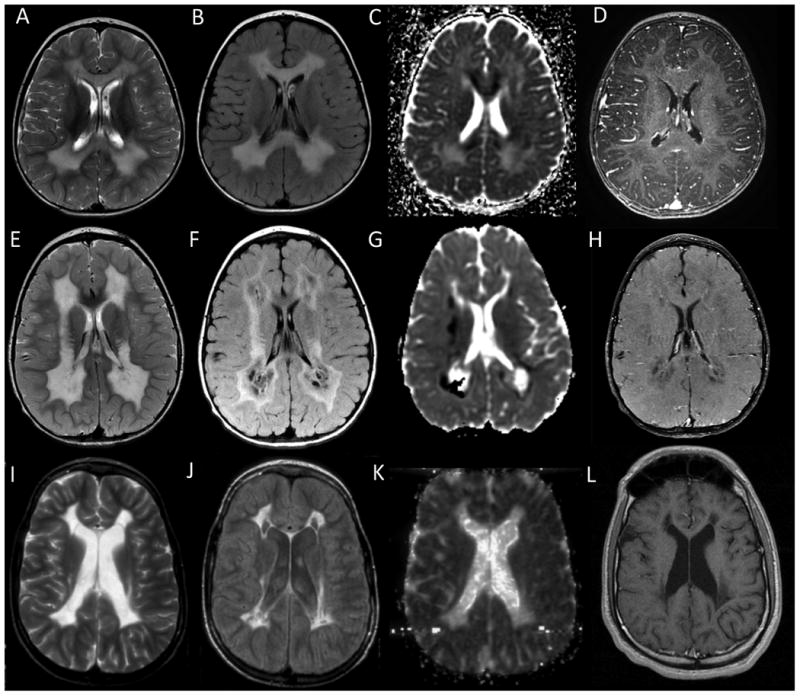
(A-D) Top Row – Early stage MRI. The abnormal white matter looks swollen, especially the corpus callosum. Myelin micro-vacuolization is suspected in areas of restricted diffusion seen on ADC maps. There is no enhancement. (E-H) Middle Row – Intermediate stage MRI. Tissue necrosis is suspected based on MRI findings with white matter rarefaction seen on FLAIR imaging and small foci of contrast enhancement. Areas of low ADC values continue to be present. (I-L) Bottom Row – Late stage MRI. Atrophy and collapse of the affected white matter is seen, with cysts. ADC values are low and no further contrast enhancement is present. (A,E,I) T2-weighted imaging from select patients. (B,F,J) FLAIR imaging from select patients. (C,G,K) Diffusion weighted imaging from select patients. (D,H,L) Contrast enhanced images from select patients.
MRIs obtained between 2 and 5 years of age were classified as representing the intermediate disease stage (n=7) (Figure 2B). All MRIs showed extensive white matter abnormalities with increased T2 signal but a less swollen appearance. For those MRIs with available FLAIR imaging, evidence of white matter rarefaction and cystic degeneration appeared at this stage of disease (5/6). There was continued evidence of restricted diffusion with low values on ADC maps, even extending into the cervical spinal cord in one patient. This phase was characterized by variable contrast enhancement in the affected white matter (Supplemental Table S4), which can be seen in the setting of subacute necrosis.
MRIs obtained between 8 and nearly 20 years of age were classified as representative of the late disease stage (n=8) (Figure 3C). At this stage, there was white matter volume loss with evidence of gliotic retraction, large areas of rarefied or cystic white matter on FLAIR (8/8), and resolution of diffusion abnormalities (4/4) (Supplemental Table S5).
Discussion
SDH is a four-subunit enzyme complex, known as complex II of the mitochondrial electron transport chain. It includes a flavoprotein subunit (Fp, encoded by SDHA) and an iron-sulphur subunit (Fe-S, encoded by SDHB), paired with two subunits that anchor the complex to the inner mitochondrial membrane (SDHC (OMIM:602413) and SDHD (OMIM:602690)). In addition, the SDH protein complex comprises four assembly factor proteins (SDHAF1, SDHAF2 (OMIM:613019), SDHAF3 (OMIM:615773), and SDHAF4).
While leukoencephalopathy is a frequent presenting symptom in individuals with mutations in subunit genes SDHA and SDHB and the assembly factor gene SDHAF17-10 leukoencephalopathy is not the only presentation caused by mutations in genes encoding components of the SDH protein complex. No mutations in the subunit genes SDHC and SDHD or assembly factor gene SDHAF2 have thus far been identified in cases of MRI confirmed leukoencephalopathy. Mutations in SDHD have recently been associated with severe neurological symptoms despite a normal MRI11 and SDHA mutations can cause cardiomyopathy (MIM:613642).12, 13 Apart from its role in cellular energy metabolism, SDH also plays a role as a tumor suppressor and mutations in a number of genes have been associated with hereditary paraganglioma (MIM:616165) (SDHA, SDHB, SDHC, SDHD and SDHAF2 mutations)14 and pheochromocytomas.15 Assembly factor genes SDHAF3 and SDHAF4 are not yet known to be associated with human disease.
Although individuals with SDH-deficiency are known to often present with a related leukoencephalopathy, no specific MRI pattern has previously been described.7, 9, 10 Of course elevated succinate on MRS is a specific finding7, 8 with a succinate peak often seen at 2.4 ppm. Our findings in this group of patients support existing evidence suggesting that MRS should be utilized in the diagnostic workup of a patient with a suspected leukoencephalopathy, especially if mitochondrial disease is high on the diagnostic differential.3, 16 It should, however, be noted that in one patient, with splice-site variants in SDHB, there was only a mild elevation of succinate and that thus this finding may not be obligate in all affected patients. MRS findings may be dependent on disease stage and placement of the voxel in the affected tissue. Also, MRS is not routinely available or consistently used in the diagnosis of leukoencephalopathies. MRI pattern recognition continues to be an important diagnostic tool in the evaluation of individuals with white matter abnormalities in the era of next generation sequencing.3 We were prompted to more completely define the MRI spectrum of SDH deficiency-related leukoencephalopathy and a distinct MRI pattern emerged (Table 2).
All MRIs examined exhibited involvement of at least a portion of the corticospinal tracts through the posterior limb of the internal capsule, the pons and the pyramids of the medulla. The corticopontocerebellar tracts were also frequently affected with signal abnormalities in the corticopontine and transverse pontine fibers and middle cerebellar peduncles. Additionally, involvement of specific thalamic nuclei, the cerebral hemispheric white matter abnormalities with sparing of the U fibers, the corpus callosum involvement with sparing of the outer blades, and the spinal cord abnormalities are strongly suggestive of SDH deficiency-related leukoencephalopathy and important in directing genetic testing for these cases.
The broad age range of our individuals allowed a study of the evolution of lesions over time. MRIs obtained within the first two years of life almost invariably show cerebral white matter signal abnormalities with restricted diffusion in different white matter areas suggesting myelin micro-vacuolization.6 Myelin micro-vacuolization is followed by an intermediate stage of variable contrast enhancement in the affected white matter, indicating breakdown of the blood brain barrier, probably by necrosis. Progressive rarefaction and cystic degeneration lead to atrophy of the white matter in later stages of the disease course with evidence of gliotic retraction and cysts on FLAIR. Neuropathologic confirmation of these findings is not yet available.
Our findings also demonstrate a consistent clinical course. All individuals had disease onset in the first two years of life, most often an episode of rapid deterioration in the context of fever or trauma. Some individuals had a stable clinical course after this regression, sometimes with partial recovery of lost skills, while another group had severe onset at birth or within that two-year period and subsequently never gained or reacquired any skills. A few patients had more than one episode of subacute deterioration. The homogeneous clinical manifestations associated with mutations in the SDH genes causing mitochondrial leukoencephalopathy is surprising given the general clinical heterogeneity of other mitochondrial disorders. Strikingly, we did not notice a consistent difference between the groups of patients with mutations in the different SDH-related genes, although the groups are small.
SDH deficiency-related leukoencephalopathy is not the only leukoencephalopathy that presents early in life with acute decompensation, often provoked by febrile illness, vaccination or injury, with associated significant brainstem involvement on MRI.17-21 Infantile presentation with brain stem abnormalities on MRI can be seen in other disorders, including other mitochondrial disorders, Alexander disease (MIM:203450), peroxisomal disorders, Leukoencephalopathy with Brain stem and Spinal cord involvement and Lactate elevation (LBSL) (MIM:611105) and Leukoencephalopathy with Thalamus and Brainstem involvement and high Lactate (LTBL) (MIM:612799). However, the thalamus and brainstem MRI pattern observed in our individuals, with signal abnormalities in the anterior nucleus, pulvinar and geniculate bodies as well as the corticopontine and transverse pontine fibers and middle cerebellar peduncles, is unique for the SDH deficiency-related leukoencephalopathy and allows differentiation from other mitochondrial leukoencephalopathies.17, 19-23
MRI pattern recognition offers a highly effective and non-invasive approach to the diagnostic workup of individuals with leukoencephalopathy. Recognition of SDH deficiency-related leukoencephalopathy provides an opportunity to avoid invasive diagnostic investigations such as muscle biopsy and immediately go for targeted gene sequencing. In the context of expanding use of next generation sequencing technologies, it is also useful to determine specific clinical and imaging phenotypes of genetic leukoencephalopathies to permit interpretation of identified gene variants. Biochemical testing was not reviewed as part of our study, but when performed and if results are suggestive of complex II deficiency, genetic sequencing of the SDH subunit genes and assembly factor genes should be performed.7, 9, 15 There appears to be a large group of individuals with a loss of SDH activity as proven by biochemical analysis, but no identifiable mutations.24 It is likely that as the use of next-generation diagnostic approaches is extended to individuals with similar MRI and clinical features, individuals with mutations in the additional SDH subunit genes will be identified.3
Supplementary Material
Acknowledgments
Guy Helman was supported by the Delman Fund for Pediatric Neurology Education. Guy Helman and Adeline Vanderver are supported by the Myelin Disorders Bioregistry Project. Marjo S. van der Knaap was supported by ZonMw TOP grant 91211005. This publication was supported by Award Number UL1TR000075 from the NIH National Center for Advancing Translational Sciences.
Footnotes
Authorship Contributions: GH, AV, and MSvdK coordinated the project. GH, LC, MTW, KB, SE, IM, KVH, JMT, RJT, AV, and MSvdK wrote, reviewed, and planned the manuscript. GH, MTW, AV, and MSvdK performed MRI review and analyzed the clinical and imaging data. LC, CS, RB, and RJT provided bioinformatics analysis. KB, SE, IM, AV, and MSvdK identified individuals eligible for the study, collected longitudinal clinical, imaging and molecular data, and reviewed the manuscript.
The members of the SDH Study Group (see supplementary files for names and affiliations) also referred individuals, and provided clinical and imaging data, provided clinical care for patients, and also reviewed the manuscript.
Conflicts of Interest: The authors report no conflicts of interest. This manuscript's contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
References
- 1.Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009 Feb 24;72(8):750–9. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Knaap MS, Valk J, de Neeling N, Nauta JJ. Pattern recognition in magnetic resonance imaging of white matter disorders in children and young adults. Neuroradiology. 1991;33(6):478–93. doi: 10.1007/BF00588038. [DOI] [PubMed] [Google Scholar]
- 3.Parikh S, Bernard G, Leventer RJ, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab. 2014 Dec 29; doi: 10.1016/j.ymgme.2014.12.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanderver A, Simons C, Helman G, et al. Whole exome sequencing in a cohort of patients with unresolved central nervous system white matter abnormalities. 2015 In Press. [Google Scholar]
- 5.van der Knaap MS, Breiter SN, Naidu S, Hart AA, Valk J. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology. 1999 Oct;213(1):121–33. doi: 10.1148/radiology.213.1.r99se01121. [DOI] [PubMed] [Google Scholar]
- 6.Depienne C, Bugiani M, Dupuits C, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. The Lancet Neurology. 2013;12(7):659–68. doi: 10.1016/S1474-4422(13)70053-X. 7// [DOI] [PubMed] [Google Scholar]
- 7.Ghezzi D, Goffrini P, Uziel G, et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009 Jun;41(6):654–6. doi: 10.1038/ng.378. [DOI] [PubMed] [Google Scholar]
- 8.Ohlenbusch A, Edvardson S, Skorpen J, et al. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet journal of rare diseases. 2012;7:69. doi: 10.1186/1750-1172-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alston CL, Davison JE, Meloni F, et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. Journal of Medical Genetics. 2012;49(9):569–77. doi: 10.1136/jmedgenet-2012-101146. 2012 September 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brockmann K, Bjornstad A, Dechent P, et al. Succinate in dystrophic white matter: A proton magnetic resonance spectroscopy finding characteristic for complex II deficiency. Annals of neurology. 2002;52(1):38–46. doi: 10.1002/ana.10232. [DOI] [PubMed] [Google Scholar]
- 11.Jackson CB, Nuoffer JM, Hahn D, et al. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J Med Genet. 2014 Mar;51(3):170–5. doi: 10.1136/jmedgenet-2013-101932. [DOI] [PubMed] [Google Scholar]
- 12.Levitas A, Muhammad E, Harel G, et al. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet. 2010;18(10):1160–5. doi: 10.1038/ejhg.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Coster R, Seneca S, Smet J, et al. Homozygous Gly555Glu mutation in the nuclear-encoded 70 kDa flavoprotein gene causes instability of the respiratory chain complex II. American Journal of Medical Genetics Part A. 2003;120A(1):13–8. doi: 10.1002/ajmg.a.10202. [DOI] [PubMed] [Google Scholar]
- 14.Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Human molecular genetics. 2010 Aug 1;19(15):3011–20. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Vranken JG, Na U, Winge DR, Rutter J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Critical reviews in biochemistry and molecular biology. 2014 Dec;9:1–13. doi: 10.3109/10409238.2014.990556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bray MD, Mullins ME. Metabolic white matter diseases and the utility of MR spectroscopy. Radiol Clin North Am. 2014 Mar;52(2):403–11. doi: 10.1016/j.rcl.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 17.Steenweg ME, Ghezzi D, Haack T, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain : a journal of neurology. 2012 May;135(Pt 5):1387–94. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- 18.Melchionda L, Haack TB, Hardy S, et al. Mutations in APOPT1, encoding a mitochondrial protein, cause cavitating leukoencephalopathy with cytochrome c oxidase deficiency. American journal of human genetics. 2014 Sep 4;95(3):315–25. doi: 10.1016/j.ajhg.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kevelam SH, Rodenburg RJ, Wolf NI, et al. NUBPL mutations in patients with complex I deficiency and a distinct MRI pattern. Neurology. 2013 Apr 23;80(17):1577–83. doi: 10.1212/WNL.0b013e31828f1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Björkman K, Sofou K, Darin N, et al. Broad phenotypic variability in patients with complex I deficiency due to mutations in NDUFS1 and NDUFV1. Mitochondrion. 2015;21(0):33–40. doi: 10.1016/j.mito.2015.01.003. 3// [DOI] [PubMed] [Google Scholar]
- 21.Kashani A, Thiffault I, Dilenge ME, et al. A homozygous mutation in the NDUFS1 gene presents with a mild cavitating leukoencephalopathy. Neurogenetics. 2014 Aug;15(3):161–4. doi: 10.1007/s10048-014-0412-2. [DOI] [PubMed] [Google Scholar]
- 22.Steenweg ME, van Berge L, van Berkel CG, et al. Early-onset LBSL: how severe does it get? Neuropediatrics. 2012 Dec;43(6):332–8. doi: 10.1055/s-0032-1329395. [DOI] [PubMed] [Google Scholar]
- 23.van der Knaap M, Valk J. Magnetic Resonance of Myelination and Myelin Disorders. 3. Berlin, Germany: Springer; 2005. [Google Scholar]
- 24.Jain-Ghai S, Cameron JM, Al Maawali A, et al. Complex II deficiency--a case report and review of the literature. American journal of medical genetics Part A. 2013 Feb;161a(2):285–94. doi: 10.1002/ajmg.a.35714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.