

In a structure as complex as the human brain a multitude of things can go wrong. The wonder is that for most people the brain functions effectively and unceasingly for more than 60 years.
— Seymour S. Kety, 1979, neuroscientist and the first director of the National Institute of Mental Health (1)
A new human genome is created with the fertilization of an egg. This single cell will undergo millions of divisions to produce an individual composed of a multitude of cell types. The cells in our bodies show a remarkable array of specialization, from cone cells that sense specific colors of light to immune cells that fight infections. However, specialization alone is not enough to form a complex organism, as these cells must function in a coordinated manner. Perhaps nowhere is this more apparent than in the human brain, where 100 billion neurons integrate to form more than 100 trillion connections. It is this specialization along with synchronization that allows our brain not only to instruct basic physiologic functions, such as breathing, but also to generate the complex thoughts and feelings that make each one of us unique. Reflecting on Seymour Kety's statement, it is natural to ask: How can something so extraordinarily complex develop consistently and reliably from a single cell?
Many of us first learned about genetics in our high school biology classes. Even today, genetics is often taught beginning with Mendel's laws of inheritance followed by Crick's “central dogma” information stored in our DNA is transcribed to messenger RNA (mRNA), which is then translated into proteins. Of course, this is only a small part of the story. Since the majority of our cells have the same 20,000 protein-coding genes, the body must use complex regulatory mechanisms to ensure that genes are turned on and off at the appropriate times and places.
Gene regulation can occur at many levels in the process of making a fully functional protein (Figure 1A). A main control point is at the level of transcription. To state the obvious: if a gene is not transcribed in a cell, then it cannot be used to make a protein in that cell. For transcription to occur, an enzyme called RNA polymerase must bind to a specific region of DNA called the promoter that is located upstream of a gene. This RNA polymerase then moves along the gene of interest, reading the DNA code and forming a complementary mRNA strand. Proteins called transcription factors play a key role in regulating this process.
Figure 1.
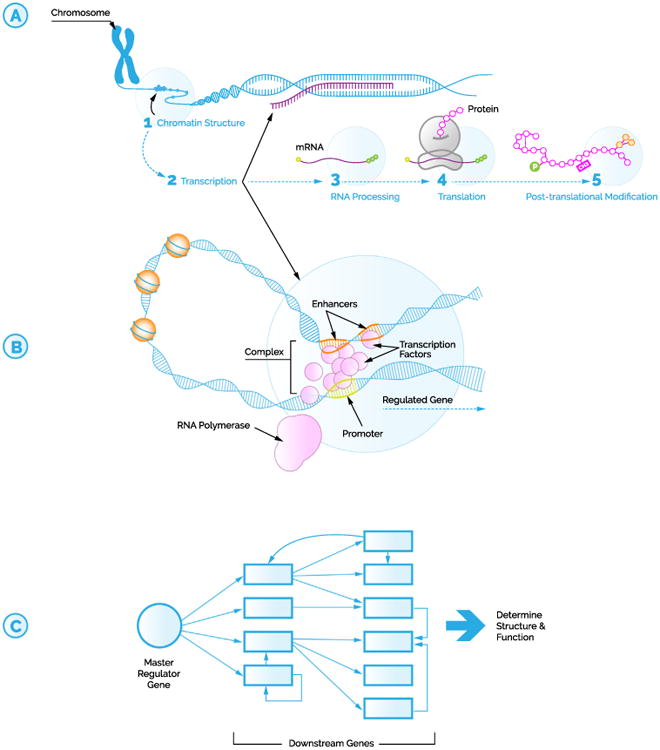
Transcription factors can serve as key master regulators influencing gene expression. (A) Multiple layers of gene regulation, including chromatin structure, transcription, RNA processing, translation, and posttranslational modification. (B) Multiple transcription factors work together to form a complex to influence gene regulation. (C) Schematic of how a single master regulator gene can influence downstream genes to drive cell structure and function. mRNA, messenger RNA.
Transcription factors are found in all living organisms and are critical for regulating gene expression. In humans, an estimated 6% of our genes encode transcription factors (2), making them perhaps the largest family of proteins in our bodies. Their core function is to cause genes to be expressed more or less frequently. They do this by binding to a specific DNA sequence adjacent to a gene of interest. Of note, this binding site may be near or relatively far away from the gene, as the three-dimensional flexibility of DNA allows distant binding sites to interact (Figure 1B). Like dance partners, a transcription factor and its DNA binding site must connect to affect expression. Further complicating the story, multiple transcription factors along with other regulatory proteins often assemble in large complexes that have a combinatorial effect (which is to say that perhaps a dance troupe is a better metaphor) (3).
Transcription factors (and other regulatory proteins) reflect one out of a vast array of mechanisms that regulate gene expression. Only approximately 1.5% of our DNA involves genes that code for specific proteins, and for many years, the other 98.5% was viewed simply as “junk DNA,” a remnant of evolution. Advances in science now suggest that the majority of our genome possesses a wide range of functional activity, including encoding a variety of noncoding RNA molecules. RNA's raison d'être extends far beyond just serving as a temporary copy of information from our genome. This includes functioning within the main enzyme involved in protein synthesis (ribosomal RNA), directing mRNA modification (small nuclear RNAs), and regulating gene expression by interacting with mRNA (microRNAs and small interfering RNAs). Beyond noncoding RNAs, each year new mechanisms of gene regulation are discovered. Recent cutting-edge lines of inquiry into gene regulation include work on superenhancers and on the three-dimensional architecture of the genome. With all these layers of control, a crucial question is, how can the cell regulate all of the regulators?
In 1979, geneticist Susumu Ohno initially hypothesized the need for a special group of genes exactly for this purpose: to provide an orderly hierarchy to ensure the proper development of complex structures. Ohno (4) reasoned that “If viewed from the bottom, any mammalian genetic regulatory system is bound to appear very complicated … yet all of these often overlapping regulatory systems must come under the control of one or the other of a small number of master regulatory genes.” Just as a maestro coordinates the timing and dynamics of each member of the orchestra, the master regulator drives the expression of downstream genes within a cell (Figure 1C). In the last 40 years, the definition of master regulators has evolved to now refer to genes that govern a specific cell's fate (5). In particular, a master regulator is often expressed at the beginning of a cell lineage and participates in specification of cell differentiation and function. As transcription factors are primary gatekeepers of gene expression, it is no surprise that the majority of identified master regulator genes code for transcription factors themselves.
So how does all this fit into our understanding of the genetics of psychiatric conditions? Interest in studying genes involved in mental illness stems from an understanding that these diseases are highly heritable: having a close family member affected with a psychiatric condition is the single strongest risk factor for disease. In the early days of psychiatric genetics research, studies focused on individual proteins hypothesized to cause mental illness. Similar to how mutations in a chloride channel cause cystic fibrosis or a mutation in hemoglobin causes sickle cell disease, researchers were optimistic that investigating critical candidate genes would unlock the mystery of psychiatric conditions. Unfortunately, many of these initial findings were not robust. More recently, genome-wide association studies enabled a departure from this narrowly focused approach. By surveying millions of genetic variants across the genome in a single experiment, genome-wide association studies have identified hundreds of distinct genetic changes significantly associated with psychiatric conditions (6). Somewhat surprisingly, emerging evidence demonstrates that the majority of significant findings from genome-wide association studies do not occur in previously hypothesized genes but actually occur in regulatory regions of the genome, including transcription factor recognition sites (7). These results provide evidence that alterations in gene regulatory networks play a critical role in disease pathogenesis.
Beyond identifying important genetic factors, scientists now have the tools to probe how regulatory elements actually influence gene expression. Initial efforts to study gene expression focused on individual genes by measuring the levels of corresponding RNAs or proteins. With the decreasing cost of sequencing, new functional genomic technologies have expanded the scope of experiments from individual genes to the examination of entire regulatory networks (8). Using chromatin immunoprecipitation sequencing, we can now measure how transcription factors and other proteins bind DNA across the genome. In addition, RNA sequencing measures levels of all mRNAs within a sample, elucidating which genes are in fact expressed. When combined with gene-editing techniques, these sequencing approaches allow scientists to determine how changing a specific transcription factor influences its binding and, ultimately, the expression of downstream genes.
In this issue of Biological Psychiatry, Michaelson et al. (9) leverage these new approaches to explore the role of two transcription factors (NPAS1 and NPAS3) in neuropsychiatric functioning. Previous work had shown that mutations in the gene encoding NPAS3 were risk factors for a range of psychiatric illnesses, including schizophrenia, bipolar disorder, major depression, and attention-deficit/hyperactivity disorder. However, the specific mechanisms by which NPAS proteins influence disease remained unclear. Using functional genomics, Michaelson et al. provide evidence that NPAS1 and NPAS3 operate as master regulators, influencing downstream genes involved in a host of core brain functions (including neurogenesis, synaptogenesis, and synaptic maintenance). This work provides an important example of how integrating multiple genomic technologies can uncover the role of master regulator genes involved in brain function.
The author Joyce Carol Oates wrote, “The ideal art, the noblest of art [is] working with the complexities of life, refusing to simplify” (10). Similarly, genetics has moved from studying single genes to thinking about intricate networks of gene regulation. As science marches forward, it is critical that we embrace the complexity of gene regulation to understand the basic mechanisms of brain function and how these processes go awry in psychiatric disease. Although there is still much that remains unknown, we are increasingly gaining insight into how something as complex as the human brain can function so “effectively and unceasingly.”
Acknowledgments
This work was supported by the National Institutes of Health (Grant Nos. R25 MH10107602S1 and R25 MH08646607S1 to DAR, in his role as co-chair of the National Neuroscience Curriculum Initiative).
This commentary was produced in collaboration with the National Neuroscience Curriculum Initiative. We thank Dr. Melissa Arbuckle for her contribution as a National Neuroscience Curriculum Initiative editor and Amanda Wang for her role in developing the figure.
Footnotes
Disclosures: The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Kety S. Disorders of the human brain. Sci Am. 1979;241:202–214. doi: 10.1038/scientificamerican0979-202. [DOI] [PubMed] [Google Scholar]
- 2.Vaquerizas JM, Kummerfeld SK, Telichmann SA, Luscombe NM. A census of human transcription factors: Function, expression, and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- 3.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohno S. Major Sex-Determining Genes. Berlin: Springer-Verlag; 1979. The number of genes in the mammalian genome and the need for master regulatory genes; pp. 17–21. [Google Scholar]
- 5.Chan SS, Kyla M. What is a master regulator? J Stem Cell Res Ther. 2013;3:114. doi: 10.4172/2157-7633.1000e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gratten J, Wray NR, Keller MC, Visscher PM. Large scale genomics unveils the genetic architecture of psychiatric disorders. Nat Neurosci. 2014;17:782–790. doi: 10.1038/nn.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maurano M, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systemic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolouri H. Modeling genomic regulatory networks with big data. Trends Genet. 2014;30:182–191. doi: 10.1016/j.tig.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Michaelson JJ, Shin MK, Koh JY, Brueggeman L, Zhang A, Katzman A, et al. Neuronal PAS domain proteins 1 and 3 are master regulators of neuropsychiatric risk genes. Biol Psychiatry. 2017;82:213–223. doi: 10.1016/j.biopsych.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oates JC. The Journal of Joyce Carol Oates: 1973-1982. New York: HarperCollins Publishers; 2007. [Google Scholar]