

Abstract
Autophagy is central to the maintenance of organismal homeostasis in both physiological and pathological situations. Accordingly, alterations in autophagy have been linked to clinically relevant conditions as diverse as cancer, neurodegeneration and cardiac disorders. Throughout the past decade, autophagy has attracted considerable attention as a target for the development of novel therapeutics. However, such efforts have not yet generated clinically viable interventions. In this Review, we discuss the therapeutic potential of autophagy modulators, analyse the obstacles that have limited their development and propose strategies that may unlock the full therapeutic potential of autophagy modulation in the clinic.
Autophagy is a highly conserved mechanism through which eukaryotic cells deliver dispensable or potentially dangerous cytoplasmic material to lysosomes for degradation1. Thus far, three major routes for the delivery of autophagic substrates to lysosomes have been characterized: microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy.
Microautophagy relies on the direct uptake of cytoplasmic material through an invagination of the lysosomal membrane2. CMA involves the lysosomal-associated membrane protein 2 (LAMP2)-dependent translocation of autophagic substrates bound to cytosolic chaperones of the heat shock protein family across the lysosomal membrane3. Macroautophagy involves specialized double-membraned vesicles known as autophagosomes, which progressively sequester autophagic cargo and — upon closure — deliver the cargo to lysosomes by membrane fusion1. The organelle that forms upon the fusion of one autophagosome and one lysosome is generally referred to as an autolysosome1. Macroautophagy is by far the best-characterized form of autophagy. For this reason, the word autophagy is used throughout this article to refer to macroautophagy, unless otherwise specified.
Autophagy is fundamental to the preservation of organismal fitness, for multiple reasons. Constitutive autophagic responses efficiently degrade products of normal cellular metabolism that can become cytotoxic upon accumulation, such as damaged mitochondria and redox-active protein aggregates4. Inducible autophagic responses promote the survival of cells that respond to perturbations of intracellular or extracellular homeostasis5. Autophagy is indeed central to adaptation to stress, as demonstrated by the fact that pharmacological or genetic inhibition of autophagy usually precipitates the demise of cells facing infections and nutritional, metabolic, physical and chemical challenges6. Furthermore, autophagy is intimately connected with cell-extrinsic circuitries that operate to maintain homeostasis and support healthy ageing at the whole-body level. For instance, autophagic responses in the liver, skeletal muscle and other tissues underlie the beneficial effects of physical exercise on whole-body glucose metabolism7. Along similar lines, autophagy induction in malignant cells that succumb to some chemotherapies and radiotherapies results in the emission of danger signals and, ultimately, the initiation of a therapeutically relevant anticancer immune response8. Autophagy can also mediate cytotoxic effects, at least in specific pathophysiological settings9, although the term <m>autophagic cell death</m> should be used with extreme caution (BOX 1). Moreover, components of the autophagic apparatus have recently been shown to participate in processes other than the degradation of cytoplasmic material. These processes include: LC3-associated phagocytosis (LAP)10 (BOX 2), migration (mainly as a result of focal adhesion turnover)11 and unconventional secretion, which is a mechanism by which cytoplasmic entities (including soluble proteins, organellar material and pathogens) are exported from the cell in a manner that does not depend on the conventional secretory route that operates between the endoplasmic reticulum and the Golgi apparatus12.
Box 1. Autophagic cell death in development and disease.
When light microscopy was the main — if not the sole — technique to study cell biology, investigators noted that the cytoplasm of dying eukaryotic cells sometimes becomes clogged with vacuoles. Soon thereafter, the term ‘autophagic cell death’ (also known as type II cell death) was introduced to indicate instances of cellular demise that are accompanied by cytoplasmic vacuolization288. This expression rapidly acquired a causal implication and has subsequently been extensively used, which led to the assumption that autophagy aetiologically contributes to cell death. With the advent of modern molecular biology techniques, however, it became clear that autophagy generally mediates cytoprotective — rather than cytotoxic — effects. Indeed, pharmacological or genetic inhibition of core components of the autophagic machinery most often accelerates — rather than retards — the death of mammalian cells that experience perturbations of homeostasis289. Thus, autophagic responses often accompany the cellular demise (as an ultimate attempt of cells to cope with stress and to survive), but rarely cause it.
However, instances of cell death that are precipitated by the autophagic machinery have been described, both in developmental scenarios and during adaptation to stress289. Various autophagy-related (Atg) genes were shown to be required for the physiological demise of cells from developing Drosophila melanogaster larvae290. The neuron-specific knockout of Atg7 limited tissue loss in a mouse model of severe neonatal hypoxia–ischaemia9. Similarly, pharmacological inhibition of autophagy with 3-methyladenine (3-MA) as well as the depletion of various components of the autophagic machinery, including beclin 1 (BECN1), protected human cancer cells from a Na+/K+-ATPase-dependent form of autophagic cell death known as ‘autosis’ (REF. 94). In line with this notion, cardiac glycosides (which are potent inhibitors of the Na+/K+-ATPase) mediated robust neuroprotective effects in a rat model of neonatal hypoxia–ischaemia94. Thus, autophagy may precipitate cell death in some circumstances. However, this possibility must be addressed experimentally with specific pharmacological and genetic tools.
Box 2. Mechanisms of autophagy.
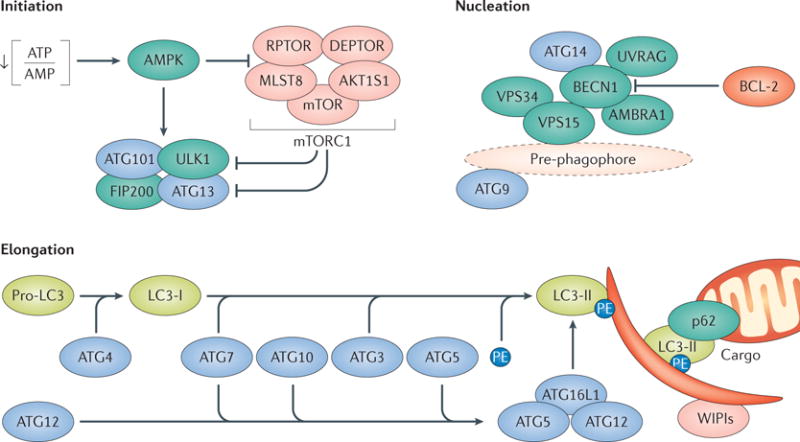
Canonical autophagy relies on two ubiquitin-like conjugation systems. One involves autophagy-related 7 (ATG7) and ATG10, and is responsible for the formation of a supramolecular protein complex containing ATG5, ATG12 and autophagy-related 16-like 1 (ATG16L1)1. The other ubiquitin-like conjugation system, which involves ATG3, ATG4 and ATG7, promotes the cleavage of members of the Atg8 protein family, including human LC3, and their conjugation to phosphatidylethanolamine (PE)1. Lipidated LC3 (LC3-II) and LC3-like molecules such as GABA type A receptor-associated protein (GABARAP) are recruited to forming autophagosomes, to operate as receptors for autophagic substrates or autophagic adaptors like p62 and have largely been exploited for monitoring autophagy in vitro and in vivo274. ATG9, another member of the ATG protein family, has a crucial function in autophagosome nucleation, which is initiated by a supramolecular complex that contains UNC-51-like autophagy-activating kinase 1 (ULK1), RB1-inducible coiled-coil 1 (RB1CC1; also known as FIP200), ATG13 and ATG101 (REF. 1). Of note, recent data indicate that the ATG conjugation systems are less important for autophagosome formation than previously thought but are crucial for the degradation of the inner autophagosomal membrane291 (see the figure).
Mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) exerts prominent autophagy-suppressing functions by catalysing the inactivating phosphorylation of ATG13 and ULK1 (REF. 276). Such an inhibition can be relieved upon the inactivation of mTORC1 by AMP-activated protein kinase (AMPK), which is sensitive to cAMP accumulation (a consequence of ATP consumption) and also catalyses the activating phosphorylation of ULK1 and beclin 1 (BECN1)292. ULK1 promotes autophagic responses by activating a multiprotein complex with phosphatidylinositol 3-kinase activity that contains BECN1, VPS34 and phosphoinositide 3-kinase regulatory subunit 4 (PIK3R4; also known as VPS15)1. The BECN1–VPS34 complex can interact with a variety of additional regulatory factors, including UV radiation resistance-associated (UVRAG) and autophagy and beclin 1 regulator 1 (AMBRA1), which stimulate the catalytic activity of VPS34, as well as BCL-2, apoptosis regulator (BCL-2), which mediates VPS34-inhibitory effects1,157. The expansion of autophagosomes in the course of canonical autophagic responses indeed relies on phosphatidylinositol-3-phosphate (PtdIns3P) production and PtdIns3P-binding proteins of the WD-repeat domain phosphoinositide-interacting (WIPI) family1 (see the figure). Finally, closed autophagosomes fuse with lysosomes to generate autolysosomes, followed by luminal acidification and consequent activation of lysosomal hydrolases1.
Several non-canonical instances of autophagy that proceed independently of specific components of the autophagic apparatus have also been described204,205,282,293. This suggests the existence of functional redundancy in the molecular mechanisms that underlie autophagic responses (at least in mammals). One of these pathways, that is, LC3-associated phagocytosis (LAP), involves the recruitment of parts of the autophagic machinery to single-membraned phagosomes that form in the context of danger signalling, followed by LC3 lipidation and delivery of phagosomes to lysosomes for degradation10,213. LAP proceeds independently of the ULK1 complex, AMBRA1 and ATG14 (which are required for canonical autophagy) but it relies on LC3, RUN and cysteine-rich domain-containing beclin 1 interacting protein (RUBCN) and NADPH oxidase 2, which are dispensable for canonical autophagy10,213. Finally, the molecular machineries for canonical autophagy and LAP share multiple components, including BECN1, VPS34, UVRAG, ATG3, ATG5, ATG7, ATG12 and ATG16L1 (REFS 10,213). Thus, the role of LAP in various processes might have been overlooked based on the pharmacological or genetic modulation of these shared factors. AKT1S1, AKT1 substrate 1; DEPTOR, DEP domain-containing mTOR-interacting protein; MLST8, mTOR-associated protein, LST8 homologue; RPTOR, regulatory-associated protein of mTOR complex 1.
The detailed description of the molecular machinery that underlies constitutive and inducible autophagic responses is beyond the scope of this Review (BOX 2). However, it should be noted that the biochemical reactions that enable the generation of autophagosomes, the recognition of autophagic substrates, their sequestration and the delivery of autophagic cargo to lysosomes for degradation involve at least 100 different proteins1. Thus, they provide multiple targets for the activation or inhibition of autophagy (FIG. 1). Although alterations in autophagy have been implicated in the aetiology of neurodegeneration, acute neuronal injury, ageing, cardiovascular conditions, hepatic and metabolic disorders, cancer, infectious diseases, inflammatory and autoimmune conditions, and other pathological conditions (as discussed below), no intervention aimed specifically at modulating autophagy is currently available for use in humans (TABLE 1). Indeed, although rapamycin (also known as sirolimus), chloroquine, hydroxychloroquine (HCQ) and other drugs that are approved for some indications stimulate or inhibit autophagy, they were not developed for this purpose. Thus, there is considerable, but still unrealized, potential for translating preclinical findings on autophagy modulation into therapeutic benefit for different patient populations13. Notably, the key role of autophagy in cell biology and its considerable therapeutic potential recently received one of the most important forms of recognition from the scientific community as the Japanese cell biologist Yoshinori Ohsumi was awarded the 2016 Nobel Prize in Physiology or Medicine for his discoveries on the mechanisms of autophagy.
Figure 1. Autophagic processes amenable to therapeutic modulation.
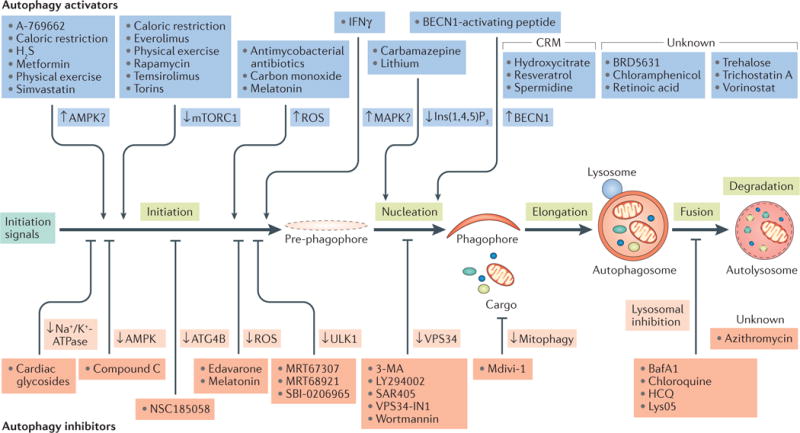
Several pharmacological and nutritional interventions are available to inhibit autophagy at the nucleation, elongation, fusion or degradation phase. In addition, several agents modulate autophagy through multipronged or hitherto uncharacterized molecular mechanisms. For additional details, please refer to TABLE 1. 3-MA, 3-methyladenine; AMPK, AMP-activated protein kinase; ATG4B, autophagy-related 4B cysteine peptidase; BafA1, bafilomycin A1; BECN1, beclin 1; CRM, caloric restriction mimetic; H2S, hydrogen sulfide; HCQ, hydroxychloroquine; IFNγ, interferon-γ; Ins(1,4,5)P3, inositol-1,4,5-trisphosphate; MAPK, mitogen-activated protein kinase; mTORC1, mechanistic target of rapamycin complex 1; ROS, reactive oxygen species; ULK1, UNC-51-like autophagy activating kinase 1.
Table 1.
Main modulators of autophagy available to date and their limitations
| Agent | Mode of action | Blood–brain barrier permeant | Status | Major limitations | Refs |
|---|---|---|---|---|---|
| Activators | |||||
| A-769662 | AMPK activation? | Unknown | In preclinical development | Unclear MOA (requires upstream AMPK-activating kinase) | 130 |
| Antimycobacterial antibiotics | Altered ROS production | Yes | Approved for treatment of mycobacterial infections | Potentially interferes with many ROS-sensitive processes | 178 |
| BECN1-derived peptide | BECN1 activator | Yes | In preclinical development | Limitations associated with chemical nature (shelf stability and potential immunogenicity) | 256 |
| BRD5631 | Unknown | Unknown | In preclinical development | Unclear MOA (independent of mTORC1) | 179 |
| Caloric restriction | Multiple | NA | NA | Potentially dangerous for subjects with weight problems (for example, patients with cachectic cancer); compliance issues | 10,54,118,128,13 1,135,148,150,16 4,180,188,231,237,240 |
| Carbamazepine | Reduction in Ins(1,4,5)P3 and inositol levels | Yes | Approved for treatment of seizures and bipolar disorders | Psychoactive agent, inhibits various neuronal functions | 134,137 |
| Carbon monoxide | Altered ROS production | Yes | Experimental agent | Potentially interferes with many ROS-sensitive processes | 182 |
| Chloramphenicol | Unknown | Yes | Approved for second-line treatment of bacterial infections | Unclear MOA and potentially mitochondriotoxic | 116 |
| Everolimus (also known as RAD-001) | mTORC1 inhibition | Yes | Approved for cancer therapy | Inhibits multiple mTORC1-dependent processes and has robust immunosuppressive effects | 219,220 |
| Hydrogen sulfide | AMPK activation? | Yes | Experimental agent | Unclear MOA and potentially toxic for the respiratory trait | 133 |
| Hydroxycitrate | CRM | Unknown | Clinically tested for treatment of diabetes, now discontinued | May cause weight loss upon chronic administration | 163 |
| IFNγ | MAPK activation? | No | In clinical trials, mainly for cancer immunotherapy | Unclear MOA (involves MAPK signalling) | 169 |
| Lithium | Reduction in Ins(1,4,5)P3 and inositol levels | Yes | Approved for treatment of bipolar disorders | Psychoactive agent, inhibits various neuronal functions | 42,50,55 |
| Melatonin* | Altered ROS production | Yes | In clinical trials for the treatment of a wide range of conditions | Potentially interferes with ROS-sensitive processes and has been associated with autophagy inhibition in some models | 97 |
| Metformin | AMPK activation? | Yes | Approved for treatment of type 2 diabetes | Mediates multiple AMPK-unrelated effects, including inhibition of respiratory complex I | 136 |
| Physical exercise | Multiple | NA | NA | Not appropriate for patients affected by cardiovascular or skeletal disorders but safe in most other cases | 7,119,129 |
| Rapamycin (also known as sirolimus) | mTORC1 inhibition | Yes | Approved for use in coronary stents (to prevent transplant rejection) and to treat a rare pulmonary disease | Inhibits multiple mTORC1-dependent processes, has robust immunosuppressive effects and may cause mTORC2 inhibition on chronic administration | 10,22,31,32,38,40, 41,43–47,50,51,56, 60,68–70,77,79,80, 90,95,96,99, 101–103,132,134, 146,180,183,184, 196,198,219,220, 223,225–228,234, 241,244,245, 255,258–260 |
| Resveratrol | CRM | Yes | Nutritional supplement that is available over the counter; in clinical trials for treatment of several disorders | Potentially causes nephrotoxicity at high concentrations | 33,132,147,229,235 |
| Retinoic acid | Unknown | Yes | Approved for cancer therapy (ATRA) | Reported to specifically inhibit CMA in some settings | 100 |
| Simvastatin | AMPK activation? | Yes | Approved for treatment of obesity | Unclear MOA (associated with AMPK activation) and potentially mitochondriotoxic | 99 |
| Spermidine | CRM | Yes | Nutritional supplement that is available over the counter | Degradation products include ROS and potentially cytotoxic aldehydes | 163,230 |
| Temsirolimus (also known as CCI-779) | mTORC1 inhibition | Yes | Approved for cancer therapy | Inhibits multiple mTORC1-dependent processes and has robust immunosuppressive effects | 51 |
| Torins | mTORC1 inhibition | Unknown | Experimental agents | Inhibit multiple mTORC1-dependent processes | 246 |
| Trehalose | Unknown | Yes | In clinical trials for treatment of bipolar disorders, dry eye syndrome and vascular ageing | Unclear MOA (independent of mTORC1) but safe | 39,53,65 |
| Trichostatin A | Unknown | No | Discontinued from clinical tests | Unclear MOA, potentially linked to transcriptional effects | 98 |
| Vorinostat | Unknown | Yes | Approved for cancer therapy | Unclear MOA, potentially linked to transcriptional effects | 115 |
| Inhibitors | |||||
| 3-MA | VPS34 inhibition | Yes | Experimental agent | Inhibits various class III PI3Ks | 10,36,63,76,78–80, 82,84–88,93,96,98, 99,105,243,255 |
| Azithromycin | Unknown | Yes | Approved for treatment of multiple bacterial infections | Unclear MOA, blocks autophagic flux | 187 |
| Bafilomycin A1 | Lysosomal inhibition | Yes | Experimental agent | Inhibitor of lysosomal functions | 36,58,79,86–88,105,180,255 |
| Cardiac glycosides | Na+/K+-ATPase inhibition | Yes | Extensively used in the past for treatment of cardiac disorders | Narrow therapeutic window but specific for autosis | 94 |
| Chloroquine | Lysosomal inhibition | Yes | Extensively used in the past as an antimalarial agent | Inhibitor of lysosomal functions | 100,104,134, 206,215,224 |
| Compound C (also known as dorsomorphin) | AMPK inhibition | Unknown | In preclinical development | Potentially interferes with AMPK-dependent processes | 82 |
| Edavarone | ROS scavenger | Unknown | Experimental agent | Potentially interferes with many ROS-sensitive processes | 89 |
| HCQ | Lysosomal inhibition | Yes | Extensively used in the past as an antimalarial agent | Inhibitor of lysosomal functions | 190,206,215 |
| LY294002 | VPS34 inhibition | Yes | In clinical trials for the treatment of refractory neuroblastoma | Exhibits improved selectivity compared with 3-MA and wortmannin but commonly considered nonspecific | 84 |
| Lys05 | Lysosomal inhibition | Yes | In preclinical development | Exhibits increased potency compared with HCQ | 294 |
| Mdivi-1 | Mitophagy inhibition | Yes | In preclinical development | Inhibitor of mitochondrial fragmentation | 250 |
| Melatonin* | Altered ROS production | Yes | In clinical trials for treatment of a wide panel of conditions | Potentially interferes with ROS-sensitive processes and has been associated with autophagy activation in some models | 73 |
| MRT67307 | ULK1 inhibition | Unknown | In preclinical development | Also inhibits ULK2, IKK and TBK1 | 299 |
| MRT68921 | ULK1 inhibition | Unknown | In preclinical development | Also inhibits ULK2 | 299 |
| NSC185058 | ATG4B inhibition | Unknown | In preclinical development | Exhibits improved selectivity for the autophagic pathway | 297 |
| SAR405 | VPS34 inhibition | Unknown | In preclinical development | Exhibits improved selectivity compared with 3-MA and wortmannin | 295 |
| SBI-0206965 | ULK1 inhibition | Unknown | In preclinical development | Exhibits improved selectivity for the autophagic pathway | 298 |
| VPS34-IN1 | VPS34 inhibition | Unknown | In preclinical development | Exhibits improved selectivity compared with 3-MA and wortmannin | 296 |
| Wortmannin | VPS34 inhibition | No | Experimental agent | Inhibits various class III PI3Ks | 99 |
3-MA, 3-methyladenine; AMPK, AMP-activated protein kinase; ATG4B, autophagy-related 4B cysteine peptidase; ATRA, all-trans-retinoic acid; BECN1, beclin 1; CMA, chaperone-mediated autophagy; CRM, caloric restriction mimetic; HCQ, hydroxychloroquine; IFNγ, interferon-γ; IKK, inhibitor of nuclear factor-κB kinase; Ins(1,4,5)P3, inositol-1,4,5-trisphosphate; MAPK, mitogen-activated protein kinase; MOA, mode of action; mTORC, mechanistic target of rapamycin complex; NA, not applicable; PI3K, phosphoinositide 3-kinase; ROS, reactive oxygen species; TBK1, TANK-binding kinase 1; ULK, UNC-51-like autophagy activating kinase.
Conflicting data exist on the ability of melatonin to modulate autophagy. Adapted with permission from REF. 9, Macmillan Publishers Limited.
Here, we discuss recent progress on the therapeutic potential of pharmacological and nutritional modulators of autophagy, dissect the obstacles that have limited the development of these interventions thus far, and propose strategies by which such hurdles may be circumvented in the near future to obtain clinically relevant interventions for a variety of human disorders.
Autophagy as a therapeutic target
Whole-body knockout studies in mice have demonstrated that specific components of the autophagic machinery are required for embryonic development or are critical for animals to survive birth and reach adulthood14–16. Three main approaches have been pursued as alternatives to the use of whole-body knockout mice to study the role of autophagy in physiology and disease: the generation of animals with partial autophagic defects at the whole-body level (such as Becn1+/− mice); the engineering of tissue-specific or inducible knockout models; and the restoration of autophagic activity in key organs (such as the central nervous system (CNS)) in animals with whole-body autophagic defects17. Results obtained with these models have implicated alterations of autophagy or autophagy-associated processes in a wide range of clinically relevant disorders (as discussed below), which supports the possibility that pharmacological modulators (activators or inhibitors) of autophagy may be beneficial for large patient populations.
Neurodegeneration
The deletion of autophagy-related 5 (Atg5) or Atg7 in the mouse CNS during embryonic development results in the delivery of viable pups that survive birth, but develop progressive motor and behavioural deficits starting at 3 weeks of age18,19. The cortex and cerebellum of these animals exhibit swelling, markers of <m>regulated cell death</m> (RCD) and ubiquitin-containing inclusions18,19, which are pathological hallmarks of various neurodegenerative disorders, including Alzheimer disease (AD), Parkinson disease (PD), dementia with Lewy bodies (DLB), Huntington disease (HD), amyotrophic lateral sclerosis (ALS) and Lafora disease20. Autophagy robustly protects neurons from RCD by preventing the accumulation of cytotoxic protein aggregates and by preserving metabolic homeostasis21. In line with this idea, markers of impaired autophagy — such as activation of mechanistic target of rapamycin (mTOR), autophagosome accumulation and limited degradation of sequestosome 1 (SQSTM1; also known as p62) — have been detected in samples from patients with multiple forms of neurodegeneration22. Moreover, many of the genes that are mutated in familiar variants of PD — including parkin RBR E3 ubiquitin protein ligase (PARK2), Parkinsonism-associated deglycase (PARK7), PTEN-induced putative kinase 1 (PINK1) and leucine-rich repeat kinase 2 (LRRK2) — are involved in <m>mitophagy or aggrephagy</m>23,24. Furthermore, AD-associated variants of <m>presenilin 1</m> (PSEN1) block autophagy as a result of impaired lysosomal acidification25; mutations in SQSTM1, optineurin (OPTN, which encodes another <m>autophagic adaptor</m>) and TANK-binding kinase 1 (TBK1, which encodes a regulator of both p62 and OPTN) are common among individuals with familial and sporadic ALS26–28; and both laforin glucan phosphatase (EPM2A) and NHL repeat-containing E3 ubiquitin protein ligase 1 (NHLRC1), which are mutated in individuals with Lafora disease, also seem to promote autophagy29. Finally, mutations in WD repeat domain 45 (WDR45) — which encodes an ATG9 interactor of the WD repeat domain phosphoinositide-interacting (WIPI) family — have been shown to be involved in the pathogenesis of static encephalopathy of childhood with neurodegeneration in adulthood, which is a rare neurological disorder30. Interventions that promote autophagy or autophagy-associated processes have been shown to mediate beneficial effects in animal models of neurodegeneration.
Alzheimer disease
Administration of the mTOR inhibitor rapamycin — which potently activates autophagy — ameliorates cognitive deficits and alleviates the accumulation of β-amyloid in mice expressing mutant <m>myloid-β precursor protein</m> (APP)31 as well as in 3xTg-AD mice (which bear three distinct genetic alterations that are associated with AD in humans)32. Along similar lines, resveratrol, which is a natural polyphenol that promotes autophagy by operating as a <m>caloric restriction mimetic</m> (CRM), decreased β-amyloid overload in mice expressing a chimeric variant of mutant APP and AD-associated human PSEN1. This effect was ascribed to the AMP-activated protein kinase (AMPK)-dependent inhibition of mTOR complex 1 (mTORC1)33 (BOX 2). Of note, behavioural alterations that develop in mice engineered to express one or several APP mutations that are linked to AD in humans could also be ameliorated by the concomitant deletion of genes that encode endogenous inhibitors of lysosomal proteases, such as cystatin B (Cstb) or cystatin C (Cst3)34,35.
Parkinsonism
The intracerebral injection of a lentiviral vector encoding ATG7 or BECN1 decreases neuronal inclusions in synuclein-α (SNCA)-expressing mice (a model of PD and DLB), and this has been associated with reduced neurodegeneration22,36. Along similar lines, the intracerebral administration of an adenoviral vector encoding BECN1 or transcription factor EB (TFEB) — which is a master transcriptional regulator of autophagy and lysosomal functions — to rats expressing SNCA in the brain limited the accumulation of SNCA aggregates within dopaminergic neurons and prevented behavioural impairment37. Comparable results have been obtained by activating autophagy with systemic or intracerebral administration of rapamycin, <m>trehalose</m> or <m>valproate</m> in several mouse models of PD and DLB, including SNCA-expressing mice22, mice expressing mutant Park2 (REF. 38), Park2−/− mice expressing human microtubule-associated protein tau (MAPT)39 as well as mice or rats that develop parkinsonism upon administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)40–42, 6-hydroxydopamine (6-OHDA)43,44 or lactacystin45. Rapamycin also alleviated neurotoxicity in Drosophila melanogaster with mutations in parkin (park; the fly orthologue of human PARK2) or Pink1 (REF. 46) as well as in flies treated with the dopaminergic toxin paraquat47. Of note, defects in late-stage autophagy leading to PD-like and DLB-like disorders in mice have also been ascribed to loss of type I interferon signalling48. Accordingly, intracerebral interferon β1 (Ifnb1) delivery by a lentiviral vector boosted autophagy and limited the loss of dopaminergic neurons in rats expressing human SNCA in the brain48. These findings identify cytokine signalling as a potential target for the treatment of PD and DLB through the induction of autophagy. Interestingly, defects in CMA may also be implicated in the development of PD and DLB, as was demonstrated recently in rats49. Whether boosting CMA ameliorates the manifestations of disease in animal models of parkinsonism remains to be elucidated.
Huntington disease
In flies and mice expressing HD-associated variants of human huntingtin, rapamycin (and other mTOR inhibitors, including temsirolimus and everolimus) alone or combined with <m>lithium</m> exerts considerable neuroprotective effects, as has been determined histologically and in behavioural tests50,51. However, the adenovirus-mediated intracerebral delivery of either of two mTORC1 activators — namely, RASD family member 2 (RASD2; also known as RHES) and RAS homologue enriched in brain (RHEB) — in a constitutively active form alleviated metabolic, histological and behavioural manifestations of the disease in HD-prone mice52. mTORC1 activation by RHEB was paradoxically associated with an increase in multiple biomarkers of autophagy52. Although this was not formally investigated, RHEB and/or RHES may be involved in the regulation of autophagy or autophagy-associated processes that are independent or downstream of mTORC1 (TABLE 2).
Table 2.
Pathologies potentially responding to autophagy-modulatory interventions
| Disease | Approach | Observations | Refs |
|---|---|---|---|
| Acute brain intoxication | Debated | • Role of autophagy in neuronal responses to toxicants has not been studied in appropriate models | 9,58–60 |
| Ageing | Activation | • Most lifespan-extending interventions activate autophagy, and their beneficial effects depend on an intact autophagic machinery in multiple model organisms | 47,229–240 |
| Atherosclerosis | Activation | • Efficient autophagic responses in macrophages from arterial lesions limit disease progression • Coronary angioplasty generally involves rapamycin-eluting stents |
111 |
| Autoimmune disorders | Debated | • LAP defects cause an SLE-like disease in mice • Autophagy activators mediate therapeutic effects in multiple autoimmune diseases • Autophagy supports cell populations or processes that underlie disease |
166,167,210–228 |
| Bacterial infections | Activation | • Elimination of intracellular bacteria relies on xenophagic responses, which couple danger signalling to autophagosome formation | 10,168–190 |
| Cancer | Debated | • Autophagy inhibition may exacerbate effect of cytotoxic therapies • Autophagy underlies the activation of therapeutically relevant immune responses |
8,15,16,152–167 |
| Cardiac stroke | Debated | • Both autophagy activators and the Becn1+/− genotype are associated with cardioprotection in models of stroke | 110,115–117,22,125 |
| Cardiomyopathy | Debated | • Autophagic defects in cardiomyocytes provoke cardiomyopathies • Autophagy activators mediate beneficial effects in multiple disease models |
107–109,112–114, 118–121,124,126 |
| Cerebral stroke | Debated | • There is conflicting literature on the impact of autophagy on disease outcome in models of 4VO, pMCAO and tMCAO | 9,73–89 |
| COPD | Inhibition | • Mitophagy may participate in the pathogenesis of cigarette smoke-associated COPD | 248–251 |
| Diabetes | Debated | • Autophagy activation improves insulin sensitivity in type 2 diabetes models • Insulin release by β-cells is negatively regulated by autophagy during fasting |
130,141,145–150 |
| Eukaryotic parasites | Debated | • LAP activation may control fungal and eukaryotic parasites | 10,200–208 |
| Hepatic disorders | Activation | • Autophagy activators limit hepatic fat accumulation and damage in models of steatosis and α1-antitrypsin deficiency-associated liver disease | 127,129,133–137, 140,142,143 |
| Lysosomal storage disorders | Debated | • The inhibition of autophagy upstream of autophagosome formation may ameliorate lysosomal overload. Similar effects may be achieved with autophagy activators that overcome lysosomal blockage | 262–265 |
| Metabolic syndrome | Activation | • Exercise links intact autophagic responses in multiple organs with leanness and improved systemic metabolism | 7,128–133,135,140, 141,144,145,151 |
| Neonatal asphyxia | Inhibition | • Pharmacological and genetic inhibition of autosis or autophagy mediates neuroprotective effects | 9,72,90–94 |
| Neurodegenerative disorders | Activation | • Autophagy is inhibited in the ageing brain, which contributes to the accumulation of pathogenic and pathognomonic neuronal inclusions | 18–20, 22–57 |
| Neurotrauma | Debated | • Autophagy activation limits functional impairment and promotes recovery in models of SCI and SAH, whereas data are conflicting in models of TBI | 95–105 |
| Pulmonary disorders other than COPD | Activation | • Autophagic defects have been documented in patients with certain pulmonary disorders. Preclinical data support the benefit of autophagy activation in mouse models of cystic fibrosis, IPF and PAH | 241–247 |
| Renal conditions | Activation | • Deletion of Atg5 or Atg7 in podocytes induces glomerulosclerosis and aggravates renal degeneration that is caused by PAN administration or kidney overload | 252–255 |
| Seizures | Activation | • Preclinical data and epidemiological studies link excitotoxicity to defective autophagic responses | 61–73 |
| Skeletal conditions | Debated | • Autophagy is crucial for bone growth • Administration of rapamycin has been associated with both beneficial and detrimental effects in mouse models of bone fracture |
256–260 |
| Viral infections | Debated | • Preclinical data link autophagic responses to improved cellular and organismal control of viruses • Some viruses exploit autophagic responses in the host |
191–199 |
| Vision disorders | Activation | • Efficient autophagic responses and LAP are required for the survival and function of retinal photoreceptors | 266–269,271–273 |
4VO, 4-vessel occlusion; Atg, autophagy-related; Becn1, beclin 1; COPD, chronic obstructive pulmonary disease; IPF, idiopathic pulmonary fibrosis; LAP, LC3-associated phagocytosis; PAH, pulmonary arterial hypertension; PAN, puromycin aminonucleoside; pMCAO, permanent middle carotid artery occlusion; SAH, subarachnoid haemorrhage; SCI, spinal cord injury; SLE, systemic lupus erythematosus; TBI, traumatic brain injury; tMCAO, transient middle carotid artery occlusion.
Amyotrophic lateral sclerosis
Activation of autophagy with caloric restriction, trehalose, valproate or lithium delays disease onset, reduces neurological deficits and prolongs survival in mice expressing an ALS-associated variant of superoxide dismutase 1 (SOD1), namely SOD1-G93A53–55. However, rapamycin had detrimental effects on motor neuron degeneration and overall survival in mice expressing SOD1-G93A56. Moreover, crossing these mice with Becn1+/− mice resulted in a paradoxical increase in lifespan that was accompanied by p62 accumulation and an unexpected interaction between SOD1-G93A and BECN1 (REF. 57). The reasons that underlie these contrasting observations remain to be elucidated. At least theoretically, when lysosomal degradation is congested, limiting the initiation of autophagy may be more beneficial than attempting to boost autophagic responses with interventions that fail to overcome the block in autophagosomal processing (see below). It remains to be determined whether this applies to ALS (TABLE 2).
Acute neuronal injury
Acute challenges to the CNS, including drug and ethanol intoxication, seizures, adult stroke, neonatal asphyxia and traumatic brain injury (TBI), have been associated with biomarkers of ongoing or blocked autophagic responses (see below), which suggests that autophagy modulators might provide therapeutic benefits9. However, a clear aetiological link between failing autophagic responses and acute neuronal injury has not yet been confirmed, not only owing to methodological issues but also owing to an inherent heterogeneity in models (see below).
Acute intoxication
Autophagic responses have been documented in the cortex of mice that were exposed to cocaine58, but other neuronal populations, including the nucleus accumbens, respond to systemic cocaine with mTORC1 activation (and hence autophagy inhibition)59. Depletion of mTOR or regulatory-associated protein of mTOR complex 1 (RPTOR; a key component of mTORC1) from specific neuronal populations reduced <m>locomotor sensitization</m> (one of the symptoms of cocaine administration) in mice59, as did rapamycin administration in rats60. By contrast, inhibition of autophagy with small interfering RNAs (siRNAs) targeting Atg5 or Becn1 limited the capacity of cocaine to trigger RCD in mouse primary cortical neurons58. Similar results were obtained in vitro with chemical inhibitors of autophagy, including 3-methyladenine (3-MA) and wortmannin, which target phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3; also known as VPS34), as well as the lysosomal inhibitor bafilomycin A1 (BafA1)58. Thus, some aspects of cocaine neurotoxicity (for example, cocaine-driven RCD) may be aggravated, whereas others (for example, locomotor sensitization) may be alleviated, by efficient autophagic responses. As the pathogenesis of acute cocaine intoxication involves multiple cell-extrinsic components (for example, neuroinflammation), great caution should be taken before extrapolating in vitro data to human disease. Likewise, numerous studies suggest that autophagic responses are beneficial in the course of acute brain intoxication with methamphetamine, ethanol or olanzapine (an antipsychotic drug)9. However, studies to elucidate the relationship between autophagy or autophagy-associated processes and acute neuronal responses to toxicants in vivo are urgently awaited to clarify the potential of modulating autophagy for therapeutic purposes in this setting (TABLE 2).
Seizures
One of the mechanisms by which seizures mediate neurotoxic effects involves the sustained depolarization of postsynaptic terminals and consequent influx of Ca2+ ions from the synaptic cleft, which is a process that is commonly known as excitotoxicity61. Cytosolic Ca2+ is a potent activator of autophagy, and several excitotoxic neurotransmitters — including glutamate, N-methyl-D-aspartate (NMDA) and kainic acid — induce the expression of autophagy biomarkers in the rodent brain, possibly as a compensatory response to damage62,63. The depletion of BECN1 by siRNAs as well as pharmacological inhibitors of autophagy aggravated (whereas rapamycin and trehalose limited) the demise of primary rodent neurons and multiple neuronal cell lines of human origin that were exposed to glutamate, NMDA or kainic acid in vitro64,65, which suggests that autophagy supports neuronal viability in the course of excitotoxic challenges. Further corroborating this idea, mutations in three different genes encoding endogenous inhibitors of mTORC1 — namely, tuberous sclerosis 1 (TSC1), TSC2 and phosphatase and tensin homologue (PTEN) — are associated with an increased predisposition to epilepsy in humans66. Accordingly, mice lacking Tsc1, Tsc2, Pten or Atg7 in neuronal or glial cell populations experienced spontaneous epileptic episodes that were associated with premature death, which is an outcome that could be limited by systemic rapamycin administration67–70. However, mice receiving an siRNA targeting Atg7 in the hippo campus were more resistant to the neurotoxic effects of kainic acid than mice receiving a control siRNA71. Moreover, the stable depletion of Becn1 or Atg7 expression as well as the administration of 3-MA, LY294002 (another VPS34-targeting agent) or lysosomal inhibitors limited the death of cultured rodent neurons from the striatum, cerebellum and cortex that were challenged with NMDA or kainic acid72,73. Further experiments are required to understand whether autophagy modulators may indeed benefit patients with seizures and to what extent this may involve autophagy-associated processes (TABLE 2).
Adult stroke
Tissues that are served by an occluded artery experience nutrient deprivation and hypoxia, which are two potent activators of autophagy, and this is potentially followed by excessive reactive oxygen species (ROS) production (at reperfusion), which has also been associated with autophagy activation9. Consistent with this, biomarkers of autophagy activation have been detected in the brain of adult rodents experiencing stroke9. The intracerebral delivery of a lentiviral vector encoding a Tsc1-targeting short hairpin RNA (shRNA) aggravated neuronal loss in rats experiencing permanent middle carotid artery occlusion (pMCAO), which supports the notion that autophagy mediates neuroprotective effects in the course of a stroke74. Similarly, inhibition of autophagy through deletion of Prkaa2 (which encodes AMP-activated, α2 catalytic subunit) or sirtuin 1 (Sirt1), or by downregulation of ATG7 or TSC1, aggravated the cytotoxicity of oxygen glucose deprivation (OGD) in primary mouse75 or rat74,76 cortical neurons. In addition, autophagy activation (with rapamycin) or inhibition (with 3-MA, BafA1 or AMPK inhibitors) improved or worsened, respectively, disease outcome in rodents that were experiencing transient middle carotid artery occlusion (tMCAO) or pMCAO76,77, as well as neuroprotection in multiple models of <m>ischaemic preconditioning</m> in vivo78–82. Stable downregulation of Becn1 in the rat brain by stereotactic injection of a lentiviral vector limited the neurotoxic effects of tMCAO83. Moreover, both 3-MA and BafA1 limited infarct size in various rodent models of tMCAO84,85, pMCAO86,87 or four-vessel occlusion88, and multiple molecules that protect neurons in adult rodents experiencing stroke (for example, melatonin and edavarone) also seem to inhibit autophagy73,89, possibly through their antioxidant effects. Thus, although autophagy seemingly participates in the pathophysiology of stroke in adults, whether therapeutic interventions should aim to activate or inhibit autophagy remains to be clarified. Although not yet demonstrated, the time at which autophagy modulators are administered to a patient experiencing stroke (for example, before versus after reperfusion) may considerably influence their therapeutic effects (TABLE 2).
Neonatal asphyxia
Although it has been suggested that autophagy would be activated as an adaptive response to neonatal asphyxia90, accumulating evidence indicates that autophagy contributes mechanistically to neuronal loss in the course of neonatal asphyxia9. Newborn rats that received a Becn1-specific shRNA intracerebrally displayed twice the amount of intact striatal tissue 24 hours after ischaemia compared with their control counterparts (that received a non-targeting shRNA)72. In addition, the neuron-specific knockout of Atg7 in mice provided a high degree of long-term protection from neonatal asphyxia to hippocampal cornu ammonis regions91. Similarly, the hippocampus, thalamus, cortex and striatum of newborn mice lacking Atg7 in neurons were considerably protected from RCD following hypoxia–ischaemia, which corresponds to a global decrease in tissue loss of approximately 40%92. Finally, newborn rats treated with intracerebroventricular 3-MA up to 4 hours after ischaemia exhibited considerably smaller lesions than did control animals93, as did newborn rats receiving the <m>cardiac glycoside</m> neriifolin, which is an inhibitor of autosis (BOX 1), immediately after carotid artery occlusion94. In summary, inhibiting autophagy is expected to limit the severity of hypoxic–ischaemic encephalopathy in newborn rodents (TABLE 2).
Neurotrauma
Consistent with the hypothesis that autophagy is beneficial, but usually impaired, in the context of neurotrauma, rapamycin and other molecules with autophagy-inducing potential (for example, melatonin and retinoic acid) limit CNS damage, support regeneration and improve the restoration of neuromuscular functions in rodents experiencing spinal cord injury (SCI)95 or subarachnoid haemorrhage (SAH)96–98. Chemical inhibitors of autophagy, including 3-MA, wortmannin and the antimalarial drug chloroquine (which blocks lysosomal degradation), aggravated neurological damage imposed by SCI or SAH96,99, and abolished the neuroprotective effects of autophagy inducers98,100. Activating autophagy with rapamycin or melatonin also had beneficial effects in rodents experiencing several forms of TBI, including closed-head injury101, weight-drop damage102 and hemicerebellectomy103, even when the drug was administered after traumatic injury. In this latter model, neuroprotection by rapamycin was lost in Becn1+/− mice, which were a priori more sensitive to hemicerebellectomy-induced damage103. Of note, melatonin has been suggested to inhibit autophagy in other settings73. Moreover, post-injury chloroquine administration limited neuronal damage and improved neurological recovery in rats that were subjected to controlled cortical impact104, as did 3-MA and BafA1 administered as a prophylactic intervention in a mouse weight-drop model105. Thus, autophagy activators may be beneficial for patients experiencing SCI or SAH, even if administered in a therapeutic (as opposed to prophylactic) setting. It remains to be clarified whether the same holds true for other forms of neurotrauma, including TBI (TABLE 2).
Cardiovascular conditions
The efficient disposal of various autophagic substrates seems to be crucial for the maintenance of cardiovascular homeostasis, in both physiological and pathological conditions106. For example, ageing Lamp2−/− mice, which display defective CMA, develop a disorder that is characterized by cardioskeletal myopathy similar to that associated with Danon disease, and patients with this condition exhibit LAMP2 defects107,108. Similarly, the temporally controlled deletion of Atg5 from mouse cardiomyocytes promoted cardiac hypertrophy109, and the hearts of Park2−/− mice, which are mitophagy-incompetent, failed to benefit from cardiac ischaemic preconditioning ex vivo110. In addition, the conditional knockout of Atg5 in macrophages of Ldlr−/− mice fed a high-fat diet (HFD) — which are prone to accumulate atherosclerotic plaques — aggravated arterial lesions (by facilitating apoptotic and necrotic RCD, and by worsening <m>efferocytosis</m>111). Furthermore, the cardiomyocyte-specific deletion of Dnase2a (which encodes a lysosomal nuclease that is involved in the autophagic degradation of mitochondrial DNA) sensitized mice to pressure overload-driven hypertrophy that was accompanied by a robust inflammatory response112. Finally, the Becn1+/− genotype aggravated the disease progression in mice overexpressing mutant crystallin αB (CRYAB) in the heart (which is a model of desmin-related cardiomyopathy)113. Conversely, the cardiomyocyte-specific overexpression of ATG7 reduced biochemical and functional biomarkers of the disease in this model, as did physical exercise (which is an established activator of autophagy)114. Consistent with this, autophagy activators — including (but not limited to) caloric restriction, physical exercise, rapamycin, <m>spermidine</m> and a peptide derived from the BECN1 region that interacts with the HIV-1 protein Nef (whose mechanism of action has not been characterized yet) — had beneficial effects in models of myocardial ischaemia reperfusion115–117, pressure overload-driven hypertrophy or heart failure118–120, and cardiac senescence121. Notably, the devices that are currently used in the clinic for <m>coronary angioplasty</m> generally deliver rapamycin122, although the underlying rationale resides in the antiproliferative activity of this drug123. Moreover, endurance exercise may preserve cardiovascular and metabolic fitness by activating autophagy in multiple organs7. Interestingly, the Becn1+/− genotype was associated with some extent of cardioprotection in the setting of myocardial ischaemia reperfusion, pressure overload-driven hypertrophy and diabetic cardiomyopathy, which the authors ascribed to reduced maladaptive autophagy124–126. Data obtained in mice subjected to the inducible cardiomyocyte-specific deletion of Atg5, however, argue against these findings109, which highlights the potential bias introduced by the organismal adaptation to life-long gene knockout (see below).
In summary, activation of autophagy seems to be a main goal not only for the preservation of cardiovascular fitness but also for the management of multiple cardiovascular disorders (TABLE 2).
Hepatic and metabolic disorders
Several reports suggest that the cell-intrinsic and cell-extrinsic functions of autophagy in the liver and pancreas are crucial not only for the maintenance of organ homeo stasis but also for the regulation of neuroendocrine circuitries that control systemic metabolism127. Thus, acutely activating autophagy with caloric restriction, physical exercise, rapamycin, CRMs (that is, <m>metformin</m> or resveratrol), AMPK-targeting agents or hydrogen sulfide limits non-alcoholic steatohepatitis (NASH) and/or improves various metabolic parameters (including body weight, circulating glucose or triglyceride levels, and insulin sensitivity) in 24-month-old rats128, rodents that had been fed a HFD129–133, mice acutely or chronically exposed to ethanol134 as well as in <m>db/db mice</m> or <m>ob/ob mice</m> (which are two genetic models of hyperphagia and thus metabolic syndrome)130,135,136. Moreover, activation of autophagy with carbamazepine considerably reduced hepatic fibrosis in a model of α1<m>-antitrypsin deficiency</m>-associated liver disease137. However, rapamycin administered according to specific schedules also causes insulin resistance as it inhibits mTORC2 (REFS 138,139). This suggests that adequate administration schedules or alternative autophagy activators are preferable for the treatment of metabolic disorders that are linked to type 2 diabetes.
An Atg7-targeting shRNA delivered by an adenoviral vector, an Atg7-targeting siRNA that was injected intravenously, the whole-body knockout of forkhead box O3 (Foxo3, which encodes a transcription factor that supports autophagic responses), or the conditional co-deletion of Prkab1 and Prkab2 (which encode the AMPK subunit β1 and the AMPK subunit β2, respectively) in adipocytes aggravated hepatic damage and metabolic dysregulation in HFD-fed mice140, ob/ob mice141 and mouse models of ethanol intoxication142,143, which further corroborates the beneficial role of autophagic responses in hepatic and metabolic conditions. The hepatocyte-specific deletion of serine/threonine kinase 11 (Stk11, which encodes the main AMPK activator and is also known as Lkb1) compromised glucose homeostasis in mice and rendered them insensitive to metformin144. Furthermore, mice expressing a non-phosphorylatable mutant BCL-2, apoptosis regulator (BCL-2) — a model in which inducible autophagy is selectively blocked — were unable to obtain metabolic benefits from physical exercise7, whereas ob/ob mice specifically lacking Atg7 in the myeloid compartment were more susceptible to obesity-induced diabetes than their autophagy-proficient littermates145. These findings are intriguing, as they suggest that systemic metabolic homeostasis may (at least partially) rely on efficient autophagic responses in compartments other than the liver, pancreas and adipose tissue. Interestingly, acute caloric restriction, rapamycin and resveratrol also attenuated multiple manifestations of the diabetic syndrome induced by <m>streptozotocin</m> — including nephropathy — in mice and rats146–148. In addition, Atg7 was required for pancreatic β-cells to develop normally and to ensure physiological glucose control149. However, autophagic responses within pancreatic β-cells seem to contribute to the physiological inhibition of insulin release by fasting150. It remains to be elucidated whether autophagy inhibitors may support insulin secretion and systemic glucose control in patients with type 1 diabetes (which is characterized by a primary defect in insulin secretion). Finally, the deletion of Atg5 or Atg12 in UCP1+ adipocytes prevented the beige-to-white fat transition in mice, hence limiting HFD-driven obesity and glucose intolerance151. This inhibition of beige-to-white fat transition suggests that — although autophagy seems to globally support metabolic fitness — the development, survival or functions of some detrimental cells (such as white adipocytes) may also rely on efficient autophagic responses.
Taken together, these observations suggest that systemic autophagy activators may mediate therapeutic activity in patients with a variety of metabolic disorders, although the inhibition of autophagic responses in specific cell compartments might amplify such a beneficial effect (TABLE 2).
Cancer
Autophagic responses contribute to preservation of homeostasis and adaptation to stress in both normal and malignant cells152. Thus, autophagy has been shown to inhibit <m>malignant transformation</m> in a variety of models and by a multitude of mechanisms152. Accordingly, Becn1+/− mice were more prone to developing spontaneous malignancies as they aged than were their wild-type littermates15,16. Mice with a systemic mosaic deletion of Atg5 or the liver-specific knockout of Atg7 spontaneously accumulated benign liver adenomas153, and the local deletion of Atg5 or Atg7 markedly accelerated the onset of KRAS-G12D-driven or BRAF-V600E-driven pancreatic or pulmonary adenomas in mice154–156. In addition, multiple <m>oncosuppressor genes</m> — including tumour protein 53 (TP53) and PTEN — support autophagic responses, whereas several <m>proto-oncogenes</m> — such as BCL2, AKT serine/threonine kinase 1 (AKT1) and epidermal growth factor receptor (EGFR) — inhibit them157–160. However, autophagy also promotes <m>tumour progression</m> and resistance to treatment, at least at the cancer cell-intrinsic level, through a multitude of mechanisms152. Thus, the conversion of early KRAS-G12D-driven or BRAF-V600E-driven pancreatic or pulmonary adenomas into advanced, invasive adenocarcinomas was attenuated in the context of local Atg5 or Atg7 deletion154–156. Along similar lines, the stable depletion of ATG5, ATG7 or BECN1 with shRNA-coding constructs limited the growth of multiple human cancer cells that were subcutaneously or orthotopically xenografted into athymic immunodeficient mice161. Furthermore, human cancer cells that were implanted in immunodeficient hosts were more sensitive to chemotherapy or radiotherapy in the presence of pharmacological inhibitors of autophagy, including 3-MA, wortmannin, chloroquine and HCQ (which is another antimalarial agent)161. Taken together, these findings suggested that autophagy inhibitors would be useful agents for the clinical management of cancer, either as a standalone intervention or as a means to sensitize malignant cells to therapy162.
Recent clinical trials testing chloroquine or HCQ (alone or combined with chemotherapy or radiotherapy) in patients with lymphoma, melanoma, glioblastoma and other solid neoplasms established the safety of this approach161, which fostered the initiation of additional phase II and phase III clinical studies in Europe and the United States (ClinicalTrials.gov). However, none of these clinical trials has formally confirmed the hypothesis that inhibiting autophagy in cancer cells provides therapeutic benefits to patients with cancer161. Moreover, preclinical findings indicate that intact autophagic responses in malignant cells are required for appropriate danger signalling (and hence for the initiation of therapeutically relevant anticancer immune responses) in tumours established in syngeneic immunocompetent hosts and treated with immunogenic chemotherapy or radiotherapy8,163. Consistent with this, caloric restriction and various CRMs enhanced (rather than limited) the therapeutic efficacy of mitoxantrone, oxaliplatin and radiotherapy in the same tumour models161,163,164. Furthermore, biomarkers of autophagic responses in malignant cells were associated with intensified <m>immunosurveillance</m> and improved disease outcome in cohorts of patients with breast carcinoma who were treated with anthracyclines165. Finally, local as well as systemic inhibition of autophagy could have short-term and long-term detrimental effects in patients with cancer for two reasons. First, autophagy is important for the survival, proliferation and effector functions of immune cell subtypes that are involved in tumour control166,167. Second, at least hypothetically, inhibiting autophagy may increase the risk of healthy tissues to undergo malignant transformation or experience other toxic effects. Thus, the activation of autophagy with safe nutritional measures stands out as a promising approach to improve the clinical efficacy of anticancer agents that operate (at least in part) by promoting tumour-specific immune responses (TABLE 2).
Infectious diseases
Autophagy is required for the cellular and organismal control of multiple pathogens, including bacterial, viral and eukaryotic parasites (such as fungi).
Bacterial infections
Autophagic responses that are specific for cytoplasmic bacteria (referred to as xenophagy) are a crucial component of the innate immune system and have been shown to restrict the growth of bacterial pathogens, including Salmonella enterica subsp. enterica serovar Typhimurium168, Mycobacterium tuberculosis169,170, Listeria monocytogenes171 and group A Streptococcus spp.172. Accordingly, many bacteria have evolved strategies to inhibit autophagic responses in the host. These include (but are not limited to) the production of cAMP-elevating toxins (Vibrio cholera and Bacillus anthracis)173, the normalization of otherwise dwindling amino acid levels at the surface of bacterium-containing vacuoles (S. Typhimurium)174, the deconjugation of microtubule-associated protein 1 light chain 3β (MAP1LC3B; also known as LC3) (Legionella pneumophila)175, the inactivation of GTPases that are required for normal vesicular trafficking (Shigella flexneri and pathogenic Escherichia coli)176 and the escape from autophagic recognition (L. monocytogenes)177. Activation of autophagy through starvation or treatment with rapamycin, a BECN1-derived peptide or other agents restricted bacterial growth and improved cellular or organismal resistance to infection caused by M. tuberculosis (in D. melanogaster and mouse macrophages)169,178, S. enterica (in human cancer cell lines)179, or pathogenic E. coli (in mice and human cancer cell lines)180,181. Moreover, autophagic responses to carbon monoxide protected mice from sepsis induced by cecal ligation and puncture182. Of note, LAP (BOX 2) is also involved in the control of intracellular bacteria, including Burkholderia pseudomallei and L. monocytogenes, by monocytes10,183,184.
Interestingly, the ability of mice to control M. tuberculosis infection was partially abrogated by the Park2−/− genotype (which imposes a selective defect on mitophagy)185 as well as by the conditional deletion of Atg5 (but not other autophagy-related genes) from monocytes and neutrophils, possibly as a consequence of exacerbated lung inflammation186. Thus, caution should be taken when extrapolating data obtained from single-gene knockouts to entire cellular processes (see below). Nevertheless, activators of autophagy, mitophagy and LAP may constitute promising tools for the clinical management of some bacterial infections, whereas molecules with unsuspected autophagy-inhibitory functions may be detrimental (such as azithromycin for patients with cystic fibrosis)187. That said, other pathogenic bacteria, including Anaplasma phagocytophilum (which causes a tick-borne disease with relatively mild symptoms) and Coxiella burnetii (which causes a severe endocarditis), stimulate autophagy in the host to support their own metabolic needs188,189. In this situation, autophagy inhibitors (including HCQ) have been shown to provide some clinical benefits190 (TABLE 2).
Viral infections
Several viruses are efficiently controlled by autophagic responses in host cells191. The autophagic degradation of viruses, which is commonly referred to as ‘virophagy’, relies on core components of the autophagic machinery, including BECN1 (REF. 192), as well as on proteins that also participate in mitophagy, such as SMAD-specific E3 ubiquitin protein ligase 1 (SMURF1) and Fanconi anaemia complementation group C (FANCC)191,193. Consistent with an important role of virophagy in the control of viral infections, some viruses evolved virulence factors that actively inhibit autophagy, such as the BECN1 inhibitor ICP34.5 from herpes simplex virus 1 (HSV-1)194. Interestingly, Becn+/− and Fancc−/− mice were more susceptible to Sindbis virus infection than their wild-type littermates, and D. melanogaster that had received an Atg18-targeting shRNA exhibited increased sensitivity to vesicular stomatitis virus infection192,193,195. Futher findings corroborate the potential therapeutic value of autophagy activators for the control of viral infections: rapamycin and a BECN1-derived peptide efficiently limited HIV-1 replication in a human lymphoblastoid cell line196, in primary human monocyte-derived macrophages197 and in peripheral blood lymphocytes (PBLs) from healthy donors196,198 as well as in severe combined immunodeficient (SCID) mice reconstituted with human PBLs (which is a model for the study of HIV-1 infection in vivo)198. The same BECN1-derived peptide restrained viral replication and improved overall survival in mouse models of West Nile virus and chikungunya virus197. Specific viruses, such as coxsackievirus B3, however, may have a replication advantage linked to autophagy activation, as was demonstrated in mice with a conditional deletion of Atg5 in pancreatic acinar cells199 (TABLE 2).
Eukaryotic parasites
Mice lacking Atg5 or Atg7 specifically in the myeloid cell compartment are more susceptible to an intravenous challenge with Candida albicans (but not with Cryptococcus neoformans) than their wild-type counterparts, possibly as a result of defective neutrophil recruitment200,201. Similarly, mice bearing Atg7-deficient or Becn1-deficient macrophages, as well as Rubcn−/− mice, were less resistant to intra-nasal Aspergillus fumigatus infection than their control litter mates10. Moreover, some fungal pathogens, such as A. fumigatus, seem to have evolved strategies to block LAP, including the production of a cell wall component that specifically inhibits the activation of NADPH oxidases at the phagosome202. Atg5, Atg7 and autophagy-related 16-like 1 (Atg16l1) in macrophages were also required for mice to control infection caused by the eukaryotic parasite Toxoplasma gondii, although such a process was not accompanied by autophagic degradation203–205. These data point to an essential contribution of autophagy and, to a greater degree, LAP in the control of fungal infections and eukaryotic parasites. Interestingly, two commonly used inhibitors of autophagy (chloroquine and HCQ) have long been used for the treatment of malaria, which is caused by the eukaryote Plasmodium falciparum206. Chloroquine and HCQ, however, inhibit both the autophagic and non-autophagic functions of lysosomes, and have been proposed to exhibit antimalarial properties through inhibition of haem polymerase, which is toxic to the parasite207. Taken together, these observations suggest that autophagy activators may be beneficial for the treatment of fungi and other eukaryotic parasites (TABLE 2). However, it should be borne in mind that autophagy is conserved across the eukaryotic kingdom, which implies that pathogenic eukaryotes may also take advantage of enhanced autophagic responses208. In this setting, specific LAP activators may represent superior therapeutic tools, and efforts should be dedicated to the development of such molecules.
Of note, autophagy engages in extensive crosstalk with Toll-like receptor signalling and plays a crucial part in antigen cross-presentation209, which together underlie optimal innate and adaptive immunity against bacteria, viruses and eukaryotic parasites. Thus, autophagy activators may also support the eradication of invading pathogens by promoting innate and adaptive immune responses.
Inflammatory and autoimmune conditions
Autophagy mediates prominent anti-inflammatory effects, which reflects its ability to degrade inflammasomes as well as to limit the availability of endogenous inflammasome activators, including ROS and mitochondrial DNA210,211. However, intact autophagic responses may also support the survival, proliferation and activity of multiple cells that contribute to the aetiology of autoimmune disorders166,167. Thus, although autophagy represents a promising target for the treatment of multiple inflammatory and autoimmune disorders, including (but not limited to) systemic lupus erythematosus (SLE), Crohn’s disease, rheumatoid arthritis and multiple sclerosis, the implementation of autophagy-modulatory interventions for the management of these pathologies may be less straightforward than was initially envisioned.
Systemic lupus erythematosus
Genetic polymorphisms in ATG5 and possibly ATG7 have been associated with SLE in multiple studies212, which suggests that autophagic defects may contribute to the pathogenesis of disease. Accordingly, mice lacking Atg5, Atg7 or Becn1 in LysM+ cells — which comprise macrophages, monocytes, some neutrophils and some dendritic cells — as well as Rubcn−/− and Cybb−/− mice (which are characterized by a specific defect in LAP) spontaneously developed an SLE-like autoimmune disorder linked to deficient phagocytic uptake of dying cells and consequent production of pro-inflammatory cytokines213. Importantly, a similar phenotype is not observed in Ulk1−/− mice and mice lacking RB1-inducible coiled-coil 1 (Rb1cc1) in LysM+ cells (which are two genetic alterations that provoke an autophagic defect but spare LAP)213. Thus, similarly to wild-type mice, autophagy-deficient but LAP-competent mice do not develop an SLE-like disease and exhibit a normal phagocytic response to dying cells coupled to the production of anti-inflammatory mediators such as interleukin-10 (IL-10)213. These findings suggest that pharmacological LAP activators may be beneficial for patients with SLE. It remains to be elucidated whether general autophagy activators may compensate for LAP defects. Of note, autophagy has been involved in the production of <m>neutrophil extracellular traps</m> (NETs), which contributes to SLE pathogenesis214. Thus, the inhibition of autophagy in specific cell populations may also provide therapeutic benefits to patients with SLE. Indeed, chloroquine and HCQ have been used for the treatment of SLE with some success215. However, this clinical activity probably reflects the immunosuppressive effects of these molecules rather than their capacity to block autophagy. Indeed, neither the cell populations nor the cellular processes that are targeted by HCQ have been formally investigated in the context of SLE (TABLE 2).
Crohn’s disease
Various non-synonymous polymorphisms that are associated with an increased susceptibility to Crohn’s disease (data on prevalence are not available for all polymorphisms and vary considerably in different studies) negatively affect the activity of proteins that participate in autophagic or xenophagic responses, such as ATG16L1, UNC-51-like autophagy-activating kinase 1 (ULK1) and nucleotide-binding oligomerization domain-containing 2 (NOD2)216,217. This reflects the multifactorial aetiology of this disorder, which involves both a microbial or epithelial and an autoimmune component218, and suggests that autophagy activators may be beneficial for patients with Crohn’s disease. Indeed, everolimus ameliorated disease severity in Il10−/− mice, which spontaneously develop a Crohn’s disease-like syndrome219. Moreover, everolimus has been used with some success in the management of specific SLE cases220. However, it remains to be determined whether the activity of everolimus in this context truly stems from the activation of autophagy or from its autophagy-independent immunosuppressive effects (TABLE 2).
Rheumatoid arthritis
There are contrasting observations about autophagy in CD4+ T cells in the context of rheumatoid arthritis. Naive CD4+ T lymphocytes from patients with rheumatoid arthritis have been reported to exhibit autophagic defects that are secondary to a metabolic reprogramming that affects glycolysis221. By contrast, CD4+ T cells from patients with rheumatoid arthritis reportedly display increased autophagic responses, hence resembling CD4+ T lymphocytes that are activated in vitro222. Whether this apparent discrepancy reflects the CD4+ T cell subset under consideration (naive versus total or activated CD4+ T cells) or a methodological bias remains unclear. Irrespectively, HCQ limits the resistance to apoptosis displayed by CD4+ T cells from patients with rheumatoid arthritis ex vivo and reduces disease severity in a mouse model of collagen-induced arthritis222. In addition, the specific deletion of Atg5 from mouse CD4+ T cells limits their proliferation and activation ex vivo222. Taken together, these observations suggest that the inhibition of autophagy in specific immune cell populations, notably CD4+ T cells, may limit disease progression in patients with rheumatoid arthritis (TABLE 2).
Multiple sclerosis
Rapamycin treatment limits skin and lung fibrosis in mouse models of multiple sclerosis induced by bleomycin administration or a hetero zygous mutation in fibrillin 1 (Fbn1), which is accompanied by reduced production of fibrogenic cytokines and decreased levels of hypergammaglobulinaemia and anti-DNA topoisomerase 1 antibodies (two circulating markers of disease)223. However, these findings were not mechanistically linked to the activation of autophagy, which implies that they could reflect the established antiproliferative and immunosuppressive activity of rapamycin. Conversely, the specific deletion of Atg7 from dendritic cells in mice ameliorates experimental autoimmune encephalomyelitis — which is a model of CD4+ T cell-dependent multiple sclerosis — by reducing the priming of autoimmune responses224. Consistent with this, chloroquine delays disease progression if administered before disease onset and reduces disease severity if administered after onset224. These findings suggest that the specific inhibition of autophagy in dendritic cells may constitute a desirable therapeutic objective for the management of some forms of multiple sclerosis (TABLE 2).
Other conditions
Autophagy may also be involved in the pathogenesis of other inflammatory and autoimmune conditions. Rapamycin administration provided beneficial effects in mouse models of autoimmune myositis225, autoimmune encephalomyelitis226 and autoimmune uveitis (only when used at high doses)227, as well as in patients with autoimmune lymphoproliferative syndrome228. Conversely, low-dose rapamycin administration aggravated experimental autoimmune uveitis in mice227. It remains to be clarified to what extent these findings relate to autophagy activation versus immuno-suppression by rapamycin (TABLE 2).
Ageing
The healthy lifespan of multiple model organisms, including Saccharomyces cerevisiae (yeast)229,230, Caenorhabditis elegans (worm)231–233, D. melanogaster (fly)47,230, Mus musculus (mouse)234–236 and Macaca mulatta (monkey)237, can be experimentally extended by autophagy-activating measures, and this lifespan extension almost invariably depends on an intact autophagic machinery238. Indeed, autophagy mediates robust homeostatic functions at both the cell-intrinsic and cell-extrinsic levels, which directly counteract several processes that are associated with ageing. These processes include (but may not be limited to): the accumulation of macromolecular damage that drives cellular senescence and RCD (which are particularly detrimental in the context of an aged stem cell compartment); systemic metabolic deregulation; chronic, mild inflammation (so-called inflammaging); and accrued oncogenesis239. It is common knowledge that a balanced dietary regimen coupled with regular exercise preserves organismal fitness and can postpone several, if not all, of the manifestations of ageing. Accumulating evidence suggests that various beneficial effects of a healthy lifestyle depend (at least in part) on the activation of autophagy240. It will therefore be interesting to see whether CRMs or other pharmacological activators of autophagy can be used to extend lifespan (TABLE 2).
Other disorders
Autophagy modulators may also be beneficial for patients that are affected by pathologies such as pulmonary, renal and skeletal diseases.
Pulmonary disorders
On the one hand, autophagic defects have been documented in the lungs of patients with idiopathic pulmonary fibrosis (IPF)241, pulmonary arterial hypertension (PAH)242 and cystic fibrosis243. Accordingly, rapamycin treatment partially protects mice from IPF caused by bleomycin241 or radiation244, as well as from hypoxia-induced PAH245. Similarly, the intranasal delivery of a Becn1-coding lentivirus successfully restores autophagy and limits inflammation in CftrF508Δ mice (a model of cystic fibrosis)243, as does the genetic inactivation of Rptor or Mtor in mouse models of hyperoxia-induced or lipopolysaccharide-induced acute lung injury246,247. Moreover, Map1lc3b−/− mice exhibit aggravated PAH upon chronic hypoxia242. Thus, pharmacological activators of autophagy may be beneficial in patients with some pulmonary disorders (TABLE 2).
On the other hand, autophagy contributes to (rather than counteracts) the aetiology of chronic obstructive pulmonary disease (COPD). Indeed, Map1lc3b−/− and Becn1+/− mice develop limited emphysema upon chronic cigarette smoke exposure compared with their wild-type littermates248,249. Similar observations were made in Pink1−/− mice as well as in mice receiving the mitophagy inhibitor Mdivi-1 (REF. 250). Interestingly, the transcription factor early growth response 1 (EGR1) seems to be mechanistically involved in detrimental autophagic responses that underlie COPD251. However, no direct links between EGR1 signalling and mitophagy have been established yet. Similarly, whether pharmacological inhibitors of EGR1 signalling may be beneficial for individuals with COPD remains to be determined (TABLE 2).
Renal conditions
Autophagy may mediate important homeostatic functions in the kidney. The podocyte-specific deletion of Atg5 or Atg7 in mice results in spontaneous glomerulosclerosis that is preceded by mitochondrial alterations (which are also documented in patients with idiopathic <m>focal segmental glomerulosclerosis</m>), excessive ROS production and podocyte loss252,253. Similarly, mice bearing Atg5−/− podocytes exhibit increased glomerular degeneration that is caused by puromycin aminonucleoside (PAN) or doxorubicin compared with their wild-type littermates252. In addition, the podocyte-specific knockout of Atg7 considerably sensitizes mice to kidney overload imposed by unilateral nephrectomy254. Comparable results were obtained in PAN-treated rats receiving chemical autophagy inhibitors (3-MA or chloroquine), whereas autophagy activation with rapamycin limited glomerular degeneration in this model255. Thus, autophagy activators may not only benefit diabetic patients who are at risk of nephropathy (see above) but also individuals with other renal conditions (TABLE 2).
Skeletal disorders
Autophagy has recently been identified as an important mediator of bone growth in response to fibroblast growth factor (FGF) signalling256. Thus, the bone growth defects that are imposed by the Fgf18+/− or Fgfr4−/− phenotype can be rescued by intraperitoneal administration of a BECN1-derived peptide256. However, the deletion of Mtor or Rptor from PRX1+ cells (which are found in the limb, cranial and interlimb mesenchymal tissues) significantly impairs skeletal growth in mouse embryos, which results in a severe phenotype that is associated with death shortly after birth257. Similarly, rapamycin administration has been found to mediate beneficial258 and detrimental259,260 effects in rodent models of bone fracture. It remains to be determined whether these apparent discrepancies reflect the pleiotropic effects of mTORC1 (which, among various functions, controls cell proliferation). Additional investigation is required to elucidate the potential benefits that are associated with the use of autophagy modulators in skeletal disorders (TABLE 2).
Lysosomal storage disorders
Lysosomal storage disorders (LSDs) are a heterogeneous group of rare inheritable conditions that originate from defects in lysosomal activity, which result in the cytotoxic accumulation of specific lysosomal substrates261. The conditional deletion of Atg5 or Atg7 from myocytes alleviates lysosomal overload and enables enzyme replacement therapy in a mouse model of Pompe disease (which is characterized by glycogen accumulation)262,263. Similarly, rapamycin is toxic for inducible pluripotent stem cells (iPSCs) from patients with Gaucher disease (which is characterized by <m>glucosylceramide</m> accumulation)264. Conversely, rapamycin, carbamazepine, trehalose and other autophagy inducers have cytoprotective effects on iPSCs from patients with Niemann–Pick type C disease (which is characterized by the accumulation of cholesterol and glycolipids)265. These observations suggest that the inhibition of autophagy upstream of autophagosome formation may be beneficial for patients with certain LSDs. By contrast, autophagy activators may have detrimental effects, unless they successfully overcome the lysosomal blockage that characterizes these disorders.
Vision disorders
Autophagy and related processes sustain healthy vision by a number of mechanisms. Retinal photoreceptor cells, especially cones, exhibit high mitochondrial turnover by mitophagy and rely on AMPK-dependent autophagic responses to cope with glucose deprivation266. Consistent with this, the cone-restricted deletion of Atg5 results in a functional decline that is accompanied by the accumulation of damaged mitochondria, an increased sensitivity to light toxicity and glucose deprivation266. Moreover, the inhibition of autophagy reportedly contributes to photoreceptor cell loss in models of <m>retinitis pigmentosa</m>267. ATG5-dependent and BECN1-dependent LAP has been involved in a process known as the ‘visual cycle’, in which shedding photoreceptor outer segments are engulfed by the retinal pigment epithelium (RPE), and all-trans-retinoic acid is converted to 11-cis-retinal for photoreceptor regeneration. Thus, mice lacking Atg5 (but not Ulk1) in the RPE display defective 11-cis-retinal generation and reduced vision, which can be transiently corrected by the administration of all-trans-retinol268. Mice with a macrophage-specific deletion of Atg5 as well as mice carrying the Atg16l1T300A mutation exhibit increased susceptibility to lipopolysaccharide-induced uveitis that is associated with increased inflammasome activation269. In humans, the ATG16L1T300A mutation is associated with Crohn’s disease (see above), which is also linked to an increased predisposition for uveitis270. Finally, mutations in OPTN as well as duplications of TBK1 are associated with normal-tension glaucoma271,272. One of these mutants (namely, OPTN-E50K) displays an increased interaction with TBK1, and mice that are engineered to express the OPTN-E50K variant develop a disease that phenocopies the human disorder273. At this point, it is unclear whether these mutations promote normal-tension glaucoma as a consequence of changes in autophagy or other TBK1-related processes. However, activators of autophagy or LAP may counteract the pathogenesis of multiple vision disorders.
Challenges in developing autophagy modulators
In spite of great potential, no interventions that are specifically aimed at modulating autophagy are currently available for use in humans. Indeed, although rapamycin, chloroquine, HCQ and several drugs that are licensed for the use in humans activate or inhibit autophagy, they were not developed for this purpose. What are the obstacles that, until now, have prevented the development of autophagy modulators for clinical use? How can we circumvent them and finally harness the entire therapeutic potential of autophagy activators and inhibitors?
Specificity
Besides being used with limited rigour (that is, at non-standardized concentrations or time points), multiple chemical agents that are currently available to activate or inhibit autophagy have limited specificity for the autophagic process, for either of two reasons. First, some of these molecules have an intrinsically low pharmacological specificity for their target. This is the case for 3-MA and wortmannin, both of which are non-selective phosphoinositide 3-kinase (PI3K) inhibitors and hence block the catalytic activity of several PI3Ks beyond VPS34 (REF. 274). Along similar lines, although acute rapamycin administration results in the relatively specific inhibition of mTORC1 through FK506-binding protein 1A (FKBP1A), chronic exposure to rapamycin promotes mTORC2 disassembly139. Second, several components of the autophagic machinery operate at the interface of multiple cellular processes, that is, they mediate autophagy-independent functions203,275. Thus, the inhibition of mTORC1 using rapamycin not only activates autophagy but also inhibits translation, cellular growth and proliferation276. Similarly, the blockage of lysosomal functions using chloroquine, HCQ or BafA1 inhibits not only the disposal of autophagosomes but also the degradation of endosomes and impairs vesicular trafficking277. In line with this notion, rapamycin and multiple <m>rapalogues</m> mediate robust immunosuppressive effects, as they block T cell proliferation278, whereas both chloroquine and HCQ exert immunomodulatory as well as antineoplastic effects that are independent of autophagy279–281. Several approaches are being explored to circumvent these specificity issues, including the development of novel autophagy modulators (BOX 3).
Box 3. Emerging autophagy modulators with increased specificity.
Considerable efforts are being dedicated to the development of agents with increased pharmacological specificity (TABLE 1), including new lysosomal inhibitors (such as Lys05)294 as well as new mechanistic target of rapamycin complex 1 (mTORC1)-targeting and VPS34-targeting agents (including SAR405 and VPS34-IN1)295,296 (see also Navitor Pharmaceuticals). In parallel, increasing attention has been dedicated to components of the autophagic machinery that seem to have limited roles in other processes, such as autophagy-related 4B cysteine peptidase (ATG4B; which can be targeted by NSC185058)297 and UNC-51-like autophagy-activating kinase 1 (ULK1; which can be targeted by SBI-0206965, MRT67307 and MRT68921)298,299, as well as to specific autophagic pathways, including mitophagy and LC3-associated phagocytocis (LAP)193,213. Lys05, SAR405, NSC185058, and SBI-0206965 have been shown to mediate anticancer effects in vitro and in vivo294,295,297. Lys05 had single-agent antineoplastic activity in xenograft models of human metastatic melanoma (1205Lu cells) and colorectal carcinoma (HT29 cells)294, as did NSC185058 in xenograft models of human osteosarcoma (Saos-2 cells)297. SAR405 and SBI-0206965 synergized with mTORC1 inhibitors in arresting the proliferation of human lung (H1299 and A549) and kidney (ACHN and 786-O) cells in vitro295,298. Conversely, the potential therapeutic activity of VPS34-IN1 (which has been characterized biochemically in human osteosarcoma U2OS cells), MRT67307 and MRT68921 (both of which have been studied for their biochemical properties in mouse embryonic fibroblasts) has not yet been tested in relevant disease models296,299. Thus, the actual therapeutic potential of these approaches remains largely unexplored.
An additional specificity-related issue derives from the complex architecture of all tissues, which contain several different cell types that are engaged in extensive homologous and heterologous interactions. Most autophagy modulators available at present are also poorly specific because they do not preferentially target one cell type. Thus, evaluating the actual impact of autophagy activators or inhibitors in disease scenarios in which autophagic responses in different tissue compartments may have opposite effects is challenging. Addressing this complexity by studying the effects of highly targeted autophagy modulators in disease models with cell-specific autophagic defects (see below), is key to the development of clinically viable strategies to activate or inhibit autophagy. Of note, this issue may predominantly concern the development of autophagy inhibitors. Indeed, there is not necessarily a downside to activating autophagy at the whole-body level for the treatment of diseases in which autophagy activation may be useful.
Biomarkers and assays
Multiple biomarkers that are routinely used to monitor autophagy in vitro and in vivo are intrinsically unsuitable to monitor autophagic flux, that is, the actual degradation of autophagosomes and their content within lysosomes274. Several of these indicators, such as the levels of lipidated LC3 (measured by immunoblotting) or the amount of GFP–LC3+ puncta (measured by fluorescence microscopy or flow cytometry upon cell permeabilization), statically monitor the size of the autophagosomal compartment, which expands not only in the course of productive autophagic responses (increased on-rate) but also when the formation or activity of autolysosomes is blocked (decreased off-rate). Such a technical concern de facto invalidates the conclusions of multiple studies in which autophagy was monitored only with static autophagosomal biomarkers9.
Besides assessing the actual degradation of autophagic substrates (which may be cumbersome and cannot be carried out in all experimental scenarios), several strategies have been developed to circumvent this issue274. One common approach relies on comparing LC3 lipidation (or the accumulation of GFP–LC3+ puncta) in the presence or the absence of lysosomal inhibitors, such as BafA1 (REF. 282). This approach can differentiate between situations of increased on-rate and decreased off-rate and can be conveniently applied to human peripheral blood mononuclear cells ex vivo274. As an alternative, tandem fluorescence-tagged versions of LC3 have been successfully used to identify cells in which autophagic flux is operational in vitro and in vivo274,283. In paraffin-fixed formalin-embedded patient samples, encouraging results have been obtained by simultaneously quantifying LC3+ puncta and p62 abundance165. We are convinced that accurately measuring autophagic flux is key to the development of clinically viable modulators of autophagy.
An additional problem relates to the lack of reliable methods for discriminating between distinct forms of autophagy (for example, general autophagy versus mitophagy), especially in vivo and in patient material274. This issue is particularly relevant, as it prevents the establishment of proof of principle (that is, the mechanistic correlation between an autophagy-modulating intervention and actual autophagy modulation) and proof of concept (that is, the mechanistic correlation between an autophagy-modulating intervention and disease outcome) for most pathologies. Moreover, despite considerable efforts towards standardization274, many assays to measure autophagy and autophagy-related processes are still implemented with consistent variability across the scientific community. Thus, new highly standardized approaches that allow for monitoring specific forms of autophagy and autophagy-related processes in vivo and in patient material are urgently awaited.
Genetic models
Many of the genetic models that have been used so far for studying the impact of autophagy on the pathogenesis of disease and developing autophagy-targeting agents suffer from limitations that are often overlooked. One of the initial obstacles was the embryonic or perinatal lethality imposed by the whole-body knockout of several components of the autophagic machinery14–16. As mentioned above, this issue has been partially circumvented by the generation of mice with partial autophagic defects at the whole-body level, such as Becn1+/− or Atg4b−/− mice15,16,284. These animals have been highly instrumental for studying the role of autophagy in multiple disorders. However, they preserve some proficiency in <m>canonical autophagy</m> and mount normal non-canonical autophagic responses in several tissues, including the liver282. This implies that observing normal disease progression or the response to treatment in Becn1+/− or Atg4b−/− mice does not formally exclude a role for autophagy.
As an alternative approach, multiple models of tissue-specific autophagic incompetence have been generated. Most of these models rely on the deletion of ‘floxed’ Atg5, Atg7 or Becn1 in selected cell types, which is based on the expression of Cre recombinase under the control of a promoter of choice285. In this scenario, gene knockout occurs upon the physiological activation of the Cre-controlling promoter, which introduces considerable variability. Thus, whereas some promoters — such as the collagen, type II, alpha 1 (Col2a1) promoter286 — impose an autophagic defect early during differentiation or embryonic development, others — such as the cyclin-dependent kinase inhibitor 2A (Cdkn2a) promoter287 — abolish autophagic proficiency in terminally differentiated cells (in this case, senescent) cells. One of the major issues in this setting is the occurrence of compensatory processes, that is, a general reorganization of cellular functions that partially (if not totally) compensate for the lack of a specific protein. This is generally favoured when gene knockout occurs early during cellular or organismal lifespan as well as when the degree of functional redundancy is high (as is the case for the autophagic machinery)1.
Two main strategies have been devised to minimize this potential source of bias: the tissue-specific administration of viral vectors coding for Cre recombinase154 or the use of Cre variants that can be activated pharmacologically (for example, by the systemic administration of tamoxifen)109. One potential problem with these models relates to the efficiency of Cre activation. Indeed, viral infection may not affect all target cells, or tamoxifen concentrations may be suboptimal in some tissue areas, resulting in partial autophagic defects. However, both these approaches generally display high efficiency and result in widespread gene knockout within a specific tissue.
All types of conditional knockout models (including those that rely on viral Cre delivery) also suffer from some degree of nonspecificity, which may impose autophagic defects in tissues that are unintentionally affected. For instance, genes that are under the control of the Col2a1 promoter are expressed not only by chondrogenic tissues but also — transiently — in non-chondrogenic tissues, including the notochord, eye, heart, epidermis and discrete areas of the brain286. Although this issue is generally marginal, it may have affected — at least to some degree — data interpretation in specific cases. Finally, it should be kept in mind that most, if not all, components of the autophagic machinery mediate known or hitherto undiscovered autophagy-unrelated functions203. This implies that great caution should be taken before attributing the phenotype that results from whole-body or conditional knockout of a single component of the autophagic machinery to autophagy as a process.
Developing new models that circumvent these persisting obstacles may be less straightforward than desirable. Nonetheless, careful consideration of the limitations that are associated with each model may enable the correct interpretation of data and hence support drug discovery and development in this field.
Future development of autophagy modulators
There seems to be considerable confusion as to how autophagy modulators should be developed and used for the treatment of several pathologies (BOX 4), which has limited the development of novel therapeutic agents. Here, we summarize some principles that must be carefully considered in the future development and use of autophagy-targeting interventions in the context of three specific scenarios.
Box 4. Improving the pharmacological audit trail for autophagy modulators.
The so-called pharmacological audit trail (PhAT) is a conceptual guide to the preclinical and clinical development of novel therapeutic agents that sequentially assesses the risk of failure at key steps of the entire process as it guides decision-making300. It is clear that therapeutic interventions aimed at modulating autophagy or autophagy-related processes cannot be developed according to a generic PhAT (see the figure, left). On the one hand, it will be imperative to identify with precision: the pathologies that are mechanistically determined or aggravated by alterations in autophagy (patient selection); the cell population or populations in which such alterations actually underlie disease, as opposed to bystander cell populations that may exhibit secondary autophagic defects (cell type identification); and how autophagy is specifically altered in such cells (for example, hyperactivation, inhibition before autophagosome formation or inhibition after autophagosome formation) (autophagic pathway characterization). This will enable the development of a therapeutic strategy that is aimed at activating or inhibiting autophagy in specific cell types (including diseased and bystander cells) with one (or more) targeted drug candidate or candidates. On the other hand, besides carrying out conventional pharmacokinetic and pharmacodynamic studies, it will be important to determine whether and how the autophagic flux changes in response to the drug candidates in both target and bystander cells, followed by a step of hypothesis validation with intermediate end points of clinical efficacy. In case of disease progression, compensatory mechanisms (related to autophagy) that are possibly operating in diseased or bystander cells will have to be assessed, which may lead to a complete re-evaluation of the initial therapeutic strategy (see the figure, right).
Autophagy activation underlies disease
For cases in which the activation of autophagy underlies disease, pharmacological inhibition in specific cell populations constitutes an obvious therapeutic objective. However, the functional outcome and actual therapeutic value of such an intervention may vary considerably depending on the stage of the autophagic cascade that is targeted. For instance, whereas inhibiting autophagy by targeting upstream modulators — such as AMPK (with compound C), ULK1 (with SBI-0206965) or Na+/K+-ATPase (with cardiac glycosides) — seems to be a potentially safe intervention, using lysosomal inhibitors may increase (rather than decrease) the detrimental effects of autophagy activation by causing the accumulation of non-functional autophagosomes and autolysosomes, and hence a general blockage in vesicular trafficking (FIG. 2a).
Figure 2. Principles of autophagy modulation.
a | When autophagy mechanistically contributes to the aetiology of the disease, inhibiting autophagy initiation in diseased cells is expected to restore normal autophagic degradation and mediate therapeutic effects, whereas blocking lysosomal degradation may favour a detrimental accumulation of non-functional autophagosomes and autolysosomes. b | In the presence of initiation defects, stimulating autophagy upstream of autophagosome formation (in diseased cells) is expected to normalize autophagic flux (at least in part) and hence mediate beneficial effects, whereas boosting lysosomal degradation may exert limited (if any) therapeutic activity. c | In the presence of degradation defects, activating autophagy upstream of autophagosome formation (in diseased cells) may aggravate disease severity by exacerbating (thick arrows) the accumulation of non-functional autophagosomes and autolysosomes. Conversely, accelerating lysosomal degradation or inhibiting initiation (in diseased cells) may exert beneficial effects, at least to some degree (dashed arrow). d | Boosting autophagic flux in cells that survived a pathological insult or in non‐diseased cells may favour functional recovery and/or mediate beneficial effects linked to improved inflammatory tissue homeostasis. Beneficial and detrimental interventions are indicated by green and red arrows, respectively.
Autophagy inhibition underlies disease
Autophagic defects can contribute to disease by two general, non-mutually exclusive mechanisms: the accumulation of potentially dangerous autophagic substrates or non-functional autophagosomes and autolysosomes, and the lack of potentially beneficial autophagic products or functions. In this situation, the activation of autophagy with nutritional or pharmacological interventions may ameliorate disease outcome. However, the exact nature of the autophagic defect determines which approach should be undertaken to obtain beneficial (rather than detrimental) effects. Autophagy can be blocked upstream of autophagosome formation, which results in a limited number of autophagosomes and reduced autophagic activity. In this case, nutritional interventions (that are mainly detected by the AMPK–mTORC1 signalling node) or molecules that promote the formation of phosphatidylinositol-3-phosphate (PtdIns3P) by the BECN1–VPS34 complex are expected to mediate positive effects, as they boost autophagy initiation (FIG. 2b). Conversely, interventions that are aimed at the acceleration of lysosomal degradation, such as the inhibition of CST3 or CSTB, may not mediate therapeutic effects. Autophagy can also be blocked downstream of autophagosome formation, for instance, as a consequence of a lysosomal defect. In this case, further boosting the generation of autophagosomes is not only expected to have little therapeutic activity but may also aggravate the homeostatic perturbations imposed by the lysosomal blockage. On the contrary, the acceleration of lysosomal degradation may relieve the blockage in the disposal of autophagosomes and autolysosomes, and mediate beneficial effects. Of note, the inhibition of autophagy upstream of autophagosome formation (by targeting AMPK or ULK1, for instance) may also mediate therapeutic activity in this setting, which reflects a relative normalization of the autophagosomal and autolysosomal compartments (FIG. 2c). Thus, clinically useful autophagy activators should be designed based on careful characterization of the autophagic defect that accompanies each specific disease. At least theoretically, the combination of upstream autophagy activators with molecules that accelerate lysosomal degradation might overcome multiple forms of autophagic blockade. However, it has not yet been formally demonstrated whether this approach mediates superior benefits in models of disease that are caused by autophagic defects.
Autophagy activation compensates for disease
In some cases, autophagy alterations have little (if any) relationship to primary disease aetiology, but autophagy activation may still support compensatory mechanisms, including the degeneration of cells and tissues. Although many preclinical models that have been used thus far do not allow discrimination between these two aspects of pathogenesis (aetiology versus recovery; see below), interventions that boost autophagic activity in cells that are not directly affected by disease may ameliorate the long-term outcome in multiple disorders, even when administered after the primary pathological insult. This is particularly true for conditions with multifactorial aetiology, such as cerebral stroke or neurotrauma. In this scenario, efficient autophagic responses in glial cells that survive stroke considerably limit RCD-driven inflammation and the consequent loss of additional neurons (which is known to participate in the pathogenesis of disease), hence mitigating long-term functional impairment9. Thus, the activation of autophagy in cells that survive the primary pathological insult or in non-diseased cells (which exhibit normal autophagic capacity) may constitute a promising therapeutic intervention for multiple disorders with complex aetiologies (FIG. 2d). It is possible that patients with some of these pathologies may benefit from a sequential approach in which autophagy is inhibited first (or selectively in diseased cells) and activated subsequently (or selectively in non-diseased cells). Future studies are required to elucidate this possibility in specific pathologies.
Conclusions and perspectives
As discussed throughout this Review, the therapeutic potential of interventions that activate or inhibit autophagy is enormous. Nonetheless, multiple obstacles of pharmacological, technical or experimental nature have hampered the straightforward implementation of autophagy modulators in the clinic. We are confident that many of these hurdles can be circumvented upon the development of more selective autophagy modulators, more precise biomarkers of the autophagic flux and more physiological models of autophagy deficiency in vivo. The very nature of autophagy imposes obstacles that must also be overcome for the development of clinically viable autophagy activators and inhibitors. Additional work is required to understand how to inhibit or activate autophagy independently of cellular processes including proliferation and RCD, and hence harness the full therapeutic potential of autophagy modulators.
Acknowledgments
Owing to space limitations, the authors apologize to the authors of several high-quality articles in the field for not being able to discuss and cite their work. L.G. is supported by the Department of Radiation Oncology of Weill Cornell Medical College (intramural funds) and Sotio a.c. J.M.B.-S.P. and G.K. are supported by the French Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR), Projets blancs; ANR under the frame of the programme E-Rare-2, the European Research Area (ERA)-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Institut Universitaire de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LeDucq Foundation; the LabEx Immuno-Oncology; the Site de Recherche Intégrée sur le Cancer (SIRIC) Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI). B.L. is supported by the National Institutes of Health, Cancer Prevention Research Institute of Texas, and the LeDucq Foundation. D.R.G. is supported by the National Institutes of Health and the American Lebanese Syrian Associated Charities (ALSAC).
Glossary
- Autophagic cell death
A variant of regulated cell death (RCD) that is precipitated by the autophagic machinery and hence can be retarded with pharmacological or genetic inhibitors of autophagy
- Regulated cell death (RCD)
A form of cell death that relies on the activation of genetically encoded machinery and hence can be retarded or accelerated with specific pharmacological or genetic interventions
- Mitophagy
Autophagic response that is specific for depolarized or otherwise damaged mitochondria.
- Aggrephagy
Autophagic response that is specific for intracellular protein aggregates, which often are highly ubiquitylated
- Presenilin 1 (PSEN1)
Component of the γ-secretase complex that contributes to the accumulation of amyloid plaques in the brain of patients with Alzheimer disease (AD).
- Autophagic adaptor
A protein that directs autophagic substrates to forming autophagosomes through its capacity to bind ubiquitylated structures and lipidated Atg8 family members
- Amyloid-β precursor protein
(APP). A protein that — upon cleavage — accounts for the majority of amyloid plaques in the brain of patients with Alzheimer disease (AD).
- Caloric restriction mimetic (CRM)
A molecule that mimics the biochemical and cellular effects of fasting, including autophagy activation and cytosolic acetyl-CoA depletion, but does not provoke a sizeable weight loss
- Trehalose
A natural α-linked disaccharide that potently activates autophagy through poorly characterized mechanisms
- Valproate
A widely used antiepileptic drug that induces autophagy by affecting myo-inositol-1,4,5-trisphosphate levels
- Lithium
An antidepressant that promotes autophagic responses by altering myo-inositol-1,4,5-trisphosphate levels
- Locomotor sensitization
Long-lasting exacerbation of a psychostimulant-induced locomotor response, which is brought about by repeated intermittent administration of the same psychoactive agent
- Ischaemic preconditioning
An experimental technique for increasing the resistance of neurons or cardiomyocytes to prolonged, severe ischaemia based on the repeated administration of short, mild ischaemic episodes
- Cardiac glycoside
A natural compound that exerts positive inotropic effects and retards some forms of autophagic cell death as it inhibits the plasma membrane Na+/K+-ATPase
- Efferocytosis
The removal of dying or dead cells by professional phagocytes
- Spermidine
A natural polyamine that potently activates autophagy by operating as a caloric restriction mimetic (CRM)
- Coronary angioplasty
A minimally invasive surgical procedure for the treatment of narrowed or weakened arteries, which consists of the insertion of a small mesh tube (stent) through the femoral artery
- Metformin
An antidiabetic agent with pleiotropic effects, including the capacity to trigger autophagy by acting as a caloric restriction mimetic (CRM)
- db/db mice
Mice homozygous for the spontaneous db (for diabetes) mutation in leptin receptor (Lepr), which causes limited leptin signalling. These animals are commonly used as models for type 2 diabetes and metabolic syndrome
- ob/ob mice
Mice homozygous for the spontaneous ob (for obesity) mutation in leptin receptor (Lepr), which causes absent leptin signalling. These animals are commonly used as models for obesity and metabolic syndrome
- Carbamazepine
A widely used antiepileptic drug that induces autophagy by altering myo-inositol-1,4,5-triphosphate levels
- α1-antitrypsin deficiency
A genetic disease that causes the defective production of serpin family A member 1 (SERPINA1; also known as α1-antitrypsin) in the lungs and liver, which results in pulmonary disorders that are often associated with hepatic symptoms.
- Streptozotocin
A naturally occurring toxin that is commonly used to generate rodent models of type 1 diabetes owing to its pronounced selectivity for pancreatic β-cells
- Malignant transformation
The conversion of a healthy, normal cell into a neoplastic cell precursor. Malignant transformation is insufficient to drive tumorigenesis
- Oncosuppressor genes
Genes mutated or silenced in familial or sporadic forms of cancer. Many of these genes encode proteins that are involved in the maintenance of cellular homeostasis or in the activation of regulated cell death (RCD)
- Proto-oncogenes
Genes overexpressed or hyperactivated in familial or sporadic forms of cancer. Many of these genes encode positive regulators of cellular proliferation or proteins that inhibit regulated cell death (RCD)
- Tumour progression
A process through which a neoplastic cell precursor acquires additional genetic or epigenetic alterations that allow it to escape cell-intrinsic and cell-extrinsic control mechanisms and form aggressive tumours
- Immunosurveillance
A process in which the immune system recognizes and eliminates a potentially dangerous entity, including invading pathogens as well as pre-malignant and malignant cells
- Inflammasomes
Supramolecular platforms that support caspase 1 activation, hence allowing for the proteolytic maturation and secretion of pro-inflammatory interleukin-1β (IL-1β) and IL-18
- Neutrophil extracellular traps (NETs)
Chromatin-based and granule protein-containing fibres that are released by neutrophils to immobilize and kill invading microorganisms.
- Cellular senescence
A permanent proliferative arrest that is generally associated with specific morphological and biochemical features, including the secretion of multiple cytokines and other biologically active factors
- Chronic obstructive pulmonary disease (COPD)
A progressive lung disease that is characterized by long-term limited airflow, which is often caused or aggravated by tobacco smoke
- Focal segmental glomerulosclerosis
A leading cause of kidney failure in adults that is characterized by the degeneration of sections of the glomerulus with a focal (as opposed to diffuse) intrarenal distribution
- Glucosylceramide
A sphingolipid that accumulates in patients with Gaucher disease (mostly in the macrophages) as a result of mutations in glucosylceramidase beta (GBA)
- Retinitis pigmentosa
An inherited, degenerative eye disease that causes severe vision impairment owing to the progressive degeneration of the rod photoreceptor cells
- Rapalogues
Rapamycin derivatives with improved pharmacodynamic and pharmacokinetic properties
- Canonical autophagy
A term commonly used to refer to an autophagic response that is dependent on autophagy-related 5 (ATG5), ATG7, beclin 1 (BECN1) and phosphatidylinositol-3-phosphate (PtdIns3P) production
Footnotes
Competing interests statement
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Noda NN, Inagaki F. Mechanisms of autophagy. Annu Rev Biophys. 2015;44:101–122. doi: 10.1146/annurev-biophys-060414-034248. This comprehensive review describes the molecular mechanisms that control autophagic responses and their regulation. [DOI] [PubMed] [Google Scholar]
- 2.Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–1136. doi: 10.1007/s00018-011-0865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92–104. doi: 10.1038/cr.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sica V, et al. Organelle-specific initiation of autophagy. Mol Cell. 2015;59:522–539. doi: 10.1016/j.molcel.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 6.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He C, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michaud M, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. This article demonstrates that autophagy is strictly required for mouse cancer cells that succumb to some chemotherapeutics to release danger signals and hence alert the immune system of a threatening situation. [DOI] [PubMed] [Google Scholar]
- 9.Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17:467–484. doi: 10.1038/nrn.2016.51. [DOI] [PubMed] [Google Scholar]
- 10.Martinez J, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17:893–906. doi: 10.1038/ncb3192. This is the first identification of molecules that are specifically required for LAP but dispensable for canonical autophagy, including RUBCN. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Kenific CM, Wittmann T, Debnath J. Autophagy in adhesion and migration. J Cell Sci. 2016;129:3685–3693. doi: 10.1242/jcs.188490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ponpuak M, et al. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116. doi: 10.1016/j.ceb.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. References 15 and 16 provide the first demonstration that the Becn1−/− genotype is embryonically lethal and that Becn1+/− mice are more susceptible to spontaneous carcinogenesis than their wild-type littermates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshii SR, et al. Systemic analysis of Atg5-null mice rescued from neonatal lethality by transgenic ATG5 expression in neurons. Dev Cell. 2016;39:116–130. doi: 10.1016/j.devcel.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 19.Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. References 18 and 19 were the first to demonstrate that neuron-specific deletion of Atg5 or Atg7 causes a progressive neurodegenerative disorder that is associated with premature mortality. [DOI] [PubMed] [Google Scholar]
- 20.Ross CA, Poirier MA. Opinion: what is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 21.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 22.Crews L, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of α-synucleinopathy. PLoS ONE. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Bang Y, Kim KS, Seol W, Choi HJ. LRRK2 interferes with aggresome formation for autophagic clearance. Mol Cell Neurosci. 2016;75:71–80. doi: 10.1016/j.mcn.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 24.Vives-Bauza C, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JH, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fecto F, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 27.Cirulli ET, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freischmidt A, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–636. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- 29.Aguado C, et al. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet. 2010;19:2867–2876. doi: 10.1093/hmg/ddq190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saitsu H, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45:445–449. doi: 10.1038/ng.2562. [DOI] [PubMed] [Google Scholar]
- 31.Spilman P, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer’s disease. PLoS ONE. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vingtdeux V, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J Biol Chem. 2010;285:9100–9113. doi: 10.1074/jbc.M109.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang DS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun B, et al. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron. 2008;60:247–257. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spencer B, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Decressac M, et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci USA. 2013;110:E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siddiqui A, et al. Mitochondrial quality control via the PGC1α-TFEB signaling pathway is compromised by parkin Q311X mutation but independently restored by rapamycin. J Neurosci. 2015;35:12833–12844. doi: 10.1523/JNEUROSCI.0109-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez-Navarro JA, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–438. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Liu K, Shi N, Sun Y, Zhang T, Sun X. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem Res. 2013;38:201–207. doi: 10.1007/s11064-012-0909-8. [DOI] [PubMed] [Google Scholar]
- 41.Dehay B, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li XZ, et al. Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced parkinsonism in mice: possible mediation through enhanced autophagy. Int J Neurosci. 2013;123:73–79. doi: 10.3109/00207454.2012.729234. [DOI] [PubMed] [Google Scholar]
- 43.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang J, Jiang J, Zuo Y, Gu Z. Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson’s disease. Int J Mol Med. 2013;31:825–832. doi: 10.3892/ijmm.2013.1280. [DOI] [PubMed] [Google Scholar]
- 45.Pan T, et al. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis. 2008;32:16–25. doi: 10.1016/j.nbd.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Tain LS, et al. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bjedov I, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11:35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ejlerskov P, et al. Lack of neuronal IFN-β-IFNAR causes lewy body- and Parkinson’s disease-like dementia. Cell. 2015;163:324–339. doi: 10.1016/j.cell.2015.08.069. This article demonstrates that defective type I interferon signalling promotes parkinsonism in mice, as it imposes an autophagic defect that is accompanied by the accumulation of senescent mitochondria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xilouri M, et al. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy. 2016;12:2230–2247. doi: 10.1080/15548627.2016.1214777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarkar S, et al. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet. 2008;17:170–178. doi: 10.1093/hmg/ddm294. [DOI] [PubMed] [Google Scholar]
- 51.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 52.Lee JH, et al. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron. 2015;85:303–315. doi: 10.1016/j.neuron.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castillo K, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9:1308–1320. doi: 10.4161/auto.25188. [DOI] [PubMed] [Google Scholar]
- 54.Zhang K, et al. Food restriction-induced autophagy modulates degradation of mutant SOD1 in an amyotrophic lateral sclerosis mouse model. Brain Res. 2013;1519:112–119. doi: 10.1016/j.brainres.2013.04.050. [DOI] [PubMed] [Google Scholar]
- 55.Feng HL, et al. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience. 2008;155:567–572. doi: 10.1016/j.neuroscience.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7:412–425. doi: 10.4161/auto.7.4.14541. [DOI] [PubMed] [Google Scholar]
- 57.Nassif M, et al. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy. 2014;10:1256–1271. doi: 10.4161/auto.28784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guha P, Harraz MM, Snyder SH. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc Natl Acad Sci USA. 2016;113:1417–1422. doi: 10.1073/pnas.1524860113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sutton LP, Caron MG. Essential role of D1R in the regulation of mTOR complex1 signaling induced by cocaine. Neuropharmacology. 2015;99:610–619. doi: 10.1016/j.neuropharm.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu J, McCallum SE, Glick SD, Huang Y. Inhibition of the mammalian target of rapamycin pathway by rapamycin blocks cocaine-induced locomotor sensitization. Neuroscience. 2011;172:104–109. doi: 10.1016/j.neuroscience.2010.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol. 2013;698:6–18. doi: 10.1016/j.ejphar.2012.10.032. [DOI] [PubMed] [Google Scholar]
- 62.Stamoula E, et al. Low dose administration of glutamate triggers a non-apoptotic, autophagic response in PC12 cells. Cell Physiol Biochem. 2015;37:1750–1758. doi: 10.1159/000430250. [DOI] [PubMed] [Google Scholar]
- 63.Wang Y, et al. An autophagic mechanism is involved in apoptotic death of rat striatal neurons induced by the non-N-methyl-D-aspartate receptor agonist kainic acid. Autophagy. 2008;4:214–226. doi: 10.4161/auto.5369. [DOI] [PubMed] [Google Scholar]
- 64.Perez-Carrion MD, et al. Dendrimer-mediated siRNA delivery knocks down Beclin 1 and potentiates NMDA-mediated toxicity in rat cortical neurons. J Neurochem. 2012;120:259–268. doi: 10.1111/j.1471-4159.2011.07556.x. [DOI] [PubMed] [Google Scholar]
- 65.Kulbe JR, Mulcahy Levy JM, Coultrap SJ, Thorburn A, Bayer KU. Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res. 2014;1542:12–19. doi: 10.1016/j.brainres.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giorgi FS, Biagioni F, Lenzi P, Frati A, Fornai F. The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J Neural Transm (Vienna) 2015;122:849–862. doi: 10.1007/s00702-014-1312-1. [DOI] [PubMed] [Google Scholar]
- 67.McMahon J, et al. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci. 2012;32:15704–15714. doi: 10.1523/JNEUROSCI.2392-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou J, et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zeng LH, et al. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang CF, et al. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation. J Pineal Res. 2012;52:312–321. doi: 10.1111/j.1600-079X.2011.00945.x. [DOI] [PubMed] [Google Scholar]
- 72.Ginet V, et al. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy. 2014;10:846–860. doi: 10.4161/auto.28264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng Y, et al. Inhibition of autophagy contributes to melatonin-mediated neuroprotection against transient focal cerebral ischemia in rats. J Pharmacol Sci. 2014;124:354–364. doi: 10.1254/jphs.13220fp. [DOI] [PubMed] [Google Scholar]
- 74.Papadakis M, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med. 2013;19:351–357. doi: 10.1038/nm.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang P, et al. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011;69:360–374. doi: 10.1002/ana.22236. [DOI] [PubMed] [Google Scholar]
- 76.Zhang X, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321–1333. doi: 10.4161/auto.25132. [DOI] [PubMed] [Google Scholar]
- 77.Buckley KM, et al. Rapamycin up-regulation of autophagy reduces infarct size and improves outcomes in both permanent MCAL, and embolic MCAO, murine models of stroke. Exp Transl Stroke Med. 2014;6:8. doi: 10.1186/2040-7378-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: involvement of PARK2-dependent mitophagy. Autophagy. 2014;10:1801–1813. doi: 10.4161/auto.32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sheng R, et al. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482–494. doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- 80.Su J, Zhang T, Wang K, Zhu T, Li X. Autophagy activation contributes to the neuroprotection of remote ischemic perconditioning against focal cerebral ischemia in rats. Neurochem Res. 2014;39:2068–2077. doi: 10.1007/s11064-014-1396-x. [DOI] [PubMed] [Google Scholar]
- 81.Jiang T, et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br J Pharmacol. 2014;171:3146–3157. doi: 10.1111/bph.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang T, et al. Ischemic preconditioning provides neuroprotection by induction of AMP-activated protein kinase-dependent autophagy in a rat model of ischemic stroke. Mol Neurobiol. 2015;51:220–229. doi: 10.1007/s12035-014-8725-6. [DOI] [PubMed] [Google Scholar]
- 83.Zheng YQ, Liu JX, Li XZ, Xu L, Xu YG. RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol Sin. 2009;30:919–927. doi: 10.1038/aps.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cui DR, et al. Propofol prevents cerebral ischemia-triggered autophagy activation and cell death in the rat hippocampus through the NF-κB/p53 signaling pathway. Neuroscience. 2013;246:117–132. doi: 10.1016/j.neuroscience.2013.04.054. [DOI] [PubMed] [Google Scholar]
- 85.Zheng C, et al. NAD+ administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci Lett. 2012;512:67–71. doi: 10.1016/j.neulet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 86.Wen YD, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 87.Kubota C, et al. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem. 2010;285:667–674. doi: 10.1074/jbc.M109.053058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xin XY, et al. 2-Methoxyestradiol attenuates autophagy activation after global ischemia. Can J Neurol Sci. 2011;38:631–638. doi: 10.1017/s031716710001218x. [DOI] [PubMed] [Google Scholar]
- 89.Liu N, Shang J, Tian F, Nishi H, Abe K. In vivo optical imaging for evaluating the efficacy of edaravone after transient cerebral ischemia in mice. Brain Res. 2011;1397:66–75. doi: 10.1016/j.brainres.2011.04.038. [DOI] [PubMed] [Google Scholar]
- 90.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 91.Koike M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xie C, et al. Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy. 2016;12:410–423. doi: 10.1080/15548627.2015.1132134. This report provides compelling evidence in support of the concept that efficient autophagic responses in the neurons of newborns contribute to the aetiology of neonatal asphyxia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- 94.Liu Y, et al. Autosis is a Na+, K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA. 2013;110:20364–20371. doi: 10.1073/pnas.1319661110. This is the first description of the mechanisms and pathophysiological relevance of autosis, which is an instance of autophagic cell death that also relies on the plasma membrane Na+/K+-ATPase and hence can be inhibited by cardiac glycosides. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang ZY, Liu WG, Muharram A, Wu ZY, Lin JH. Neuroprotective effects of autophagy induced by rapamycin in rat acute spinal cord injury model. Neuroimmunomodulation. 2014;21:257–267. doi: 10.1159/000357382. [DOI] [PubMed] [Google Scholar]
- 96.Jing CH, et al. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;213:144–153. doi: 10.1016/j.neuroscience.2012.03.055. [DOI] [PubMed] [Google Scholar]
- 97.Chen J, et al. Melatonin-enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J Pineal Res. 2014;56:12–19. doi: 10.1111/jpi.12086. [DOI] [PubMed] [Google Scholar]
- 98.Shao A, et al. Enhancement of autophagy by histone deacetylase inhibitor Trichostatin A ameliorates neuronal apoptosis after subarachnoid hemorrhage in rats. Mol Neurobiol. 2016;53:18–27. doi: 10.1007/s12035-014-8986-0. [DOI] [PubMed] [Google Scholar]
- 99.Zhao H, et al. Role of autophagy in early brain injury after subarachnoid hemorrhage in rats. Mol Biol Rep. 2013;40:819–827. doi: 10.1007/s11033-012-2120-z. [DOI] [PubMed] [Google Scholar]
- 100.Zhou Y, et al. Retinoic acid prevents disruption of blood-spinal cord barrier by Iinducing autophagic flux after spinal cord injury. Neurochem Res. 2015;41:813–825. doi: 10.1007/s11064-015-1756-1. [DOI] [PubMed] [Google Scholar]
- 101.Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 102.Song Q, Xie D, Pan S, Xu W. Rapamycin protects neurons from brain contusioninduced inflammatory reaction via modulation of microglial activation. Mol Med Rep. 2015;12:7203–7210. doi: 10.3892/mmr.2015.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Viscomi MT, et al. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2012;8:222–235. doi: 10.4161/auto.8.2.18599. [DOI] [PubMed] [Google Scholar]
- 104.Cui CM, et al. Chloroquine exerts neuroprotection following traumatic brain injury via suppression of inflammation and neuronal autophagic death. Mol Med Rep. 2015;12:2323–2328. doi: 10.3892/mmr.2015.3611. [DOI] [PubMed] [Google Scholar]
- 105.Luo CL, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54–63. doi: 10.1016/j.neuroscience.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 106.Martinet W, Knaapen MW, Kockx MM, De Meyer GR. Autophagy in cardiovascular disease. Trends Mol Med. 2007;13:482–491. doi: 10.1016/j.molmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 107.Tanaka Y, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 108.Nishino I, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 109.Nakai A, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 110.Huang C, et al. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liao X, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tannous P, et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci USA. 2008;105:9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bhuiyan MS, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xie M, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sala-Mercado JA, et al. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation. 2010;122:S179–S184. doi: 10.1161/CIRCULATIONAHA.109.928242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sciarretta S, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Finckenberg P, et al. Caloric restriction ameliorates angiotensin II-induced mitochondrial remodeling and cardiac hypertrophy. Hypertension. 2012;59:76–84. doi: 10.1161/HYPERTENSIONAHA.111.179457. [DOI] [PubMed] [Google Scholar]
- 119.Bostrom P, et al. C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shirakabe A, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eisenberg T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428–1438. doi: 10.1038/nm.4222. This paper shows that the CRM spermidine extends lifespan in mice as it mediates autophagy-dependent cardioprotective effects, and that a spermidine-rich diet is associated with a lower incidence of cardiovascular disorders in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Garg S, Bourantas C, Serruys PW. New concepts in the design of drug-eluting coronary stents. Nat Rev Cardiol. 2013;10:248–260. doi: 10.1038/nrcardio.2013.13. [DOI] [PubMed] [Google Scholar]
- 123.Marx SO, Marks AR. Bench to bedside: the development of rapamycin and its application to stent restenosis. Circulation. 2001;104:852–855. doi: 10.1161/01.cir.104.8.852. [DOI] [PubMed] [Google Scholar]
- 124.Zhu H, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Matsui Y, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 126.Xu X, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077–18092. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schneider JL, Cuervo AM. Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol. 2014;11:187–200. doi: 10.1038/nrgastro.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sequea DA, Sharma N, Arias EB, Cartee GD. Calorie restriction enhances insulin-stimulated glucose uptake and Akt phosphorylation in both fast-twitch and slow-twitch skeletal muscle of 24-month-old rats. J Gerontol A Biol Sci Med Sci. 2012;67:1279–1285. doi: 10.1093/gerona/gls085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marcinko K, et al. High intensity interval training improves liver and adipose tissue insulin sensitivity. Mol Metab. 2015;4:903–915. doi: 10.1016/j.molmet.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cool B, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 131.Hatori M, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–860. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Leontieva OV, Paszkiewicz G, Demidenko ZN, Blagosklonny MV. Resveratrol potentiates rapamycin to prevent hyperinsulinemia and obesity in male mice on high fat diet. Cell Death Dis. 2013;4:e472. doi: 10.1038/cddis.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sun L, et al. Hydrogen sulfide reduces serum triglyceride by activating liver autophagy via the AMPK-mTOR pathway. Am J Physiol Endocrinol Metab. 2015;309:E925–E935. doi: 10.1152/ajpendo.00294.2015. [DOI] [PubMed] [Google Scholar]
- 134.Lin CW, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–999. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim KE, et al. Caloric restriction of db/db mice reverts hepatic steatosis and body weight with divergent hepatic metabolism. Sci Rep. 2016;6:30111. doi: 10.1038/srep30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Song YM, et al. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. 2015;11:46–59. doi: 10.4161/15548627.2014.984271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hidvegi T, et al. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 138.Lamming DW, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schreiber KH, et al. Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell. 2015;14:265–273. doi: 10.1111/acel.12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mottillo EP, et al. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab. 2016;24:118–129. doi: 10.1016/j.cmet.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ding WX, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ni HM, Du K, You M, Ding WX. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol. 2013;183:1815–1825. doi: 10.1016/j.ajpath.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shaw RJ, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee HY, et al. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy. 2016;12:1390–1403. doi: 10.1080/15548627.2016.1184799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sakaguchi M, et al. Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem Biophys Res Commun. 2006;340:296–301. doi: 10.1016/j.bbrc.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 147.Ding DF, et al. Resveratrol attenuates renal hypertrophy in early-stage diabetes by activating AMPK. Am J Nephrol. 2010;31:363–374. doi: 10.1159/000300388. [DOI] [PubMed] [Google Scholar]
- 148.Tikoo K, Tripathi DN, Kabra DG, Sharma V, Gaikwad AB. Intermittent fasting prevents the progression of type I diabetic nephropathy in rats and changes the expression of Sir2 and p53. FEBS Lett. 2007;581:1071–1078. doi: 10.1016/j.febslet.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 149.Jung HS, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 150.Goginashvili A, et al. Insulin granules. Insulin secretory granules control autophagy in pancreatic beta cells. Science. 2015;347:878–882. doi: 10.1126/science.aaa2628. This article provides compelling evidence in support of the concept that insulin granules are degraded by autophagy in pancreatic β-cells as a mechanism that negatively regulates insulin secretion under fasting conditions. [DOI] [PubMed] [Google Scholar]
- 151.Altshuler-Keylin S, et al. Beige adipocyte maintenance is regulated by autophagy-induced mitochondrial clearance. Cell Metab. 2016;24:402–419. doi: 10.1016/j.cmet.2016.08.002. This article shows that efficient autophagic responses are required for the beige-to-white fat transition, which is generally coupled to HFD-induced obesity and insulin resistance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Galluzzi L, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Takamura A, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rao S, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 155.Strohecker AM, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3:1272–1285. doi: 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rosenfeldt MT, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. References 154–156 provide solid genetic data demonstrating that autophagy inhibits malignant transformation but accelerates tumour progression in the setting of KRAS-driven or BRAF-driven pulmonary or pancreatic carcinogenesis. [DOI] [PubMed] [Google Scholar]
- 157.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 158.Wei Y, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154:1269–1284. doi: 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Maiuri MC, et al. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 160.Wang RC, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956–959. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2016;14:247–258. doi: 10.1038/nrclinonc.2016.183. This recent review summarizes preclinical and clinical data indicating that autophagy activators, rather than inhibitors, may potentiate the therapeutic effects of anticancer agents that promote, or at least are compatible with, tumour-targeting immune responses. [DOI] [PubMed] [Google Scholar]
- 162.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pietrocola F, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30:147–160. doi: 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Di Biase S, et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell. 2016;30:136–146. doi: 10.1016/j.ccell.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ladoire S, et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy. 2015;11:1878–1890. doi: 10.1080/15548627.2015.1082022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Puleston DJ, et al. Autophagy is a critical regulator of memory CD8+ T cell formation. eLife. 2014;3:e03706. doi: 10.7554/eLife.03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 169.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 170.Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 171.Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3:117–125. doi: 10.4161/auto.3618. [DOI] [PubMed] [Google Scholar]
- 172.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 173.Shahnazari S, Namolovan A, Mogridge J, Kim PK, Brumell JH. Bacterial toxins can inhibit host cell autophagy through cAMP generation. Autophagy. 2011;7:957–965. doi: 10.4161/auto.7.9.16435. [DOI] [PubMed] [Google Scholar]
- 174.Tattoli I, et al. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. 2012;11:563–575. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 175.Choy A, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012;338:1072–1076. doi: 10.1126/science.1227026. This paper identifies a protein from L. pneumophila that operates as a virulence factor by inhibiting autophagy in host cells upon LC3 deconjugation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dong N, et al. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell. 2012;150:1029–1041. doi: 10.1016/j.cell.2012.06.050. [DOI] [PubMed] [Google Scholar]
- 177.Yoshikawa Y, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 178.Kim JJ, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 2012;11:457–468. doi: 10.1016/j.chom.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 179.Kuo SY, et al. Small-molecule enhancers of autophagy modulate cellular disease phenotypes suggested by human genetics. Proc Natl Acad Sci USA. 2015;112:E4281–E4287. doi: 10.1073/pnas.1512289112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. 2012;14:791–807. doi: 10.1111/j.1462-5822.2012.01768.x. [DOI] [PubMed] [Google Scholar]
- 181.Miao Y, Li G, Zhang X, Xu H, Abraham SNA. TRP channel senses lysosome neutralization by pathogens to trigger their expulsion. Cell. 2015;161:1306–1319. doi: 10.1016/j.cell.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lee S, et al. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal. 2014;20:432–442. doi: 10.1089/ars.2013.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gong L, et al. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS ONE. 2011;6:e17852. doi: 10.1371/journal.pone.0017852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy. 2013;9:985–995. doi: 10.4161/auto.24406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Manzanillo PS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kimmey JM, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–569. doi: 10.1038/nature16451. This report demonstrates that ATG5, but not several other components of the canonical autophagic machinery, in monocytes is required for mice to normally control M. tuberculosis infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Renna M, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest. 2011;121:3554–3563. doi: 10.1172/JCI46095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gutierrez MG, et al. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol. 2005;7:981–993. doi: 10.1111/j.1462-5822.2005.00527.x. [DOI] [PubMed] [Google Scholar]
- 189.Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc Natl Acad Sci USA. 2012;109:20800–20807. doi: 10.1073/pnas.1218674109. This study provides robust evidence in support of the notion that A. phagocytophilum actively promotes BECN1-dependent autophagic responses in host cells to acquire nutrients for growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Raoult D, et al. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med. 1999;159:167–173. doi: 10.1001/archinte.159.2.167. [DOI] [PubMed] [Google Scholar]
- 191.Orvedahl A, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Liang XH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sumpter R, Jr, et al. Fanconi anemia proteins function in mitophagy and immunity. Cell. 2016;165:867–881. doi: 10.1016/j.cell.2016.04.006. This is the first demonstration that members of the FANC protein family participate not only in the DNA damage response but also in virophagy and mitophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Orvedahl A, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 195.Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30:588–598. doi: 10.1016/j.immuni.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Roy J, Paquette JS, Fortin JF, Tremblay MJ. The immunosuppressant rapamycin represses human immunodeficiency virus type 1 replication. Antimicrob Agents Chemother. 2002;46:3447–3455. doi: 10.1128/AAC.46.11.3447-3455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shoji-Kawata S, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Nicoletti F, et al. Inhibition of human immunodeficiency virus (HIV-1) infection in human peripheral blood leucocytes-SCID reconstituted mice by rapamycin. Clin Exp Immunol. 2009;155:28–34. doi: 10.1111/j.1365-2249.2008.03780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Alirezaei M, Flynn CT, Wood MR, Whitton JL. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell Host Microbe. 2012;11:298–305. doi: 10.1016/j.chom.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nicola AM, et al. Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect Immun. 2012;80:3065–3076. doi: 10.1128/IAI.00358-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kanayama M, et al. Autophagy enhances NFκB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun. 2015;6:5779. doi: 10.1038/ncomms6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Akoumianaki T, et al. Aspergillus cell wall melanin blocks LC3-associated phagocytosis to promote pathogenicity. Cell Host Microbe. 2016;19:79–90. doi: 10.1016/j.chom.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 203.Zhao Z, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Choi J, et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity. 2014;40:924–935. doi: 10.1016/j.immuni.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Selleck EM, et al. A noncanonical autophagy pathway restricts Toxoplasma gondii growth in a strain-specific manner in IFN-γ-activated human cells. mBio. 2015;6:e01157–15. doi: 10.1128/mBio.01157-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Flannery EL, Chatterjee AK, Winzeler EA. Antimalarial drug discovery — approaches and progress towards new medicines. Nat Rev Microbiol. 2013;11:849–862. doi: 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sullivan DJ, Jr, Gluzman IY, Russell DG, Goldberg DE. On the molecular mechanism of chloroquine’s antimalarial action. Proc Natl Acad Sci USA. 1996;93:11865–11870. doi: 10.1073/pnas.93.21.11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Roetzer A, Gratz N, Kovarik P, Schuller C. Autophagy supports Candida glabrata survival during phagocytosis. Cell Microbiol. 2010;12:199–216. doi: 10.1111/j.1462-5822.2009.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Dang J, et al. Gene–gene interaction of ATG5, ATG7, BLK and BANK1 in systemic lupus erythematosus. Int J Rheum Dis. 2016;19:1284–1293. doi: 10.1111/1756-185X.12768. [DOI] [PubMed] [Google Scholar]
- 213.Martinez J, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533:115–119. doi: 10.1038/nature17950. This report elegantly proves that LAP defects, but not defects in canonical autophagy, are associated with impaired efferocytosis, secretion of pro-inflammatory cytokines and spontaneous onset of an SLE-like disorder in mice. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 214.Remijsen Q, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Lee SJ, Silverman E, Bargman JM. The role of antimalarial agents in the treatment of SLE and lupus nephritis. Nat Rev Nephrol. 2011;7:718–729. doi: 10.1038/nrneph.2011.150. [DOI] [PubMed] [Google Scholar]
- 216.Rioux JD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Henckaerts L, et al. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm Bowel Dis. 2011;17:1392–1397. doi: 10.1002/ibd.21486. [DOI] [PubMed] [Google Scholar]
- 218.Kaul A, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039. doi: 10.1038/nrdp.2016.39. [DOI] [PubMed] [Google Scholar]
- 219.Matsuda C, et al. Therapeutic effect of a new immunosuppressive agent, everolimus, on interleukin-10 gene-deficient mice with colitis. Clin Exp Immunol. 2007;148:348–359. doi: 10.1111/j.1365-2249.2007.03345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Dumortier J, et al. Everolimus for refractory Crohn’s disease: a case report. Inflamm Bowel Dis. 2008;14:874–877. doi: 10.1002/ibd.20395. [DOI] [PubMed] [Google Scholar]
- 221.Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. 2013;210:2119–2134. doi: 10.1084/jem.20130252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.van Loosdregt J, et al. Increased autophagy in CD4+ T cells of rheumatoid arthritis patients results in T-cell hyperactivation and apoptosis resistance. Eur J Immunol. 2016;46:2862–2870. doi: 10.1002/eji.201646375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yoshizaki A, et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 2010;62:2476–2487. doi: 10.1002/art.27498. [DOI] [PubMed] [Google Scholar]
- 224.Bhattacharya A, Parillon X, Zeng S, Han S, Eissa NT. Deficiency of autophagy in dendritic cells protects against experimental autoimmune encephalomyelitis. J Biol Chem. 2014;289:26525–26532. doi: 10.1074/jbc.M114.575860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Prevel N, Allenbach Y, Klatzmann D, Salomon B, Benveniste O. Beneficial role of rapamycin in experimental autoimmune myositis. PLoS ONE. 2013;8:e74450. doi: 10.1371/journal.pone.0074450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Esposito M, et al. Rapamycin inhibits relapsing experimental autoimmune encephalomyelitis by both effector and regulatory T cells modulation. J Neuroimmunol. 2010;220:52–63. doi: 10.1016/j.jneuroim.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 227.Zhang Z, et al. Low dose rapamycin exacerbates autoimmune experimental uveitis. PLoS ONE. 2012;7:e36589. doi: 10.1371/journal.pone.0036589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Teachey DT, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145:101–106. doi: 10.1111/j.1365-2141.2009.07595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Howitz KT, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 230.Eisenberg T, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 231.Morselli E, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Melendez A, et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 233.Lapierre LR, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun. 2013;4:2267. doi: 10.1038/ncomms3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. This is the first demonstration that administering rapamycin late in life extends the lifespan of mice independently of their genetic background. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Baur JA, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Pyo JO, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Colman RJ, et al. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun. 2014;5:3557. doi: 10.1038/ncomms4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125:85–93. doi: 10.1172/JCI73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lopez-Otin C, Galluzzi L, Freije JM, Madeo F, Kroemer G. Metabolic control of longevity. Cell. 2016;166:802–821. doi: 10.1016/j.cell.2016.07.031. This recent review dissects the intimate relationship between metabolism and ageing, highlighting several points of intervention at which strategies that support metabolic fitness may extend healthy lifespan in humans. [DOI] [PubMed] [Google Scholar]
- 240.Yang L, et al. Long-term calorie restriction enhances cellular quality-control processes in human skeletal muscle. Cell Rep. 2016;14:422–428. doi: 10.1016/j.celrep.2015.12.042. [DOI] [PubMed] [Google Scholar]
- 241.Patel AS, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE. 2012;7:e41394. doi: 10.1371/journal.pone.0041394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Lee SJ, et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2011;183:649–658. doi: 10.1164/rccm.201005-0746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Luciani A, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. 2010;12:863–875. doi: 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- 244.Chung EJ, et al. Mammalian target of rapamycin inhibition with rapamycin mitigates radiation-induced pulmonary fibrosis in a murine model. Int J Radiat Oncol Biol Phys. 2016;96:857–866. doi: 10.1016/j.ijrobp.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wang W, et al. mTORC1 is involved in hypoxia-induced pulmonary hypertension through the activation of Notch3. J Cell Physiol. 2014;229:2117–2125. doi: 10.1002/jcp.24670. [DOI] [PubMed] [Google Scholar]
- 246.Sureshbabu A, et al. Inhibition of regulatory-associated protein of mechanistic target of rapamycin prevents hyperoxia-induced lung injury by enhancing autophagy and reducing apoptosis in neonatal mice. Am J Respir Cell Mol Biol. 2016;55:722–735. doi: 10.1165/rcmb.2015-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hu Y, et al. Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy. 2016;12:2286–2299. doi: 10.1080/15548627.2016.1230584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Chen ZH, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA. 2010;107:18880–18885. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lam HC, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123:5212–5230. doi: 10.1172/JCI69636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mizumura K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124:3987–4003. doi: 10.1172/JCI74985. This paper shows that mitophagic responses participate in the pathogenesis of COPD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Chen ZH, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE. 2008;3:e3316. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Hartleben B, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kawakami T, et al. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol. 2015;26:1040–1052. doi: 10.1681/ASN.2013111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Oliva Trejo JA, et al. Transient increase in proteinuria, poly-ubiquitylated proteins and ER stress markers in podocyte-specific autophagy-deficient mice following unilateral nephrectomy. Biochem Biophys Res Commun. 2014;446:1190–1196. doi: 10.1016/j.bbrc.2014.03.088. [DOI] [PubMed] [Google Scholar]
- 255.Zeng C, et al. Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J Pathol. 2014;234:203–213. doi: 10.1002/path.4382. [DOI] [PubMed] [Google Scholar]
- 256.Cinque L, et al. FGF signalling regulates bone growth through autophagy. Nature. 2015;528:272–275. doi: 10.1038/nature16063. [DOI] [PubMed] [Google Scholar]
- 257.Chen J, Long F. mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis. Development. 2014;141:2848–2854. doi: 10.1242/dev.108811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Yang GE, et al. Rapamycin-induced autophagy activity promotes bone fracture healing in rats. Exp Ther Med. 2015;10:1327–1333. doi: 10.3892/etm.2015.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Alvarez-Garcia O, et al. Rapamycin induces growth retardation by disrupting angiogenesis in the growth plate. Kidney Int. 2010;78:561–568. doi: 10.1038/ki.2010.173. [DOI] [PubMed] [Google Scholar]
- 260.Holstein JH, et al. Rapamycin affects early fracture healing in mice. Br J Pharmacol. 2008;154:1055–1062. doi: 10.1038/bjp.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–565. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 262.Raben N, et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder — murine Pompe disease. Autophagy. 2010;6:1078–1089. doi: 10.4161/auto.6.8.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Raben N, et al. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 2008;17:3897–3908. doi: 10.1093/hmg/ddn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Awad O, et al. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum Mol Genet. 2015;24:5775–5788. doi: 10.1093/hmg/ddv297. [DOI] [PubMed] [Google Scholar]
- 265.Maetzel D, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann–Pick Type C patient-specific iPS cells. Stem Cell Rep. 2014;2:866–880. doi: 10.1016/j.stemcr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zhou Z, et al. Autophagy supports color vision. Autophagy. 2015;11:1821–1832. doi: 10.1080/15548627.2015.1084456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Rodriguez-Muela N, et al. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015;22:476–487. doi: 10.1038/cdd.2014.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kim JY, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154:365–376. doi: 10.1016/j.cell.2013.06.012. This article was the first to demonstrate that LAP is essential for the degradation of photoreceptor outer segments phagocytosed by the retinal epithelium in the course of the visual cycle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Santeford A, et al. Impaired autophagy in macrophages promotes inflammatory eye disease. Autophagy. 2016;12:1876–1885. doi: 10.1080/15548627.2016.1207857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Selmi C. Diagnosis and classification of autoimmune uveitis. Autoimmun Rev. 2014;13:591–594. doi: 10.1016/j.autrev.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 271.Rezaie T, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295:1077–1079. doi: 10.1126/science.1066901. [DOI] [PubMed] [Google Scholar]
- 272.Fingert JH, et al. Copy number variations on chromosome 12q14 in patients with normal tension glaucoma. Hum Mol Genet. 2011;20:2482–2494. doi: 10.1093/hmg/ddr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Chi ZL, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet. 2010;19:2606–2615. doi: 10.1093/hmg/ddq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Lee IH, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225–228. doi: 10.1126/science.1218395. This study characterizes one of the autophagy-independent functions of ATG7, namely, the regulation of p53 activity in the course of metabolic stress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Laplante M, Sabatini D. M mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Rodriguez-Boulan E, Kreitzer G, Musch A. Organization of vesicular trafficking in epithelia. Nat Rev Mol Cell Biol. 2005;6:233–247. doi: 10.1038/nrm1593. [DOI] [PubMed] [Google Scholar]
- 278.Calne RY, et al. Rapamycin for immunosuppression in organ allografting. Lancet. 1989;2:227. doi: 10.1016/s0140-6736(89)90417-0. [DOI] [PubMed] [Google Scholar]
- 279.Taherian E, Rao A, Malemud CJ, Askari AD. The biological and clinical activity of anti-malarial drugs in autoimmune disorders. Curr Rheumatol Rev. 2013;9:45–62. doi: 10.2174/1573397111309010010. [DOI] [PubMed] [Google Scholar]
- 280.Maes H, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. 2014;26:190–206. doi: 10.1016/j.ccr.2014.06.025. This report elegantly demonstrates that chloroquine exerts robust anticancer effects through its ability to promote tumour vessel normalization independent of autophagy. [DOI] [PubMed] [Google Scholar]
- 281.Eng CH, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA. 2016;113:182–187. doi: 10.1073/pnas.1515617113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Niso-Santano M, et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015;34:1025–1041. doi: 10.15252/embj.201489363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kaizuka T, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64:835–849. doi: 10.1016/j.molcel.2016.09.037. [DOI] [PubMed] [Google Scholar]
- 284.Cabrera S, et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy. 2013;9:1188–1200. doi: 10.4161/auto.24797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Barinaga M. Knockout mice: round two. Science. 1994;265:26–28. doi: 10.1126/science.8016653. [DOI] [PubMed] [Google Scholar]
- 286.Blaney Davidson EN, van de Loo FA, van den Berg WB, van der Kraan PM. How to build an inducible cartilage-specific transgenic mouse. Arthritis Res Ther. 2014;16:210. doi: 10.1186/ar4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Baker DJ, et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Kroemer G, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Galluzzi L, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73. doi: 10.1038/cdd.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Tsuboyama K, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–1041. doi: 10.1126/science.aaf6136. [DOI] [PubMed] [Google Scholar]
- 292.Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Nishida Y, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. This was the first characterization of a non-canonical, ATG5-independent and ATG7-independent autophagic response in mouse cells. [DOI] [PubMed] [Google Scholar]
- 294.McAfee Q, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci USA. 2012;109:8253–8258. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ronan B, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. 2014;10:1013–1019. doi: 10.1038/nchembio.1681. [DOI] [PubMed] [Google Scholar]
- 296.Bago R, et al. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem J. 2014;463:413–427. doi: 10.1042/BJ20140889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Akin D, et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10:2021–2035. doi: 10.4161/auto.32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Egan DF, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Petherick KJ, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290:11376–11383. doi: 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Yap TA, Sandhu SK, Workman P, de Bono JS. Envisioning the future of early anticancer drug development. Nat Rev Cancer. 2010;10:514–523. doi: 10.1038/nrc2870. [DOI] [PubMed] [Google Scholar]