

Abstract
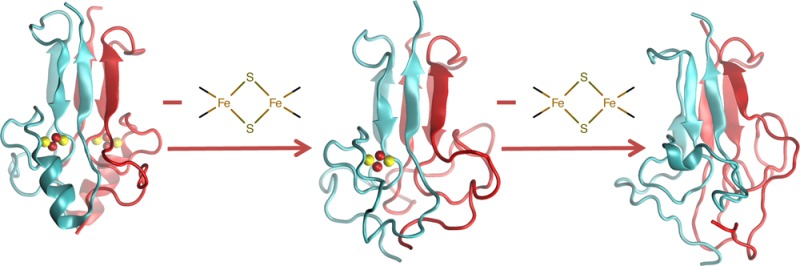
The NEET proteins are a novel family of iron–sulfur proteins characterized by an unusual three cysteine and one histidine coordinated [2Fe–2S] cluster. Aberrant cluster release, facilitated by the breakage of the Fe–N bond, is implicated in a variety of human diseases, including cancer. Here, the molecular dynamics in the multi-microsecond timescale, along with quantum chemical calculations, on two representative members of the family (the human NAF-1 and mitoNEET proteins), show that the loss of the cluster is associated with a dramatic decrease in secondary and tertiary structure. In addition, the calculations provide a mechanism for cluster release and clarify, for the first time, crucial differences existing between the two proteins, which are reflected in the experimentally observed difference in the pH-dependent cluster reactivity. The reliability of our conclusions is established by an extensive comparison with the NMR data of the solution proteins, in part measured in this work.
1. Introduction
The MitoNEET (mNT) and NAF-1 proteins, members of the so-called NEET protein family, regulate apoptosis, autophagy, iron, and reactive oxygen species (ROS) homoeostasis.1−5 Localized to the outer mitochondrial membrane (mNT),6 and to the endoplasmic reticulum and the mitochondrial associated membranes (NAF-1),3 these membrane-anchored proteins contain a conserved CDGSH domain,6,7 featuring a unique “NEET fold.” They harbor a novel redox-active and labile 3Cys:1His [2Fe–2S] cluster.6,8,9 Abnormal cluster release in NAF-1 is associated with cancer progression,10 and it might also be involved in neurodegenerative diseases.3,8,11−15 Hence, NEET proteins are emerging as a promising pharmaceutical target.10
NEET proteins fold into a homodimeric structure organized into two distinct domains. The β-cap domain, which is composed of an intertwined β-sheet structure and the cluster-binding domain, that harbors the two [2Fe−2S] clusters in the homodimer.9 The histidine ligand (His87 in mNT and His114 in NAF-1), which binds the metal ion via its Nδ, is the primary cause of the labile nature of the cluster and its transfer to apo-acceptor protein(s).3,6,15−18 The presence of a single histidine ligand, rare across iron–sulfur proteins, is likely to be largely responsible for their important function in human health and diseases.3,18 Intriguingly, the cluster reactivity, as a function of pH, is larger for mNT than NAF-1.8 The structural features determining this difference in reactivity are not clearly defined.
All-atom molecular simulations can assist in identifying the molecular determinants and the mechanisms involved in the function of proteins containing Fe–S clusters, their associated co-factors. They are complementary to structure-based methods, which may be used to infer structural information for Fe–S proteins, such as mNT19 and ferredoxin.20 In fact, all atom molecular dynamics (MD) simulations were used already a quarter century ago to refine the structure of the multinuclear Fe–S proteins HiPIP in aqueous solution,21 proving to be in agreement with the NMR data.a
Here, by using quantum mechanical (QM) methods, we have defined a representative force field for the cluster/cluster-binding domain of human NEET proteins. Using this force field, we have applied a force-field-based MD21 and enhanced sampling MD27 simulations. The accuracy of the MD simulations was established by a comparison with the NMR experiments, some of which were performed here. Our results show that the loss of the cluster(s) is associated with a dramatic increase in protein disorder and provide a molecular basis for the larger cluster pH sensitivity of mNT compared to NAF-1.
2. Methods
2.1. NEET Proteins Cluster-Binding Domain QM Parametrization
The force field of the [2Fe–2S] cluster of NEET proteins in the oxidized state was built so as to be consistent with the AMBER force field.28,29 Quantum mechanical (QM) calculations on two geometry-optimized models of the metal site were implemented, following ref (30). Given the structural similarity between NAF-1 and mNT cluster-binding domains,9,31 we have used the same parametrization for both systems.
Model I (Figure S1A) consisted of the metal ions, the inorganic sulfur atoms, and residues C72, C74, C83, and H87, along with R73, N84, G85, and A86 backbone unitsb (Figure S1A). The N- and C-terminals were acetylated and methylated, respectively (Figure S1A). Here, the two Fe3+ ions are tetrahedrally coordinated. One of the two metal ion (Fex in Figure S1A) binds to cysteine sulfur atoms (SA and SB) and two bridging sulfur atoms S1 and S2. The other ion (Fey) binds to C83 sulfur atom (SC) and H87-Nδ (NX).
Model II was the same as I, except that residues R73, N84, G85, and A86 were lacking, and the coordinating side chains were represented by methyl groups (Figure S1B).
For both models, we considered both the Nε-protonated and the Nε-deprotonated states of the His residue nearby the cluster (His87 in mNT and His114 in NAF-1). The total charge of the models was −1 in the His:Nε-protonated state and −2 in the His:Nε-deprotonated. The models were built using the Metal Center Parameter Builder.32
In the protein oxidized state, each Fe3+ is in its high spin state (S = 5/2) and the cluster total spin is S = 0.6 To describe the iron ions’ antiferromagnetic state, we defined four monomers (the so-called Gaussian09 fragments33), consisting of the following atoms and groups: (1) the ion Fex with its cysteine ligands; (2) Fey with its histidine and cysteine ligands; (3) and (4) the two inorganic sulfur atoms. The spin multiplicity and total charge of each monomer were set taking into account the spin and oxidation states of each atom, respectively.
The models underwent geometry optimization at the B3LYP level of theory,34 using the 6-311G++ (2d,2p) basis set. We used the Gaussian09 program.33
The relatively inexpensive calculations of the RESP atomic partial charges were carried out for the extended model I. The charges on the backbone atoms were set to the standard values of the AMBER99sb-ILDN28,29 force field. The van der Waals parameters of the iron atoms were those of ref (35), whereas those of the other atoms were taken from the AMBER99sb-ILDN force field.28,29 The calculations of bonded parameters (stretching and bending force constants) were carried out on the reduced model II. The calculations were based on the Hessian matrix, using the Seminario’s method30 (Tables S1–S4). The torsion force constants were set to 0 as in ref (30).
2.2. Simulation Protocols
The mNT (pdbID:2QH7,9 residues 43:108) and NAF-1 (pdbID:4OO7,31 residues 69:135) protein X-ray structures were embedded in water boxes. Both His:Nε-protonated and His:Nε-deprotonated states were considered (Table 1). The size of the boxes was such that the distance of the proteins to the border was 1.4 nm or larger. The total charge of the systems, ranging from +4 to −2 (Table 1), was neutralized by adding Cl– or Na+ ions. Na+ and Cl– ions were finally added so as to reach a ionic strength of ∼0.1 mM (Table 1).
Table 1. Information on the Four Systems Simulated Herea.
| protein | state | protein [e] | Na+ | Cl– |
|---|---|---|---|---|
| NAF-1 | protonated | +4 | 22 | 26 |
| NAF-1 | deprotonated | +2 | 22 | 24 |
| mNT | protonated | 0 | 24 | 24 |
| mNT | deprotonated | –2 | 26 | 24 |
The protonation states of His87 in mNT and His114 in NAF-1, the number of Na+ and Cl– ions, and the total charge of the proteins are reported.
The AMBER force-field 99SB-ILDN28,29 was used for the protein frame (i.e., all of the protein except the metal cluster and coordinating residues) and for the ions. The [2Fe–2S] cluster and the coordinating residues parameters were taken from the QM parametrization. The TIP3P model was used for water molecules.
Periodic boundary conditions were applied. The electrostatic interactions were treated using particle mesh Ewald (PME) summation.36 The cutoff for the real part of the summation was 1.2 nm. The grid in the reciprocal space had a spacing of 0.1 nm–1. The cutoff for the van der Waals interactions was set to 1.2 nm. All of the bonds involving hydrogen atoms were constrained using the LINCS algorithm.37 The leap-frog integrator was used to integrate the equations of motions.38 Constant temperature and pressure conditions were obtained using the Nose–Hoover thermostat39,40 and Parrinello–Rahman barostat,41 respectively.
The systems were first energy-minimized using 20,000 steps of steepest descent algorithm. Then, water and the proteins side chains were gently heated up to 300 K in 2 ns molecular dynamics (MD).42,43 The backbone was also allowed to move. Overall, 2.5 μs MD simulations at room conditions (T = 300 K, P = 1 atm) were performed in the NPT ensemble for each system. The last 1.0 μs were collected for analysis.
Replica Exchange Solute Tempering (REST) in its new variant (REST2)44 was performed on mNT and NAF-1 without one or both [2Fe–2S] clusters and mNT with both [2Fe–2S] clusters. The setup was the same as that used for the MD except that the water box was larger, as the systems are expected to experience larger mobility. It was chosen to provide a space of 1.6 nm between the protein and the edges. The number of replicas we used is 32 for each calculation. The replica temperatures were chosen between 300 and 465 K to get the highest exchange rate in the first steps of the REST2. The same temperature set was chosen for the two proteins. The exchange between the replicas was attempted every thousand steps. We performed 0.45 and 0.40 μs long REST2 simulations for NAF-1 and mNT derivate models, respectively, whereas mNT with two [2Fe–2S] clusters was simulated for 80 ns. All of the simulations were carried out with the GROMACS-2016.336,45,46 program.
2.3. Data Analysis
MD representative structures were identified using the gmx cluster program36,45,46 applying the gromos algorithm.36,45,46 The cutoff distance, defined as the maximum allowed root mean square distances (RMSD) values between two structures belonging to the same cluster, were 0.095 and 0.150 nm for MD and REST2, respectively.
The protein angular dispersion (PAD) along the proteins torsion angles was calculated as in ref (47). H-bonds and salt bridges were identified using the Cpptraj tool from the ambertools-17 program.48 The standard deviation of the distance between the Cα of residues i,j through the simulations formed the i,j element of the standard deviation matrix (SDM).
We calculated the chemical shifts (CS) of the N, H, Cα, C′, and Cβ atoms of the proteins using the SHIFTX + tool of the SHIFTX249 package. The influence of each physical and geometrical parameter on the CS is available in the SI of ref (49).
The Fe–Nδ bond polarization was investigated in terms of Boys orbitals50 centroids. The quantum problem was here solved by using unrestricted B3LYP/g calculations51,52 and the 6-311G(2df,2pd) basis set. These estimations were performed on 150 frames extracted from the MD trajectories. From each frame, we selected Cys99 (in NAF-1, 72 in mNT), Cys101 (74), Cys110 (83), and His114 (87) side chains, along with the FeS cluster. Two water molecules H-bonding to Cys110 and His114 residues emerged from our simulations (see Results section). These molecules were included. At times, Lys81(55) replaced the water molecule H-bonding the histidine. Hence, this residue was included instead of the water molecule in additional models. The side chains were capped with H atoms in place of the Cαs.
The calculations were carried out in the absence and in the presence of the protein electrostatic field. The latter was calculated by using AMBER partial atomic charges28,29 placed on the atomic centres. The ORCA program was used.51,52
The Nδ–Fe bond polarization53 was also calculated using the natural bond orbital (NBO) theory.53 We used the same models as those used for the Boys orbitals, except that the H-bonding partners of the solvent exposed residues were not considered at the quantum level. The quantum problem was solved within unrestricted B3LYP34 calculations, with the 6-31g(df) basis set. Each bond orbital is the linear combination of atomic orbitals.53 The polarization coefficient of a bond (Δ) monitors the sharing of the electronic density between the atoms participating in the bond.53 Δ ranges from 0 to 100, and it depends on which atom is taken as reference.54 Δ ∼ 50 means that the NBO is covalent. Hence, the averaged values of the bond order are reported. The GAUSSIAN program33 was used. The Boys orbitals and NBO analysis were performed on 150 MD frames. The average values are here reported.
2.4. NMR Experimental
Uniformly, 15N-, 13C-labeled NAF-1 H114C mutant soluble domain was expressed and purified as described previously,31 with the exception that the cells were grown in M9 minimal media supplemented with 15N ammonium chloride and 13C glucose. All of the NMR experiments were performed on a Bruker Avance 600 MHz spectrometer equipped with a triple-resonance cryoprobe at 25 °C. The NMR sample contained 460 μM protein in a buffer 25 mM sodium phosphate pH 7 with 50 mM sodium chloride and 10% (v/v) D2O. A 1H–15N HSQC spectrum was collected followed by collection of a standard set of triple-resonance experiments used for backbone resonance assignments: HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, and HNCO. The NMR data were processed using NMRPipe55 and analyzed using Sparky.56
3. Results
3.1. Calculated Structural Features and Flexibility of NAF-1 and mNT Proteins: Comparison with Experimental Data
The force fields for mNT and NAF-1 [2Fe–2S] clusters are currently not available. They were here constructed to be compatible with the AMBER force field28 (see Sections 2.1 and S2.1). We have considered the more labile oxidized state of the cluster, containing two Fe(III) ions,16 present under oxidative stress conditions,15,17 often found in disease conditions. The histidine ligand, crucial for cluster release,8,9,16,57,58 can be protonated or deprotonated.58 Indeed, the pKa of the system is similar to that measured in rieske non-heme–iron protein in which the deprotonated state of the coordinating histidine was found at high pH.59 The pKa has been measured to be 6.9.60 Hence, we constructed a force field for the protonated and deprotonated states of the cluster-coordinating histidine (Tables S1–S3). The resulting parameters turned out to be rather similar for the two states.
To test the accuracy of our force field, we compared 2.5 μs canonical MD simulations of NAF-1 and mNT in both protonated states in explicit solvent (see Section 2.2) with X-ray and NMR data. The identity between simulated and X-ray structures’ secondary structure elements31 was high, ranging from 81 to 86% (Figure 1A). The secondary structure analyses and the root mean square distances (RMSD) of the Cα with respect to the X-ray structures,9,31 as a function of simulated time, supported these results (Figures S2 and S3). The calculated bond lengths and bond angles of the cluster compared well with those of the X-ray structures9,31 (Table S4 and Figure 1A). Consistently, the RMSD of the [2Fe–2S] cluster atoms were low (0.010 ± 0.005 nm for the cluster, and 0.020 ± 0.006 nm for cluster with its coordinating atoms). The NMR chemical shifts (CS) of backbone’s and Cβ’s atoms, calculated using the SHIFTX2 code49 from the MD trajectory, compared well with the corresponding experimental values (Figure 1B, see Section 2.4 for NMR experimental setup) for both protonation states. Analogously satisfactory results were obtained for mNT (Figure S4).61,62
Figure 1.

Structural and flexibility determinants of the NAF-1 protein in the His114:Nε protonated state. (A) Superposition of the main representative MD structure (cyan), as obtained by clusterization (see Section 2.3), with the corresponding X-ray structure (green, pdbID:4OO7). (B) Calculated N, H, Cα, and Cβ chemical shifts for monomers A (violet) and B (green) and corresponding experimental values. The differences were lower than the uncertainties associated with the root mean square error (error bars) of the program used for the chemical shift prediction (SHIFTX249). (C) Protein angular dispersion (PAD) values47 overlaid on the experimental Debye–Waller factors.31 Monomers A and B are colored in red and in blue, respectively. Similar results were obtained for the other systems simulated here (see Section S2.2 and Figure S5).
The proteins’ flexibility was described using the so-called MD-based Protein Angular Dispersion (PAD,47 see Figure 1C, for NAF-1 in its protonated state, Figure S5 for the three other systems). The PAD values are calculated for each peptide unit, and they range between 0 and 180°.47 The larger the values, the higher the local fluctuations. The local flexibilities of the two proteins turned out to be similarly low over most of their regions, including the cluster (Figure 1C). However, loop L2 (NAF-1: amino acid (aa) 89–93; mNT: aa 62–66) and the C-terminal domains (NAF-1: aa 132–135 and mNT: aa 105–108), feature high local flexibly (PAD values 40° or larger). This finding is consistent with the relatively high temperature factors reported in the X-ray structure.31 A comparison of NAF-1 MD structure with the X-ray one31 emphasized these two regions (Figure 2A). The structural fluctuations are also described by the standard deviation map (SDM) of the distances between the Cα of each pair residues of both monomers (Figure 2B). The SDM confirmed that the two regions experience relatively high local fluctuations. In contrast, the local flexibility of the N-terminal domain of NAF-1 was lower than that of these two domains (Figure 1C).
Figure 2.

Structure and conformational fluctuations of NAF-1 (A, B), semi-holo NAF-1 (C, D), and apo NAF-1 (E, F). (A, C, E) Superimposition of the X-ray structures (transparent) with several MD representatives, color coded as follows: blue: relatively rigid structure, PAD < 20°; white: low flexibility, 20° < PAD < 80°; red: high flexibility, PAD > 80°. (B, D, F) Standard deviation matrices of the three proteins. (C) The [2Fe–2S] cluster has been removed from monomer “B”. (F) A loss of off-diagonal blue squares pattern points to a partial unfolding of β-sheets (aa 84–88; 94–98; 128–132).
3.2. Effect of Cluster Absence on NAF-1’s Structure and Flexibility
With a high confidence in our simulation setup, we proceeded toward understanding the impact of the cluster(s) presence/absence on the protein’s global structure and conformational fluctuations. To this aim, comparison was made with sub-microsecond enhanced sampling REST244,63 simulations for the protein without the cluster in one monomer (monomer B in Figure 2C,D, “semi-holo NAF-1”) and without the clusters in both monomers (apo NAF-1, see Figure 2E,F). We used an analogous setup as for the previous MD (see Section 2.2). The histidine ligand was considered protonated. This condition is more representative for the proteins in acidic environment, which may favor cluster release in vivo.3
The removal of the cluster from monomer B led to (i) a complete unfolding of the α-helix in that monomer B (A) (aa 113–121), along with a large rearrangement of the L1 domain of monomer A. This is shown pictorially in Figure 2C. The secondary structure assignment confirmed that structural changes on one monomer could affect the structure of the other monomer (Figure S7C). This is probably due to intermonomer interactions. (ii) A higher flexibility of monomer B (in particular, of its cluster-binding domain), relative to that of monomer A, is shown by the SDM map (Figure 2D).
The absence of both clusters introduced additional structural disorder and flexibility (Figures 2E,F and S8). In particular, (i) the helical content decreased dramatically in both monomers (Figure 2E, Table S5) and (ii) the β-sheets (aa 84–88; 94–98; 128–132) experienced partial unfolding (Figures 2F and S8, Table S5).c The α-helices domains, along with the L1 loop, were the most flexible domains (Figure 2F). Similar results were obtained with mNT protein, in its semi-holo and apo states (see Section S2.3 and Figures S9 and S10).
3.3. Hydration of the Histidine Ligand and the His:Nδ–Fe Bond
We next focus our attention on the coordinating histidine. This residue, in its protonated state, forms an H-bond with a water molecule in both proteins (see chart in Figure 3A for NAF-1), as established by a calculation of the integral of His:Nε–water oxygen radial distribution functions (rdfs, see Figure S11). In the deprotonated states, instead, the analysis of the integral of rdfs along with the MD trajectory (Figure S11) shows that the H-bonded water molecule is replaced, at times, by a nearby lysine (Lys81 in NAF-1 and Lys 55 in mNT). However, the resulting residence times of the histidine ligand–water H-bond are very different (19 and 51%, for NAF-1 and mNT, respectively). An analysis of the interactions formed by the coordinating histidine (Tables S5, S6 and S9 and Figure S11) provided the molecular basis for this difference: in NAF-1, the lysine side chain is also engaged in an H-bond with the Asn115 side chain (Figure 4). This anchors the Lys residues nearby the histidine ligand, making it very poorly hydrated. This interaction does not exist in mNT (Figure 4), rendering the histidine a much better solvent exposed residue (see Section S2.4 for further details). We then studied the polarity and the strength of the His:Nδ–Fe bond. The latter is crucial for the labile nature of cluster.3,6,15−18 Bond polarity might play a key role for its reactivity, as we expect that the more polar the bond, the easier it is for it to break from the cluster. Bond polarity is here described in terms of location of the Boys orbitals centroids (BOCs),50,64 as calculated by the density functional theory (DFT) in the presence of the protein electric field (see Section 2.3). The closer the BOCs are to the Nδ donor atom, the more polar the bond is (Figure 3). The bond is clearly more polar in the protonated form of NAF-1. Notice that different BOCs values were obtained if some of the interacting partners (water molecules and/or Lys81) and/or the electrostatic fields from the protein frame were not included (table in Figure 3), pointing to the key and nontrivial role of the environment in tuning the electronic properties of the cluster (see Section S2.4). We describe bond strength in terms of bond orders,53,65 also calculated by DFT (see Tables S6 and S9). More facile ligand exchange in [2Fe–2S] proteins is observed in weaker bonds.66 Consistently with the BOCs analysis, the Fe–Nδ bond orders decreases (from 0.61 to 0.49, see Table S6) upon histidine protonation. Similar results are also obtained for mNT (Tables S7–S9).
Figure 3.

His:Nδ–Fe bond polarity in NAF-1. It is estimated from the distance between Nδ and BOCs, as obtained by spin-polarized DFT calculations (see Section 2.3). His114:Nε forms an H-bond with either water (B) or Lys81 (C), whereas His114:Nε–H forms H-bond only with water (A). Table (D) contains the BOCs distances from Nδ (Val) in the presence (yes) and the absence (no) of coordinating histidine Nε proton (His:Nε–H), nearest water molecules to the coordinating sites (Wat), Lys81 (Lys), and protein environment (Env). The distances are reported for both α and β electron populations (Pop). The H-bond column (where specified) indicates the H-bonding partner (Lys81 or water) of the Nε.
Figure 4.

Histidine hydration in NAF-1 (A) and mNT (B). The protonated histidine ligand forms a H-bond with water molecule. The deprotonated histidine residue forms an H-bond with a lysine side chain or with water (not shown). The lysine forms an additional H-bond with a residue close by the cluster (Asn115) in NAF-1, but it can do so neither with the equivalent structural position (Thr88) nor with other residues nearby the cluster.
4. Discussion and Conclusions
Our simulated structures show that the cluster-binding domain is fairly rigid (Figure 1C). However, in the β-cap domain, both the C-terminal and L2 domains are highly flexible. Our results (i) are consistent with the available experimental X-ray and NMR data (Figure 1) and (ii) show that the His:Nδ–Fe bond is more polar (and hence possibly more reactive) and weaker in the protonated state of both proteins, consistent with the experimentally observed increase in cluster liability upon protonation of the coordinating histidine.31,67 Overall, these results validated our simulation methodologies.
The absence of one cluster from one domain induced an unfolding of the cluster-binding domain and affected the L1 domain of the other monomer. The removal of the second cluster led the protein toward a loss of secondary structure folding, consistent with the observed loss in chemical shift dispersion and change in far-UV circular dichroism signal.61
Most importantly, our simulations identified the difference between NAF-1 Asn115 and mNT Thr88 as a key factor for the experimentally observed larger pH sensitivity of the mNT cluster relative to the NAF-1 one.8 Asn115 anchors Lys81 nearby the cluster, decreasing the solvent exposure of the histidine ligand. The electrostatic potential generated by the Lys residue is likely to prevent proton access, affecting the protonation reaction of the histidine. In mNT, because of the replacement of Asn115 with Thr88, the lysine residue is more mobile and the histidine is more hydrated.
Preventing aberrant cluster loss using small molecules is of paramount importance for a variety of pharmaceutical applications. The knowledge of the key structural modifications upon the loss of the clusters and of the structural determinants playing a role in cluster pH sensitivity, as emerging from this study, are instrumental for the design of future drugs that differentially target these clusters. Based on our molecular dynamics setup, cluster-docking ligands might be developed using free energy approaches (such as metadynamics). By affecting NEET proteins’ cluster lability/stability, these ligands may interfere with cell derangement associated to a variety of diseases, including cancer.
Acknowledgments
R.N., R.M., and P.A.J. acknowledge the support of NSF-MCB-1613462 (R.M.), BSF Grant 2015831 (R.N.), and NIH Grant GM101467 (P.A.J.). We acknowledge the computing time granted by JARA-HPC and very fruitful discussions with Prof. Maria Ramos.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.7b10584.
Parametrization of the NEET cluster/binding domain, additional results on NEET structures by MD and REST2 and additional details on the cluster-binding domain (PDF)
The authors declare no competing financial interest.
Footnotes
The corresponding residues for NAF-1 are Cys99, Cys101, Cys110, and His114 for the coordination, and the Arg100, Asn111, Gly112, and Ser113 are considered as glycine. This provides us with the same model as that for mNT.
The structural properties in the two domains differ (Figure 2F,E). This was ascribed to a lack of full convergence of the simulation of the highly flexible apo NAF-1. In spite of this caveat, simulations did provide a clear-cut information: the large unfolding of the protein.
Supplementary Material
References
- Chang N. C.; Nguyen M.; Germain M.; Shore G. C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010, 29, 606–618. 10.1038/emboj.2009.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley S. E.; Murphy A. N.; Ross S. A.; van der Geer P.; Dixon J. E. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 5318–5323. 10.1073/pnas.0701078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir S.; Paddock M. L.; Darash-Yahana-Baram M.; Holt S. H.; Sohn Y. S.; Agranat L.; Michaeli D.; Stofleth J. T.; Lipper C. H.; Morcos F.; Cabantchik I. Z.; Onuchic J. N.; Jennings P. A.; Mittler R.; Nechushtai R. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 1294–1315. 10.1016/j.bbamcr.2014.10.014. [DOI] [PubMed] [Google Scholar]
- Sohn Y. S.; Tamir S.; Song L. H.; Michaeli D.; Matouk I.; Conlan A. R.; Harir Y.; Holt S. H.; Shulaev V.; Paddock M. L.; Hochberg A.; Cabanchick I. Z.; Onuchic J. N.; Jennings P. A.; Nechushtai R.; Mittler R. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 14676–14681. 10.1073/pnas.1313198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt S. H.; Darash-Yahana M.; Sohn Y. S.; Song L. H.; Karmi O.; Tamir S.; Michaeli D.; Luo Y. T.; Paddock M. L.; Jennings P. A.; Onuchic J. N.; Azad R. K.; Pikarsky E.; Cabantchik I. Z.; Nechushtai R.; Mittler R. Activation of apoptosis in NAF-1-deficient human epithelial breast cancer cells. J. Cell Sci. 2016, 129, 155–165. 10.1242/jcs.178293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley S. E.; Paddock M. L.; Abresch E. C.; Gross L.; van der Geer P.; Nechushtai R.; Murphy A. N.; Jennings P. A.; Dixon J. E. The outer mitochondrial membrane protein mitoNEET contains a novel redox-active 2Fe-2S cluster. J. Biol. Chem. 2007, 282, 23745–23749. 10.1074/jbc.C700107200. [DOI] [PubMed] [Google Scholar]
- Inupakutika M. A.; Sengupta S.; Nechushtai R.; Jennings P. A.; Onuchic J. N.; Azad R. K.; Padilla P.; Mittler R. Phylogenetic analysis of eukaryotic NEET proteins uncovers a link between a key gene duplication event and the evolution of vertebrates. Sci. Rep. 2017, 7, 42571 10.1038/srep42571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlan A. R.; Axelrod H. L.; Cohen A. E.; Abresch E. C.; Zuris J.; Yee D.; Nechushtai R.; Jennings P. A.; Paddock M. L. Crystal structure of Miner1: The redox-active 2Fe-2S protein causative in Wolfram Syndrome 2. J. Mol. Biol. 2009, 392, 143–153. 10.1016/j.jmb.2009.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddock M. L.; Wiley S. E.; Axelrod H. L.; Cohen A. E.; Roy M.; Abresch E. C.; Capraro D.; Murphy A. N.; Nechushtai R.; Dixon J. E.; Jennings P. A. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 14342–14347. 10.1073/pnas.0707189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darash-Yahana M.; Pozniak Y.; Lu M.; Sohn Y. S.; Karmi O.; Tamir S.; Bai F.; Song L.; Jennings P. A.; Pikarsky E.; Geiger T.; Onuchic J. N.; Mittler R.; Nechushtai R. Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 10890–10895. 10.1073/pnas.1612736113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L.; Youdim M. B. H.; Riederer P.; Connor J. R.; Crichton R. R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Valko M.; Morris H.; Cronin M. T. D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- Zuo L.; Motherwell M. S. The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene 2013, 532, 18–23. 10.1016/j.gene.2013.07.085. [DOI] [PubMed] [Google Scholar]
- Uttara B.; Singh A. V.; Zamboni P.; Mahajan R. T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir S.; Zuris J. A.; Agranat L.; Lipper C. H.; Conlan A. R.; Michaeli D.; Harir Y.; Paddock M. L.; Mittler R.; Cabantchik Z. I.; Jennings P. A.; Nechushtai R. Nutrient-deprivation autophagy factor-1 (NAF-1): biochemical properties of a novel cellular target for anti-diabetic drugs. PLoS One 2013, 8, e61202 10.1371/journal.pone.0061202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicus M. M.; Conlan A.; Nechushtai R.; Jennings P. A.; Paddock M. L.; Britt R. D.; Stoll S. Binding of Histidine in the (Cys)3(His)1-Coordinated [2Fe-2S] Cluster of Human mitoNEET. J. Am. Chem. Soc. 2010, 132, 2037–2049. 10.1021/ja909359g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuris J. A.; Harir Y.; Conlan A. R.; Shvartsman M.; Michaeli D.; Tamir S.; Paddock M. L.; Onuchic J. N.; Mittler R.; Cabantchik Z. I.; Jennings P. A.; Nechushtai R. Facile transfer of [2Fe-2S] clusters from the diabetes drug target mitoNEET to an apo-acceptor protein. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 13047–13052. 10.1073/pnas.1109986108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai F.; Morcos F.; Sohn Y. S.; Darash-Yahana M.; Rezende C. O.; Lipper C. H.; Paddock M. L.; Song L.; Luo Y.; Holt S. H.; Tamir S.; Theodorakis E. A.; Jennings P. A.; Onuchic J. N.; Mittler R.; Nechushtai R. The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 as chemotherapeutic targets in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 3698–3703. 10.1073/pnas.1502960112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter E. L.; Zuris J. A.; Wang C.; Phu L. T. V.; Herbert L. A.; Cohen A. E.; Paddock M. L.; Nechushtai R.; Onuchic J. N.; Jennings P. A. Allosteric control in a metalloprotein dramatically alters function. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 948–953. 10.1073/pnas.1208286110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtai R.; Lammert H.; Michaeli D.; Eisenberg-Domovich Y.; Zuris J. A.; Luca M. A.; Capraro D. T.; Fish A.; Shimshon O.; Roy M.; Schug A.; Whitford P. C.; Livnah O.; Onuchic J. N.; Jennings P. A. Allostery in the ferredoxin protein motif does not involve a conformational switch. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 2240–2245. 10.1073/pnas.1019502108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Carloni P.; Luchinat C.; Orioli P. L. Molecular dynamics simulations on HiPIP from Chromatium vinosum and comparison with NMR data. J. Am. Chem. Soc. 1992, 114, 10683–10689. 10.1021/ja00053a001. [DOI] [Google Scholar]
- Dance I.Computational Methods for Metal Sulfide Clusters. In Transition Metal Sulfur Chemistry: Biological and Industrial Significance; Stiefel E. I., Matsumoto K., Eds.; Exxon Research & Engineering, 1996; Vol. 653, pp 135–152. [Google Scholar]
- Stich T. A.; Seravalli J.; Venkateshrao S.; Spiro T. G.; Ragsdale S. W.; Brunold T. C. Spectroscopic studies of the corrinoid/iron-sulfur protein from Moorella thermoacetica. J. Am. Chem. Soc. 2006, 128, 5010–5020. 10.1021/ja054690o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K. P.; Ooi B. L.; Christensen H. E. M. Computational Chemistry of Modified MFe3S4 and M2Fe2S4 Clusters: Assessment of Trends in Electronic Structure and Properties. J. Phys. Chem. A 2008, 112, 12829–12841. 10.1021/jp8014782. [DOI] [PubMed] [Google Scholar]
- Tran V. T.; Hendrickx M. F. A. Assignment of the Photoelectron Spectra of FeS3- by Density Functional Theory, CASPT2, and RCCSD(T) Calculations. J. Phys. Chem. A 2011, 115, 13956–13964. 10.1021/jp208824b. [DOI] [PubMed] [Google Scholar]
- Marinoni E. N.; de Oliveira J. S.; Nicolet Y.; Raulfs E. C.; Amara P.; Dean D. R.; Fontecilla-Camps J. C. (IscS-IscU)2 Complex Structures Provide Insights into Fe2S2 Biogenesis and Transfer. Angew. Chem., Int. Ed. 2012, 51, 5439–5442. 10.1002/anie.201201708. [DOI] [PubMed] [Google Scholar]
- Liu P.; Kim B.; Friesner R. A.; Berne B. J. Replica exchange with solute tempering: a method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 13749–13754. 10.1073/pnas.0506346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorin E. J.; Pande V. S. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys. J. 2005, 88, 2472–2493. 10.1529/biophysj.104.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho A. T.; Teixeira A. F.; Ramos M. J. Parameters for molecular dynamics simulations of iron-sulfur proteins. J. Comput. Chem. 2013, 34, 1540–1548. 10.1002/jcc.23287. [DOI] [PubMed] [Google Scholar]
- Tamir S.; Eisenberg-Domovich Y.; Conlan A. R.; Stofleth J. T.; Lipper C. H.; Paddock M. L.; Mittler R.; Jennings P. A.; Livnah O.; Nechushtai R. A point mutation in the [2Fe-2S] cluster binding region of the NAF-1 protein (H114C) dramatically hinders the cluster donor properties. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 1572–1578. 10.1107/S1399004714005458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M. B.; Yang Y.; Wang B.; Fuesti-Molnar L.; Weaver M. N.; Merz K. M. Jr. Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. 10.1021/ct1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision E.01; Gaussian, Inc.: Wallingford, CT, 2009.
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Giammona D. A. Ph.D. Thesis, University of California, Davis, 1984. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. . [DOI] [Google Scholar]
- Hockney R. W.; Goel S. P.; Eastwood J. W. Quiet high-resolution computer models of a plasma. J. Comput. Phys. 1974, 14, 148–158. 10.1016/0021-9991(74)90010-2. [DOI] [Google Scholar]
- Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. 10.1080/00268978400101201. [DOI] [Google Scholar]
- Hoover W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. 10.1103/PhysRevA.31.1695. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Černý V. Thermodynamical approach to the traveling salesman problem: An efficient simulation algorithm. J. Optim. Theory Appl. 1985, 45, 41–51. 10.1007/BF00940812. [DOI] [Google Scholar]
- Kirkpatrick S.; Gelatt C. D.; Vecchi M. P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- Wang L.; Friesner R. A.; Berne B. J. Replica exchange with solute scaling: a more efficient version of replica exchange with solute tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. 10.1021/jp204407d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Vanderspoel D.; Vandrunen R. GROMACS - A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Caliandro R.; Rossetti G.; Carloni P. Local Fluctuations and Conformational Transitions in Proteins. J. Chem. Theory Comput. 2012, 8, 4775–4785. 10.1021/ct300610y. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Cerutti D. S.; Cheatham T. III; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Greene D.; Homeyer N.; Izadi S.; Kovalenko A.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Mermelstein D.; Merz K. M.; Monard G.; Nguyen H.; Omelyan I.; Onufriev A.; Pan F.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Simmerling C. L.; Botello-Smith W. M.; Swails J.; Walker R. C.; Wang J.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 2017; University of California: San Francisco, 2017.
- Han B.; Liu Y. F.; Ginzinger S. W.; Wishart D. S. SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 2011, 50, 43–57. 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster J. M.; Boys S. F. Canonical Configurational Interaction Procedure. Rev. Mod. Phys. 1960, 32, 300–302. 10.1103/RevModPhys.32.300. [DOI] [Google Scholar]
- Arif W.; Xu S.; Isailovic D.; Geldenhuys W. J.; Carroll R. T.; Funk M. O. Complexes of the outer mitochondrial membrane protein mitoNEET with resveratrol-3-sulfate. Biochemistry 2011, 50, 5806–5811. 10.1021/bi200546s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neese F. The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Reed A. E.; Curtiss L. A.; Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. 10.1021/cr00088a005. [DOI] [Google Scholar]
- Reed A. E.; Weinhold F. Natural bond orbital analysis of near-Hartree-Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. 10.1063/1.445134. [DOI] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Goddard T. D.; Kneller D. G.. SPARKY 3; University of California: San Francisco, 2008.
- Bak D. W.; Elliott S. J. Conserved hydrogen bonding networks of MitoNEET tune Fe-S cluster binding and structural stability. Biochemistry 2013, 52, 4687–4696. 10.1021/bi400540m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuris J. A.; Halim D. A.; Conlan A. R.; Abresch E. C.; Nechushtai R.; Paddock M. L.; Jennings P. A. Engineering the Redox Potential over a Wide Range within a New Class of FeS Proteins. J. Am. Chem. Soc. 2010, 132, 13120–13122. 10.1021/ja103920k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirrell T. F.; Paddock M. L.; Conlan A. R.; Smoll E. J. Jr.; Nechushtai R.; Jennings P. A.; Kim J. E. Resonance Raman studies of the (His)(Cys)3 2Fe-2S cluster of MitoNEET: comparison to the (Cys)4 mutant and implications of the effects of pH on the labile metal center. Biochemistry 2009, 48, 4747–4752. 10.1021/bi900028r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak D. W.; Zuris J. A.; Paddock M. L.; Jennings P. A.; Elliott S. J. Redox characterization of the FeS protein MitoNEET and impact of thiazolidinedione drug binding. Biochemistry 2009, 48, 10193–10195. 10.1021/bi9016445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Lin J.; Feng Y.; Wang J. Binding of reduced nicotinamide adenine dinucleotide phosphate destabilizes the iron-sulfur clusters of human mitoNEET. Biochemistry 2010, 49, 9604–9612. 10.1021/bi101168c. [DOI] [PubMed] [Google Scholar]
- Jennings P. A. Personal communication, 2016.
- Bussi G. Hamiltonian replica exchange in GROMACS: a flexible implementation. Mol. Phys. 2014, 112, 379–384. 10.1080/00268976.2013.824126. [DOI] [Google Scholar]
- Alber F.; Folkers G.; Carloni P. Dimethyl phosphate: Stereoelectronic versus environmental effects. J. Phys. Chem. B 1999, 103, 6121–6126. 10.1021/jp9901038. [DOI] [Google Scholar]
- Mayer I. Bond order and valence indices: A personal account. J. Comput. Chem. 2007, 28, 204–221. 10.1002/jcc.20494. [DOI] [PubMed] [Google Scholar]
- Bergner M.; Roy L.; Dechert S.; Neese F.; Ye S.; Meyer F. Ligand Rearrangements at Fe/S Cofactors: Slow Isomerization of a Biomimetic [2Fe-2S] Cluster. Angew. Chem., Int. Ed. 2017, 56, 4882–4886. 10.1002/anie.201612621. [DOI] [PubMed] [Google Scholar]
- Conlan A. R.; Paddock M. L.; Homer C.; Axelrod H. L.; Cohen A. E.; Abresch E. C.; Zuris J. A.; Nechushtai R.; Jennings P. A. Mutation of the His ligand in mitoNEET stabilizes the 2Fe-2S cluster despite conformational heterogeneity in the ligand environment. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 516–523. 10.1107/S0907444911011577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.