

The precise placement of the PTPK motif leads to relaxed substrate specificity and double methylation capability of PaTrm5a.
Abstract
The wyosine derivatives present at position 37 in transfer RNAs (tRNAs) are critical for reading frame maintenance. The methyltransferase Trm5a from Pyrococcus abyssi (PaTrm5a) plays a key role in this hypermodification process in generating m1G37 and imG2, two products of the wyosine biosynthetic pathway, through two methyl transfers to distinct substrates, but the mechanism is currently unknown. We report two cocrystal structures of PaTrm5a in complex with tRNAPhe and reveal the structural basis for substrate recognition, which was supported by in vitro activity assays. The crystal structures showed that the D1 domain of the enzyme undergoes large conformational changes upon the binding of tRNA. The deletion of this domain greatly reduces the affinity and activity of PaTrm5a toward its RNA substrate, indicating that the enzyme recognizes the overall shape of tRNA. Using the small-angle x-ray scattering technique and crystallographic analysis, we discovered that PaTrm5a adopts distinct open conformations before and after the binding of tRNA. Last, through structure comparison with its ortholog Methanococcus jannaschii Trm5b (MjTrm5b), we propose a reaction mechanism for the double methylation capability of this unique enzyme.
INTRODUCTION
Mature transfer RNA (tRNA) contains many modified nucleosides, and tRNA is the most extensively modified RNA type. These diverse modifications serve various purposes including ensuring that translational fidelity and are functionally important (1–3). In tRNA, positions 34 and 37 usually contain highly modified nucleosides (hypermodifications), and their generation usually requires a series of enzymatic steps (4, 5). The tricyclic wyosine derivatives are an example of these hypermodifications in eukaryotes and archaea, and they are found exclusively at position G37 of tRNAPhe, 3′ adjacent to the anticodon (6, 7). These modification products are important in reading frame maintenance during protein synthesis, because the hypomodification on G37 leads to elevated error rates in frameshifting (8). In eukarya and archaea, the first biosynthetic step of wyosine derivatives is initiated by a key enzyme named Trm5, which produces m1G37 (9, 10). Trm5 is an S-adenosine-l-methionine (SAM)–dependent tRNA methyltransferase, and it belongs to the class I methyltransferase family (11). In eukarya, the second step is catalyzed by the flavin-binding enzyme Tyw1, which uses a radical-dependent mechanism to form imG-14. The following steps are completed by other members named Tyw2 to Tyw4 from the Tyw enzyme family, which generate yW-86, yW-72, and the final product yW, respectively (fig. S1). Archaea also contain wyosine derivatives, but the modification scenarios and diversity of the products are much greater than those of eukaryotes (Fig. 1A) (9, 12), and the corresponding pathways are still poorly understood.
Fig. 1. The overall structure of the PaTrm5a-tRNAPhe-SAH ternary complex and the multiple sequence alignment of the representatives from other species.
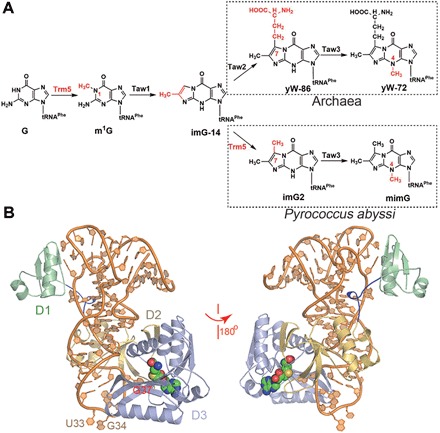
(A) Biosynthetic pathways for wyosine derivatives in archaea including P. abyssi. Note that the same enzyme Trm5 performs two methyltransfers and is marked in red. (B) The front and back views of the complex in ribbon rendition. The D1, D2, and D3 domains are colored pale green, yellow orange, and light blue, respectively. The active site SAM is shown as spheres, whereas tRNA is in orange. The linker connecting D1 and D2 is colored blue.
Phylogenetic distribution analyses of the trm5 genes revealed that three Trm5 subfamilies exist in archaea, and they are subsequently named Trm5a, Trm5b, and Trm5c. Recently, Trm5a from the archaeon Pyrococcus abyssi (PaTrm5a) has been identified and characterized (9). PaTrm5a, but not PaTrm5b, is a unique enzyme in that it catalyzes two methyltransfer reactions on G37 for the tRNAPhe hypermodifications: In the first reaction, it transfers a methyl group to the N1 atom of G37 to produce m1G, whereas in the second reaction, it further methylates the C7 atom of imG-14 to generate imG2, an intermediate along the pathway (Fig. 1A) (12). Therefore, PaTrm5a replaces the enzyme Tyw2 in eukarya and plays double roles in the mimG biosynthesis. The structural basis of the bifunctional substrate specificity of PaTrm5a is of interest but unclear at present.
The first crystal structures of Trm5 were of the Trm5b subfamily, from the methanogenic archaeon Methanococcus jannaschii (MjTrm5b). MjTrm5b in complex with the methyl donor analog sinefungin or adenosine displays three structural domains named D1, D2, and D3 [Protein Data Bank (PDB) ID: 2YX1 and 3AY0] (13, 14). D3 displays the signature Rossmann fold at the C terminus for catalysis and makes contacts with both D1 and D2. In contrast, D1 is poorly conserved and is connected to D2 by a flexible linker. There is a consensus NPPY motif located in the fourth β strand of the Rossmann fold, which is considered to play a role in positioning the nitrogen atom of G37 (14). Goto-Ito et al. (15) followed up on this work and determined the crystal structures of MjTrm5b complexed with the substrates tRNALeu and tRNACys (PDB ID: 2ZZM and 2ZZN). In the tRNA-bound structures, D1 acts like a clamp and makes contacts with the outer corner of tRNA. The catalytic domain (composed of D2 and D3) catalyzes the methyltransfer reaction by recognizing G37 and the anticodon loop. In particular, D3 stabilizes the methyl donor SAM during its binding and interacts with SAM at the active site by hydrogen bonds using Asp223, Asp251, and Val252, whereas Phe203 and Ile224 play an additional role by stacking interactions (15). MjTrm5b recognizes tRNAs through their L-shaped tertiary structures rather than specific sequences (15). Therefore, Trm5 has been speculated to be a checkpoint enzyme in the tRNA maturation process.
We previously determined the crystal structures of apo-PaTrm5a and PaTrm5a in complex with various ligands at high resolution (16). We discovered that PaTrm5a in these structures displays an extended conformation (or the “open” state), distinct from the well-folded MjTrm5b. To find out whether this extended conformation is the natural conformation in solution, we conducted the fluorescence resonance energy transfer (FRET) experiment, which was consistent with the crystal structures that we determined (16). However, these tRNA-free structures provided limited insights into the methylation mechanism of PaTrm5a, especially on the recognition of tRNA substrate. Here, we cocrystallized PaTrm5a in complexes with tRNAPhe in the presence of the methyl donor SAM and its demethylated product S-adenosyl-l-homocysteine (SAH), which reveal the structural basis of PaTrm5a for tRNA recognition. The interaction details were further supported by methyltransfer activity assays and electrophoretic mobility shift assay (EMSA) experiments. Using the small-angle x-ray scattering (SAXS) technique, we confirmed that PaTrm5a constantly adopts the open conformation in solution, a result that may be attributed to the general flexibility of D1. Last, by structure comparison of PaTrm5a with MjTrm5b, we proposed a reaction mechanism of the double methylation capability of this unique enzyme.
RESULTS AND DISCUSSION
Overall structure of the PaTrm5a-tRNAPhe-SAH complex
In the complex structure of PaTrm5a-tRNAPhe-SAH, there are two protein monomers in the asymmetric unit, each bound by a molecule of tRNA and SAH (PDB ID: 5WT1). Both the protein and tRNA molecules are free of internal disorders. In the final model, the two protein monomers are visible from Met1 to Leu332 (chain A and chain B), whereas the two tRNA molecules are structured from G4-C69 (chain C) and G3-C69 (chain D) (Fig. 1B and fig. S2). The two ternary complexes are very similar to each other, and they are related to each other by a translation of ~64 Å (fig. S3). The tRNA molecules maintain the regularly inverted “L” conformation. PaTrm5a makes contacts with tRNA mainly through two areas: The D1 domain (Met1-Pro60; fig. S4) interacts with the elbow region of tRNA, whereas the D3 domain (Lys163-Ser333) interacts with the anticodon loop region. Of these contacts, most interactions are lysine- or arginine-mediated salt bridges, which form electrostatic interactions with the backbone of tRNA (table S1).
Enzyme-substrate interactions in the catalytic domain
The D3 domain features the characteristic SAM-binding active site (the Rossmann fold) and contains the necessary functional elements for the methyltransfer reaction. SAH occupies a similar position to the tRNA-free structure (PDB ID: 5HJK) (16), and the N1 and N6 atoms of the adenine moiety each form a hydrogen bond with conserved Asp243 and Val244 from D3, respectively (Fig. 2A). The base ring is inserted between Phe191 and Ile214, and it is almost perpendicular to the phenyl ring of Phe191. The 2′ and 3′ hydroxyls of the ribose are fixed by three hydrogen bonding contacts with the carboxylate oxygens of Glu213. In addition, the amino group of the methionyl moiety donates a hydrogen bond to the main-chain carbonyl of Phe191, whereas the carboxyl group forms a unidentate salt bridge with Arg174.
Fig. 2. Substrate interactions with the catalytic region in the PaTrm5a-tRNAPhe-SAH ternary complex.
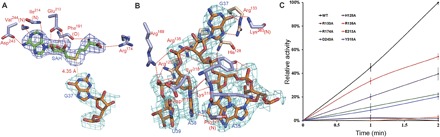
(A) The recognition pattern of SAH. The residues participating in ligand recognition are depicted in a ball-and-stick model and labeled. The hydrogen bonds are shown by the red dashed lines (distance, <3.6 Å). The distance of the S atom of SAH to N1 of G37 is shown by the yellow dashed line. (B) The recognition of the anticodon bases A35-U39. (C) Time course of the relative methyltransfer activities of PaTrm5a and mutants that are involved in substrate recognition. The measurements were made at 1- and 2-min time points. The activity of wild-type (WT) PaTrm5a at the 2-min time point was normalized to 100%, and the readings at time point zero were used as blanks. Error bars represent SD calculated from at least three measurements. All the 2Fo − Fc maps are contoured at 1σ.
The anticodon interactions mostly concentrate on the A36-G37-A38 triplet, burying a surface area of 1913.6 Å2. Except for specific contacts on the G37 and A38 bases, all the other interactions are on the backbone or the riboses of tRNA. U33, G34, and G37 are flipped out, and the backbone of tRNA experiences unwinding to a certain extent. Conformations of these bases are quite different from their counterparts in free, unmodified tRNAPhe of Escherichia coli (PDB ID: 3L0U) (17), which stay in the anticodon loop. Notably, G34 forms base-base contacts with the SAH molecule from a symmetry-related molecule. There are two consecutive adenosines (A35 and A36) adjacent to the to-be-modified G37, and these two adenine rings stack onto each other (Fig. 2B). A36 forms three nonspecific hydrogen bonds with Tyr318, with its 2′ hydroxyl group contacting the main-chain carbonyl and its nonbridging phosphate groups interacting with the side-chain hydroxyl of Tyr318. The specific recognition of G37 is mainly accomplished by three hydrogen bonds: While the O6 atom of the guanine ring accepts two hydrogen bonds from the Lys263 backbone and the Nε atom from Arg133, the N7 atom of the guanine ring forms another hydrogen bond with the NH2 atom of Arg133. On the other hand, the nonspecific recognition of G37 is mediated by His128 and Arg135: The former makes two hydrogen bonds with the phosphate oxygen and the 2′ hydroxyl group of G37, respectively, and the latter forms two salt bridges with the nonbridging oxygens of G37 and A38, respectively. His128, Arg135, and Arg125 partially stack onto each other. Their side chains are parallel, and each residue interacts with adjacent phosphate oxygens of tRNA (Fig. 2B).
To validate our structural model, we performed methyltransfer activity assays to assess the contribution of each individual residue to enzymatic activity. We first discovered that in the presence of 200 nM WT PaTrm5a, the initial velocity rate remains linear for up to 5 min under our assay conditions. Therefore, we chose two time points (1 and 2 min) for measurements so that the reaction stage is well within the linear range. Compared to WT, whose activity was normalized to 100% for the 2-min time point, all the mutants showed reduced activities to various extents. Particularly, the mutation of Arg133 to an alanine eliminated activity, indicating the absolute importance of specific recognition of G37 to catalysis (Fig. 2C). Mutation of Glu213 and Asp243—the two residues for SAH recognition to alanines—left the enzyme with only 2 to 3% activity of that of WT at the 2-min reaction state, suggesting that the recognition of the methyl donor is also key for the reaction to proceed. Arg174 only forms a single hydrogen bond with the terminal carboxylate of SAH, and its mutation led to less severe impairment to activity (22%). Compared to the specific interactions, the phosphate backbone interactions appear to play a less significant role. Except for Tyr318, which retains 20% activity, the other mutants generally retained 30 to 50% activity. The least important residue is Arg135, which makes two hydrogen bonds with the phosphate oxygens of G37 and A38, and its mutation only resulted in ~50% activity loss.
Conformational changes
The overall fold of the protein in the ternary complex is quite different from our previously solved tRNA-free structures (PDB ID: 5HJK) (16) as a result of protein-RNA interactions. First of all, the D1 domain exhibits the largest movements. The entire domain moves as a rigid body without changing its general shape (Fig. 3A). These conformational changes allow D1 to “grab” tRNA by its elbow region through specific and nonspecific interactions, which will be detailed in the next section. In addition, the Lys319-Pro320 dipeptide in D3 moves away to avoid the steric clashes with the C32-A38 base pair due to the insertion of the tRNA (Fig. 3A). Meanwhile, the D2 domain also moves correspondingly, but the shift is relatively small. The disordered Ser129Gly130 dipeptide in the apo structure becomes well ordered upon the formation of the complex and shows clear density for this region. G37 flips out and inserts itself into a narrow hydrophobic pocket formed by Tyr318, Phe165, Phe284, Met170, and Pro262, which leads to the rearrangements of the side chain of Arg133 (Fig. 3A).
Fig. 3. Structural changes upon the formation of complex.
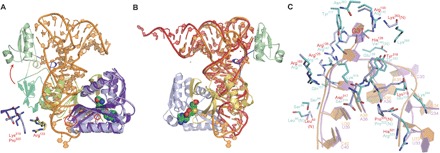
(A) Structure comparison of the enzyme in complex with tRNA (PDB ID: 5WT1; coloring scheme as in Fig. 1A) with the tRNA-free form (PDB ID: 5HJK; the three domains are colored green, yellow, and purple). The structural changes for Lys319Pro320 and Arg133 before and after the binding of tRNA are indicated by the red circles and enlarged in the lower left corner. The tRNA molecule is depicted in orange. (B) tRNA structural changes compared to yeast tRNAPhe. PatRNAPhe is superimposed onto the canonical, free yeast tRNAPhe (PDB ID: 4TNA). The view angle is rotated 180° with respect to that in (A). (C) A detailed comparison of the structural differences between the MjTrm5b-tRNACys (cyan, PDB ID: 2ZZN) and the PaTrm5a-tRNAPhe (orange, PDB ID: 5WT1) complexes.
In terms of tRNA, the anticodon stem undergoes stretching to some extent, and the loop unwinds as well, when compared to the normal anticodon structure of free tRNAPhe (Fig. 3B). The purpose of these rearrangements is to reduce the steric clashes with nucleotides A35-G37 and also to maximize the contacts at the enzyme active site.
To compare the tRNA-recognition differences between PaTrm5a and MjTrm5b, we superimposed the two tRNA-bound structures (PDB ID: 2ZZN and 5WT1) (15), and the two proteins could be aligned with a root mean squared deviation of 2.55 Å over 283 Cαs. We discovered that although the two structures resemble each other, the tRNAs are quite different in positions 33 to 35 within the anticodon loop region. All the three bases of U33-C35 in tRNACys (complexed with MjTrm5b) are flipped out (Fig. 3C). U33 and G34 of tRNAPhe in our structure are both capable of forming base-specific contacts with part of another symmetry mate including the SAH ligand (fig. S5). In tRNAPhe, all four bases at positions 34 to 37 are purines, and A35 and A36 stack onto each other. Although both tRNAPhe and tRNACys harbor an adenine at positions 36, only A36 in tRNAPhe can form a hydrogen bond with the phosphate of G34. This G34A35A36 motif is similar to the tetraloop GNRA that exists in the group I intron, except that the GA base pairing in the latter is replaced by the PO43−(G34)-A hydrogen bond interactions. Therefore, the adenine ring of A36 is locked within the loop along with A35 because of their stacking interactions. In contrast, the base at position 35 of tRNACys in M. jannaschii is a cytosine, which is much weaker in forming stacking interactions with A36. Consequently, C35 is flipped out of the loop in the tRNACys-bound complex. On the other hand, the guanine at position 37 in tRNACys preserves the same type of interactions with the conserved arginine-Arg145 from D2 (or Arg133 in PaTrm5a), but G37 makes two extra hydrogen bonds by interacting with Asn263 and Tyr177 (Fig. 3C). These hydrogen bonding interactions are lost because of the replacement by an isoleucine and a phenylalanine, respectively (fig. S4).
One should note that tRNA transcript is used in this study. Complicated modifications present in real tRNAs may influence their structures and recognition modes by tRNA modification enzymes. Although PaTrm5a modifies unmodified PatRNAPhe transcript quite well, minor differences due to the lack of tRNA modifications might still exist.
The flexibility of the D1 domain
The D1 domain is an α/β-fold, formed by a three-stranded antiparallel β sheet packed against two α helices, burying a surface area of 896.0 Å2. The contacts are mainly on the G19-C56 base pair and the G20 base. The two bases both form hydrogen bonds with the enzyme: C56 forms hydrogen bonds with the backbone of Arg8 and Lys12, whereas G19 forms three hydrogen bonds with Arg32 and Lys12 (Fig. 4A). In addition, there are also two salt bridges on the bridging and nonbridging oxygen of C19 (table S1). G20 adds eight more hydrogen bonds, six of which are base-specific contacts.
Fig. 4. The flexibility of the D1 domain.
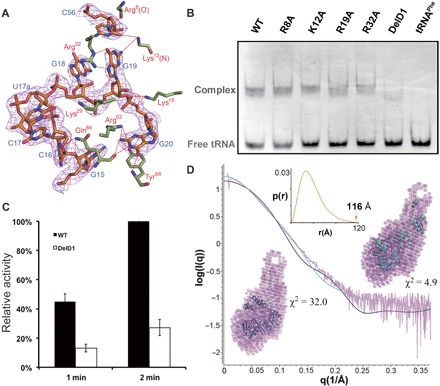
(A) The specific interactions involving the G19-C56 base pair at the outer corner of tRNA. The 2Fo − Fc map is contoured at 1σ. (B) EMSA of the WT, DelD1, and single mutants with tRNAPhe. (C) The methyltransfer activity of the DelD1 mutant. (D) SAXS results of the apoprotein, including the simulated SAXS profiles of the tRNA-free MjTrm5b structure (blue, PDB ID: 2YX1) and the tRNA-free PaTrm5a structure (cyan, PDB ID: 5HJJ), along with the experimental SAXS data (pink). The MjTrm5b (blue) and the PaTrm5a structures (cyan) were superimposed onto their respective DR model (pink) using PyMOL. The P(r) distance distribution function is shown in the inset.
The D1 domain of MjTrm5b transitions from its original state to an open state upon the binding of tRNA. The α1 helix grabs the outer corner of tRNA and holds it in place. We deleted the D1 domain of PaTrm5a, carried out the EMSA assay, and discovered that the binding affinity of the truncated protein (DelD1) was much lower than that of the full-length WT protein (~60%), whereas the single-point mutants R8A, K12A, K19A, and R32A showed no significant difference (Fig. 4B). Furthermore, our activity measurements mirrored the EMSA results, with about one-quarter activity being retained in the methyltransfer reaction for the deletion mutant (Fig. 4C). The large affinity loss as evidenced by the D1 deletion, as compared to the insignificant decrease in affinity exhibited by the specific single mutations, indicated that Trm5 recognizes the overall shape by contacting its outer corner, consistent with the notion of the enzyme being a checkpoint protein.
We previously proved through bulk FRET experiments that PaTrm5a is in an extended state (16). To further support our results, we conducted SAXS studies on apo-PaTrm5a. The pure apoprotein without the 6× His tag was passed through a size exclusion column to ensure the monodispersity of PaTrm5a before the data collection. The data collection and structural parameters are presented in table S2, and the P(r) distance distribution function is shown in the inset of Fig. 4D. After calculation of the SAXS profile using the program DAMMIF, the dummy residue (DR) models of the PaTrm5a ensemble with the open conformation (PDB ID: 5HJJ, cyan curve, Fig. 4D) (16) and the curve computed from the atomic structure of the MjTrm5b ensemble with the closed D1 conformation (PDB ID: 2YX1, blue curve, Fig. 4D) (14) were superimposed. The results showed that the discrepancy (χ2) values between the experimental SAXS and the two crystal structures were 4.9 and 32.0, respectively, indicating that the open-state model but not the closed-state model is the major form in solution. Therefore, the SAXS agrees with our previous FRET data. Further fitting of the SAXS data with the MultiFoxS program (18) suggested that PaTrm5a may adopt multiple conformations ranging from partially to fully open conformations in solution. This is a more plausible scenario in solution, given the flexibility of this domain. Together, we conclude that PaTrm5a molecules form an ensemble with various open conformations that might undergo interchangeable conversions in solution.
Methyltransfer activity assays on tRNA truncation mutants
To evaluate the significance of each individual region in tRNA to substrate recognition, we generated a series of tRNA truncation mutants by removing the D-region, T-region, or both regions, or by retaining only the anticodon stem-loop from PatRNAPhe (Fig. 5A). The purity and folding of each tRNA variant have been checked by Urea-PAGE (polyacrylamide gel electrophoresis) and size exclusion chromatography (fig. S6). All the tRNA variants displayed a symmetric shape on the Superdex 75 column (GE Healthcare), suggesting no major issues with their folding. Meanwhile, methyltransfer activity assays showed that deletion of the D-region (DelD) strongly impaired enzymatic activity, whereas deletion of the T-region (DelT) left the enzyme with ~20% activity (Fig. 5B). Removal of both regions (ASL-12, which consists of the acceptor stem and the anticodon stem-loop only) reduced the activity even further. These results indicated that PaTrm5a is very sensitive to the D-region truncation and is less sensitive to the T-region truncation, consistent with the number of interactions between each individual region and the enzyme (Fig. 4A and table S1). Furthermore, the enzyme was completely inactive toward ASL-5 (the anticodon region of PatRNAPhe) or ASL-5Δ32 and ASL-5Δ34 (single-base deletion mutants of ASL-5), in which the deleted bases have very few interactions with the enzyme (table S1). These findings are in agreement with the studies conducted by Christian and Hou (19) and also supported our cocrystal structure.
Fig. 5. Recognition of tRNA truncation mutants by PaTrm5a.
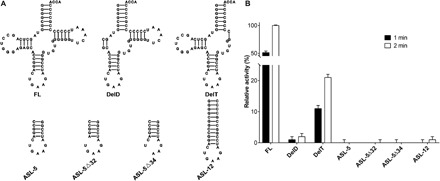
(A) Cartoon drawings showing the sequences and predicted secondary structures of the full-length and truncation mutants of the PatRNAPhe transcript. (B) The methyltransfer activity assays of tRNA truncation mutants by PaTrm5a.
The crystal structure of the PaTrm5a-tRNAPhe-MTA complex
Because SAH is not the methyl donor, the PaTrm5a-tRNAPhe-SAH ternary complex structure described above does not reflect the authentic initial state of the chemical reaction. Consequently, we determined the cocrystal structure of PaTrm5a in the presence of SAM and tRNAPhe (PDB ID: 5WT3). In this 3.2 Å cocrystal structure, there is only one ternary complex in the asymmetric unit. The SAM-bound complex is nearly identical to the SAH-bound structure. Whereas the protein and tRNA backbones are essentially the same between the two structures (Fig. 6A), many side chains of the protein are barely visible in the electron density map and thus not modeled. Despite the lower resolution of the structure, two notable differences could not be overlooked. First, SAM was hydrolyzed during crystallization as shown by the electron density around the ligand-binding site, and we could fit the degradation product 5′-methylthioadenosine (MTA) into the map (Fig. 6B). MTA has been observed in our previous cocrystals of PaTrm5a in the presence of SAM (16). The MTA molecule is highly similar to SAH in terms of positions and orientation and maintains the same type of interactions involving the base and the ribose (Fig. 6C). Second, we resolved three more base pairs at the acceptor arm of the tRNA molecule, with the electron density extending to the G1-C72 base pair. Close examination revealed that the more ordered acceptor end is due to crystal contacts, in which two tRNA molecules form a “head-to-head” packing pattern (Fig. 6C).
Fig. 6. The structure of the PaTrm5a-tRNAPhe-MTA ternary complex.
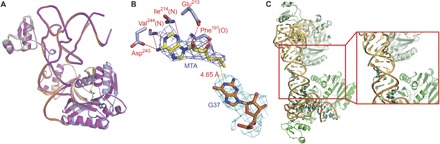
(A) The overlay of the two cocrystal structures. The coloring scheme for the SAH cocrystal structure is as in Fig. 1A, whereas the MTA cocrystal structure is in magenta. The SAH and MTA molecules present at the active site are shown in a ball-and-stick model. (B) The recognition pattern of MTA. The 2Fo − Fc map is contoured at 1σ. The distance of the sulfur atom of MTA to N1 of G37 is indicated by the number near the yellow dashed line. (C) The tRNA-mediated crystal packing. The box on the right is in the close-up view.
Proposed reaction mechanism of Trm5a
By overlaying the structure of the fully modified yeast tRNAPhe (with wybutosine at position 37, PDB ID: 4TNA) (20) onto that of our complex, we can derive the structure of the PaTrm5a-tRNAPhe (imG-14)–SAH ternary complex (Fig. 7A), with the imG-14 coordinates taken from Y37 of tRNAPhe (PDB ID: 4TNA) (20). In this model, imG-14 is flipped into a hydrophobic pocket formed by Phe165, Phe284, Tyr318, Met170, Tyr197, and the 260PTPK263 fragment. The SAH molecule is nearby, and the distances of the sulfur atom to N1 and C7 are 4.4 and 3.8 Å, respectively. Lys324 forms a hydrogen bond with the N2 atom of imG-14 to maintain its orientation (with their distance of 3.5 Å) and possibly helps to neutralize the negative charge of the carbanion at position 7 of imG-14, once the connected proton is deprotonated by Glu173. In addition, Phe284 makes edge-to-face π-π interactions with the tricyclic substrate (Fig. 7A). Therefore, once G or imG-14 is bound to the enzyme, SAM is ready to donate its methyl group to the substrate (N1 of G or C7 of imG-14). In this aspect, the chemistry for PaTrm5a is identical to that of MjTrm5b. But why is MjTrm5b incapable of methylating imG-14 like PaTrm5a? This is probably due to the unique substrate-recognition motif 260PTPK263 present in PaTrm5a, which deviates from the conserved NPPY motif among Trm5s. In previously proposed catalytic models of amino methyltransferases, the carboxyl oxygen of the first proline of NPPY makes hydrogen bonds with the nucleophilic nitrogen of the substrate, which pulls the target nitrogen toward the SAM (15, 21–23). In MjTrm5b, the equivalent NLPK motif is present at positions 265 to 268. In our superimposed model, the side chain of Asn265 of MjTrm5b (and also possibly other residues from this motif) causes steric clashes with the additional five-membered ring of the tricyclic imG-14 produced after Taw1 modification, with their closest distance being only 1.1 Å (Fig. 7B). This asparagine residue is absolutely conserved in Trm5bs, and therefore, further methylation at the C7 position is impossible because of the steric hindrance. In addition, MjTrm5b is unable to form the hydrogen bond involving Lys324 or the hydrophobic interactions involving Phe284 as observed in PaTrm5a, because both residues at these positions in MjTrm5b are isoleucines. In contrast, PaTrm5a substitutes the asparagine with a proline (the PTPK motif), which accommodates the tricyclic substrate (Fig. 7B). Consequently, the variation to the consensus motif confers enzyme promiscuity and expands the substrate spectrum to imG-14–containing tRNAs. Another case in point is PaTrm5b, which has an NLPK motif at the corresponding position. The bulky side chains of leucine and asparagine will likely disallow the efficient binding of imG-14 and thus eliminate its double-methylation capability as well.
Fig. 7. Structural basis for the bifunctional methyltransfer activity of PaTrm5a.
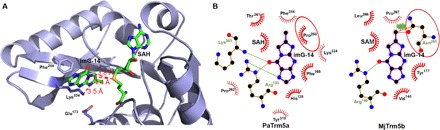
(A) The PaTrm5a-tRNAPhe-SAH ternary complex model containing the modified base. The modified tRNA is omitted for clarity except for the imG-14 base at position 37. The distances of the sulfur atom of SAH to N1 and C7 of imG2 are shown (units in angstroms), along with the distance of Lys324 to N5 of imG-14. (B) Active site comparison of the PaTrm5a in the ternary complex with the MjTrm5b complex in cartoon representation. The residues participating in ligand recognition through hydrogen bonds and hydrophobic contacts are depicted in sticks and eyelashes, respectively. The equivalent hydrophobic residues in the two models are compared, and the two key residues are circled. The tricyclic imG-14 substrate is blocked by the highly conserved NLPK motif in MjTrm5b, and the second methylation reaction is thus prevented. For clarity, the tRNA molecules are omitted.
MATERIALS AND METHODS
Cloning, protein expression, and purification
The PaTrm5a gene was cloned into the pET-28a (+) vector as described by Wang et al. (16). The resulting construct included full-length Trm5 and an N-terminal 6× His tag. Mutants of PaTrm5a were generated using the QuikChange method (Stratagene) (24). The expression and purification of all constructs followed the protocol as described previously (16).
In vitro transcription of tRNA
The full-length P. abyssi tRNAPhe (FL-PatRNAPhe) was prepared after the previous protocol (25). tRNA truncation mutants were based on FL-PatRNAPhe, by intentionally omitting specific regions. Prepared tRNA variants were redissolved in the TE buffer [40 mM tris-HCl (pH 7.0) and 1 mM EDTA] to a concentration of 2.5 mg/ml and allowed to anneal.
Crystallization, data collection, and structure determination
The screens for cocrystals were set up at room temperature using the sitting-drop vapor diffusion method. For the formation of the complex, protein (with the uncleaved 6× His tag) was mixed with tRNAPhe at a molar ratio of 1:0.4 in a buffer containing 20 mM tris-HCl (pH 8.0), 150 mM NaCl, 1 mM dithiothreitol (DTT), and 1.5 mM SAH or SAM. The final concentration of the protein was 6.0 mg/ml. The drops were equilibrated against a reservoir solution containing 45% 2-methyl-2,4-pentanediol (MPD), 100 mM MES (pH 6.5), and 200 mM NH4OAc. The cocrystals of the ternary complex appeared after a week. Fully grown crystals were flash-frozen in liquid nitrogen after being soaked in a cryoprotectant containing the reservoir solution supplemented with 10% ethylene glycol (v/v).
Diffraction data were collected using beamline 17U (BL17U1) and beamline 19U (BL19U1) at the Shanghai Synchrotron Radiation Facility (SSRF; Shanghai, People’s Republic of China) and were processed with the program HKL2000 (26). The PaTrm5a-tRNAPhe-SAH cocrystals belong to the P2 space group with 2.60 Å resolution, whereas the PaTrm5a-tRNAPhe-SAM cocrystals belong to C2221 with 3.20 Å resolution. The SAM data set was predicted to contain one protein and one tRNA molecule each in the asymmetric unit, whereas the SAH data set was predicted to contain two copies of enzyme and two tRNAs in the asymmetric unit. Molecular replacement was first performed on the SAH data set with Phenix (27) using the known complex structure of the MjTrm5b-tRNACys-SAM complex (PDB ID: 2ZZN) (15) as the search model. After a plausible solution was obtained, the model was manually built by COOT (28) according to the electron density map. The rebuilt model was fed to the phenix.refine (27), and multiple cycles of refinement alternated with model rebuilding. The final model was validated by SFCHECK (29). The Rfree/Rwork factors were 0.243 and 0.274 for the PaTrm5a-tRNAPhe-SAH complex, respectively. The SAM-complex structure, which was solved using the PaTrm5a-tRNAPhe-SAH structure, was the search model. At the late stage of the refinement, it was discovered that an MTA molecule (a degradation product of SAM) was bound at the active site. The Rfree/Rwork factors for the PaTrm5a-tRNAPhe-MTA complex were 0.246 and 0.256, respectively (Table 1). The structures of PaTrm5a (PDB ID: 5HJJ) and MjTrm5b (PDB ID: 2YX1) were prepared by PyMOL (http://pymol.org/). The cartoons representing the interaction network as shown in Fig. 7B were generated by LIGPLOT (30).
Table 1. Data collection and refinement statistics.
| Crystals | PaTrm5a-tRNAPhe-MTA (5WT3) | PaTrm5a-tRNAPhe-SAH (5WT1) |
| Data collection | ||
| Beamlines | SSRF-BL19U1 | SSRF-BL17U1 |
| Space group | C2221 | P2 |
| a, b, c (Å) | 58.1, 212.3, 130.0 | 102.1, 57.0, 115.8 |
| α, β, γ (°) | 90, 90, 90 | 90, 101.5, 90 |
| Resolution (Å) | 50–3.20 (3.31–3.20)* | 50–2.60 (2.69–2.60) |
| Rmerge† | 0.079 (0.174) | 0.077 (0.911) |
| I/σ(I) | 18.9 (8.5) | 15.6 (1.67) |
| Completeness (%) | 99.6 (98.0) | 99.5 (99.9) |
| Redundancy | 6.2 (5.9) | 3.7 (3.8) |
| Refinement | ||
| Resolution (Å) | 50–3.20 (3.45–3.20) | 40.22–2.60 (2.66–2.60) |
| Number of reflections | 13,481 | 40,281 |
| Rwork‡/Rfree§ | 0.246/0.256 | 0.247/0.274 |
| Number of atoms | ||
| Protein | 2,268 | 4,966 |
| tRNA | 1,565 | 2,895 |
| Ligand | 20 (MTA) | 52 (SAH) |
| Water molecules | 13 | 60 |
| B factors (Å2) | ||
| Protein | 107.3 | 70.9 |
| tRNA | 93.6 | 68.2 |
| Ligand | 129.1 (MTA) | 55.0 (SAH) |
| Water molecules | 76.6 | 55.7 |
| Root-mean-squared deviations | ||
| Bond lengths (Å) | 0.005 | 0.005 |
| Bond angles (°) | 0.94 | 0.97 |
| Ramachandran favored (%) | 96.36 | 98.63 |
| Allowed | 3.33 | 1.37 |
| Outliers (%) | 0.3 | 0 |
*Values in parentheses are for the highest-resolution shell.
†Rmerge = Σ|(I − 〈I〉)|/σ(I), where I is the observed intensity.
‡Rwork = Σhkl||Fo| − |Fc||/Σhkl|Fo|, calculated from working data set.
§Rfree is calculated from 5.0% of data randomly chosen and not included in refinement.
Electrophoretic mobility shift assay
The binding reactions of tRNA and PaTrm5a in 1:1 molar ratio (containing 1 mM DTT) were incubated on ice for 30 min. Two identical sets of binding reactions were set up, for staining with ethidium bromide and coomassie brilliant blue in parallel. Samples were mixed with an equal volume of 0.5× tris-borate EDTA (TBE) buffer (pH 8.3) containing 5% glycerol and loaded onto a 7% nondenaturing polyacrylamide gel. Electrophoresis was performed for 1.5 hours at 100 V after prerunning the gel for 30 min at 4°C, with 0.5× TBE buffer as the running buffer.
SAXS measurement and data processing
The relatively pure PaTrm5a protein was further purified by size exclusion chromatography with a Superdex 75 column (10/30, GE Healthcare), and the protein was eluted with a buffer of 20 mM tris-HCl (pH 7.0), 150 mM NaCl, and 1 mM DTT. The protein fraction was collected and concentrated to three concentrations of 3.0, 5.0, and 10.0 mg/ml. The SAXS data of PaTrm5a were collected at the beamline 19U2 (BL19U2) of SSRF with a wavelength of 1.03 Å. The data were analyzed using the ATSAS package following the standard procedures (31). After subtracting the scattering signals from buffer, the data from the 3.0-mg/ml sample were scaled and merged using PRIMUS. GNOM was then used to estimate the particle maximum dimension (Dmax) and to calculate the pair distance distribution function (PDDF). The radius of gyration (Rg) of the protein was derived in real space using PDDF. Chain-compatible DR models of PaTrm5a were constructed ab initio with DAMMIF. The structures of PaTrm5a (PDB ID: 5HJJ) and MjTrm5b (PDB ID: 2YX1) were superimposed onto the DR models by PyMOL. The data collection and structural parameters are presented in table S2, and the P(r) distance distribution function is shown in the inset of Fig. 4D.
Methyltransfer assay
The methyltransfer assay mixture contained 100 mM tris-HCl (pH 8.0), 100 mM KCl, 6 mM MgCl2, 2 mM DTT, 250 nM [3H]-SAM (PerkinElmer, 82.7 Ci/mmol), and 2 μM annealed PatRNAPhe. PaTrm5a (200 nM) or mutants were added to initiate the reaction. All the activity assays were carried out at 37°C, and 4-μl aliquots were removed at the designated time points, spotted onto 5% trichloroacetic acid (TCA)–soaked filter pads, and washed twice with 5% cold TCA and 95% ethanol. The filter pads were dried, and the radioactivity was measured by scintillation counting.
Accession numbers
The atomic coordinates and structure factors were deposited in the PDB with accession numbers 5WT1 for the PaTrm5a-tRNAPhe-SAH complex and 5WT3 for the PaTrm5a-tRNAPhe-MTA complex structures.
Supplementary Material
Acknowledgments
We thank BL17U1, BL19U1, and BL19U2 beamlines (National Center for Protein Sciences Shanghai) at SSRF for assistance during data collection. We are grateful to Z. Zhang for helpful discussion on SAXS data analysis. Funding: This work was supported by the Fundamental Research Funds for the Central Universities (16lgjc76) and the Science and Technology Program of Guangzhou (201504010025 and 201605030012). Author contributions: W.X. conceived and designed the research. C.W., Q.J., J.Z., and R.C. performed the research. W.X. analyzed the data and wrote the paper. All authors reviewed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/12/e1700195/DC1
fig. S1. Biosynthesis of wyosine derivatives in eukaryotic tRNAPhes.
fig. S2. The overall structure of the PaTrm5a-tRNAPhe-SAH ternary complex.
fig. S3. The asymmetric unit contents in the cocrystals of the PaTrm5a-tRNAPhe-SAH ternary complex.
fig. S4. The multiple sequence alignment of Trm5 sequences from different model organisms.
fig. S5. The interaction mode of U33/G34 in the PaTrm5a-tRNAPhe-SAH ternary complex.
fig. S6. Purity test of tRNA truncation mutants by size exclusion chromatography and Urea-PAGE.
table S1. Specific interactions between the enzyme and the tRNA substrate.
table S2. Statistics on SAXS data collection, analysis, and modeling.
REFERENCES AND NOTES
- 1.Limbach P. A., Crain P. F., McCloskey J. A., Summary: The modified nucleosides of RNA. Nucleic Acids Res. 22, 2183–2196 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirino Y., Mourelatos Z., Mouse Piwi-interacting RNAs are 2′-O-methylated at their 3′ termini. Nat. Struct. Mol. Biol. 14, 347–348 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Ohara T., Sakaguchi Y., Suzuki T., Ueda H., Miyauchi K., Suzuki T., The 3′ termini of mouse Piwi-interacting RNAs are 2′-O-methylated. Nat. Struct. Mol. Biol. 14, 349–350 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Jühling F., Mörl M., Hartmann R. K., Sprinzl M., Stadler P. F., Pütz J., tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 37, D159–D162 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Machnicka M. A., Milanowska K., Oglou O. O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K. M., Helm M., Bujnicki J. M., Grosjean H., MODOMICS: A database of RNA modification pathways—2013 update. Nucleic Acids Res. 41, D262–D267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith C., Schmidt P. G., Petsch J., Agris P. F., Nuclear magnetic resonance signal assignments of purified [13C]methyl-enriched yeast phenylalanine transfer ribonucleic acid. Biochemistry 24, 1434–1440 (1985). [DOI] [PubMed] [Google Scholar]
- 7.Droogmans L., Grosjean H., Enzymatic conversion of guanosine 3′ adjacent to the anticodon of yeast tRNAPhe to N1-methylguanosine and the wye nucleoside: Dependence on the anticodon sequence. EMBO J. 6, 477–483 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.S. Yokoyama, S. Nishimura, Modified nucleosides and codon recognition, in tRNA: Structure, Biosynthesis and Function, D. Söll, U. L. Rajbhandary, Eds. (ASM Press, 1995), pp. 207–223. [Google Scholar]
- 9.de Crécy-Lagard V., Brochier-Armanet C., Urbonavičius J., Fernandez B., Phillips G., Lyons B., Noma A., Alvarez S., Droogmans L., Armengaud J., Grosjean H., Biosynthesis of wyosine derivatives in tRNA: An ancient and highly diverse pathway in Archaea. Mol. Biol. Evol. 27, 2062–2077 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Björk G. R., Jacobsson K., Nilsson K., Johansson M. J. O., Byström A. S., Persson O. P., A primordial tRNA modification required for the evolution of life? EMBO J. 20, 231–239 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christian T., Evilia C., Williams S., Hou Y.-M., Distinct origins of tRNA (m1G37) methyltransferase. J. Mol. Biol. 339, 707–719 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Urbonavičius J., Meškys R., Grosjean H., Biosynthesis of wyosine derivatives in tRNAPhe of archaea: Role of a remarkable bifunctional tRNAPhe:m1G/imG2 methyltransferase. RNA 20, 747–753 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahoud G., Goto-Ito S., Yoshida K.-i., Ito T., Yokoyama S., Hou Y.-M., Differentiating analogous tRNA methyltransferases by fragments of the methyl donor. RNA 17, 1236–1246 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goto-Ito S., Ito T., Ishii R., Muto Y., Bessho Y., Yokoyama S., Crystal structure of archaeal tRNA (m1G37) methyltransferase aTrm5. Proteins 72, 1274–1289 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Goto-Ito S., Ito T., Kuratani M., Bessho Y., Yokoyama S., Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat. Struct. Mol. Biol. 16, 1109–1115 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Wang C., Jia Q., Chen R., Wei Y., Li J., Ma J., Xie W., Crystal structures of the bifunctional tRNA methyltransferase Trm5a. Sci. Rep. 6, 33553 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrne R. T., Konevega A. L., Rodnina M. V., Antson A. A., The crystal structure of unmodified tRNAPhe from Escherichia coli. Nucleic Acids Res. 38, 4154–4162 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneidman-Duhovny D., Hammel M., Tainer J. A., Sali A., FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 44, W424–W429 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christian T., Hou Y.-M., Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J. Mol. Biol. 373, 623–632 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hingerty B., Brown R. S., Jack A., Further refinement of the structure of yeast tRNAPhe. J. Mol. Biol. 124, 523–534 (1978). [DOI] [PubMed] [Google Scholar]
- 21.Christian T., Evilia C., Hou Y.-M., Catalysis by the second class of tRNA (m1G37) methyl transferase requires a conserved proline. Biochemistry 45, 7463–7473 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klimasauskas S., Kumar S., Roberts R. J., Cheng X., Hhal methyltransferase flips its target base out of the DNA helix. Cell 76, 357–369 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Morrone S. R., Wang T., Constantoulakis L. M., Hooy R. M., Delannoy M. J., Sohn J., Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl. Acad. Sci. U.S.A. 111, E62–E71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H., Naismith J. H., An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C., Guo Y., Tian Q., Jia Q., Gao Y., Zhang Q., Zhou C., Xie W., SerRS-tRNASec complex structures reveal mechanism of the first step in selenocysteine biosynthesis. Nucleic Acids Res. 43, 10534–10545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otwinowski Z., Minor W., Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaguine A. A., Richelle J., Wodak S. J., SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Wallace A. C., Laskowski R. A., Thornton J. M., LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I., ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Cryst. 39, 277–286 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/12/e1700195/DC1
fig. S1. Biosynthesis of wyosine derivatives in eukaryotic tRNAPhes.
fig. S2. The overall structure of the PaTrm5a-tRNAPhe-SAH ternary complex.
fig. S3. The asymmetric unit contents in the cocrystals of the PaTrm5a-tRNAPhe-SAH ternary complex.
fig. S4. The multiple sequence alignment of Trm5 sequences from different model organisms.
fig. S5. The interaction mode of U33/G34 in the PaTrm5a-tRNAPhe-SAH ternary complex.
fig. S6. Purity test of tRNA truncation mutants by size exclusion chromatography and Urea-PAGE.
table S1. Specific interactions between the enzyme and the tRNA substrate.
table S2. Statistics on SAXS data collection, analysis, and modeling.