

Abstract
Ubiquinone (Q) is an isoprenoid quinone that functions as membrane electron carrier in mitochondria and bacterial organisms belonging to the alpha, beta, and gamma class of proteobacteria. The biosynthesis of Q follows various biochemical steps catalyzed by diverse proteins that are, in general, homologous in mitochondria and bacteria. Nonorthologous proteins can also contribute to some biochemical steps as originally uncovered in Escherichia coli, which is the best studied organism for Q biosynthesis in prokaryotes. However, the origin of the biosynthetic pathway of Q has remained obscure. Here, I show by genome analysis that Q biosynthesis originated in cyanobacteria and then diversified in anaerobic alpha proteobacteria which have extant relatives in members of the Rhodospirillaceae family. Two distinct biochemical pathways diverged when ambient oxygen reached current levels on earth, one leading to the well-known series of Ubi genes found in E. coli, and the other containing CoQ proteins originally found in eukaryotes. Extant alpha proteobacteria show Q biosynthesis pathways that are more similar to that present in mitochondria than to that of E. coli. Hence, this work clarifies not only the origin but also the evolution of Q biosynthesis from bacteria to mitochondria.
Keywords: ubiquinone, menaquinone, plastoquinone, quinone biosynthesis, evolution of mitochondria
Introduction
Ubiquinone (Q) is an isoprenoid substituted benzoquinone which shuttles electrons between membrane-bound respiratory dehydrogenases and oxidoreductases that ultimately reduce oxygen, either directly or indirectly via other redox components (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Aussel et al. 2014; Zhi et al. 2014; Marreiros et al. 2016; Ravcheev and Thiele 2016). The electron carrier function of Q is equivalent to that of other membrane quinones such as menaquinone (MQ) and plastoquinone (PQ), which are involved in diverse electron transport chains producing protonmotive reactions to drive ATP synthesis (Meganathan and Kwon 2009; Nowicka and Kruk 2010). Most deep branching bacteria and some Archaea have protonmotive systems based upon MQ or its derivatives (Nowicka and Kruk 2010; Marreiros et al. 2016). PQ is restricted to cyanobacteria and plant plastids, whereas Q is present in the phylum proteobacteria among prokaryotes, as well as in all eukaryotes (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Marreiros et al. 2016).
Two major differences distinguish Q from MQ and its derivatives: 1) the midpoint redox potential, which is significantly higher for Q; 2) the biosynthetic pathway of Q, which is predominantly membrane-associated and requires oxygen in multiple steps. Both differences have their evolutionary origin in the great oxygenation event brought about by oxygenic photosynthesis in primordial earth (Nowicka and Kruk 2010; Degli Esposti 2016; Marreiros et al. 2016; Pelosi et al. 2016). How the complex biosynthetic pathway of Q was put together, presumably assembling different steps from those used for the biosynthesis of pre-existing MQ (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Zhi et al. 2014; Ravcheev and Thiele 2016), remains completely unknown. What is generally assumed is that Q biosynthesis evolved in the common ancestor of alpha, beta, and gamma proteobacteria (Nowicka and Kruk 2010; Degli Esposti 2016; Marreiros et al. 2016; Pelosi et al. 2016), organisms diversely adapted to different levels of ambient oxygen. However, it is unclear which of extant proteobacteria may be close to the ancestral organism which “invented” Q. This work was aimed to unveil the origin of Q biosynthesis and identify extant relatives of the ancestral organisms that evolved the pathways for Q biosynthesis.
I found that complete pathways of Q biosynthesis originated in alpha proteobacteria related to current members of the Rhodospirillaceae family, probably as a diversification of the pathway for PQ biosynthesis in cyanobacteria. These pathways then evolved in chemolithoautotrophic Fe2+-oxidizing organisms of the recently described class of zetaproteobacteria (Emerson et al. 2007) to form the dominant biosynthetic system in beta and gamma proteobacteria, including E. coli. Other pathways evolved within alpha proteobacteria and led to the system of Q biosynthesis that is characteristic of mitochondria. Hence, the origin of Q biosynthesis is discussed also in its possible evolutionary trajectory from prokaryotes to eukaryotes.
Challenges in Deducing the Biosynthetic Pathways of Membrane Quinones
To deduce in silico the biosynthetic pathway of a membrane metabolite is never an easy task, but in the case of Q it is particularly challenging. A fundamental reason for this is that eukaryotic Q biosynthesis, confined to mitochondria, is still partially unknown (Nowicka and Kruk 2010; Kawamukai 2015); the same applies to Q biosynthesis in the bacterial lineage from which mitochondria evolved, alpha proteobacteria (Degli Esposti 2016; Pelosi et al. 2016). Another reason is that current knowledge of the bacterial pathway for Q biosynthesis is essentially restricted to the model organism, Escherichia coli—and is not fully complete yet (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Aussel et al. 2014; Ravcheev and Thiele 2016). Escherichia coli belongs to the large order of Enterobacterales, which is a recently evolved lineage within the highly diversified class of gamma proteobacteria (Almagro et al. 2015), therefore phylogenetically distant from the roots of Q biosynthesis (Kawamukai 2015; Pelosi et al. 2016; Degli Esposti and Martinez-Romero 2017). Moreover, the genes of the proteins essential for Q biosynthesis are much less associated in syntenic groups than those involved in MQ biosynthesis (Aussel et al. 2014; Ravcheev and Thiele 2016). These genes often code for proteins that are poorly conserved, especially across different classes of proteobacteria. Finally, even structurally conserved enzymes such as the prenyl transferase UbiA can accept biosynthetically different substrates in diverse groups of prokaryotes and eukaryotes (Cluis et al. 2011; Aussel et al. 2014; Cheng and Li 2014; Pfaff et al. 2014; Kawamukai 2015). However, comprehensive knowledge of Q and other membrane quinones can be combined with bioinformatic analysis exploiting the ever increasing wealth of sequence information now available (Degli Esposti 2016; Degli Esposti and Martinez-Romero 2017) to simplify and partially resolve the problems just mentioned. Following this rationale, I have developed the strategy detailed below for uncovering the bacterial pathways of Q biosynthesis.
Strategy
To guide genome-wide analyses of the biosynthetic pathways for membrane isoprenoid quinones, I developed a research strategy based upon the following points: 1) reduce the biosynthetic pathways to the minimal steps that are indispensable for the biochemical assembly of Q as in the biosynthesis of plastoquinone (PQ) in cyanobacteria (Pfaff et al. 2014; fig. 1 and table 1); 2) analyze the available genomes of proteobacterial organisms which have been reported to produce both Q and MQ (Schoepp-Cothenet et al. 2009; Marreiros et al. 2016; Degli Esposti and Martinez-Romero 2017) and deduce the most complete pathway possible for the biosynthesis of either quinone; 3) run BLAST searches of all the proteins thus identified against progressively wider sets of taxa to cover the complete phylogenetic span of proteobacteria, including all unclassified organisms with at least 1,000 coded proteins identified in metagenomic studies (Nielsen et al. 2014; Kantor et al. 2015; Anantharaman et al. 2016; Emerson et al. 2016; Probst et al. 2017); 4) build a distribution list of the homologues of the biosynthetic proteins for Q and MQ in all proteobacterial taxa examined; 5) evaluate the presence of functional Q-reacting enzymes (Marreiros et al. 2016; Ravcheev and Thiele 2016; Degli Esposti and Martinez-Romero 2017) in the taxa of the list just built; 6) examine the completeness of the genomes of organisms which appear to have incomplete sets of proteins for Q biosynthesis while possessing functional Q-reacting enzymes, and then scan the same genomes for potential nonorthologous proteins that may complete the biosynthetic pathway; 7) compare the combinations of Q biosynthesis enzymes found in bacterial genomes to that known in eukaryotes (Kawamukai 2015); 8) coherently classify different types of biosynthetic pathways for Q by integrating the information obtained in steps 4–7.
Fig. 1.
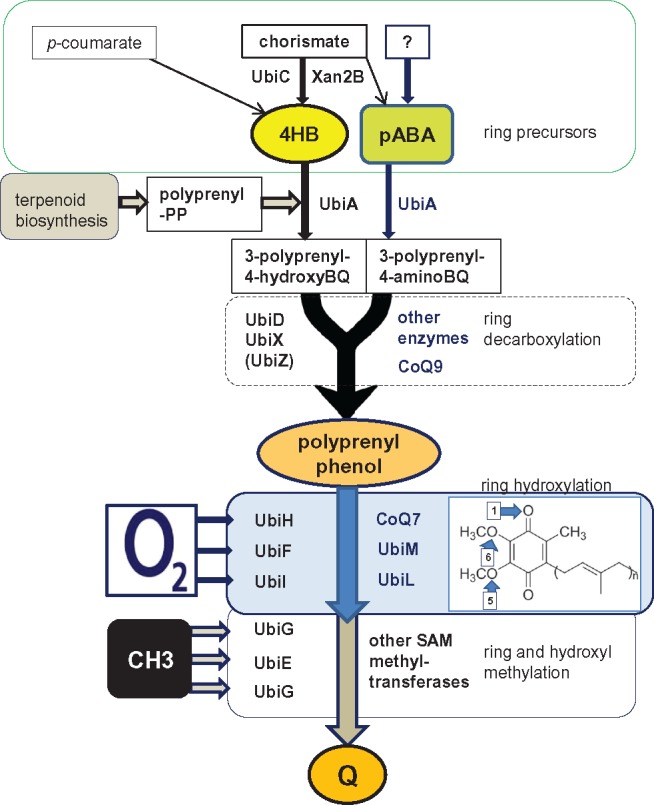
—Simplified scheme for Q biosynthesis in bacteria. The scheme has been rationalized on the basis of the pathway for PQ biosynthesis in cyanobacteria (Pfaff et al. 2014) and taking into consideration previously reported schemes (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Aussel et al. 2014; Zhi et al. 2014; Kawamukai 2015; Ravcheev and Thiele 2016). The major steps in the pathways of bacterial Q biosynthesis are represented in boxed blocks, starting from the production of two major ring precursors, 4HB and pABA, from known and unknown metabolites. These precursors follow parallel pathways of ring prenylation via UbiA isoforms that produce different substrates (3-polyprenyl-4-hydroxy BenzoQuinones and 3-polyprenyl-4-aminoBenzoQuinones) for the subsequent step of ring decarboxylation, which can be carried out by different, nonhomologous enzymes as indicated (see also table 1). The two pathways thus converge in polyprenylphenols, which then undergo a series of three hydroxylation and three methylation steps carried out by various enzymes: the indicated variants of FAD-dependent hydroxylases plus the ferritin-based CoQ7 for ring hydroxylation, and the indicated types of SAM-dependent methyl transferases for ring and hydroxyl methylation. In general, each hydroxylation step is followed by a methylation step (Aussel et al. 2014), but variations in this sequence are possible as in the mitochondrial pathway of Q biosynthesis (Nowicka and Kruk 2010; Kawamukai 2015).
Table 1.
Fundamental Aspects and Genes for the Biosynthesis of Ubiquinone in Selected Taxa of Bacteria and Eukaryotes
| Organism | Genome | Q Ring Precursor (Source) | Ring Prenylation | Ring Decarboxylation | Ring Hydroxylation | Other | Methylation | Pathway, Other Q | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C5 | C1 | C6 | O- | C- | |||||||
| Synechocystis sp. PCC 6803 | complete | chorismate-4HB | UbiAa | UbiD, UbiX | - | UbiH | - | - | UbiG | UbiE | PQ, c ancestral |
| Escherichia coli str. K-12 substr. MG1655 | complete | chorismate-4HB (p-coumarate) | UbiAa | UbiD, UbiX | UbiI | UbiH | UbiF | - | UbiG | UbiE | (a), c—CoQ7 and MQ |
| Magnetococcus marinus | complete | chorismate-4HB | UbiAa | UbiD, UbiX | - | - | - | - | UbiG | UbiE | a |
| Phaeospirillum fulvum | 97.3%c | ?, pABA | UbiA | UbiD, UbiX | - | - | - | CoQ9 | UbiG | UbiE | a and MQ |
| Rhodospirillum centenum | complete | ?, pABA | UbiA | UbiD, UbiX | UbiM | UbiL | CoQ7 | CoQ9 | UbiG | UbiE | m + DX |
| Granulibacter bethesdensis | complete | chorismate-4HBb | UbiA | - - | UbiM | UbiL | CoQ7 | CoQ9 | UbiG | UbiE | m (XanB2) |
| Inquilinus | 98.0%c | ?, pABA | UbiA | - - | UbiM | UbiL | CoQ7 | CoQ9 | UbiG | UbiE | m |
| Micavibrio | complete | ?, pABA | UbiA | - - | UbiM | UbiL | UbiM | CoQ9 | UbiG | UbiE | m—CoQ7 |
| Bosea vaviloviae Vaf18 | complete | ?, pABA | UbiA | - - | UbiM | UbiL | CoQ7 | - | UbiG | UbiE | m—CoQ9 |
| Aureimonas sp. AU40 | complete | ?, pABA | UbiA | - - | UbiM | UbiL | UbiM | - | UbiG | UbiE | m—CoQ7&9 |
| Proteobacteria bacterium CG1_02_64_396 | 94.6%c | chorismate-4HB | UbiAa | UbiD, UbiX | UbiI | UbiH | CoQ7 | - | UbiG | UbiE | c |
| Sideroxydans lithotrophicus | complete | chorismate-4HBb | UbiAa | UbiD, UbiX | UbiI | UbiH | CoQ7 | - | UbiG | UbiE | c |
| Neisseria meningitidis FAM18 | complete | chorismate-4HB | UbiAa | UbiD, UbiX | UbiM | UbiM | UbiM | - | UbiG | UbiE | c—CoQ7 |
| Acinetobacter junii SH205 | complete | chorismate-4HB | UbiAa | UbiZ - | UbiI | UbiH | UbiF | - | UbiG | UbiE | c—CoQ7 & X |
| Endosymbiont of Ridgeia piscesae | 100.0%c | chorismate-4HB | UbiAa | UbiD - | UbiI | UbiH | CoQ7 | - | UbiG | UbiE | c—X and MQ |
| Eukaryotes | |||||||||||
| Yeast | complete | pABA,? | CoQ2 | ? ? | CoQ6 | ? | CoQ7 | CoQ9 | CoQ3 | CoQ5 | m |
| Plants | complete | pABA,? | CoQ2, PPT1 | ? ? | CoQ6 | ? | CoQ7 | CoQ9 | CoQ3 | CoQ5 | m |
UbiA isoform associated with UbiC cf. figure 2.
Using XanB2 instead of UbiC cf. Zhou et al (2013).
Obtained with BUSCO, program for deducing the degree of completeness of genomes (Simão et al, 2015).
Of note, steps 3, 4, 7, and 8 of this research strategy are novel, because previous studies have analyzed only subsets of bacterial taxa (Zhi et al. 2014; Marreiros et al. 2016; Pelosi et al. 2016; Ravcheev and Thiele 2016) and required the presence of nearly all proteins known in E. coli for the in silico reconstruction of the biosynthetic pathways of Q and MQ. Moreover, I have taken into consideration also the possibility of anaerobic pathways of Q biosynthesis, which are known to be present in E. coli and other proteobacteria (Alexander and Young 1978; Meganathan and Kwon 2009; Schoepp-Cothenet et al. 2009; Cluis et al. 2011; Aussel et al. 2014), but not yet identified in molecular terms. This possibility, never considered in genome-wide searches before, is very important for identifying ancestral pathways of Q biosynthesis, which must have originated in organisms well-adapted to the anaerobic conditions that were prevalent at the beginning the great oxygenation event (Schoepp-Cothenet et al. 2009; Degli Esposti 2016).
Materials and Methods
Experimentally, the biosynthetic pathways for Q and menaquinone have been deduced using very detailed BLAST searches of proteins known (or suspected) to participate in such pathways. At difference with previous studies, which have systematically used potentially arbitrary cut-off values (Zhi et al. 2014; Pelosi et al. 2016; Ravcheev and Thiele 2016), I have extended the protein searches to all bacterial organisms for which partial or complete genomes are available, initially without applying cut-off values. A cut-off <1e−10 was used as in previous works (Ravcheev and Thiele 2016) when the searched protein showed a high degree of conservation across the wide phylogenetic span of the proteobacterial organisms that are currently available. However, several proteins involved in the biosynthetic pathways of membrane quinones and their metabolites show low levels of conservation, sometimes even in related taxa. This is particularly the case for chorismate lyase or UbiC, generally the first enzyme for Q biosynthesis (Gallagher et al. 2001; Zaitseva et al. 2006; Aussel et al. 2014; Ravcheev and Thiele 2016), which often has few hits in extended BLAST searches with any given protein query, because its level of amino acid identity is very limited—only the signature conserved domain (CDD; Marchler-Bauer et al. 2015) can be recognized across unrelated taxa. Multiple UbiC proteins maintaining the signature CDD of chorismate lyase (cl01230) were thus used in iterative BLAST searches extended to partially overlapping sets of taxa, so as to progressively cover the whole phylogenetic span of proteobacteria and uncover also potential instances of Lateral Gene Transfer (LGT). Clear cases of LGT were inferred from the absence of homologous proteins in related taxa and the clustering together with proteins belonging to another class of proteobacteria having similar ecological properties. For instance, the UbiC proteins present in various strains of the pathogen Bartonella, which belongs to the Rhizobiales order of alpha proteobacteria, did not cluster together with UbiC proteins present in other Rhizobiales, namely those from organisms of the genus Methylobacterium, but were closely related to those of pathogenic taxa of gamma Enterobacterales, such as Serratia and Salmonella. A manually curated alignment of diverse UbiC proteins was produced to assist the proteins searches and refine the results of phylogenetic affinity. Comparable iterative searches and sequence analysis were undertaken for UbiA, UbiD, UbiX, UbiH, and MenF of MQ biosynthesis, as well as all CoQ proteins identified so far in the eukaryotic pathway of Q biosynthesis (Kawamukai 2015).
Conversely, the fundamental steps of ring methylation and hydroxylation are carried out by members of two large superfamilies of proteins (fig. 1 and table 1): S-adenosylmethionine-dependent methyltransferases (SAM or AdoMet-MTase) and flavin mono-oxygenases with the common Rossmann-fold of NAD(P)(+)-binding proteins, frequently defined as Ubi-OHases (Aussel et al. 2014; Pelosi et al. 2016). Consequently, it is often difficult to discern the homology of a member of such super-families from paralogues having close structural resemblance. To narrow the BLAST searches to the proteins that genuinely have the closest structural homology to a protein query, Neighbour Joining (NJ) distance trees were derived from the BLAST searches and then carefully examined (Degli Esposti 2016). Close homologues were then considered when proteins clustered in the same monophyletic branch that showed the signature CDD of the query in the majority of its leaves. Whenever NJ trees were insufficiently resolved to allow such a selection, manually curated alignments and Maximum Likelihood (ML) trees were produced and examined as previously described (Degli Esposti 2016; Degli Esposti et al. 2016). See the legend of figures 2 and 4 and supplementary figures S1–S3, Supplementary Material online, for more details.
Fig. 2.

—UbiA proteins have different molecular signatures whether they are associated with UbiC or not. (A) Phylogenetic tree of UbiA proteins from various bacterial groups. The NJ tree was obtained after manual refinement of a CLUSTALW alignment of 26 UbiA sequences from phylogenetically diverse taxa using the program MEGA5 (Tamura et al. 2011). The percentage value of 500 bootstraps is shown for each node. After removing all positions containing gaps and missing data, the final data set contained a total of 244 positions. Clearly, the proteins from alphaproteobacteria (black squares) which are not associated with UbiC form a sister group to that including zeta (black triangles), gamma (empty circles), and beta proteobacteria (empty diamonds). Note the precursor position of the cyanobacterial Synechocystis (black circle) and the outgroup position of the Archaean Aeropyrum (white triangle). White squares indicate distant homologous proteins from delta proteobacteria. Similar results were obtained with ML trees. (B) Alignment of conserved regions of UbiA proteins. The same alignment of UbiA proteins used in (A) was reduced to 22 sequences and additionally included the yeast CoQ2 homologue of UbiA. The alignment block shown includes the functionally important regions II and III that were previously deduced (Ohara et al. 2009) and later verified by the 3D structure of UbiA (Cheng and Li 2014). Highlighted in pale blue are the residues conserved among UbiA proteins from taxa without UbiC (indicated as no UbiC) that lie close to the active site at the negative side of the membrane and are structurally different from those of UbiA proteins associated with UbiC, following the numeration of Aeropyrum UbiA (Cheng and Li 2014). The regions corresponding to alpha helices in the 3D structure of Aeropyrum UbiA (Cheng and Li 2014) are highlighted in gray (transmembrane, TM) or light blue.
Fig. 4.

—Phylogenetic trees of FAD-dependent Q ring hydroxylases indicate cyanobacterial ancestry. A selection of each variant of FAD-dependent UbiOHases that have been recently categorized (Pelosi et al. 2016) has been chosen to cover the phylogenetic span used to analyze other enzymes involved in Q biosynthesis, for example, UbiA (fig. 2A), whereas matching as much as possible the topology of NJ trees obtained from all the results of wide BLASTP searches, as described earlier (Degli Esposti 2016). The percentage of replicate NJ trees in which the associated taxa clustered together in the bootstrap test is shown next to the branches. The rate variation among sites was modeled with a gamma distribution (shape parameter = 2) and all positions containing gaps were eliminated, for a final data set of 326 amino acid positions. Note how the UbiOHases of proteobacteria clearly originate from UbiH proteins of cyanobacteria (black circles), segregating into two sister groups (cf. Pelosi et al. 2016): the top group contains UbiL and UbiM homologues that are predominantly present in alphaproteobacteria (black squares) and a few betaproteobacteria (empty diamonds), whereas the bottom group includes UbiH and the closely related UbiI and UbiF proteins, which are present in zeta (black triangles) ans gamma proteobacteria (empty circles), but generally absent from alpha proteobacteria. Similar trees showing the same separation of groups of Ubi hydroxylases have been obtained with the ML method and different numbers of bootstrap replicates. The original annotation of most proteins has been modified for providing a homogeneous representation of the various types of FAD-dependent hydroxylases.
Taxonomic position and relationships of unclassified proteobacterial organisms was evaluated by the reciprocal BLAST approach of all coded proteins, as previously used to define genome chimaerism (Esser et al. 2007; Ku et al. 2015; Ji et al. 2017). The taxonomic affiliation at the family level was evaluated by statistical analysis of the top hits (5–10) of all the proteins of either unclassified or classified organisms against the whole nonredundant (NR) database. A simplified version of this method was developed and showed high correlation with the results of genomic chimaerism obtained previously (Esser et al. 2007; Ji et al. 2017). This method is based on the computation of the top five hits obtained with smartBLAST [https://blast.ncbi.nlm.nih.gov/smartblast/] for about 100 proteins essential for membrane and energy metabolism (the list of these proteins will be made available upon request). The results obtained with the above approaches were then compared with those derived from the analysis of ribosomal proteins or 16 rRNA as frequently undertaken in metagenomic studies (Nielsen et al. 2014; Anantharaman et al. 2016; Probst et al. 2017). Genome completeness was evaluated with the program BUSCO applied to proteins (Simão et al. 2015).
Chemical Nomenclature and Abbreviation of Genes/Proteins
In this work, menaquinone is abbreviated as MQ (Degli Esposti and Martinez-Romero 2017) instead of the common MK acronym (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Aussel et al. 2014; Zhi et al. 2014; Ravcheev and Thiele 2016), which refers also to vitamin K—a quinone of no interest in the present study. With regard to the numeration of the ring positions of Q and its metabolites, I have systematically followed that used by Aussel et al. (2014): position 1 corresponds to the carboxylate of the chorismate precursor, whereas the isoprenoid tail of subsequent metabolites is at position 3 (fig. 1). This numeration differs from that normally used for the ring of Q or PQ, in which the isoprenoid tail is at position 6, whereas position 1 corresponds to the hydroxyl group in position 4 of chorismate (Nowicka and Kruk 2010). The latter numeration has been essentially ignored in order to avoid confusion in referring to specific biochemical steps in Q biosynthesis. The enzymes carrying the same step in Q biosynthesis are annotated with different names in bacteria and eukaryotes (Aussel et al. 2014; Kawamukai 2015): in bacteria, they follow the discovery of the Ubi genes in early studies with E. coli (Aussel et al. 2014; Pelosi et al. 2016), whereas in eukaryotes they are named as CoQ1-11 following pioneering studies in yeast (Kawamukai 2015). In the presentation of phylogenetic trees I have homogenized the original annotations of the Ubi proteins, which are often annotated by the biochemical reaction associated with the protein coded by the Ubi genes. Intriguingly, some genes initially found in yeast turn out to be present in proteobacteria too, sometimes with different annotations. This applies to methoxy-producing CoQ7, recently found to be widespread among proteobacteria (Pelosi et al. 2016), as well as CoQ9, which I show here to be present in alpha proteobacteria too, but often annotated as RpsU-divergently transcribed protein. The complicated picture of gene nomenclature applies also to MQ biosynthesis, owing to the presence of two alternative pathways for its production in bacteria (Zhi et al. 2014; Ravcheev and Thiele 2016). Here, I have considered the futalosine pathway only cursorily, because it is generally not present in proteobacteria that produce Q (Zhi et al. 2014).
Statistical Analysis
Whenever needed, statistical analysis was undertaken with either parametric of nonparametric tests as recently described (Degli Esposti and Martinez-Romero 2017).
Results and Discussion
Simplified View of Q Biosynthesis
Although the biosynthetic pathways of Q and MQ are usually represented as a series of consecutive steps that appear to be of equal importance (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Aussel et al. 2014; Zhi et al. 2014; Ravcheev and Thiele 2016), some biochemical reactions are definitely more important than others. In particular, the addition of the prenyl tail to the ring of Q precursors, a reaction catalyzed by the integral membrane protein UbiA, is the most critical and often rate-liming in the whole pathway of Q biosynthesis (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Cluis et al. 2011; Aussel et al. 2014; Cheng and Li 2014; Kawamukai 2015). UbiA has a broad substrate specificity (Meganathan and Kwon 2009; Nowicka and Kruk 2010), particularly for its prenyl substrate which can have different numbers of isoprenoid units in different organisms (Collins and Jones 1981; Nowicka and Kruk 2010; Cluis et al. 2011; Aussel et al. 2014; Cheng and Li 2014; Pfaff et al. 2014; Kawamukai 2015). Hence Q-8 is the dominant Q species in E. coli because its UbiA protein prefers octaprenyl as its substrate (Aussel et al. 2014), whereas Rhodobacter and other alpha proteobacteria have Q-10, as in human mitochondria, because their UbiA protein preferentially reacts with the decaprenyl substrate (Nowicka and Kruk 2010; Cluis et al. 2011; Kawamukai 2015). Such a substrate versatility of the critical UbiA enzyme was overlooked in the past, when the isoprenoid tail of Q was considered to have taxonomic value across proteobacterial classes (Collins and Jones 1981). Conversely, UbiA proteins can be rather selective for their benzene ring substrate as in the case of E. coli, which normally accepts only 4-hydroxy-benzoate (4HB) for ring prenylation (Cluis et al. 2011; Aussel et al. 2014; Xie et al. 2015). However, UbiA of alpha proteobacteria can accept p-amino-benzoic acid (pABA) as the substrate for this reaction (Cluis et al. 2011; Aussel et al. 2014; Xie et al. 2015), similarly to the CoQ2 homologue of UbiA in eukaryotes (Kawamukai 2015). Effectively, chorismate is not the source of ring precursors in mitochondria, because they do not have homologues of E. coli chorismate lyase (UbiC), the enzyme generally assumed to initiate Q biosynthesis (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Aussel et al. 2014; Zhi et al. 2014; Ravcheev and Thiele 2016). Chorismate is the initial metabolite in the pathways of MQ biosynthesis too (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Zhi et al. 2014; Ravcheev and Thiele 2016), which suggests that Q biosynthesis might have emerged from a modification of such pathways (Ravcheev and Thiele 2016). However, the classical pathway of MQ biosynthesis has only one reaction in common with Q biosynthesis, the C-methylation of the prenylated ring, which is carried out by the same enzyme called bifunctional UbiE (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Zhi et al. 2014; Ravcheev and Thiele 2016). The attachment of the prenyl group to the ring precursor is one of the last steps in MQ biosynthesis, whereas it is the second step in Q biosynthesis (Meganathan and Kwon 2009), which subsequently occurs in association with the membrane (Meganathan and Kwon 2009; Kawamukai 2015). These are large differences that can be hardly reconciled with the possibility that Q biosynthesis originated from a variation of MQ pathways, as recently suggested (Ravcheev and Thiele 2016).
Here, I present the fundamental steps of Q biosynthesis in blocks (fig. 1 and table 1), with a bottleneck junction representing the funneling of prenylated precursors into the multiple steps of hydroxylation and methylation of the ring (fig. 1). Prior to ring prenylation, there is uncertainty in the incompletely known and potentially diverse steps that can produce ring precursors (Xie et al. 2015), even if 4HB could be considered the fundamental metabolite to start Q biosynthesis in many proteobacteria (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Aussel et al. 2014; Xie et al. 2015; Ravcheev and Thiele 2016), as well as PQ biosynthesis in cyanobacteria (Pfaff et al. 2014). Hence, the steps leading to 4HB and other potential ring precursors constitute a variable part of the entire pathway, which is partially illustrated by the question mark at the top of the scheme in figure 1. Decarboxylation of the benzoate ring constitutes the fundamental step in Q biosynthesis following ring prenylation. In E. coli, this decarboxylation is carried out by the coordinated activity of UbiD and UbiX (Aussel et al. 2014) as in cyanobacteria (Pfaff et al. 2014). The same reaction occurs later in mitochondrial Q biosynthesis, catalyzed by proteins which remain unknown—even if potential homologues of UbiD and UbiX are present in fungi and other eukaryotes (Nowicka and Kruk 2010; Kawamukai 2015). Several alpha proteobacteria also lack homologous of the E. coli decarboxylases, suggesting the possibility that UbiD and UbiX may be replaced by nonorthologous proteins (collectively defined as other decarboxylases in fig. 1), as recently proposed for the gamma proteobacterium Acinetobacter junii (Ravcheev and Thiele 2016; table 1, middle). Alternatively, bacterial organisms lacking UbiD and UbiX in their genome may have a Q biosynthesis pathway similar to that of mitochondria (see below).
Subsequent to the above-mentioned UbiE-mediated C-methylation of the ring, there are three consecutive ring hydroxylation steps of prenyl Q metabolites (table 1). In contrast, there is a single hydroxylation at the C-1 position in the ring of PQ, which is carried out by a homologue of E. coli UbiH (Pfaff et al. 2014). UbiH belongs to a superfamily of FAD-dependent hydroxylases which insert a hydroxyl group in one or more positions of the Q ring (Pelosi et al. 2016). Overall, these enzymes are often labeled UbiOHases and include the nonorthologous CoQ7—a ferritin-like protein initially discovered in yeast genetic screens (Kawamukai 2015; Pelosi et al. 2016) that is specific for last ring hydroxylation at position 6 (Pelosi et al. 2016). Of note, UbiB has also been considered among such hydroxylases, and often remains annotated as 2-polyphenol hydroxylase in databases. However, Pierrel and coworkers (Hajj Chehade et al. 2013) have demonstrated that UbiB does not carry out the first hydroxylation step of Q ring precursors, as previously considered (Meganathan and Kwon 2009; Zhi et al. 2014; Ravcheev and Thiele 2016). This reaction is catalyzed instead by UbiI, a close relative of UbiF, the gene of which precedes that of UbiH in E. coli (Hajj Chehade et al. 2013; Aussel et al. 2014). UbiB has a conserved domain of protein kinases and therefore may be involved in the regulation of Q hydroxylation (Hajj Chehade et al. 2013; Aussel et al. 2014). Finally, UbiG carries out the O-methylation of the hydroxyls introduced at positions 5 and 6 of the Q ring by UbiOHases (table 1). This reaction can be catalyzed also by other SAM methyl-transferases, which are often found in multiple forms within the genomes of metagenomic organisms, including those that lack bona fine homologues of UbiG. Again, this step of Q biosynthesis can be considered indeterminate, because it can be carried out by a variety of SAM methyl-transferases and not a single critical enzyme (fig. 1).
From the information presented earlier, it follows that the minimal combination of enzymes that can produce effective pathways of Q biosynthesis includes UbiA, UbiE, and one or more UbiOHases—notably, the recently described UbiM can attach an OH group to positions 1, 5, and 6 of the ring (Pelosi et al. 2016).Therefore, it is not necessary to require the presence of homologous for all the Ubi proteins of E. coli (Marreiros et al. 2016; Ravcheev and Thiele 2016) to deduce complete pathways for Q biosynthesis in bacteria. The carboxylation step may be carried out by proteins yet unknown as in eukaryotes, whereas O-methylation can be carried out by UbiG or other members of the SAM superfamily. The resulting simplified combination of biosynthetic steps (fig. 1) leads to different pathways that can be deduced from genomic analysis (table 1).
Anaerobic Pathways for Q Biosynthesis in Proteobacteria
One interesting consequence of the simplified view for the pathways of Q biosynthesis elaborated here is that organisms which completely lack known ring hydroxylases, but have all the other fundamental elements of the pathway, could be considered to produce Q even in the presence of low or no oxygen. An anaerobic pathway of this kind was initially inferred from the residual production of Q in E. coli mutants lacking ring hydroxylases and the consistent presence of large complements of Q in E. coli strains grown under strictly anaerobic conditions (Alexander and Young 1978; Meganathan and Kwon 2009). Similar findings have been subsequently reported in the gamma proteobacterium Halorhodospira (Schoepp-Cothenet et al. 2009) and the alpha proteobacterium Paracoccus (Matsumura et al. 1983). Here, I have deduced the likely presence of anaerobic pathways of Q biosynthesis, labelled a in table 1, in two deep branching alpha proteobacteria: Magnetococcus marinus (Schübbe et al. 2009) and Phaeospirillum fulvum (Hiraishi and Hoshino 1984; Sizova et al. 2007). The genome of Magnetococcus is complete (Schübbe et al. 2009), whereas that of Phaeospirillum is at least 97% complete as deduced with the program BUSCO (Simão et al. 2015). Hence, the absence of UbiH, UbiL, CoQ7, or other ring hydroxylases in these genomes would indicate a situation similar to that found in E. coli under anaerobic conditions (Alexander and Young 1978), namely the insertion of hydroxy groups in the Q ring could follow reactions catalyzed by aromatic degrading enzymes, which occur under anaerobic conditions (Alexander and Young 1978; Xie et al. 2015). The alternative possibility that H2O can be used as a source of ring hydroxylation as in the case of the MenF-catalyzed reaction of MQ (Meganathan and Kwon 2009) is unlikely for Magnetococcus, because this organism does not possess MenF or any other enzyme for MQ metabolism, like its close relative Magnetofaba (Morillo et al. 2014). Conversely, the genome of P. fulvum contains a complete set of genes for MQ biosynthesis, consistent with the presence of MQ in this organism as determined biochemically (Hiraishi and Hoshino 1984). However, P. fulvum has no homologue of UbiC as all its relatives of the Rhodospirillaceae family (table 1). Hence, it is unlikely that chorismate functions as ring precursor for Q biosynthesis in P. fulvum. An alternative enzyme for the chorismate lyase reaction producing 4HB is coded by the Xan2B gene (Zhou et al. 2013), which is also absent in P. fulvum but common in taxa of the Acetobacteraceae family of the same Rhodospirillales order, for example, Granulibacter (table 1).
The requirement for Q biosynthesis in Magnetococcus and Phaeospirillum is strongly supported by the autotrophic nature of these organisms, which have multiple Q reacting enzymes—from complex I to the bc1 complex—that are fundamental for their energy metabolism (table 2). I additionally found a potential pathway a for Q biosynthesis in the metagenomic proteobacterium, alphaproteobacteria bacterium GWF2_58_20. The genome of this organism has been retrieved from shotgun metagenomics of a subterranean aquifer (Anantharaman et al. 2016) and contains an environmental homologue of [FeFe]-hydrogenase (table 2). Previously, the genes for [FeFe]-hydrogenases (HydA in table 2) and their maturases have been found in a few free-living proteobacteria, including Phaeospirillum, and metagenomic organisms from human gut microbiota which do not have genes for Q biosynthesis (Degli Esposti et al. 2016). In contrast, alphaproteobacteria bacterium GWF2_58_20 contains the same set of Q biosynthesis proteins as that of P. fulvum (tables 1 and 2) and two Q-reacting enzymes: an incomplete complex I and a homologue of the mitochondrial alternative ubiquinol oxidase, AOX (table 2). AOX is a ferritin-like domain protein that has been previously found in alpha proteobacteria, suggesting its direct transmission to proto-mitochondria (Atteia et al. 2004). Hypothetically, alphaproteobacteria bacterium GWF2_58_20 might be related to the ancestral lineage leading to the proto-mitochondrial organism that contained both AOX and anaerobic traits such as [FeFe]-hydrogenase, as well as Q biosynthesis. The genome of this organism does not contain homologues for UbiC or XanB2 but shows the peculiar synteny UbiJ-UbiD-UbiX (table 2). Such as synteny looks an ancestral feature, because UbiD and UbiX are involved in the same decarboxylation reaction in Q biosynthesis (fig. 1). In turn UbiJ, which is the least frequent Ubi gene among those associated with Q biosynthesis, is often part of another syntenic group with UbiB and UbiE (table 2, cf. Aussel et al. 2014). This synteny is typically found in E. coli (Aussel et al. 2014) and may also have an ancestral origin, because it is present in the genome of deep-branching proteobacteria such as Magnetococcus (table 2).
Table 2.
Selection of Taxa Having Different Pathways for Q Biosynthesis and Q-Reacting Enzymes
| Q-Reacting Enzymes |
Anaerobic |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Organism | Genome | Ubi Genes for Q Biosynthesis | Q Pathway | Complex I | Complex II | bc1 | bd Oxidase | AOX | FeFe-HydA |
| Alpha | |||||||||
| Alphaproteobacteria bacterium GWF2_58_20 | 97.3%a | UbiA, JDX, E, B, G | a | incomplete | no | no | no | yes | yes |
| Phaeospirillum fulvum | 97.3%a | UbiA, D, X, E, B, G | a + CoQ9 | yes, split operon | yes | yes | yes | no | yes |
| Magnetococcus marinus | complete | UbiC, A, D, X, EJB, G | a | yes, ancestral | yes | yes | no | no | no |
| Magnetospirillum magneticum AMB-1 | complete | UbiA, D, X, E, B, G, 2L | m + DX | yes | yes | yes | yes | no | no |
| Ca. Endolissoclinum faulkneri L2 | complete | UbiA, E, B, G, 2L | m—CoQ7 | yes | yes | yes | no | no | no |
| Rhizobium phaseoli Brasil 5 | complete | UbiA, E, B, G, L, M | m—CoQ9c | yes, plus green | yes | yes | yes | no | no |
| Zeta | |||||||||
| Proteobacteria bacterium CG1_02_64_396 | 94.6%a,b | UbiCA, D, X, EJB, G, HI | c | yes | yes | yes | no | no | no |
| Mariprofundus ferrooxydans PV-1 | 94.6%a | UbiCA, D, X, E, J, B, G, H, I | c | yes | yes | yes | yes | no | no |
| Beta | |||||||||
| Gallionella capsiferriformans ES-2 | complete | UbiC, A, D, X, E, J, B, G, I, H | c | yes | yes | yes | yes | no | no |
| Rhodoferax ferrireducens T118 | complete | UbiC, A, D, X, E, J, B, G, I, H, L, M | c | yes | yes | yes | yes | no | no |
| Gamma | |||||||||
| Thiolapillus brandeum | complete | UbiC, 3A, D, X, E, B, G, F, H, I | c—CoQ7 | yes | yes | yes | no | no | no |
| Escherichia coli str. K-12 substr. MG1655 | complete | UbiCA, D, X, EJB, G, F, HI | c—CoQ7 | yes, Nuo13 | yes | no | yes | no | no |
Note.—Ubi genes are contiguous, that is, form a synteny, when not separated by a comma.
Estimated with program BUSCO using proteins Arturo 28 May BUSCO, (Simão et al, 2015).
Estimated to be 98% complete by another method, Probst et al. (2017).
Rare combination found only in another strain of R. phaseoli and also Bosea vaviloviae Vaf18 (table 1) among taxa with complete genome.
Detailed phylogenetic analysis (see Materials and Methods) indicates that alphaproteobacteria bacterium GWF2_58_20 could be classified within the family of Rhodospirillaceae, with Azospirillum as its possible closest genus. Hence, the ancestral anaerobic pathways for Q biosynthesis might have evolved in organisms related to current taxa of Rhodospirillaceae, even if their origin may be older, as discussed below.
Alpha Proteobacteria Contain Homologues of Eukaryotic CoQ9 and Pathways for Q Biosynthesis Different from That of Escherichia coli
A novel finding of the present study is that Phaeospirillum and other alphaproteobacteria have close homologues of the CoQ9 gene (table 1). CoQ9 was originally found in yeast screens for Q biosynthesis defects and is indispensable for this biosynthesis in eukaryotes (Lohman et al. 2014; He et al. 2015; Ozeir et al. 2015). The protein coded by the CoQ9 gene assists the hydroxylase reaction of CoQ7 within the membrane complex that carries out Q biosynthesis in mitochondria (Lohman et al. 2014; Kawamukai 2015). Recently, CoQ9 has been shown to be specifically involved in the hydroxylation of amino precursors of Q derived from pABA (He et al. 2015; Kawamukai 2015). Presumably, bacterial homologues of CoQ9 have the same function, given that they are present in alpha proteobacterial organisms which do not have UbiC and thus rely on pABA as a ring precursor (table 1). Interestingly, CoQ9 is present also in bacteria that do not have CoQ7 or completely lack UbiOHases such as P. fulvum (table 1), thereby suggesting a functional involvement with amino Q precursors and yet unknown enzymes capable of Q hydroxylation in the absence of oxygen. CoQ9 may thus represent an ancestral protein of biosynthetic Q pathways, which underwent differential loss among alpha proteobacteria after being transmitted to proto-mitochondria, in a way similarly to AOX (Atteia et al. 2004). Indeed, it is absent in several, generally late-diverging organisms of the Rhizobiales order such as Rhizobium phaseoli (tables 1 and 2).
Notably, the absence of UbiC implies a likely usage of pABA as a ring precursor (Cheng and Li 2014; Xie et al. 2015). Hence, if the taxonomic distribution of CoQ9 reflects the use of pABA as ring precursor, one would expect to find phylogenetic features and molecular signatures in the UbiA proteins that could be related to their capacity of reacting with the same Q precursor. To test this possibility, I have examined UbiA sequences in detail and found that UbiA proteins belonging to taxa that do not have UbiC form a sister clade to that including UbiA proteins from taxa possessing UbiC (fig. 2A). Detailed inspection of sequence alignments indicated the presence of molecular signatures that distinguish the UbiA proteins belonging to taxa that do not have UbiC from those belonging to taxa possessing UbiC (highlighted in fig. 2B). These signatures are close to the conserved residues lying at the negative (cytoplasmic) side of the membrane that are known to be involved in the binding of both the prenyl-pyrophosphate and 4HB substrate of UbiA (fig. 2B) (Ohara et al. 2009; Cheng and Li 2014).
Such new properties of UbiA proteins define a bifurcation in the bacterial biosynthesis of Q into two pathways, as illustrated in the scheme of figure 3. One pathway fundamentally relies on chorismate as ring precursor, and consequently is characterized by UbiC and the associated variant of UbiA. I have labelled it pathway c, for chorismate. The other pathway does not contain UbiC and often includes CoQ9, therefore relying on pABA for ring precursors. This pathway, which I have labelled m for its similarity with that of mitochondria, fundamentally derives from the anaerobic pathways found in Rhodospirillaceae and is probably the only system for producing Q that works in alpha proteobacteria. Two lines of evidence sustain such a possibility. First, all the UbiA proteins of alpha proteobacteria have the molecular signatures of those that lack the UbiC proteins, even in the case of Rhodobacter vinaykumarii (fig. 2B), the genome of which uniquely shows the combination of UbiC with CoQ9. Secondly, the UbiC proteins found in the genomes of alpha proteobacteria turn out to be likely cases of LGT from other proteobacterial classes (cf. supplementary fig. S1, Supplementary Material online). In the case of R. vinaykumarii, a photosynthetic member of the Rhodobacteraceae family of alpha proteobacteria, phylogenetic trees suggest a likely LGT from beta proteobacteria that are also photosynthetic, such as Polynucleobacter (Degli Esposti and Martinez-Romero 2017).
Fig. 3.

—Scheme for the possible evolution of the biosynthetic pathways of Q. The flow scheme illustrates the hypothetical evolution of the pathways of Q biosynthesis presented in this work: a (in gray box on the left), c (top), and m (bottom), all originating from the ancestral pathway of PQ biosynthesis in cyanobacteria (Pfaff et al. 2014). The scheme includes only the enzymes for Q biosynthesis that vary in presence and combination within the genome of proteobacterial taxa; their symbols are presented in the boxed caption at the bottom of the illustration. UbiA is implied to be present in all combinations for its crucial function (see fig. 1 and table 1), whereas UbiB and UbiJ are not considered because their role in Q biosynthesis is not essential and partially unknown (Nowicka and Kruk 2010; Aussel et al. 2014). Gene loss and acquisition is represented by a red thin arrow and thin black line, respectively. A dark gray square indicates the absence of a gene in the combination of enzymes deduced from genomic information. The dashed arrows in the middle indicate a minor alternative route (or intermediate) in pathway c that seems to be present only in deep branching gamma proteobacteria such as Salinisphaera (Pelosi et al. 2016). A genus or species characteristic for each combination of Q biosynthesis enzymes is indicated at the bottom of each set of symbols (cf. tables 1 and 2). Acquisition of other CoQ genes specific to mitochondria (Kawamukai 2015) is not represented at the end of pathway m, bottom right of the scheme. Q in gray circle indicates anaerobically synthesized ubiquinone (pathway a, within the gray square), whereas aerobically synthesized Q is represented as in figure 1. RQ in red circle indicates the Q derivative rhodoquinone that is found in Rhodosprillum and very few other proteobacteria such as Rhodoferax, as well as in anaerobically adapted mitochondria of invertebrates (Hiraishi and Hoshino 1984; Nowicka and Kruk 2010; Müller et al. 2012). α GWF2, alphaproteobacterium GWF2_58_20 (Anantharaman et al. 2016).
The Escherichia coli Pathway of Q Biosynthesis Comes from Cyanobacteria via Zeta Proteobacteria
The bifurcation of bacterial Q biosynthesis into two separate pathways has deep phylogenetic consequences, because only pathway m leads to that known in eukaryotes, whereas pathway c is present in various proteobacterial classes (table 1) but not alpha proteobacteria (fig. 3). The well-known pathway of Q biosynthesis in E. coli, therefore, represents a late stage of pathway c and cannot be considered a model for mitochondrial Q biosynthesis. Where is pathway c coming from? The answer can be deduced from the observation that pathway c is very similar to that for the biosynthesis of PQ in cyanobacteria (Dähnhardt et al. 2002; Pfaff et al. 2014). The latter pathway includes all the Ubi genes of E. coli (Pfaff et al. 2014) except UbiJ, UbiI, and UbiF, as verified here. Although UbiJ is not indispensable for Q biosynthesis (Aussel et al. 2014), UbiI and UbiF derive from UbiH (Pelosi et al. 2016), which is already present in cyanobacteria to carry out C1 hydroxylation of the ring (Dähnhardt et al. 2002; Pfaff et al. 2014). Detailed phylogenetic trees indicate that the UbiH proteins of cyanobacteria may be the ancestors of all FAD-dependent UbiOHases present in proteobacteria (fig. 4).
An evolutionary connection between the prokaryotic biosynthesis of PQ and that of Q has been unveiled in the plant field (Dähnhardt et al. 2002; Pfaff et al. 2014), but has been recently overlooked in microbiological and biomedical fields (Meganathan and Kwon 2009; Nowicka and Kruk 2010; Aussel et al. 2014; Zhi et al. 2014; Kawamukai 2015; Marreiros et al. 2016; Ravcheev and Thiele 2016). Presumably, the different pathway of PQ biosynthesis in plant chloroplasts (Nowicka and Kruk 2010; Pfaff et al. 2014) has hampered appreciation of the strong similarity between the biosynthesis of the same quinone in cyanobacteria and that of prokaryotic Q. Results obtained in the present study indicate that the origin for the biosynthetic pathway of Q stems from cyanobacteria. Besides the UbiH protein (fig. 4), several other Ubi proteins from cyanobacteria consistently lie in deep branching position with respect to those of proteobacteria in extended phylogenetic trees (see fig. 2 for UbiA and supplementary fig. S1 for UbiC and fig. S2 for UbiD, Supplementary Material online). Although such data are in partial agreement with previous results (Pfaff et al. 2014), their interpretation is different. Pfaff et al. (2014) considered that cyanobacteria once had Q and then adapted its biosynthesis to that of PQ. However, such a proposition is untenable in phylogenetic terms, because cyanobacterial proteins are in ancestral position versus their proteobacterial homologues in the great majority of trees examined here (figs. 2A and 4, and supplementary figs. S1 and S2, Supplementary Material online). It is also untenable considering that Q biosynthesis evolved as a consequence of, and hence after the oxygenation event generated by cyanobacteria on ancestral earth (Schoepp-Cothenet et al. 2009; Degli Esposti 2016), additionally requiring two oxygen-dependent steps with respect to the biosynthesis of PQ.
Intriguingly, these phylogenetic analyses uncovered homologous of the UbiC, UbiA, and UbiD proteins also in the genome of unclassified delta proteobacteria (fig. 2 and supplementary figs. S1 and S2, Supplementary Material online), a class of predominantly anaerobic organisms having MQ as their typical membrane quinone (Nowicka and Kruk 2010; Degli Esposti and Martinez-Romero 2017). Some members of this class may synthesize Q (Spain et al. 2016) via anaerobic pathways, because no homologues of UbiH or other UbiOHases could be detected among delta proteobacteria (cf. fig. 4). The scattered presence of some Ubi proteins, UbiC in particular (supplementary fig. S1, Supplementary Material online), among deltaproteobacteria suggests that the pathway of PQ biosynthesis in cyanobacteria might have been vertically transmitted to delta and other proteobacteria, until additional enzymes allowed the novel synthesis of Q under aerobic conditions. The bacterial taxa in which the complete c pathway emerged along evolution appear to be represented by the metagenomic bacterium, Proteobacteria bacterium CG1_02_64_396 (tables 1 and 2). This organism was found in a subterranean aquifer (Probst et al. 2017) and can be squarely classified among the recently introduced class of zeta proteobacteria, which now have multiple representatives in freshwater environments (McBeth et al. 2013; Probst et al. 2017). The taxonomic placement among zeta proteobacteria derives from a genome-wide comparison of all genes, including various proteins involved in Q biosynthesis or reactions (tables 1 and 2). Most of such proteins cluster with those of zeta proteobacteria including Mariprofundus, the type genus of the class (Emerson et al. 2007; Barco et al. 2015; Makita et al. 2017) (fig. 4 and supplementary fig. S3, Supplementary Material online).
Proteobacteria bacterium CG1_02_64_396, which will be called proteozeta1 hereafter, has the same syntenic groups of Ubi genes as E. coli, including the UbiI-UbiH diad (table 2). This diad suggests an early process of gene duplication of the ancestral UbiH of cyanobacteria (cf. fig. 4). In addition, the genome of proteozeta 1 contains the CoQ7 gene, as in all zeta and many gamma proteobacteria (tables 1 and 2). Hence, the original pathway c for Q biosynthesis included CoQ7, as indicated in table 1 and figure 3. Interestingly, zeta proteobacteria have multiple Q-reacting enzymes involved in bioenergetics (table 2) plus another, the Rnf complex, which is typically found in facultatively anaerobic, chemolithotrophic organisms such as Magnetococcus. Zeta proteobacteria and Magnetococcus share similar ecological niches with low or gradient concentrations of oxygen (Emerson et al. 2007; Schübbe et al. 2009; McBeth et al. 2013), thereby using both aerobic and anaerobic pathways of energy conservation for their survival (Emerson et al. 2007; Schübbe et al. 2009; Barco et al. 2015). Indeed, some Q biosynthetic proteins of Magnetococcus and its relatives often cluster with those of proteozeta 1 (fig. 2 and supplementary fig. S1, Supplementary Material online).
All CoQ Proteins of Eukaryotes Have Bacterial Homologues
The finding of bacterial homologues of eukaryotic Q biosynthesis proteins such as CoQ7 (Stenmark et al. 2001; Pelosi et al. 2016) and CoQ9 (tables 1 and 2) stimulated a systematic search of bacterial homologous for the remaining CoQ proteins that do not have orthologs in E. coli, namely CoQ4, CQ10, and CoQ11 (Kawamukai 2015). CoQ4 was found to be widespread in alpha, beta, gamma, and also zeta proteobacteria, besides its previously reported presence in cyanobacteria (Kawamukai 2015). Interestingly, cyanobacterial CoQ4 proteins are in precursor position with respect to eukaryotic and proteobacterial homologues, which form sister clades. CoQ10 showed a widespread distribution in proteobacteria but not in cyanobacteria, where it is present in a few organisms such as Mastigocladus laminosus likely due to LGT. Finally, CoQ11 has only paralogue proteins in proteobacteria, which are related to a sort chain dehydrogenase normally forming a subunit of complex I. Hence, all eukaryotic CoQ proteins appear to have homologous or similar proteins in proteobacteria that produce Q—and in the case of CoQ4 among cyanobacteria too. Consequently, their absence from E. coli is not due to substantial differences in bacterial Q metabolism, but from gene loss leading to the derived pathway of Q biosynthesis that is present in this bacterium (fig. 3).
Conclusion
This work presents the first comprehensive evaluation of the pathways of Q biosynthesis currently present in prokaryotes and their evolution from ancestral, facultatively anaerobic organisms that have extant relatives in Phaeospirillum fulvum and Magnetococcus (fig. 3). The evolutionary trajectory of these pathways would emerge as follows. The earliest set of Q biosynthetic enzymes was present in cyanobacteria and used for the biosynthesis of PQ (fig. 3 and table 1), which lacks the two methoxy groups of Q at positions 5 and 6. Hence, Q biosynthesis required the development of new UbiOHases capable of hydroxylating these positions of the ring. Under anaerobic conditions, the UbiH protein inherited from cyanobacteria (Pfaff et al. 2014) was lost and ring hydroxylation was carried out by alternative, yet unidentified enzymes in pathway a (fig. 3, left). Under aerobic conditions, cyanobacterial UbiH was instead duplicated, as the UbiI-UbiH synteny suggests (table 2), subsequently undergoing diversification in UbiL, UbiM, and UbiF—the latter typical of gamma proteobacteria (Pelosi et al. 2016). The diversification into the enzymatically versatile UbiL and UbiM must have occurred in ancestral alpha proteobacteria, because no close homologue of UbiH is found among current members of this class (cf. [7]), nor in delta proteobacteria (fig. 4).
Conversely, homologous of UbiC, UbiA, and UbiD were probably inherited from cyanobacteria (Pfaff et al. 2014) in parallel to some delta proteobacteria, given their scattered distribution in the genome of unclassified delta proteobacteria (fig. 2 and supplementary figs. S1 and S2, Supplementary Material online). Ancestral UbiD homologous such that of Deferrisoma (supplementary fig. S2, Supplementary Material online) can actually function in the decarboxylation steps of the futalosine pathway of MQ biosynthesis (Zhi et al. 2014; Ravcheev and Thiele 2016), which is widespread in delta proteobacteria (Zhi et al. 2014). The hypothesis that the futalosine pathway may have contributed to the evolution of Q biosynthesis (Ravcheev and Thiele 2016) is not sustained by the results obtained here, which indicate instead that the biosynthetic pathway of PQ in cyanobacteria is the likely ancestor of the proteins used by proteobacteria for the anabolism of Q (fig. 4 and supplementary figs. S1 and S2, Supplementary Material online). Of note, cyanobacteria also have ancestors of eukaryotic Q biosynthesis proteins such as CoQ4 (Kawamukai 2015), as confirmed in this study.
Functionally linked CoQ7 and CoQ9 fundamentally distinguish Q biosynthesis from that of either PQ or MQ. Originally discovered in yeast, these small proteins originated in ancestral alpha proteobacteria, as reported here for CoQ9 (table 1) and earlier for CoQ7 (Stenmark et al. 2001; Pelosi et al. 2016). Intriguingly, CoQ7 is present in diverse pathways of Q biosynthesis, whereas CoQ9 is present only in those of alpha proteobacteria, including anaerobic pathway a of P. fulvum (table 1). Hence, the strict dependence of CoQ7 on CoQ9 that is found in eukaryotes (Lohman et al. 2014; He et al. 2015; Kawamukai 2015) may result from a late evolutionary variation in the involvement of these proteins in Q biosynthesis. What was the original function of CoQ9 in bacteria is unclear. However, it is clear that this protein was “invented” by ancestral alpha proteobacteria, probably after the loss of enzymes producing ring precursors from chorismate, concomitantly with the duplication and diversification of cyanobacterial UbiH (fig. 3). Additional studies are now required to further clarify the function and relevance of bacterial CoQ proteins in the pathways for Q biosynthesis of prokaryotes.
In sum, the present study has rationalized the increasing complexity of Q biosynthesis in bacteria, clarifying that current biosynthetic pathways leading to that present in eukaryotes originated in the Rhodospirillaceae family of alpha proteobacteria (fig. 3). Notably, the same pathway is present in organisms producing the Q derivative, rhodoquinone (RQ, fig. 3), which is also found in anaerobically adapted mitochondria (Müller et al. 2012). The data and considerations discussed here will thus pave the way to discover the enigmatic enzymes (Nowicka and Kruk 2010; Müller et al. 2012) that are responsible for RQ biosynthesis.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
I thank Esperanza Martinez-Romero (CCG UNAM) for her support and advice. Luis Lozano and Arturo Ponce de Leon are thanked for their technical help in some aspects of the analyses. This work was sponsored by CONACyT grants No. 263876 and 253116 to Esperanza Martinez-Romero.
Literature Cited
- Alexander K, Young IG.. 1978. Alternative hydroxylases for the aerobic and anaerobic biosynthesis of ubiquinone in Escherichia coli. Biochemistry 17(22):4750–4755.http://dx.doi.org/10.1021/bi00615a024 [DOI] [PubMed] [Google Scholar]
- Almagro G, et al. 2015. Comparative genomic and phylogenetic analyses of Gammaproteobacterial glg genes traced the origin of the Escherichia coli glycogen glgBXCAP operon to the last common ancestor of the sister orders Enterobacteriales and Pasteurellales. PLoS One 10(1):e0115516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman K, et al. 2016. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 7:13219.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atteia A, et al. 2004. Identification of prokaryotic homologues indicates an endosymbiotic origin for the alternative oxidases of mitochondria (AOX) and chloroplasts (PTOX). Gene 330:143–148.http://dx.doi.org/10.1016/j.gene.2004.01.015 [DOI] [PubMed] [Google Scholar]
- Aussel L, et al. 2014. Biosynthesis and physiology of coenzyme Q in bacteria. Biochim Biophys Acta 1837(7):1004–10011.http://dx.doi.org/10.1016/j.bbabio.2014.01.015 [DOI] [PubMed] [Google Scholar]
- Barco RA, et al. 2015. New insight into microbial iron oxidation as revealed by the proteomic profile of an obligate iron-oxidizing chemolithoautotroph. Appl Environ Microbiol. 81(17):5927–5937.http://dx.doi.org/10.1128/AEM.01374-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Li W.. 2014. Structural insights into ubiquinone biosynthesis in membranes. Science 343(6173):878–881.http://dx.doi.org/10.1126/science.1246774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluis CP, et al. 2011. Identification of bottlenecks in Escherichia coli engineered for the production of CoQ(10). Metab Eng 13(6):733–744.http://dx.doi.org/10.1016/j.ymben.2011.09.009 [DOI] [PubMed] [Google Scholar]
- Collins MD, Jones D.. 1981. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol Rev. 45(2):316–354. 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dähnhardt D, et al. 2002. The hydroxyphenylpyruvate dioxygenase from Synechocystis sp. PCC 6803 is not required for plastoquinone biosynthesis. FEBS Lett. 523(1–3):177–181. [DOI] [PubMed] [Google Scholar]
- Degli Esposti M. 2016. Late mitochondrial acquisition, really? Genome Biol Evol. 8(6):2031–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti M, et al. 2016. Alpha proteobacterial ancestry of the [Fe-Fe]-hydrogenases in anaerobic eukaryotes. Biol Direct. 11:34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti M, Martinez-Romero E.. 2017. The functional microbiome of arthropods. PLoS One 12(5):e0176573.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson D, et al. 2007. A novel lineage of proteobacteria involved in formation of marine Fe-oxidizing microbial mat communities. PLoS One 2(7):e667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JB, et al. 2016. Metagenomic analysis of a high carbon dioxide subsurface microbial community populated by chemolithoautotrophs and bacteria and archaea from candidate phyla. Environ Microbiol. 18(6):1686–1703.http://dx.doi.org/10.1111/1462-2920.12817 [DOI] [PubMed] [Google Scholar]
- Esposti MD, et al. 1993. Mitochondrial cytochrome b: evolution and structure of the protein. Biochim Biophys Acta 1143(3):243–271.http://dx.doi.org/10.1016/0005-2728(93)90197-N [DOI] [PubMed] [Google Scholar]
- Esser C, Martin W, Dagan T.. 2007. The origin of mitochondria in light of a fluid prokaryotic chromosome model. Biol Lett. 3(2):180–184.http://dx.doi.org/10.1098/rsbl.2006.0582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher DT, et al. 2001. The crystal structure of chorismate lyase shows a new fold and a tightly retained product. Proteins 44(3):304–311.http://dx.doi.org/10.1002/prot.1095 [DOI] [PubMed] [Google Scholar]
- Hajj Chehade M, et al. 2013. ubiI, a new gene in Escherichia coli coenzyme Q biosynthesis, is involved in aerobic C5-hydroxylation. J Biol Chem. 288(27):20085–20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CH, et al. 2015. Yeast Coq9 controls deamination of coenzyme Q intermediates that derive from para-aminobenzoic acid. Biochim Biophys Acta 1851(9):1227–1239.http://dx.doi.org/10.1016/j.bbalip.2015.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraishi A, Hoshino Y.. 1984. Distribution of rhodoquinone in Rhodospirillaceae and its taxonomic implication. J Gen Appl Microbiol. 30(6):435–448.http://dx.doi.org/10.2323/jgam.30.435 [Google Scholar]
- Ji B, et al. 2017. The chimeric nature of the genomes of marine magnetotactic coccoid-ovoid bacteria defines a novel group of Proteobacteria. Environ Microbiol. 19(3):1103–1119.http://dx.doi.org/10.1111/1462-2920.13637 [DOI] [PubMed] [Google Scholar]
- Kantor RS, et al. 2015. Bioreactor microbial ecosystems for thiocyanate and cyanide degradation unravelled with genome-resolved metagenomics. Environ Microbiol. 17(12):4929–4941.http://dx.doi.org/10.1111/1462-2920.12936 [DOI] [PubMed] [Google Scholar]
- Kawamukai M. 2015. Biosynthesis of coenzyme Q in eukaryotes. Biosci Biotechnol Biochem. 80(1):23–33. [DOI] [PubMed] [Google Scholar]
- Ku C, et al. 2015. Endosymbiotic gene transfer from prokaryotic pangenomes: inherited chimerism in eukaryotes. Proc Natl Acad Sci U S A. 112(33):10139–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman DC, et al. 2014. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc Natl Acad Sci U S A. 111(44):E4697–E4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita H, et al. 2017. Mariprofundus micogutta sp. nov., a novel iron-oxidizing zetaproteobacterium isolated from a deep-sea hydrothermal field at the Bayonnaise knoll of the Izu-Ogasawara arc, and a description of Mariprofundales ord. nov. and Zetaproteobacteria classis nov. Arch Microbiol. 199(2):335–346.http://dx.doi.org/10.1007/s00203-016-1307-4 [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. 2015. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43(Database issue):D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marreiros BC, et al. 2016. Exploring membrane respiratory chains. Biochim Biophys Acta 1857(8):1039–1067.http://dx.doi.org/10.1016/j.bbabio.2016.03.028 [DOI] [PubMed] [Google Scholar]
- Matsumura M, Kobayashi T, Aiba S.. 1983. Anaerobic production of ubiquinone-10 by Paracoccus denitrificans. Eur J Appl Microbiol Biotechnol. 17(2):85–89.http://dx.doi.org/10.1007/BF00499856 [Google Scholar]
- McBeth JM, et al. 2013. The transition from freshwater to marine iron-oxidizing bacterial lineages along a salinity gradient on the Sheepscot River, Maine, USA. Environ Microbiol Rep. 5(3):453–463.http://dx.doi.org/10.1111/1758-2229.12033 [DOI] [PubMed] [Google Scholar]
- Meganathan R, Kwon O.. 2009. Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q). EcoSal Plus 3: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillo V, et al. 2014. Isolation, cultivation and genomic analysis of magnetosome biomineralization genes of a new genus of South-seeking magnetotactic cocci within the Alphaproteobacteria. Front Microbiol. 5:72.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, et al. 2012. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol Mol Biol Rev. 76(2):444–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen HB, et al. 2014. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 32(8):822–828.http://dx.doi.org/10.1038/nbt.2939 [DOI] [PubMed] [Google Scholar]
- Nowicka B, Kruk J.. 2010. Occurrence, biosynthesis and function of isoprenoid quinones. Biochim Biophys Acta 1797(9):1587–1605.http://dx.doi.org/10.1016/j.bbabio.2010.06.007 [DOI] [PubMed] [Google Scholar]
- Ohara K, et al. 2009. Functional characterization of LePGT1, a membrane-bound prenyltransferase involved in the geranylation of p-hydroxybenzoic acid. Biochem J. 421(2):231–241. [DOI] [PubMed] [Google Scholar]
- Ozeir M, et al. 2015. Coq6 is responsible for the C4-deamination reaction in coenzyme Q biosynthesis in Saccharomyces cerevisiae. J Biol Chem. 290(40):24140–24151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelosi L, et al. 2016. Evolution of ubiquinone biosynthesis: multiple proteobacterial enzymes with various regioselectivities to catalyze three contiguous aromatic hydroxylation reactions. mSystems 1(4):e00091-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff C, et al. 2014. Chorismate pyruvate-lyase and 4-hydroxy-3-solanesylbenzoate decarboxylase are required for plastoquinone biosynthesis in the cyanobacterium Synechocystis sp. PCC6803. J Biol Chem. 289(5):2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst AJ, et al. 2017. Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO(2) concentrations. Environ Microbiol. 19(2):459–474.http://dx.doi.org/10.1111/1462-2920.13362 [DOI] [PubMed] [Google Scholar]
- Ravcheev DA, Thiele I.. 2016. Genomic analysis of the human gut microbiome suggests novel enzymes involved in quinone biosynthesis. Front Microbiol. 7:128.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp-Cothenet B, et al. 2009. Menaquinone as pool quinone in a purple bacterium. Proc Natl Acad Sci U S A. 106(21):8549–8554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schübbe S, et al. 2009. Complete genome sequence of the chemolithoautotrophic marine magnetotactic coccus strain MC-1. Appl Environ Microbiol. 75(14):4835–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, et al. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212. [DOI] [PubMed] [Google Scholar]
- Sizova MV, et al. 2007. Novel facultative anaerobic acidotolerant Telmatospirillum siberiense gen. nov. sp. nov. isolated from mesotrophic fen. Syst Appl Microbiol. 30(3):213–220.http://dx.doi.org/10.1016/j.syapm.2006.06.003 [DOI] [PubMed] [Google Scholar]
- Spain EM, et al. 2016. Identification and differential production of ubiquinone-8 in the bacterial predator Bdellovibrio bacteriovorus. Res Microbiol. 167(5):413–423. [DOI] [PubMed] [Google Scholar]
- Stenmark P, et al. 2001. A new member of the family of di-iron carboxylate proteins. Coq7 (clk-1), a membrane-bound hydroxylase involved in ubiquinone biosynthesis. J Biol Chem. 276(36):33297–33300. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28(10):2731–2739.http://dx.doi.org/10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie LX, et al. 2015. Resveratrol and para-coumarate serve as ring precursors for coenzyme Q biosynthesis. J Lipid Res. 56(4):909–919.http://dx.doi.org/10.1194/jlr.M057919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva J, et al. 2006. Two crystal structures of the bisochorismate pyruvate lyase from Pseudomonas aeruginosa. J Biol Chem. 281(44):33441–33449. [DOI] [PubMed] [Google Scholar]
- Zhi XY, et al. 2014. The futalosine pathway played an important role in menaquinone biosynthesis during early prokaryote evolution. Genome Biol Evol. 6(1):149–160.http://dx.doi.org/10.1093/gbe/evu007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, et al. 2013. The diffusible factor synthase XanB2 is a bifunctional chorismatase that links the shikimate pathway to ubiquinone and xanthomonadins biosynthetic pathways. Mol Microbiol. 87(1):80–93.http://dx.doi.org/10.1111/mmi.12084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.