

Abstract
Mitochondrial genomes (mitogenomes) are remarkably diverse in genome size and organization, but the origins of dynamic mitogenome architectures are still poorly understood. For instance, the mutational burden hypothesis postulates that the drastic difference between large plant mitogenomes and streamlined animal mitogenomes can be driven by their different mutation rates. However, inconsistent trends between mitogenome sizes and mutation rates have been documented in several lineages. These conflicting results highlight the need of systematic and sophisticated investigations on the evolution and diversity of mitogenome architecture. This study took advantage of the strikingly variable mitogenome size among different yeast species and also among intraspecific strains, examined sequence dynamics of introns, GC-clusters, tandem repeats, mononucleotide repeats (homopolymers) and evaluated their contributions to genome size variation. The contributions of these sequence features to mitogenomic variation are dependent on the timescale, over which extant genomes evolved from their last common ancestor, perhaps due to a combination of different turnover rates of mobile sequences, variable insertion spaces, and functional constraints. We observed a positive correlation between mitogenome size and the level of genetic drift, suggesting that mitogenome expansion in yeast is likely driven by multiple types of sequence insertions in a primarily nonadaptive manner. Although these cannot be explained directly by the mutational burden hypothesis, our results support an important role of genetic drift in the evolution of yeast mitogenomes.
Keywords: genome size, mitogenome, intron, GC-cluster, tandem repeats, genetic drift
Introduction
Since their origin of an alpha-proteobacterial endosymbiont, mitochondrial genomes (mitogenomes) have undergone substantial reduction in gene content, but adopted radically different shapes and sizes with highly variable intronic and intergenic sequences (Burger et al. 2003; Shao et al. 2009; Sloan et al. 2012; Freel et al. 2014; Del Vasto et al. 2015; Smith and Keeling 2015). Studies on these radically different mitogenomes are important for mitochondrial genetics and evolutionary biology, but also important for the understanding of mitochondrial DNA mutation and maintenance, which are crucial in areas such as pathogenicity, ageing, and diseases. There are several evolutionary genetic mechanisms proposed to explain the evolution of mitogenomes. Mitogenomes in animals may have undergone recurrent adaptive evolution (Bazin et al. 2006). In plants, the evolution of mitogenomes can be driven by changes in recombinational processes (Sloan et al. 2012) and differences in DNA repair efficiency (Christensen 2013). The mutational burden hypothesis (MBH) proposed a potentially unifying explanatory framework arguing for a central role for nonadaptive processes such as mutation and genetic drift in the evolution of organelle genome size (Lynch et al. 2006). According to the MBH, introns and noncoding DNAs increase the size of the mutational target and are mutational liabilities, which have a greater tendency to accumulate in a mitogenome with a low mutation rate as opposed to one with a high rate of mutation. The MBH has gained support in a number of genomic studies (Smith and Lee 2010; Boussau et al. 2011; Chong and Mueller 2013; Smith et al. 2013), whereas inconsistency to the MBH has also been reported (Alverson et al. 2010; Whitney et al. 2010).
A lesson learned from recent studies is that the variation of genome architecture can be driven by distinct molecular mechanisms. For instance, larger eukaryotic genomes are subject to expansion of presumably deleterious transposable elements and remnants due to strong genetic drift (Lynch and Conery 2003). In bacterial genomes, in which sequence deletion is prevalent (Mira et al. 2001), genetic drift tends to fix deleterious genome reduction in lineages with smaller effective population sizes (Kuo et al. 2009), resulting a genome size trend different from that seen in eukaryotes (Daubin and Moran 2004). Furthermore, genetic mechanisms underlying the variation of genome architecture may differ over different evolutionary timescales. In pneumococcal bacteria, short-term within-lineage genomic variation is characterized by movement of phages and shuffling of variable restriction-modification systems, whereas different lineages are distinguished by infrequent transfers of stable loci (Croucher et al. 2014). In fungal nuclear genomes, genetic drift is shown to be responsible only for large-scale genome expansions, whereas small-scale modifications in genome size are independent of drift (Kelkar and Ochman 2012). Thus, to better understand the evolution of mitogenome architecture, it is important to recognize and reconcile conflicting genomic data.
As genetic variation is the substrate of evolution, study of mitogenome architecture and complexity among closely related mitogenomes of significant variation would be ideal for understanding their underlying mechanisms. This study took advantage of the yeast family Saccharomycetaceae, as their mitogenomes differ over 5-fold in size, ranging from 20.1 kb in Candida glabrata (Koszul et al. 2003) to 107.1 kb in Nakaseomyces bacillisporus (Bouchier et al. 2009). These mitogenomes exhibit not only striking interspecific divergence but also great intraspecific (within-species) diversity. For instance, mitogenomes among closely related species can vary substantially in intron presence/absence and sequence variation, and in the distribution of GC-rich transposable elements (GC-clusters) (Jung et al. 2012; Wu and Hao 2014, 2015; Wolters et al. 2015; Wu et al. 2015). Such enriched diversity and divergence in yeast mitogenomes, for which many genomic resources are available, provide an excellent opportunity to directly test the MBH and whether genetic drift plays a central role in evolution of mitogenome architecture.
To obtain broader mitogenomic divergence and higher resolution intraspecific variation, we generated additional 17 complete mitogenomes from eight yeast species. We examined mitogenome size variation among different species and also among intraspecific strains, and compared mitochondrial sequence contents and characteristics at different evolutionary scales. Our results show that rapid turnover of mobile sequences (e.g., introns and GC-clusters) can lead to significant genome size variation, but likely occur at limited insertion sites; whereas expansion and contraction of small repeats per event may cause only subtle genomic variation, but take place at a persistent manner. Such differences can sometimes lead to Simpson’s paradox, that is, reverse trends between intraspecific and interspecific comparisons. We also discuss the role of genetic drift and relaxed functional constraints on the evolution of mitogenome size in yeast.
Materials and Methods
Mitogenome Sequencing, Assembly, and Annotation
The Candida bracarensis (n = 1), Candida nivariensis (n = 1), Naumovozyma dairenensis (n = 1), Naumovozyma castellii (n = 4), Kazachstania unispora (n = 1), Torulaspora delbrueckii (n = 4), Torulaspora microellipsoides (n = 1), and Zygosaccharomyces mellis (n = 1) strains were obtained from the National Center of Agricultural Utilization Research (IL). Three Torulaspora quercuum strains were kindly provided by Prof. Feng-Yan Bai (Chinese Academy of Sciences). Genomic DNA of each strain was extracted from a 2-day culture of a single colony inoculation. Seventeen mitogenomes were sequenced at read depths ranging from 338 to 3,484× by the Illumina MiSeq platform (Paired-End 250 bp, or PE250), and assembled using SPAdes v3.7.1 (Bankevich et al. 2012) with k-mers of 55, 75, 89, 97, and 127. Each assembly was validated by visualizing mapping of raw reads versus assembled genome using BWA-MEM v0.7.12 (Li and Durbin 2009) and Integrative Genomics Viewer (IGV v2.3.60) (Robinson et al. 2011). All 17 mitogenomes were assembled into single, circularized genomes of length 24.3–46.4 kb (supplementary table S1, Supplementary Material online). The genomes were annotated using MFannot (Nadimi et al. 2012), followed by manual correction of intron boundaries. These mitogenomes have been deposited to GenBank with accession numbers of KU920675–KU920682 and KY989226–KY989234. They were analyzed together with additional 167 yeast mitogenomes downloaded from GenBank (supplementary table S1, Supplementary Material online).
Phylogenetic Construction
The yeast phylogenetic relationship was constructed based on the concatenated sequences of seven mitochondrial protein genes (atp6, atp8, atp9, cob, cox1, cox2, and cox3). The var1 (or rps3) gene was excluded due to its extremely high level of sequence variation. Homologous nucleotide sequences were aligned based on their amino acid sequences using MUSCLE (Edgar 2004) implemented in SEAVIEW v4.5.3 (Gouy et al. 2010). Phylogenetic construction was performed using PhyML v3.1 (Guindon et al. 2010) under a GTR + Γ+I nucleotide substitution model with 100 bootstraps.
Calculating the πa/πs and Ka/Ks Ratios among Intraspecific Strains
The ratio of nonsynonymous nucleotide diversity to synonymous nucleotide diversity (Ka/Ks for two strains, πa/πs for more than two strains) was calculated on the concatenated alignments (5,375–5,520 sites in length) of seven mitochondrial genes for species having sequence data in two or more strains using DnaSP v5.10.01 (Rozas et al. 2003). In Saccharomycetaceae, 12 species currently have complete mitogenomes in two or more strains (supplementary table S1, Supplementary Material online). Kazachstania unispora and Kazachstania servazzii have available sequences for the seven core mitochondrial genes in two or more strains generated in our unpublished study. The sequence alignments in each of these 14 species were analyzed and the data are available at http://haolab.wayne.edu/. Seven species that had sufficient synonymous nucleotide diversity (πs > 0.02 or Ks > 0.02 based on the Ks divergence between the closest interspecific pair Torulaspora franciscae and T. pretoriensis being 0.043) were subject to subsequent analyses. These seven species are Eremothecium gossypii (Ego), Kazachstania servazzii (Kse), Kazachstania unispora (Kun), Lachancea kluyveri (Lkl), Saccharomyces cerevisiae (Sce), Saccharomyces paradoxus (Spa), and Torulaspora globosa (Tgl).
Analysis of Mitochondrial Intron Turnover
Intron presence/absence and turnover rates were analyzed as previously done in the analyses of GC-clusters (Wu and Hao 2015). Briefly, pairwise diversity in intron presence/absence polymorphism was calculated as . Intron gain and loss at homologous sites were modeled as a two-state continuous-time Markov process, with states 0 (absence) and 1 (presence) on a phylogeny. Intron turnover rates were measured by the R package DiscML (Kim and Hao 2014) using tree branch lengths as estimators of relative time scale, and the rates are expressed as the number of gains/losses per site per nucleotide substitution (Hao and Golding 2006; Wu and Hao 2014). To allow direct and meaningful comparisons, we focused our intron analyses on the same set of S. cerevisiae (n = 18) and S. paradoxus (n = 15) genomes using the same procedures as in our previous study on GC-clusters (Wu and Hao 2015). Due to the low divergence among intraspecific strains, concatenated sequences of 630 nuclear-encoded single-copy genes present in all 18 S. cerevisiae and 15 S. paradoxus strains were used to construct intraspecific phylogenies as done previously (Wu and Hao 2015).
Identification of GC Clusters, Tandem, and Homopolymer Repeats
Our previous study (Wu and Hao 2015) classified GC clusters in mitogenomes from disperse repeats detected by a GUI (graphical user interface) -based UGene program (Okonechnikov et al. 2012). To perform batch analysis on 184 mitogenomes in this study, we conducted BLASTN searches (Camacho et al. 2009) using command line to identify disperse repeats with parameters “-W 7 -r 1 -q -4 -G 60 -E 40 -F F.” Since these yeast mitogenomes are highly AT rich (GC content ranging from 10.9% to 26.7%), A/T nucleotides at the ends of disperse repeats were trimmed off and GC clusters were required to have >50% GC content and >20 nucleotides in length.
Tandem repeats were identified using the TRF (tandem repeats finder) program with default parameters (Benson 1999). Overlapped tandem repeat sequences in genomic locations were merged for further analyses. Homopolymer runs were scanned using an in-house PERL script, and the total nucleotides in homopolymer runs (n≥5 and n≥6) were counted (supplementary table S1, Supplementary Material online). Unless mentioned otherwise, only homopolymer runs (n≥5) were used in interspecific and intraspecific comparisons.
Ancestral Mitogenome Size Reconstruction
Ancestral mitogenome sizes (with 95% confidence interval) were reconstructed using maximum likelihood under a Brownian motion model by the ACE function of the R package APE (Paradis et al. 2004). When multiple intraspecific mitogenomes were available in an extant species, the average genome size was used in the reconstruction.
High-Resolution Analysis of Short Indels between Mitogenomes
In order to best determine fine-scale indel differences, we focused on intraspecific mitogenomes that have identical intron distribution and constructed intraspecific whole genome alignments using the Mauve program (Darling et al. 2004). Since short indels between genome sequences can potentially be confounded by PCR bias in library preparation, sequencing, and assembly errors (Hao et al. 2012; Fungtammasan et al. 2015; Schirmer et al. 2015), we took advantage of the long PE250 reads generated from PCR free library preparation in this study and thoroughly examined four pairs of intraspecific mitogenomes, where short indels can be unambiguously validated within high-coverage PE250 reads by visual inspection of the mapped raw reads. Naumovozyma castellii Y12630 and Y12631 have only two single-guanine (G) indels but no nucleotide substitutions. Indel information for the remaining three mitogenome pairs is summarized in table 1, and detailed information is provided in supplementary table S2, Supplementary Material online, for Torulaspora delbrueckii Y1535 and Y11634, supplementary table S3, Supplementary Material online, for Torulaspora quercuum XZ-46 A and XZ-129 and supplementary table S4, Supplementary Material online, for Naumovozyma castellii Y664 and Y12630.
Table 1.
Small Indels between the Mitogenomes of Torulaspora quercuum (Tqu) XZ-46 A and XZ-129, Naumovozyma castellii (Nca) Y664 and Y12630, Torulaspora delbrueckii (Tde) Y1535 and Y11634
| Type of Indels | Number of Indels | Nucleotide Differences |
||
|---|---|---|---|---|
| A/T | G/C | |||
| Tqu XZ-46A and Tqu XZ-129 | Homopolymers | 9 | 6 | 4 |
| Tandem repeats | 11 | 84 | 9 | |
| Other indels | 0 | 0 | 0 | |
| Nca Y664 and Nca Y664 | Homopolymers | 3 | 0 | 3 |
| Tandem repeats | 3 | 20 | 2 | |
| Other indels | 2 | 79 | 54 | |
| Tde Y1535 and Tde Y11634 | Homopolymers | 48 | 8 | 76 |
| Tandem repeats | 8 | 65 | 2 | |
| Other indels | 5 | 33 | 82 | |
Note.—Details are shown in supplementary tables S2–S4, Supplementary Material online.
Statistical Analyses
All statistical tests were conducted in R v3.3.2 (R Development Core Team 2016) and the R scripts are available at http://haolab.wayne.edu/. As the data of genomic features are not necessarily normally distributed, nonparametric Spearman’s ρ values were calculated for the correlation coefficient.
Results and Discussions
Striking Interspecific Mitogenome Variation Driven by Intergenic Sequences
Mitogenome size varies substantially in many phylogenetic clades (fig. 1). For instance, Nakaseomyces bacillisporus and Candida glabrata belong to the Nakaseomyces clade (Gabaldon et al. 2013), and their mitogenome sizes differ by 87 kb (107.1 kb vs. 20.1 kb). The mitogenome size of Saccharomyces cerevisiae is, on an average, 18.3 kb larger than that of Saccharomyces eubayanus (82.3 kb vs. 64.0 kb). Mitogenome size also differs 20.6 kb (25.8 kb vs. 46.4 kb) between Naumovozyma castellii and Naumovozyma dairenensis. Mitogenome size ranges from 23.8 kb to 51.7 kb among the Lachancea species. On the yeast phylogeny in figure 1, 23 branches (17 external and 6 internal) show difference >4 kb from their respective recent ancestral mitogenomes. Among these 23 branches, 10 of them underwent genome expansion, whereas 13 branches underwent genome contraction (not significantly different in preference). Thus, both genome expansion and genome contraction are common during the evolution of modern mitogenomes.
Fig. 1.—

Variable mitogenomes among 33 yeast species. The phylogenetic tree is based on seven core mitochondrial genes: cob, cox1, cox2, cox3, atp6, atp8, and atp9. Thick internal branches signify bootstrap values >80%. Mitogenome sizes of extant species, and estimated ancestral mitogenome sizes (with 95% confidence interval) are shown to scale. The average genome size is shown, when multiple conspecific genomes are available in the same extant species. The branches whose ancestral and descendant nodes have >4 kb difference in mitogenome size are colored (red for expansion, blue for reduction).
The mitogenome expansion and contraction events cannot be simply explained by different evolutionary rates among lineages. The MBH predicts smaller sizes for mitogenomes with high mutation rates (Lynch et al. 2006). Consistent with the MBH, the branch leading to Eremothecium gossypii is substantially long, suggesting accelerated evolution in E. gossypii, and its mitogenome is 23.5 kb in size, smaller than any mitogenomes from related Lachancea or Kluyveromyces species (fig. 1). There is, however, no evidence that the 20.1 kb Candida glabrata mitogenome is under accelerated evolution, nor is the 107.1 kb Nakaseomyces bacillisporus mitogenome slow evolving. In fact, the branch length leading to Nakaseomyces bacillisporus is longer than the branch length leading to its sister species Candida castellii, whose mitogenome is 56.8 kb smaller than the N. bacillisporus mitogenome. Thus, it is unlikely that the expansion and contraction of yeast mitogenomes are primarily determined by different mutation rates.
All mitogenomes in Saccharomycetaceae encode eight core mitochondrial protein genes, SSU and LSU rRNAs, 22–24 tRNAs. Unlike many nonyeast fungal species, none of these mitogenomes encode the respiratory-chain NADH dehydrogenase (complex I) (Dujon 2010; Freel et al. 2014). The major variation of yeast mitogenome architecture has been shown to be due to dynamic introns and variable intergenic regions (Bouchier et al. 2009; Freel et al. 2014). We have carefully examined introns and known variable sequences (e.g., short tandem repeats and mobile GC-clusters) in intergenic regions. Among different yeast species, tandem repeats show the highest variance in sequence length and introns show the second highest variance (fig. 2A). If any genomic feature plays a major role in driving mitogenome size variation, one would expect such a genomic feature to be overrepresented in large genomes, and conversely, underrepresented in small genomes (Lynch and Conery 2003). Although intron sequences can influence mitogenome size variation (Joardar et al. 2012; Zhang et al. 2015; Kanzi et al. 2016), we found no significant overrepresentation of introns in large yeast mitogenomes at the interspecific level (fig. 2B), and no significant overrepresentation of introns was observed (P value =0.63) even after the removal of the large intron-lacking Nakaseomyces bacillisporus. These findings suggest that introns are not the main sources of mitogenome expansion at a long evolutionary time scale. Similar results have been reported in green algae that more introns do not always result in mitogenome expansion (Del Vasto et al. 2015). Instead, GC-clusters and tandem repeats are significantly overrepresented in large mitogenomes (fig. 2C and D), suggesting important roles of GC-clusters and tandem repeats in mitogenome expansion during yeast evolution.
Fig. 2.

—Contribution of introns, GC-clusters, tandem-, and homopolymer-repeats to mitogenome variation among 33 yeast species. (A) Beeswarm plot (with variances) of sequence lengths of introns, GC-clusters, tandem-, and homopolymer (HP)-repeats. (B–E) Scatter plots (with Spearman’s correlation coefficient ρ and P value) of mitogenome size against the proportion of (B) introns, (C) GC-clusters, (D) tandem-, and (E) homopolymer-repeats in each mitogenome. To ease readability, significantly positive correlations are colored in red, whereas nonsignificant (NS) correlations are in black.
Interspecific and Intraspecific Variation in Mitogenome Size Is Driven by Different Factors
To understand mitogenomic dynamics on short evolutionary timescales, we compared mitogenomes among intraspecific strains. Our analyses were performed on three species that have more than or equal to nine complete intraspecific mitogenomes, that is, Saccharomyces cerevisiae, Saccharomyces paradoxus, and Lachancea thermotolerans. The results of intraspecific strains are strikingly different from those of interspecific comparison. 1) Introns show the highest variance in sequence length among intraspecific mitogenomes (fig. 3), whereas tandem repeats show the highest variance in length among species (fig. 2A). 2) Intron sequences are significantly overrepresented in large intraspecific mitogenomes in Saccharomyces cerevisiae, and Lachancea thermotolerans (fig. 4), but not among 33 different species (fig. 2B). 3) Although GC-clusters and tandem repeats are significantly overrepresented in large genomes among the 33 yeast species (fig. 2B), such a trend is not directly evident among intraspecific genomes (fig. 4). The genomic fraction of tandem-repeat sequences is even negatively correlated with mitogenome size in S. cerevisiae. These findings suggest that different genomic features impact mitogenome variation on different evolutionary timescales.
Fig. 3.

—Beeswarm plots (with variances) of sequence lengths of introns, GC-clusters, tandem-, and homopolymer (HP)-repeats among conspecific mitogenomes in Saccharomyces cerevisiae (n = 109), Saccharomyces paradoxus (n = 15), and Lachancea thermotolerans (n = 9), respectively.
Fig. 4.

—Scatter plots (with Spearman’s correlation coefficient, ρ, and P value) of genome size against the proportion of introns, GC-clusters, tandem-, and homopolymer (HP)-repeats among conspecific mitogenomes in Saccharomyces cerevisiae (n = 109), Saccharomyces paradoxus (n = 15), and Lachancea thermotolerans (n = 9), respectively. Significant correlations are colored, that is, positive in red and negative in blue.
Given the fact that introns are the most variable sequences among intraspecific mitogenomes and may overshadow variations involving other genomic features, we further examined GC-clusters, tandem-, and homopolymer-repeats in a fraction of genome sequences after the removal of introns (excluding-introns). Our results show that GC-clusters become significantly overrepresented in a fraction of large mitogenomes after the removal of introns in S. cerevisiae and S. paradoxus, and homoplolymer repeats are overrepresented in S. cerevisiae (fig. 5). The observed negative correlation between mitogenome size and genomic fraction of tandem repeats among intraspecific strains (fig. 4) holds in S. cerevisiae and becomes significant in S. paradoxus after the removal of introns (fig. 5). These results suggest that introns and GC-clusters are important genomic features driving intraspecific mitogenomic variation in yeast. Unlike in interspecific genomic variation, tandem repeats do not play a major role in driving intraspecific mitogenomic variation.
Fig. 5.

—Scatter plots of modified genome size against the proportion of GC-clusters, tandem-, and homopolymer (HP)-repeats among conspecific mitogenomes in Saccharomyces cerevisiae, Saccharomyces paradoxus, and Lachancea thermotolerans, respectively. Genome sizes after excluding introns are shown as “Excluding-introns.” Significant correlations are colored, that is, positive in red and negative in blue.
GC-Biased Homopolymer Indels and AT-Biased Tandem-Repeat Indels in Genome Alignments of Intraspecific Strains
We performed genome-wide comparison to determine small indels and nucleotide substitutions. The number of nucleotides involving small indels is 2 for Naumovozyma castellii Y12630 and Y12631, 103 for Torulaspora quercuum XZ-46A and XZ-129 (supplementary table S2, Supplementary Material online), 155 for Naumovozyma castellii Y664 and Y12630 (supplementary table S3, Supplementary Material online), and 266 for Torulaspora delbrueckii Y1535 and Y11634 (supplementary table S4, Supplementary Material online). In comparison, the number of nucleotide substitutions between each genome pair is 0, 5, 22, and 41, respectively. Thus, small indels lead to significantly more nucleotide differences in mitogenome variation than nucleotide substitutions.
All four genome pairs contain indels in homopolymer regions, which are biased toward poly-G or poly-C [(G)n or (C)n] regions. The only two different nucleotides between Naumovozyma castellii Y12630 and Y12631 are both in (G)n regions. Three homopolymer indels between Naumovozyma castellii Y664 and Y12630 are all in (G)n or (C)n regions (supplementary table S3, Supplementary Material online). Between Torulaspora delbrueckii Y1535 and Y11634, 76-nucleotide differences are in (G)n or (C)n homopolymer indels, whereas only 8-nucleotide differences are in (A)n or (T)n homopolymer indels (supplementary table S4, Supplementary Material online). Between Torulaspora quercuum XZ-46A and XZ-129, (G)n or (C)n homopolymer indels lead to 4-nucleotide differences, (A)n or (T)n homopolymer indels lead to 6-nucleotide differences (supplementary table S2, Supplementary Material online). Since yeast mitogenomes are highly AT-rich, the total nucleotides in (A)n or (T)n homopolymer regions greatly outnumber those in (G)n or (C)n homopolymer regions. For instance, in Torulaspora quercuum XZ-46A, there are in total 18,684 nucleotides in (A)n≥2 or (T)n≥2 regions, only 2,020 nucleotides in (G)n≥2 or (C)n≥2 regions. The observed four nucleotides in (G)n≥2 or (C)n≥2, compared with six nucleotides in (A)n≥2 or (T)n≥2 homopolymer indels in Torulaspora quercuum are a significant overrepresentation of (G)n≥2 or (C)n≥2 nucleotides (P value = 0.012, Fisher’s exact test). These results suggest that G or C homopolymer regions in mitogenomes are particularly susceptible to small indel mutations. Such bias has been previously observed in bacterial genomes (Sagher et al. 1999; Dettman et al. 2016), nuclear genomes in yeast (Gragg et al. 2002), Caenorhabditis elegans (Denver et al. 2004), Daphnia pulex (Sung et al. 2010), and mammalian cells (Boyer et al. 2002), possibly due to a higher level of DNA polymerase slippage in C/G than in A/T homopolymer regions (Schlotterer and Tautz 1992; Gragg et al. 2002).
All genome pairs but the nearly identical Naumovozyma castellii pair (Y12630 and Y12631) have indels in short tandem repeats. These indels in short tandem repeats show a strong bias toward A/T nucleotides over G/C nucleotides, for example, 84 As/Ts—9 Gs/Cs in Torulaspora quercuum, 20 As/Ts—2 Gs/Cs in Naumovozyma castellii, 65 As/Ts—2 Gs/Cs in Torulaspora delbrueckii (table 1). The total GC content of these indels is only 7.1% (169 As/Ts—13 Gs/Cs), much lower than the lowest genomic GC content (18.2% of Torulaspora quercuum) among the three genome pairs (P value = 0.003, χ2 test). Furthermore, indels in tandem repeats have significantly more nucleotides than homopolymer indels in Torulaspora quercuum (supplementary table S2, Supplementary Material online) and in Naumovozyma castellii (supplementary table S3, Supplementary Material online). Thus, extremely AT-rich short tandem repeats are a major source of small indels. Small indels in tandem repeats are generally believed to arise from DNA replication slippage (Levinson and Gutman 1987; Ellegren 2004), unequal crossover or biased gene conversion (Smith 1976; Richard and Paques 2000). Given the shape contrast between G/C biased homopolymer indels and A/T biased tandem-repeat indels, the molecular mechanisms underlying DNA replication slippage in homopolymer indels and tandem-repeat indels are perhaps very different. In this study, pairwise genome comparison cannot determine the directionality of DNA slippage events. During genome evolution, the expansion and contraction of repeats are likely driven by properties of the repeat sequences, genetic drift, and selection. It has been observed in humans that short microsatellite repeats tend to expand, whereas long microsatellite repeats tend to contract (Lai and Sun 2003); and changes in repeat units can have fitness consequences, on which selection will act (Wei et al. 2014).
Small indels are also present in disperse repeats, for example, GC-clusters, and nonrepeat sequences. There is a 132-nucleotide indel between Naumovozyma castellii Y664 and Y12630 (starting at alignment position 94). The first 46 nucleotides of this indel are highly AT-rich (GC 8.7%) and show no detectable homology to any other sequences in Naumovozyma castellii, whereas the remaining 86 nucleotides of the same indel are relatively GC-rich (GC 57.0%) and share 81 identical nucleotides (94.2%) with the immediate upstream sequence from alignment positions 8–93 (supplementary table S3, Supplementary Material online). The three GC-rich indels between Torulaspora delbrueckii Y1535 and Y11634 all have paralogs within Torulaspora delbrueckii, and the two long indels share 41 common nucleotides (82% of the 50-nucleotide alignment) (supplementary table S4, Supplementary Material online). These results are consistent with previous reports that GC-clusters are of dynamic mobility in mitogenomes (Weiller et al. 1989; Lang et al. 2014; Wu and Hao 2015).
Contrast Correlations between GC Content and Mitogenome Size on Different Evolutionary Timescales
Given the strikingly skewed GC-content of homopolymer indels, tandem-repeat indels, and GC-clusters, we assessed their impacts on genomic GC-content. Among different yeast species, there is a negative correlation (Spearman’s ρ = −0.46, P value = 0.007) between mitogenome size and genomic GC-content (fig. 6A). An opposite trend, however, is evident among intraspecific mitogenomes in S. cerevisiae (ρ = 0.45, P value = 9.6 × 10−7) and in S. paradoxus (ρ = 0.60, P value = 0.018). In other words, when comparing different species, larger mitogenomes have lower GC-contents; in contrast, within either S. cerevisiae or S. paradoxus, larger mitogenomes tend to have higher GC-contents (fig. 6B). Such a sharp contrast between interspecific and intraspecific comparisons could be due to different dynamics of introns, GC-clusters, GC-rich homopolymer indels, and AT-rich tandem-repeat indels on different evolutionary timescales. AT-rich tandem repeats can vary substantially among different species (fig. 2A) and lead to mitogenomic GC-content variation. For instance, the largest mitogenome of Nakaseomyces bacillisporus is of the most tandem repeats and lowest genomic GC-content (at 10.9%). Among intraspecific strains, mitogenomic variation is predominantly driven by introns (fig. 3), whose GC-content is typically higher than the genomic GC-content (supplementary table S1, Supplementary Material online). Furthermore, even though GC-clusters and tandem repeats seemed to have comparable variance of sequences and play comparable roles in intraspecific genome length variation; GC-rich clusters, compared with AT-rich tandem repeats, lead to much more dramatic alternation of GC-content in the already very AT-rich mitogenomes in intraspecific comparisons.
Fig. 6.

—Conflicting trends between genome size and GC-content on different evolutionary timescales. (A), there is a negative correlation among 33 yeast species. (B), there are significantly positive correlations among conspecific strains within Saccharomyces cerevisiae and Saccharomyces paradoxus, respectively, whereas nonsignificant among conspecific Lachancea thermotolerans strains.
Rapid Intron and GC-Cluster Turnover, and Different Sequence Insertion Spaces
Introns and GC-clusters are mobile and of presence/absence polymorphism among intraspecific mitogenomes (Wu and Hao 2014, 2015; Wu et al. 2015). Given the fact that the variance of intron sequences among intraspecific mitogenomes is over an order of magnitude higher than that of GC-clusters (fig. 3), we sought to address whether introns undergo higher rates of sequence turnover than GC-clusters. We determined intron presence/absence patterns in the same set of S. cerevisiae (n = 18) and S. paradoxus (n = 15) genomes, in which GC-clusters have been identified and mapped (Wu and Hao 2015). Both introns and GC42, the most abundant GC-cluster in S. cerevisiae and S. paradoxus, show much higher pairwise diversity per site (per intron-insertion-site or per GC42-insertion-site) than synonymous nucleotide substitution; however, introns do not show higher pairwise diversities than GC42 in either S. cerevisiae or S. paradoxus (fig. 7). We then separately calculated turnover rates of GC42 and introns in S. cerevisiae and S. paradoxus, respectively (table 2). The turnover rates of introns, and GC42 are all about two orders of magnitude higher than nucleotide substitution rates. A higher turnover rate of introns over GC42 is only evident in S. cerevisiae, but not in S. paradoxus. Thus, the consistently higher intraspecific variance of intron sequences than GC-clusters is likely due to other reasons, for example, the long sequence length of introns.
Fig. 7.
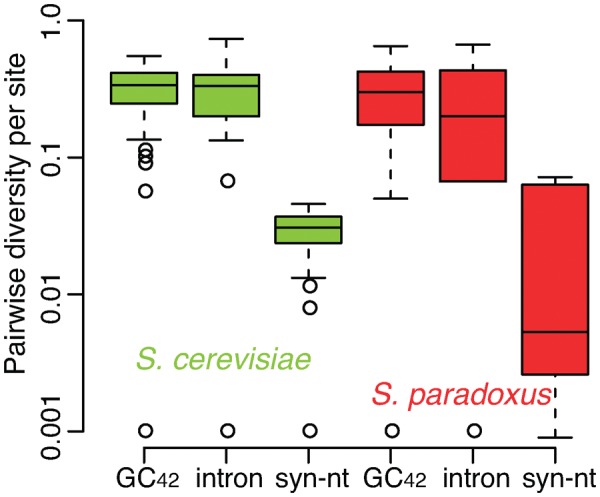
—Highly dynamic introns and GC-clusters, in comparison with synonymous nucleotide (syn-nt) substitutions in mitogenomes. Pairwise diversity for GC42 and introns in Saccharomyces cerevisiae (n = 18), Saccharomyces paradoxus (n = 15) was calculated as . GC42 is the most abundant GC-cluster in S. cerevisiae and S. paradoxus, and the distribution data of GC42 are obtained from Wu and Hao (2015).
Table 2.
Turnover Rates (±SE) of GC42 and Introns Estimated in Saccharomyces cerevisiae and Saccharomyces paradoxus
| Introns | GC42 | |
|---|---|---|
| Saccharomyces cerevisiae | 282.9 ± 46.8 | 165.1 ± 9.8 |
| Saccharomyces paradoxus | 330.4 ± 57.2 | 489.3 ± 64.9 |
Note.—The rate unit is the number of gains/losses per site per nucleotide substitution (see Materials and Methods for details). The presence–absence patterns of introns and GC42 were from the same set of mitogenomes in each species.
Intron variation dominates mitogenomic variation among intraspecific strains, but becomes nonsignificant among different species as divergence increases. This is likely because introns undergo rapid sequence turnover but have very limited insertion sites. Previously, we have analyzed 40 mitogenomes in 21 yeast species and identified only 17 homologous intron positions in three genes, cox1, cob, and 21 S rRNA (Wu et al. 2015). In this study, we have examined 184 mitogenomes in 33 species, with just one exception of an atp9 intron in Kluyveromyces marxianus, all introns are in cox1, cob, or 21 S rRNA (supplementary table S1, Supplementary Material online). In comparison, GC-clusters among different species often have very different insertion sites, and share little or no similarity at the sequence level (de Zamaroczy and Bernardi 1986; Wu and Hao 2015). GC-clusters thus should have larger insertion space than introns (illustrated in fig. 8). Consistently, GC-clusters are overrepresented in larger genomes in both interspecific (fig. 2) and intraspecific comparisons (in S. cerevisiae and in S. paradoxus, fig. 5). Homopolymer repeats and tandem repeats are also believed to have reasonably large insertion spaces, that is, many repeat units at various genomic regions. Even though slippage mutations are often in regions of high repeat units (Xu et al. 2000; Sung et al. 2010), the minimum number of repeat units in the indels observed in this study can be as low as only 1–2 (table 1; supplementary tables S2 and S3, Supplementary Material online).
Fig. 8.
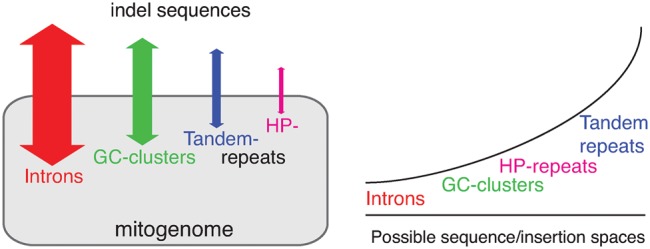
—Different tempo and mode during the evolution of introns, GC-clusters, tandem-, and homopolymer (HP)-repeats in mitogenomes. The insertion/deletion of introns may impact the most number of nucleotides per event due to longer intron length, but intron insertion sites are very limited. On the other hand, tandem repeats may only change a small number of nucleotide per slippage event, due to the high abundance of tandem repeats, the insertion space is thus relatively open for tandem repeats.
Genetic Drift and Mitogenome Size
We investigated the role of genetic drift on mitogenome size evolution using the ratio of nonsynonymous over synonymous rates as a proxy for the level of genetic drift (Daubin and Moran 2004; Kuo et al. 2009; Lefebure et al. 2017). Among seven yeast species that have sufficient intraspecific diversity, there is a significant positive correlation between the genome-wide Ka/Ks (or πa/πs) and mitogenome size (ρ = 0.82, P = 0.034) (fig. 9). This suggests an important role of genetic drift on yeast mitogenome expansion. This finding is also consistent with the previously suggested notion that mitochondrial introns, GC-clusters, and repeats are mostly deleterious (Bernardi 2005; Wu and Hao 2014, 2015; Wu et al. 2015). The largely deleterious mitochondrial introns, GC-clusters, tandem-, and homopolymer-repeats are more likely accumulated and fixed under stronger genetic drift. In addition, the functional constraint on mitochondria can be different among yeast species. For instance, mitochondrial function in yeast species after whole (nuclear-) genome duplication might have undergone relaxation (Jiang et al. 2008). The petite positive phenotype, the ability to tolerate the loss of mtDNA, is sporadically distributed among yeast species (Fekete et al. 2007).
Fig. 9.
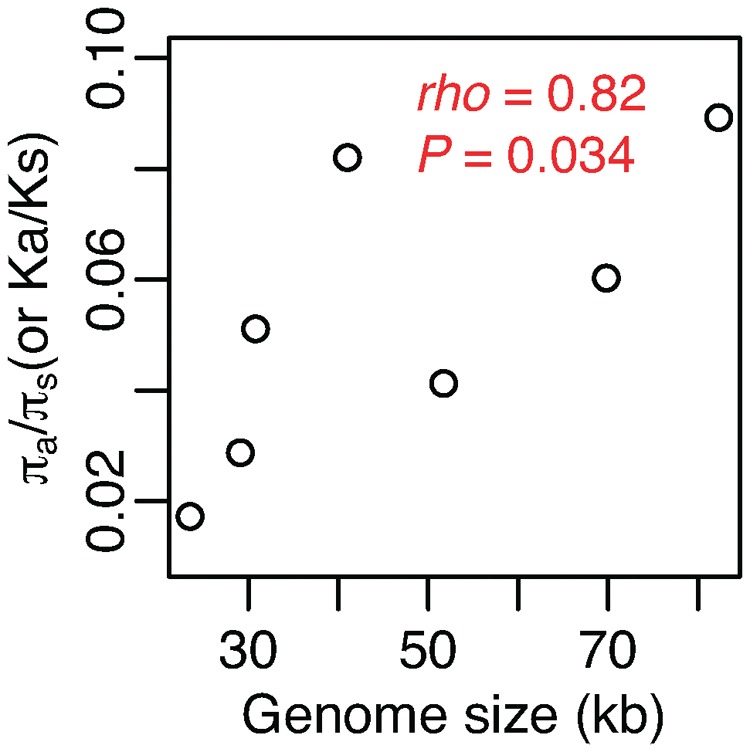
—Correlation between the level of genetic drift and mitogenome size.
It is important to note the limitation of our analyses. First, there are only seven yeast species of sufficient nucleotide diversity among their respective intraspecific mitogenomes. The two most extreme species with respect to mitogenome size, Nakaseomyces bacillisporus and Candida glabrata, each has only a single complete mitogenome, and was not included in the analysis. To further assess the robustness of the observed trend, abundant intraspecific mitogenomes from a variety of divergent species are desired. Secondly, phylogenetic independent contrast has been previously recommended (Whitney et al. 2010; Kelkar and Ochman 2012), but our analysis was not completely phylogenetically independent. In our analysis, however, the effect of phylogenetic dependence is believed to be minimal, as genome size varies substantially among closely related mitogenomes and many dramatic genome size alterations have taken place on the external branches (fig. 1).
Conclusion
Our results show that mitogenomic variation at different evolutionary time scales, for example, intraspecific versus interspecific, is dominated by different types of noncoding sequences, which have different insertion spaces in mitogenomes. The insertion and expansion of different sequences, thus increases in mitogenome size, are generally deleterious and likely to take place when the level of genetic drift is high. Unlike what the MBH would predict, there is no direct evidence that mutation rates play an important role on the evolution of yeast mitogenome sizes. Our findings, however, fit within a broader framework of genome evolution through primarily nonadaptive processes. Noncoding sequences are mutational liabilities in yeast mitogenomes, not simply because they increase the target for degenerative mutations (i.e., the mutational burden), but because the expansion and proliferation of noncoding sequences (via rapid sequence insertions and persistent DNA slippages) are the main selective cost.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors are grateful for the grid computing service from Computing & Information Technology of Wayne State University. This work was supported by funds from Wayne State University (to W.H.).
Literature Cited
- Alverson AJ, et al. 2010. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol. 276:1436–1448.http://dx.doi.org/10.1093/molbev/msq029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 195:455–477.http://dx.doi.org/10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazin E, Glemin S, Galtier N.. 2006. Population size does not influence mitochondrial genetic diversity in animals. Science 3125773:570–572.http://dx.doi.org/10.1126/science.1122033 [DOI] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 272:573–580.http://dx.doi.org/10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi G. 2005. Lessons from a small, dispensable genome: the mitochondrial genome of yeast. Gene 354:189–200.http://dx.doi.org/10.1016/j.gene.2005.03.024 [DOI] [PubMed] [Google Scholar]
- Bouchier C, Ma L, Créno S, Dujon B, Fairhead C.. 2009. Complete mitochondrial genome sequences of three Nakaseomyces species reveal invasion by palindromic GC clusters and considerable size expansion. FEMS Yeast Res. 98:1283–1292. [DOI] [PubMed] [Google Scholar]
- Boussau B, Brown JM, Fujita MK.. 2011. Nonadaptive evolution of mitochondrial genome size. Evolution 659:2706–2711.http://dx.doi.org/10.1111/j.1558-5646.2011.01322.x [DOI] [PubMed] [Google Scholar]
- Boyer JC, et al. 2002. Sequence dependent instability of mononucleotide microsatellites in cultured mismatch repair proficient and deficient mammalian cells. Hum Mol Genet. 116:707–713.http://dx.doi.org/10.1093/hmg/11.6.707 [DOI] [PubMed] [Google Scholar]
- Burger G, Gray MW, Lang BF.. 2003. Mitochondrial genomes: anything goes. Trends Genet. 1912:709–716.http://dx.doi.org/10.1016/j.tig.2003.10.012 [DOI] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong RA, Mueller RL.. 2013. Evolution along the mutation gradient in the dynamic mitochondrial genome of salamanders. Genome Biol Evol. 59:1652–1660.http://dx.doi.org/10.1093/gbe/evt119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AC. 2013. Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol Evol. 56:1079–1086.http://dx.doi.org/10.1093/gbe/evt069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, et al. 2014. Diversification of bacterial genome content through distinct mechanisms over different timescales. Nat Commun. 5:5471.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AC, Mau B, Blattner FR, Perna NT.. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 147:1394–1403.http://dx.doi.org/10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubin V, Moran NA.. 2004. Comment on “The origins of genome complexity.” Science 3065698:978; author reply 978. [DOI] [PubMed] [Google Scholar]
- de Zamaroczy M, Bernardi G.. 1986. The GC clusters of the mitochondrial genome of yeast and their evolutionary origin. Gene 411:1–22.http://dx.doi.org/10.1016/0378-1119(86)90262-3 [DOI] [PubMed] [Google Scholar]
- Del Vasto M, et al. 2015. Massive and widespread organelle genomic expansion in the green algal genus Dunaliella. Genome Biol Evol. 73:656–663.http://dx.doi.org/10.1093/gbe/evv027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, et al. 2004. Abundance, distribution, and mutation rates of homopolymeric nucleotide runs in the genome of Caenorhabditis elegans. J Mol Evol. 585:584–595.http://dx.doi.org/10.1007/s00239-004-2580-4 [DOI] [PubMed] [Google Scholar]
- Dettman JR, Sztepanacz JL, Kassen R.. 2016. The properties of spontaneous mutations in the opportunistic pathogen Pseudomonas aeruginosa. BMC Genomics 17:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. 2010. Yeast evolutionary genomics. Nat Rev Genet. 117:512–524.http://dx.doi.org/10.1038/nrg2811 [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 325:1792–1797.http://dx.doi.org/10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. 2004. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 56:435–445.http://dx.doi.org/10.1038/nrg1348 [DOI] [PubMed] [Google Scholar]
- Fekete V, Cierna M, Poláková S, Piskur J, Sulo P.. 2007. Transition of the ability to generate petites in the Saccharomyces/Kluyveromyces complex. FEMS Yeast Res. 78:1237–1247. [DOI] [PubMed] [Google Scholar]
- Freel KC, Friedrich A, Hou J, Schacherer J.. 2014. Population genomic analysis reveals highly conserved mitochondrial genomes in the yeast species Lachancea thermotolerans. Genome Biol Evol. 610:2586–2594.http://dx.doi.org/10.1093/gbe/evu203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fungtammasan A, et al. 2015. Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res. 255:736–749.http://dx.doi.org/10.1101/gr.185892.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldon T, et al. 2013. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics 14:623.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O.. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 272:221–224.http://dx.doi.org/10.1093/molbev/msp259 [DOI] [PubMed] [Google Scholar]
- Gragg H, Harfe BD, Jinks-Robertson S.. 2002. Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol Cell Biol. 2224:8756–8762.http://dx.doi.org/10.1128/MCB.22.24.8756-8762.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 593:307–321.http://dx.doi.org/10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Hao W, et al. 2012. Phylogenetic incongruence in E. coli O104: understanding the evolutionary relationships of emerging pathogens in the face of homologous recombination. PLoS One 74:e33971.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Golding GB.. 2006. The fate of laterally transferred genes: life in the fast lane to adaptation or death. Genome Res. 165:636–643.http://dx.doi.org/10.1101/gr.4746406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Guan W, Pinney D, Wang W, Gu Z.. 2008. Relaxation of yeast mitochondrial functions after whole-genome duplication. Genome Res. 189:1466–1471.http://dx.doi.org/10.1101/gr.074674.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joardar V, et al. 2012. Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics 13:698.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung PP, Friedrich A, Reisser C, Hou J, Schacherer J.. 2012. Mitochondrial genome evolution in a single protoploid yeast species. G3 (Bethesda) 2:1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzi AM, Wingfield BD, Steenkamp ET, Naidoo S, van der Merwe NA.. 2016. Intron derived size polymorphism in the mitochondrial genomes of closely related chrysoporthe species. PLoS One 116:e0156104.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar YD, Ochman H.. 2012. Causes and consequences of genome expansion in fungi. Genome Biol Evol. 41:13–23.http://dx.doi.org/10.1093/gbe/evr124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Hao W.. 2014. DiscML: an R package for estimating evolutionary rates of discrete characters using maximum likelihood. BMC Bioinformatics 151:320..http://dx.doi.org/10.1186/1471-2105-15-320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koszul R, et al. 2003. The complete mitochondrial genome sequence of the pathogenic yeast Candida (Torulopsis) glabrata. FEBS Lett. 534(1–3):39–48. [DOI] [PubMed] [Google Scholar]
- Kuo CH, Moran NA, Ochman H.. 2009. The consequences of genetic drift for bacterial genome complexity. Genome Res. 198:1450–1454.http://dx.doi.org/10.1101/gr.091785.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Sun F.. 2003. The relationship between microsatellite slippage mutation rate and the number of repeat units. Mol Biol Evol. 2012:2123–2131.http://dx.doi.org/10.1093/molbev/msg228 [DOI] [PubMed] [Google Scholar]
- Lang BF, et al. 2014. Massive programmed translational jumping in mitochondria. Proc Natl Acad Sci U S A. 11116:5926–5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebure T, et al. 2017. Less effective selection leads to larger genomes. Genome Res. 276:1016–1028.http://dx.doi.org/10.1101/gr.212589.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson G, Gutman GA.. 1987. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol. 4:203–221. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2514:1754–1760.http://dx.doi.org/10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS.. 2003. The origins of genome complexity. Science 3025649:1401–1404.http://dx.doi.org/10.1126/science.1089370 [DOI] [PubMed] [Google Scholar]
- Lynch M, Koskella B, Schaack S.. 2006. Mutation pressure and the evolution of organelle genomic architecture. Science 3115768:1727–1730.http://dx.doi.org/10.1126/science.1118884 [DOI] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA.. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 1710:589–596.http://dx.doi.org/10.1016/S0168-9525(01)02447-7 [DOI] [PubMed] [Google Scholar]
- Nadimi M, Beaudet D, Forget L, Hijri M, Lang BF.. 2012. Group I intron-mediated trans-splicing in mitochondria of Gigaspora rosea and a robust phylogenetic affiliation of arbuscular mycorrhizal fungi with Mortierellales. Mol Biol Evol. 299:2199–2210.http://dx.doi.org/10.1093/molbev/mss088 [DOI] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M, team U.. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 288:1166–1167.http://dx.doi.org/10.1093/bioinformatics/bts091 [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 202:289–290.http://dx.doi.org/10.1093/bioinformatics/btg412 [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2016. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Richard GF, Paques F.. 2000. Mini- and microsatellite expansions: the recombination connection. EMBO Rep. 12:122–126.http://dx.doi.org/10.1093/embo-reports/kvd031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, et al. 2011. Integrative genomics viewer. Nat Biotechnol. 291:24–26.http://dx.doi.org/10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R.. 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 1918:2496–2497. [DOI] [PubMed] [Google Scholar]
- Sagher D, Hsu A, Strauss B.. 1999. Stabilization of the intermediate in frameshift mutation. Mutat Res. 423(1–2):73–77. [DOI] [PubMed] [Google Scholar]
- Schirmer M, et al. 2015. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 436:e37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotterer C, Tautz D.. 1992. Slippage synthesis of simple sequence DNA. Nucleic Acids Res. 202:211–215.http://dx.doi.org/10.1093/nar/20.2.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R, Kirkness EF, Barker SC.. 2009. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 195:904–912.http://dx.doi.org/10.1101/gr.083188.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, et al. 2012. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 101:e1001241.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, et al. 2013. Organelle genome complexity scales positively with organism size in volvocine green algae. Mol Biol Evol. 304:793–797.http://dx.doi.org/10.1093/molbev/mst002 [DOI] [PubMed] [Google Scholar]
- Smith DR, Keeling PJ.. 2015. Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc Natl Acad Sci U S A. 11233:10177–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Lee RW.. 2010. Low nucleotide diversity for the expanded organelle and nuclear genomes of Volvox carteri supports the mutational-hazard hypothesis. Mol Biol Evol. 2710:2244–2256.http://dx.doi.org/10.1093/molbev/msq110 [DOI] [PubMed] [Google Scholar]
- Smith GP. 1976. Evolution of repeated DNA sequences by unequal crossover. Science 1914227:528–535.http://dx.doi.org/10.1126/science.1251186 [DOI] [PubMed] [Google Scholar]
- Sung W, Tucker A, Bergeron RD, Lynch M, Thomas WK.. 2010. Simple sequence repeat variation in the Daphnia pulex genome. BMC Genomics 11:691.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei KH, Grenier JK, Barbash DA, Clark AG.. 2014. Correlated variation and population differentiation in satellite DNA abundance among lines of Drosophila melanogaster. Proc Natl Acad Sci U S A. 11152:18793–18798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiller G, Schueller CM, Schweyen RJ.. 1989. Putative target sites for mobile G + C rich clusters in yeast mitochondrial DNA: single elements and tandem arrays. Mol Gen Genet. 2182:272–283. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Garland T, Moran NA Jr. 2010. Did genetic drift drive increases in genome complexity? PLoS Genet. 68:e1001080.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolters JF, Chiu K, Fiumera HL.. 2015. Population structure of mitochondrial genomes in Saccharomyces cerevisiae. BMC Genomics 16:451.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Buljic A, Hao W.. 2015. Extensive horizontal transfer and homologous recombination generate highly chimeric mitochondrial genomes in yeast. Mol Biol Evol. 3210:2559–2570.http://dx.doi.org/10.1093/molbev/msv127 [DOI] [PubMed] [Google Scholar]
- Wu B, Hao W.. 2014. Horizontal transfer and gene conversion as an important driving force in shaping the landscape of mitochondrial introns. G3 (Bethesda) 44:605–612.http://dx.doi.org/10.1534/g3.113.009910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Hao W.. 2015. A dynamic mobile DNA family in the yeast mitochondrial genome. G3 (Bethesda) 56:1273–1282.http://dx.doi.org/10.1534/g3.115.017822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Peng M, Fang Z.. 2000. The direction of microsatellite mutations is dependent upon allele length. Nat Genet. 244:396–399.http://dx.doi.org/10.1038/74238 [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. 2015. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet Biol. 77:95–107.http://dx.doi.org/10.1016/j.fgb.2015.04.009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.