


Abstract
Prokaryotic genome annotation is highly dependent on automated methods, as manual curation cannot keep up with the exponential growth of sequenced genomes. Current automated methods depend heavily on sequence composition and often underestimate the complexity of the proteome. We developed RibosomeE Profiling Assisted (re-)AnnotaTION (REPARATION), a de novo machine learning algorithm that takes advantage of experimental protein synthesis evidence from ribosome profiling (Ribo-seq) to delineate translated open reading frames (ORFs) in bacteria, independent of genome annotation (https://github.com/Biobix/REPARATION). REPARATION evaluates all possible ORFs in the genome and estimates minimum thresholds based on a growth curve model to screen for spurious ORFs. We applied REPARATION to three annotated bacterial species to obtain a more comprehensive mapping of their translation landscape in support of experimental data. In all cases, we identified hundreds of novel (small) ORFs including variants of previously annotated ORFs and >70% of all (variants of) annotated protein coding ORFs were predicted by REPARATION to be translated. Our predictions are supported by matching mass spectrometry proteomics data, sequence composition and conservation analysis. REPARATION is unique in that it makes use of experimental translation evidence to intrinsically perform a de novo ORF delineation in bacterial genomes irrespective of the sequence features linked to open reading frames.
INTRODUCTION
In recent years, the advent of next generation sequencing has led to an exponential growth of sequenced prokaryotic genomes. As curation-based methods cannot keep pace with the increase in the number of available bacterial genomes, researchers have reverted to the use of computational methods for prokaryotic genome annotation (1,2). However, advances in genome annotation should entail more than simply relying on automatic gene prediction or the transfer of genome annotation, as these often introduce and propagate inconsistencies (1). Moreover, the dependence on sequence composition of an open reading frame (ORF) by automatic methods often introduce biases in gene prediction, as studies have shown that translation can occur irrespective of the sequence composition of the ORF (3,4). Further, gene prediction methods that depend solely on the genomic template often lack the capabilities to capture the true complexity of the translation landscape (4), overall stressing the need for non in silico-based gene prediction approaches.
Ribosome profiling (5) (Ribo-seq) has revolutionized the study of protein synthesis in a wide variety of prokaryotic and eukaryotic species. Ribo-seq provides a global measurement of translation in vivo by capturing translating ribosomes along an mRNA. More specifically, ribosome protected mRNA footprints (RPFs) are extracted and converted into a deep sequencing complementary DNA library. When aligned to a reference genome, these RPFs provide a genome-wide snapshot of the positions of translating ribosomes along the mRNA at the time of sampling (5). This genome-wide positional information of translating ribosomes allows for the delineation of translated regions.
With the advent of Ribo-seq, numerous computational methods have been developed to detect putatively translated regions in eukaryotes, all taking advantages of inherent Ribo-seq-based metrics to identify translated ORFs. In the studies of Lee et al. (6) and Crappé et al. (7), a rule based peak detection algorithm was used to identify translation initiation sites (TIS), while Bazzini et al. (8) and Calviello et al. (9) take advantage of the triplet periodicity property of Ribo-seq data to delineate translated ORFs. Fields et al. (4) and Chew et al. (10) developed ensemble classifiers that aggregate multiple features to predict putative coding ORFs. In addition to ORF delineating tools, Michel et al. (11) developed a Ribo-seq quality control toolbox including RUST (12) and RiboSeqR (13) available in Galaxy (RiboGalaxy). However, all these methods focus mainly on eukaryote genomes with a pre-defined transcriptome and are not directly transferable to prokaryotes genomes viewing the characteristics of the features used in addition to experimental variations (14) and differences in footprint properties between pro- and eukaryotes.
So far, no computational method has yet been reported to systematically delineate protein coding ORFs in prokaryotic genomes based on Ribo-seq data. In this work, we aimed at developing an algorithm that makes use of experimental evidence of translation from Ribo-seq to perform de novo ORF delineations in prokaryotic genomes. Our algorithm, RibosomeE Profiling Assisted (Re-)AnnotaTION (REPARATION) trains an ensemble classifier to learn Ribo-seq patterns from a set of confident protein coding ORFs for a de novo delineation of translated ORFs in bacterial genomes. REPARATION deduces intrinsic characteristics from the data and thus can be applied to Ribo-seq data targeting elongating ribosomes. We evaluated the performance of REPARATION on three annotated bacterial species. REPARATION was able to identify a multitude of putative coding ORFs corresponding to previously annotated protein coding regions next to ORFs residing in so-called non-protein coding regions, ORFs corresponding to variants of annotated ORFs (i.e. in-frame truncations or 5′ extensions) and intergenic ORFs. Further, we validated our findings using matching proteomics data, sequence composition and phylogenetic conservation analyses.
MATERIALS AND METHODS
REPARATION performs de novo ORF delineation by training a random forest classifier to learn Ribo-seq patterns exhibited by protein coding ORF. A random forest model was chosen over other algorithms for training because of its robustness to outliers, low bias and its optimal performance with few parameter tuning (15). The REPARATION pipeline (Figure 1A) starts by traversing the entire prokaryotic genome sequence to generate all possible ORFs that have an arbitrary user defined length and start codon(s). In this study, only ORF initiating with either an ATG, GTG or TTG codon (the most frequently used start codons in a variety of prokaryotic species (16)) until the next in-frame stop codon were considered (the choice of start codons is a user defined parameter). REPARATION then generates a training set to train a random forest classifier for putative translated ORFs prediction.
Figure 1.

RibosomeE Profiling Assisted (Re-)AnnotaTION (REPARATION) pipeline for de novo open reading frame (ORF) delineation in prokaryotes. (A) REPARATION workflow diagram. The entire prokaryotic genome is traversed and all possible NTG-starting ORFs are generated. Next, ORF-specific positional Ribo-seq signal information is calculated based on the metagene profile (B). To discard spurious ORFs, the minimum log RPKM and ORF ribosome protected mRNA footprint (RPF) coverage thresholds are estimated using a four-parameter logistic sigmoid curve (S-curve) (C). (B) Metagene profile of salmonella data indicating read accumulation at the start and stop of ORFs (stitched together in the middle for visualization purposes). (C) S-curve with fitted four parameters logistic curve (red) and indication of predicted ORFs with support from N-terminal proteomics data (green) in the case of Escherichia coli.
Training sets
The set of positive examples is constructed by a comparative genomic approach. The algorithm uses Prodigal V2.6.3 (17) or glimmer (18) to generate an ORF set, this set is then BLAST searched against a database of curated protein sequences (e.g. UniprotKB-SwissProt). The BLAST search is performed using the UBLAST algorithm from the USEARCH package (19). ORFs that match at least one known protein coding sequence with a minimum E-value of 10−5 and a minimum identity of 75% are selected for the positive set. The negative set consist of ORFs starting with the codon CTG (the choice of the start codon for the negative set is a user definable parameter) viewing its infrequent occurrence as translation start codon (<0.01%) in the annotations of the interrogated species (Supplementary Table T1) and with a minimum ORF length corresponding to the shortest ORF in the positive set. We then grouped all CTG ORFs sharing the same in frame stop codon into an ‘ORF family’. Per ORF family we select the longest ORF as a representative member of that ‘ORF family’.
Feature construction
The metagene profile shown in Figure 1B illustrates a Ribo-seq signal pattern reminiscent to patterns previously reported for protein coding transcripts in prokaryotic Ribo-seq data targeting elongating ribosomes (20). To train the random forest classifier we constructed six features, five based on the Ribo-seq profiles of translated ORFs and the sixth being the ribosome binding energy (21). The profile exhibits read accumulation within the first 40–50 nts downstream of the start and a slight increase just before the stop codon. The six features are defined as follows:
Start region read density (start RPKM). We defined a start region of an ORF by taking 3 nt upstream (to account for any error in P-site assignment) and 45 nt downstream of the ORF start position. The start RPKM is the read density in RPKM within the defined start region. To ensure comparable read densities across ORFs with different expression levels, prior to calculating the start region read density, the RPF read count for each nucleotide position within the ORF is divided by the total number of RPF reads of the entire ORF (4). All ORFs with start region read density equal to zero are discarded from further analysis.
Stop region read density (stop RPKM). The stop region of an ORF represents the last 21 nts region upstream of the stop. The stop region read density is calculated similarly to the start region read density but within the last 21 nt of the ORF. Of note, for ORFs shorter than 63 nts we used the first 70% and last 25% of the ORF length to model the start and stop regions of the ORF.
ORF coverage. The ORF coverage represents the proportion of nucleotide positions that are covered by RPF reads relative to the length of the ORF.
ORF start coverage. The ORF start coverage refers to the coverage within the ORF start region.
-
Read accumulation proportion. This feature measures the ratio of the average RPF reads accumulated in the start region (first 45 nt) relative to the average RPF reads within the rest of the ORF as defined by the following equation;

(1) We reasoned that since Ribo-seq reads tend to accumulate within the start region of a translated ORF relative to the rest of the ORF, correctly delineated ORFs will tend to have score >1. Spurious ORFs that overlap at the start or stop of translated ORFs will score lower as their non-overlapping regions would tend to have no reads, hence resulting to accumulation proportion <1.
Ribosome binding site (RBS) energy (SD score). The interaction between Shine–Dalgarno (SD) sequence and its complementary sequence in the 16S rRNA (anti-SD), referred to as SD ribosome binding site (RBS) was proven to be very important in the recruitment of the ribosome for translation initiation in a wide variety of bacterial species (22). As such, and to aid in the prediction of SD/anti-SD-dependent translation events, the ribosome’s free binding energy or RBS energy was included as a user-defined feature in the model. The RBS energy, representative of the probability that the ribosome will bind to a specific mRNA and thus proportional to the mRNA’s translation initiation rate, was calculated using the distance dependent probabilistic method and based on the anti-SD (aSD) sequence GGAGG as described in Suzek et al. (21). The inclusion of the RBS energy features in the prediction model as well as the aSD sequence are user defined parameters to allow for bacterial species where non aSD/SD dependent translation events have been reported (17,22,23).
Sigmoid (S)-curve model
Since REPARATION was developed to allow for the identification of relatively short ORFs, the number of potential ORFs increases exponentially with decreasing ORF length. To ensure the algorithm is tractable, we defined minimum threshold values to eliminate spurious ORFs. To do this we take advantage of the sigmoid curve (S-curve) relationship observed between ORF RPF coverage and the ORF natural log read density (RPKM) as depicted in Figure 1C and Supplementary Figure S1. The fitted logistic curve (red), modeled by a four-parameter logistic regression (Equation 2) describing the relationship between ribosome density and RPF coverage. This relationship was used to estimate the minimum read density and ORF RPF coverage to allow for correct ORF delineation. We estimated the lower bend point of the fitted four-parameter logistic regression using the method described in (24) and implemented this in the R Package Sizer (25).
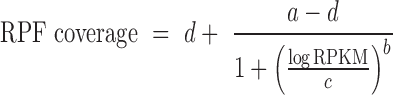 |
(2) |
Where, d represents the ORF coverage at infinite read density (RPKM) and a the ORF coverage at RPKM equal to zero while b and c represents the slope of the curve and the RPKM value at (d – a)/2 respectively.
Post-processing random forest predicted ORFs
We implement a rule-based post-processing algorithm to eliminate false positives which share overlapping regions with actual coding ORFs (Supplementary Figure S2). First, considering the simplified assumption that bacterial genes can have only one possible translation start site, we group all predicted ORFs sharing the same in frame stop codon into an ‘ORF family’. Supplementary Figure S2A depicts an ORF family with two predicted starts, if start S1 has more reads than S2 then we select S1 as the gene start. If there are no Ribo-seq reads between S1 and S2 then we select S2 as the gene start since S1 adds no extra information to the gene profile. If S1 has more reads than S2 and if S1 falls within the coding region of an out-of-frame upstream predicted ORF on the same strand, we select S1 as the most likely start if there is a peak (i.e. kurtosis > 0) within a window of −21 to + 21 around S1.
Next, we only consider two overlapping ORFs on different frames as depicted in Supplementary Figure S2B, if the read density and RPF coverage of the non-overlapping region of F1 are less than the S-curve estimated thresholds, then F1 is dropped in favor of F2 and vice versa. If both non-overlapping regions have a read density and RPF coverage greater than the minimum, then we assume both are expressed. Finally, we discard internal out-of-frame ORFs falling completely within another predicted ORF (Supplementary Figure S2C).
RESULTS
To assess the performance and utility of our REPARATION algorithm (Figure 1A), besides two publicly available bacterial Ribo-seq datasets from Escherichia coli K12 strain MG1655 and Bacillus subtilis subsp. subtilis strain 168 (26), we generated ribosome profiling data and matching RNA-seq data from a monosome and polysome enriched fraction of Salmonella enterica serovar Typhimurium strain SL1344 (experimental details in Supplementary Material). To apply our REPARATION algorithm on Ribo-seq data originating from these three species, we defined an arbitrary minimum ORF length of 10 codons (30 nts), all initiating from either an ATG, GTG or TTG start codon. The positive examples were generated using prodigal (17) and BLAST searched against a set of bacterial protein sequences obtained from UniProtKB-SwissProt. Sequences with E-values < 10−5 and a minimum identity of 75% were selected for the positive set. The negative set consisted of ORFs starting with a CTG codon and a minimum ORF length corresponding or exceeding the shortest ORF length in their respective positive sets, i.e. 87 nt in the Salmonella and E. coli samples while in Bacillus it was 105 nt. All ORFs with read density and ORF coverage below the S-curve estimated minimum thresholds were discarded from further analysis (Supplementary Table T2). When trained on these sets, the random forest classifier achieved on average 74, 76 and 81% 10-fold cross validation precision with area under the precision-recall curve values of 0.74, 0.80 and 0.89 (at probability threshold 0.5) in Salmonella, E. coli and Bacillus, respectively (Supplementary Figure S3A). Of the three species evaluated, REPARATION mapped putative coding ORFs corresponding to regions annotated as protein coding regions in addition to non-coding and intergenic regions.
REPARATION-predicted ORFs predominantly match to or overlap with annotated ORFs and follow the reference model of start codon usage
Viewing the previously reported similarities in the translation properties of monosomes and polysomes (27) and the high correlation observed between the two samples (Supplementary Figure S3C), we considered the Salmonella monosome and polysome samples as replicate samples for the purpose of translated ORF delineation.
REPARATION predicted a total of 3957 and 3881 putative ORFs in the Salmonella monosome and polysome sample respectively. Of these, 3421 (88%) ORFs found common in both datasets were considered as the high confident ORF set (Supplementary file F1). For E. coli, a high confident set of 3202 (90%) was selected based on the 3594 and 3569 predicted ORFs in replicate samples 1 and 2 respectively (Supplementary file F2). Third, in the Bacillus sample, 3435 putative coding ORFs were predicted (Supplementary file F3). In all three species analyzed, REPARATION misses out on predicting 45, 49 and 28 (variants of the) annotated ORFs that pass the S-curve estimated thresholds in Salmonella, E. coli and Bacillus respectively. While nonetheless exceeding the set threshold, these minimal set mainly represent lowly expressed and/or lowly covered ORFs. The expression levels of other annotated ORFs all fall below the minimum thresholds estimate from the S-curve and are thus likely very poorly or not expressed in the respective samples.
From the high confident set of predicted ORFs in Salmonella, E. coli and Bacillus, respectively 82% (2822), 89% (2855) and 80% (2733) ORFs correspond to previously annotated ORFs. While 14, 8 and 14% of predicted ORFs in the Salmonella, E. coli and Bacillus samples (respectively), correspond to variants of previously annotated ORFs, potentially giving rise to N-terminally truncated or extended protein variants referred to as N-terminal proteoforms (28). Consequently, in Salmonella and E. coli, 3% belong to novel putative coding regions while in case of Bacillus, 7% belong to novel ORFs (Figure 2).
Figure 2.

Proportion of REPARATION predicted ORFs per ORF category for the high confident ORF sets in case of Salmonella, Escherichia coli and Bacillus predictions.
On average, the truncations were 26, 26 and 51 codons downstream of the annotated starts while the average extensions where extensions of 18, 13 and 9 codons for Salmonella, E. coli and Bacillus (respectively). Of note, 60, 53 and 77 of the predicted variants display only 1 codon shift from the annotated starts in Salmonella, E. coli and Bacillus respectively (Supplementary Table T3). Overall, 71, 74 and 77% (including the variants) of all ENSEMBL annotated protein coding ORFs in Salmonella, E. coli and Bacillus (respectively) were predicted by REPARATION.
In our evaluation of REPARATION, we allow for the three commonly used start codons in prokaryotes ATG, GTG and TTG as translation initiation triplets. Of note however, REPARATION was designed without any bias in start-codon selection for ORF prediction. Nonetheless, the hierarchy of start codon usage over all predicted ORFs are consistent with the standard model for translation initiation in the ENSEMBL annotation of the corresponding species as in case of Salmonella and E. coli, a preference of ATG over GTG and TTG, and in case of Bacillus, a preference of ATG over TTG and GTG could be observed (Table 1).
Table 1. Start codon usage distribution of the predicted putative coding ORFs.
| ENSEMBL annotation | All predictions | Matching annotated | Extensions | Truncations | Novel | |
|---|---|---|---|---|---|---|
| Salmonella | ||||||
| ATG | 4093 (88.0%) | 2942 (86%) | 2576 (91.2%) | 166 (58%) | 139 (64%) | 61 (62%) |
| GTG | 429 (9.20%) | 344 (10%) | 206 (7.4%) | 64 (24%) | 52 (24%) | 18 (18%) |
| TTG | 126 (2.70%) | 135 (4%) | 40 (1.4%) | 51 (18%) | 25 (12%) | 19 (20%) |
| E. coli | ||||||
| ATG | 3747 (90.1%) | 2776 (87%) | 2591 (91%) | 76 (42%) | 49 (70%) | 60 (62%) |
| GTG | 386 (9.2%) | 284 (9%) | 209 (7%) | 44 (25%) | 9 (13%) | 22 (22%) |
| TTG | 71 (2.0%) | 142 (4%) | 55 (2%) | 59 (33%) | 12 (17%) | 16 (16%) |
| Bacillus | ||||||
| ATG | 3253 (77.7%) | 2502 (73%) | 2176 (80%) | 141 (41%) | 75 (56%) | 110 (49%) |
| GTG | 386 (9.2%) | 413 (12%) | 237 (9%) | 108 (31%) | 29 (22%) | 39 (17%) |
| TTG | 529 (12.6%) | 520 (15%) | 320 (12%) | 94 (29%) | 30 (22%) | 76 (34%) |
The predicted ORFs in all three species follow the starts codon usage distributions of the corresponding species annotation. In case of Salmonella and E. coli, only ORFs from the high confident set were considered.
Interestingly, we observe that novel and variant ORFs are enriched for being initiated at near-cognate start codons when compared to annotated ORFs. In case of variants, this bias is most likely due to the preference of automatic gene prediction methods to select a neighboring ATG as the start codon (17,29).
Novel ORFs are evolutionary conserved and display similar amino acid sequence patterns as compared to annotated ORFs
To gain insight into the novel predictions, we analyzed and compared their evolutionary conservation pattern to that of predicted annotations. Novel and extended ORFs exhibit similar conservation patterns to annotated ORFs, with higher nucleotide conservation from the start codon onward and within the upstream ribosomal binding site or SD region positioned from −15 to −5 nt upstream of the predicted start (Figure 3), a region aiding in translation initiation by its base pairing with the 3′ end of rRNA (21,22). The higher conservation and triplet periodicity observed upstream of the truncations is likely because in some cases multiple forms of the gene (i.e. N-terminal proteoforms) are (co-)expressed (Supplementary Table T4). A manual inspection of the alignments indeed indicates that different forms of the genes are expressed across different species. Of the 66 truncations used in the Salmonella conservation analysis, 45% shows evidence of the existence of multiple forms across different bacterial species, while in case of E. coli and Bacillus these percentages were 40% and 28% from 26 and 25 truncations respectively.
Figure 3.

Conservation pattern of REPARATION predicted ORFs. Nucleotide conservation scores are calculated using the Jukes cantor conservation matrix for nucleotides. Site conservation scores are calculated using the rate4site algorithm and displayed for a ±25 nt window around the predicted start site. The site conservation score was calculated only for ORFs with at least five orthologous sequences from a collection of randomly selected bacteria protein sequences from species within the same family as Salmonella/Escherichia coli and Bacillus and outside the family. A total of 833 annotated, 161 extensions, 99 truncations and 12 novel ORFs had at least five orthologous sequences in case of Salmonella, while the E. coli profile consisted of 2359 annotated ORFs, 70 extensions, 26 truncations and 18 novel ORFs. In the case of Bacillus, 1886 annotated, 112 extensions, 19 truncations and 2 novel ORFs were considered.
Of the 98 novel ORFs predicted in Salmonella, 48% (30) had at least one reported orthologous sequence (Supplementary file F1). While 59% (58 out of 98) and 19% (42 out of 225) in E. coli and Bacillus (respectively) have at least one orthologous sequence (Supplementary files F2 and 3).
To further confirm that the newly identified ORFs do not represent random noise, we compared the amino acid composition of predicted annotations to that of novel putative coding ORFs. In all three species, we observe a very high correlation (≥0.80) between the amino acid compositions of novel and annotated ORFs (Supplementary Figure S4). While a generally poor correlation (≤0.19) was observed when comparing novel or annotated ORFs against a set of randomly generated amino acid sequences.
Since evolutionarily conserved significant biases in protein N- and C-termini were previously reported for pro- as well as eukaryotes, often with pronounced biases at the second amino acid positions (31,32), we next investigated whether the amino acid usage frequency at second position of the novel and re-annotated ORFs exhibited a similar pattern to that of annotated ORFs. Compared to the amino acid frequency in the species matching proteomes, clearly the overall distribution is similar for the two ORF categories. More specifically, a significant enrichment of Lys (about 3-fold) at the second amino acid position was observed in case of all three species analyzed. For Salmonella and E. coli, Ser and Thr at the second amino acid position was equally enriched while in case of Bacillus, Asn was slightly more frequent in the second position while other amino acids are clearly under-represented (i.e. Trp and Tyr) (Supplementary Figure S5). All observations are very well in line with previous N-terminal biases observed (32).
Proteomics assisted validation of REPARATION predicted ORFs
To validate our predicted ORFs we generated N-terminal and shotgun proteomics data from matching E. coli and Salmonella samples respectively. While N-terminomics enables the specific isolation of N-terminal peptides, making it appropriate for the validation of translation initiation events, shotgun proteomics provides a more global assessment of the expressed proteome. Three different proteome digestions were performed in the shotgun experiment to increase proteome coverage. The shotgun and N-terminal proteomics data were searched against a six-frame translation database of the E. coli and Salmonella genomes. In both experiments, the longest non-redundant peptide sequences identified were aggregated and mapped onto the REPARATION predictions.
In case of Salmonella, 10 751 unique peptides belonging to 2235 ORFs in the six-frame translation database were identified by means of shotgun proteomics. Of these, 92% (9891) correspond to 1794 REPARATION predicted ORFs (Figure 4A), the 9% missed by REPARATION mostly correspond to poorly expressed ORFs (Supplementary Figure S6A). While most shotgun peptides support previously annotated regions (Figure 4B), we additionally identified peptides in support of novel ORFs and ORF reannotations (i.e. N-terminal protein extensions). More specifically, supportive evidence was found in case of 8 novel ORFs and 21 extensions having at least one identified peptide with a start position upstream of the annotated start (Supplementary file F1).
Figure 4.

Mass spectrometry (MS) validation of REPARATION predicted ORFs. (A) Overlap between the protein sequences identified from shotgun proteomics and the REPARATION predicted ORFs in Salmonella. (B) The number of ORFs per category with at least one identified peptide for the high confident set of Salmonella predicted ORFs. (C) Overlap between ORFs with N-terminal peptide support and REPARATION predicted ORFs in Escherichia coli. (D) Number of predicted ORFs for each category with N-terminal peptide support in the E. coli high confident set. (E) Overlap between REPARATION predicted ORFs and the Ecogene verified E. coli ORFs.
For E. coli, N-terminal proteomics identified a total of 785 blocked N-terminal peptides that are compliant with the rules of initiator methionine processing (see Supplementary Methods) originating from 781 ORFs. Under the assumption that none of these ORFs have multiple initiation sites we selected the most upstream N-terminal peptides and overlapped these with the REPARATION predictions. Of the 781 ORFs with peptide support, 725 passed the S-curve estimated minimum thresholds. A total of 86% (620) match REPARATION predicted N-termini (Figure 4C and D), while in 6% of the cases, a different translation start was predicted either downstream (i.e. 10 cases with an average distance of 8 codons) or upstream (i.e. 40 cases with an average distance of 39 codons) from the TIS matching the identified N-terminal peptides. The remaining 8%, not predicted by REPARATION, mainly represent N-termini originating from poorly expressed ORFs (Supplementary Figure S6B). Most of the N-terminal supported ORFs matched previous annotations, while 21 correspond to re-annotations or novel ORFs (13 extensions, 6 truncations and 2 novel). We also assessed the predicted ORFs against the 917 E. coli K-12 Ecogenes verified protein coding sequences, a set consisting of proteins sequences with their mature N-terminal residues sequenced using N-terminal Edman sequencing (33). Of these, 893 pass the estimated minimum thresholds of which 89% (792) matched REPARATION predicted ORFs (Figure 4E). REPARATION predicted a different start site in case of 54 of Ecogene verified ORFs, including 45 upstream (average distance of 8 codons) and 9 downstream TISs (average distance of 35 codons).
To evaluate the influence of the SD score in the prediction performance of REPARATION, models were trained using intrinsic features from Ribo-seq data only. While a substantial portion of the ORFs with matching N-terminal peptides from N-terminomics and Ecogene verified N-terminal could be identified when excluding the SD score from the model, inclusion of the SD score improves the model prediction by 10 and 18%, respectively (Supplementary Table T5).
REPARATION in the aid of genome (re)-annotation
In the three species-specific translatomes analyzed, REPARATION uncovered novel putative coding genes in addition to extensions and truncations of previously annotated genes with supporting proteomics data and conservation evidence. More specifically, in the case of the gene adhP (Salmonella), REPARATION predicts that translation initiates 27 codons upstream of the annotated start, an ORF extension supported by an N-terminal peptide identification (Figure 5A) and of which the corresponding sequence is conserved (Supplementary Figure S7A). N-terminal peptide support, next to the clear lack of Ribo-seq reads in the region between the novel and annotated start (Figure 5B) of gene yidR (E. coli), also points to translation initiating 11 codons downstream of the annotation start as predicted by REPARATION. A novel putative coding gene was found corresponding to the intergenic region Chromosome: 2 819 729-2 820 325 (Salmonella) with matching Ribo- and RNA-seq signals complemented by two unique peptide identifications (Figure 5C).
Figure 5.

REPARATION assisted reannotation of bacterial genomes. The figures depict the Ribo-seq and RNA-seq profiles of the ORFs with the x-axis representing the nucleotide positions. Features of reading frames are depicted as ORF plots in the upper panel, here all potential start (green (ATG) and blue (TTG, GTG)) and stop (red) codons are indicated as vertical bars across all three reading frames. (A) REPARATION predicted an in-frame 5′ extension of the adhP gene (Salmonella) with supportive N-terminal peptide data. (B) Gene yidR (Escherichia coli) predicted as a 5′ truncation with N-terminal peptide support and Ribo-seq reads starting downstream of the annotated start. (C) Novel putative coding intergenic ORF in the region Chromosome: 2 819 729-2 820 325 (Salmonella) with supportive peptide evidence. (D) Evidence of translation within pseudogene sugR (Salmonella), with three matching peptide identifications.
Of note, there are currently 72, 182 and 70 annotated pseudogenes in the current ENSEMBL annotations of Salmonella, E. coli and Bacillus (respectively). REPARATION predicted conserved putative coding ORFs within 12, 35 and 11 pseudogene regions leading to 15, 47 and 11 predicted ORFs for Salmonella, E. coli and Bacillus (respectively). Since ‘genuine’ pseudogenes in bacteria are typically modified/removed rapidly during evolution, coupled with the fact that only uniquely mapped reads were allowed, the observed conservation with the existence of (truncated) functional orthologs points to the genuine coding potential of these loci and thus functional importance of their translation product (34,35). One representative example is the identified putative coding ORF in the sugR pseudogene (Salmonella) which is supported by three unique peptide identifications (Figure 5D). Further investigation is needed to clarify the status of these pseudogenes as a comprehensive analysis of disrupted protein coding genes requires cases to be investigated individually (36).
Interestingly, in case of fdoG (E. coli), REPARATION predicted two juxta positioned ORFs, both contained within a previously annotated ORF holding a selenocysteine insertion event (Supplementary Figure S8A) (37). In E. coli, selenocysteine insertion has been reported in case of fdnG and fdhF (38). In case of fdoG, the Ribo-seq read density before the selenocysteine insertion site is about 100-fold higher as compared to the region after the selenocysteine insertion while only a 3-fold difference in RNA-seq density could be observed. The so-called 3′ and 5′ truncations of the current annotation as predicted by REPARATION have been delineated separately due to the algorithm not allowing for stop codon recoding events such as selenocysteine insertion, read through or ribosomal frameshifting events. A similar trend was observed in case of its Salmonella ortholog (Supplementary Figure S8B). Interestingly, changes in the efficiency of fdoG selenocysteine insertion could be observed when inspecting the data from Baek et al. (39) (REPARATION prediction data not shown). In this study, Ribo-seq was performed on Salmonella enterica Typhimurium 14028s assaying four different growth conditions (Luria-Bertani, morpholinepropanesulfonic acid (MOPS) rich defined medium and two infection-relevant conditions grown at a low Mg2+ concentration (10 μM) and low pH (5.8)). While a low recoding efficiency was observed in case of MOPS as reflected by the 27-fold decrease in the read density downstream of the selenocysteine insertion event, a higher recoding efficiency was observed in the infection relevant conditions (i.e. only ∼2-fold decrease in the read density downstream of the selenocysteine insertion event could be observed) (Supplementary Table T6 and Figure S8C) indicative of possible regulation of the efficiency of selenocysteine insertion.
REPARATION in the aid of small ORFeome annotation
Small ORFs have historically been ignored in most in silico predictions because of the assumption that they can easily occur by chance due to their small size (17). As 71 codons is the average length when considering the length of the 5% shortest annotated ORFs in the three species analyzed, we here arbitrarily define a sORF as a translation product with a length of ≤71 codons. In Salmonella, REPARATION predicted 119 putative coding sORFs. Of these, 61 (51%) matched annotations and respectively 17 (14%) and 41 (34%) represent re-annotations (i.e. 3 extensions and 14 truncations) and novel ORFs. Supportive proteomics data were found for 29 predicted sORFs. While in E. coli and Bacillus the algorithm predicted 125 (95 (76%) matching annotations, 1 extension, 4 truncations and 26 novel) and 395 (161 (41%) matching annotations, 8 extensions, 29 truncations and 197 (50%) novel) (s)ORFs, respectively. An interesting example of a possible re-annotation of gene yfaD (E. coli) is the REPARATION predicted 56 codon sORF, representative of a truncated form (Figure 6A). In line with transcriptional data pointing to transcription of an mRNA not encompassing the annotated ORF, Ribo-seq hints to expression of a smaller ORF of which the start of the gene is located 243 codons downstream of the annotated start. Other representative examples include the intergenic 47 codons long sORF Chromosome: 2 470 500-2 470 643 (E. coli) (Figure 6B), the 30 codons long sORF located on the opposite strand encoding the fre gene (Salmonella) (Figure 6C) and a 57-codon long intergenic Bacillus sORF that overlaps with the CDS of the sORF-encoding the hfq gene (Figure 6D).
Figure 6.

Novel sORFs predicted by REPARATION. Features of reading frames are depicted as ORF plots in the upper panel, here all potential start (green (ATG) and blue (TTG, GTG)) and stop (red) codons are indicated as vertical bars across all three reading frames. Corresponding Ribo- and RNA-seq profiles indicate expression of (A) a truncated form of yfaD (Escherichia coli) gene (B) a 47 codons sORF matching the region; Chromosome: 2 470 500-2 470 643 (E. coli.) (C) a sORF encoded on the reverse strand encoding the fre gene (Salmonella). (D) a sORF Chromosome: 1 867 485-1 867 655 (Bacillus) that partially overlaps the annotated hfq sORF (Bacillus). The Ribo-seq profiles indicate hfq out-of-frame translation initiation.
DISCUSSION
Experimental sequencing data from ribosome profiling exhibit patterns across protein coding ORFs which can be exploited to accurately delineate translated ORFs. Although Ribo-seq is not completely standardized (40) and certain experimental procedures such as treatments (e.g. no treatment versus antibiotic treatment) tend to have a noticeable influence on the translation patterns observed (9), we here developed an algorithm that enables a de novo delineation of translated ORFs in bacterial genomes. Our algorithm delineates putative protein coding ORFs in bacterial genomes using experimental information deduced from Ribo-seq, aiming to minimize biases inherent to in silico prediction methods.
While several methods using ribosome profiling data to delineate open reading frames in eukaryotes have been reported, due to peculiarities in prokaryotic genomes such as high gene density, occurrence of multiple overlapping genes (41) and the requirement of methodological adaptations to perform ribosome profiling experiments in bacteria, these tools are not directly transferable to ORF predictions in prokaryotes. Among them, and as cautioned by the authors, ORF-RATER is currently not applicable for use in prokaryotes as it performs its analysis on individual genes while prokaryotic genomes may not be divisible into distinct genes (4). Another method proposed by Chew et al. (10) define metrics calculated from RPF read discrepancies between the 5′ and/or 3′ untranslated regions and the relative coding region of the transcript, metrics which cannot be properly defined in bacterial genomes due to their polycistronic nature. Alternative available software packages enabling the delineation of eukaryotic ORFs such as riboseqR (13) and RiboTaper (9) take advantage of triplet periodicity in Ribo-seq data to infer translated ORFs. The former is a tool for parsing and inferring reading frames and transcript specific behavior of Ribo-seq data while the later explores the triplet periodicity across the three frames within an annotated coding region to infer all possible coding ORFs. Due to the breakdown of triplet periodicity because of read accumulation at the start of the ORFs (Figure 1B) coupled with the fact that only ATG starting ORFs are considered, applying RiboTaper on the Salmonella bacterial Ribo-seq data predominantly resulted in the prediction of 5′ truncated ORFs (Supplementary Table T7). As such and while some triplet periodicity can be observed, the strict reliance of RiboTaper on this feature makes it evidently not suited for TIS prediction and ORF delineation in bacteria. Further, all the above tools heavily rely on an existing genome annotations and transcriptome structures which are often not available in the case of prokaryotic genomes. The limitations of the current existing methods coupled with the peculiarities of prokaryotic genome structures stresses the need for a dedicated method to delineate ORFs in prokaryotes.
We applied REPARATION on three annotated bacterial species to illustrate its wide-applicability and ability to predict putative coding regions. Multiple lines of evidence, including proteomics data, evolutionary conservation analysis and sequence composition suggest that the REPARATION-predicted ORFs represent bona fide translation events. As expected, most predicted ORFs agreed with previous annotations, but additionally we could detect a multitude of ORF updates next to novel translated ORFs mainly within intergenic and pseudogene regions. While we clearly observed a shift toward near-cognate versus cognate start selection for the novel predictions, we nonetheless observe that the order of start codon usage follows the standard model in the respective species. Perhaps unsurprisingly viewing the difficulty to predict short ORF using classical gene predictions, the novel ORFs predicted by REPARATION are predominantly shorter than those previously annotated. Our predictions also point to possible errors in the current start site annotation of some genes, resulting in the identification of N-terminal truncations and extensions. The predicted extensions exhibit a similar conservation pattern to annotated ORFs while a higher conservation and triplet periodicity upstream of the truncated predictions (Figure 3) is likely due to the expression of multiple proteoforms across species. Interestingly, the identification of multiple TIS-indicative N-termini in our E. coli N-terminomics dataset point to the existence of multiple translation initiation sites in at least 11 genes (Supplementary Table T4 and Figure S7B). This number is likely an under-representation since only N-terminally formylated and thus TIS-indicative N-termini were considered irrespective of their low steady-state levels. The former observation is in line with the recently revealed and until then highly underestimated occurrence of alternative translation events in eukaryotes (42,43). In case of REPARATION however, only a single ORF is selected per ORF family, therefore representing a bias against the discovery of multiple proteoforms.
A substantial portion of the novel ORFs, with at least one identified orthologous gene, overlaps with known pseudogene loci. By virtue of the fact that pseudogenes in bacteria tend to be (sub)genus-specific and are rarely shared even among closely related species (34,35), it is likely that (part of) these genes have retained (part of) their protein coding potential, a finding that is further corroborated by proteomics data. The relatively fewer peptide identifications corresponding to the translation products of novel ORFs may in part be due to the difficulty of identifying these by mass spectrometry (MS), mainly because of their predominantly shorter nature and thus likely lower number of identifiable peptides (Fields et al. (4)). An in silico analysis of the identifiable tryptic peptide coverage shows that on average 85% of the annotated protein sequences can be covered by identifiable tryptic peptides while on average only 69% of the novel ORFs can be covered by identifiable tryptic peptides. Furthermore, bacterial translation products of sORFs have previously been shown to be more hydrophobic in nature and therefore extraction biases might also contribute to their under-representation in our proteomics datasets (44).
Historically sORFs have been neglected both in eukaryotes as well as prokaryotes. However, recently renewed interest has been directed toward the identification and characterization of sORFs (45,46,8). Small proteins represent a particularly difficult problem because they often yield weak statistics when performing computational analysis, making it difficult to discriminate protein coding from non-protein coding small ORFs (47,30). Exemplified by the identification of tens of sORFs (with supportive metadata), REPARATION’s utilization of Ribo-seq signal pattern at least in part alleviates the pitfalls of traditional bacterial gene prediction algorithms with reference to the identification of sORFs.
Based on matching N-terminal proteomics evidence and the sequenced N-termini from Ecogene, REPARATION accurately predicts 86 and 89% of the ORFs with experimental evidence. Noteworthy, the high correlative second amino acid frequency patterns observed when comparing annotated versus re-annotated/new ORFs provide further proof of the accuracy and resolution of start-codon selection in case of REPARATION predicted ORFs. Nonetheless, start-site selection by REPARATION resulted in a loss of 6% of the N-terminal supported gene starts which exceeded the S-curve thresholds. While the existence of multiple N-terminal proteoforms in bacteria in contrast to the single ORF selection by REPARATION is likely the main explanatory reason for this inconsistency, the discrepancy between predicted and N-terminally supported start might (especially in the case of short truncations) also (in part) be due to the lack of accuracy in start-codon selection.
The assumption of a single start position per ORF in REPARARTION thus limits its ability to identify cases of alternative proteoforms simultaneously expressed and thus the identification of alternative translation sites. In our evaluation of REPARATION, only ATG, GTG and TTG start codons were used for ORF prediction and thus misses out on cases such as initiation of IF3 translation at the previously characterized ATT initiation codon in infC (48). In case of the later we predicted a truncated GTG-initiated ORF, while nonetheless ribo-seq and proteomics data clearly indicate expression of the annotated variant. The choice of the start codon(s) to be used for prediction is open to the user but the use of other start codons might lead to an increase in the number of false positive predictions. Finally, REPARATION does not consider non-canonical translation events such as frameshifting, stop codon read-throughs and recoding. REPARATION could potentially take advantage of improved measures or features to increase the prediction power of the classifier. At present, REPARATION is the first attempt to perform a de novo putative ORF delineation in prokaryotic genomes that relies on Ribo-seq data. With automated bacterial gene prediction algorithms estimated to have false prediction rate of up to 30% (49), machine learning algorithms that learn properties from Ribo-seq experiments such as REPARATION pave the way for a more reliable (re-)annotation of prokaryotic genomes, a much desired need in the current era of (prokaryotic) genome sequencing.
DATA AVAILABILITY
An implementation of the REPARATION software is freely available via the GitHub development platform at https://github.com/Biobix/REPARATION.
Ribo- and RNA-seq sequencing data reported in this paper have been deposited in NCBI’s Gene Expression Omnibus with the accession number GSE91066. All MS proteomics data and search results have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005844 for the Salmonella typhimurium SL1344 datasets and PXD005901 for the E. coli K12 strain MG1655 dataset.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lorenzo Calviello for assistance with the use of RiboTaper and Prof Kris Gevaert for financial support of this research (Research Foundation–Flanders (FWO-Vlaanderen), project number G.0440.10).
Author contributions: E.N., A.G., E.V., G.M. and P.V.D. conceived the study; E.N., G.M. and P.V.D. wrote the manuscript; E.N. performed the computational analysis; P.V.D. performed the proteomics experiments, E.N. and P.V.D. performed the proteomics analyses; V.J. and P.V.D. prepared the Ribo-seq libraries. G.M. and P.V.D. supervised the research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research Foundation–Flanders (FWO-Vlaanderen) [G.0269.13N. to P.V.D.]; Research Foundation–Flanders (FWO-Vlaanderen) Postdoctoral Fellowship (to G.M.). Funding for open access charge: Commissie Wetenschappelijk Onderzoek (CWO) UGent.
Conflict of interest statement. None declared.
REFERENCES
- 1. Richardson E.J., Watson M.. The automatic annotation of bacterial genomes. Brief. Bioinform. 2013; 14:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Land M., Hauser L., Jun S.-R., Nookaew I., Leuze M.R., Ahn T.-H., Karpinets T., Lund O., Kora G., Wassenaar T. et al. . Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genomics. 2015; 15:141–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Michel A.M., Choudhury K.R., Firth A.E., Ingolia N.T., Atkins J.F., Baranov P. V. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res. 2012; 22:2219–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fields A.P., Rodriguez E.H., Jovanovic M., Stern-Ginossar N., Haas B.J., Mertins P., Raychowdhury R., Hacohen N., Carr S.A., Ingolia N.T. et al. . A regression-based analysis of ribosome-profiling data reveals a conserved complexity to mammalian translation. Mol. Cell. 2015; 60:816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee S., Liu B., Lee S., Huang S.X., Shen B., Qian S.B.. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crappé J., Ndah E., Koch A., Steyaert S., Gawron D., De Keulenaer S., De Meester E., De Meyer T., Van Criekinge W., Van Damme P. et al. . PROTEOFORMER: deep proteome coverage through ribosome profiling and MS integration. Nucleic Acids Res. 2015; 43:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bazzini A.A., Johnstone T.G., Christiano R., MacKowiak S.D., Obermayer B., Fleming E.S., Vejnar C.E., Lee M.T., Rajewsky N., Walther T.C. et al. . Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014; 33:981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calviello L., Mukherjee N., Wyler E., Zauber H., Hirsekorn A., Selbach M., Landthaler M., Obermayer B., Ohler U.. Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods. 2015; 13:1–9. [DOI] [PubMed] [Google Scholar]
- 10. Chew G.-L., Pauli A., Rinn J.L., Regev A., Schier A.F., Valen E.. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013; 140:2828–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michel A.M., Mullan J.P.A., Velayudhan V., O’Connor P.B.F., Donohue C.A., Baranov P.V.. RiboGalaxy: a browser based platform for the alignment, analysis and visualization of ribosome profiling data. RNA Biol. 2016; 13:316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O’Connor P.B.F., Andreev D.E., Baranov P. V. Comparative survey of the relative impact of mRNA features on local ribosome profiling read density. Nat. Commun. 2016; 7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung B.Y., Hardcastle T.J., Jones J.D., Irigoyen N., Firth A.E., Baulcombe D.C., Brierley I.. The use of duplex-specific nuclease in ribosome profiling and a user-friendly software package for Ribo-seq data analysis. RNA. 2015; 21:1731–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohammad F., Woolstenhulme C.J., Green R., Buskirk A.R.. Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep. 2016; 14:686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hastie T., Tibshirani R.F.. The elements of statistical learning. Methods. 2009; 1:305–317. [Google Scholar]
- 16. Panicker I.S., Browning G.F., Markham P.F.. The effect of an alternate start codon on heterologous expression of a PhoA fusion protein in mycoplasma gallisepticum. PLoS One. 2015; 10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hyatt D., Chen G.-L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Delcher A.L., Harmon D., Kasif S., White O., Salzberg S.L.. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999; 27:4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010; 26:2460–2461. [DOI] [PubMed] [Google Scholar]
- 20. Woolstenhulme C.J., Guydosh N.R., Green R., Buskirk A.R.. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 2015; 11:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suzek B.E., Ermolaeva M.D., Schreiber M., Salzberg S.L.. A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics. 2001; 17:1123–1130. [DOI] [PubMed] [Google Scholar]
- 22. Shultzaberger R.K., Bucheimer R.E., Rudd K.E., Schneider T.D.. Anatomy of Escherichia coli ribosome binding sites. J. Mol. Biol. 2001; 313:215–228. [DOI] [PubMed] [Google Scholar]
- 23. Omotajo D., Tate T., Cho H., Choudhary M.. Distribution and diversity of ribosome binding sites in prokaryotic genomes. BMC Genomics. 2015; 16:604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lutz W.K., Lutz R.W.. Statistical model to estimate a threshold dose and its confidence limits for the analysis of sublinear dose-response relationships, exemplified for mutagenicity data. Mutat. Res. 2009; 678:118–122. [DOI] [PubMed] [Google Scholar]
- 25. Sonderegger D.L., Wang H., Clements W.H., Noon B.R., Sonderegger D.L., Wang H., Clements W.H., Noon B.R.. Using SiZer to detect thresholds in ecological data. Front. Ecol. Environ. 2009; 7:190–195. [Google Scholar]
- 26. Li G.W., Burkhardt D., Gross C., Weissman J.S.. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014; 157:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heyer E.E., Moore M.J.. Redefining the translational status of 80S monosomes. Cell. 2016; 164:757–769. [DOI] [PubMed] [Google Scholar]
- 28. Gawron D., Gevaert K., Van Damme P.. The proteome under translational control. Proteomics. 2014; 14:2647–2662. [DOI] [PubMed] [Google Scholar]
- 29. Salzberg S.L., Deicher A.L., Kasif S., White O.. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998; 26:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pauli A., Valen E., Schier A.F.. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 2015; 37:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Damme P., Hole K., Pimenta-Marques A., Helsens K., Vandekerckhove J., Martinho R.G., Gevaert K., Arnesen T.. NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 2011; 7:e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Palenchar P.M. Amino acid biases in the N- and C-termini of proteins are evolutionarily conserved and are conserved between functionally related proteins. Protein J. 2008; 27:283–291. [DOI] [PubMed] [Google Scholar]
- 33. Krug K., Carpy A., Behrends G., Matic K., Soares N.C., Macek B.. Deep coverage of the Escherichia coli proteome enables the assessment of false discovery rates in simple proteogenomic experiments. Mol. Cell. Proteomics. 2013; 12:3420–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goodhead I., Darby A.C.. Taking the pseudo out of pseudogenes. Curr. Opin. Microbiol. 2015; 23:102–109. [DOI] [PubMed] [Google Scholar]
- 35. Lerat E., Ochman H.. Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res. 2005; 33:3125–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deshayes C., Perrodou E., Gallien S., Euphrasie D., Schaeffer C., Van-Dorsselaer A., Poch O., Lecompte O., Reyrat J.-M.. Interrupted coding sequences in Mycobacterium smegmatis: authentic mutations or sequencing errors. Genome Biol. 2007; 8:R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Plunkett G., Burland V., Daniels D.L., Blattner F.R.. Analysis of the Escherichia coli genome. III. DNA sequence of the region from 87.2 to 89.2 minutes. Nucleic Acids Res. 1993; 21:3391–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y., Gladyshev V.N.. An algorithm for identification of bacterial selenocysteine insertion sequence elements and selenoprotein genes. Bioinformatics. 2005; 21:2580–2589. [DOI] [PubMed] [Google Scholar]
- 39. Baek J., Lee J., Yoon K., Lee H.. Identification of unannotated small genes in salmonella. G3. 2017; 7:983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Diament A., Tuller T.. Estimation of ribosome profiling performance and reproducibility at various levels of resolution. Biol. Direct. 2016; 11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huvet M., Stumpf M.P.. Overlapping genes: a window on gene evolvability. BMC Genomics. 2014; 15:721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ingolia N.T., Lareau L.F., Weissman J.S.. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011; 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Van Damme P., Gawron D., Van Criekinge W., Menschaert G.. N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Mol. Cell. Proteomics. 2014; 13:1245–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hemm M.R., Paul B.J., Schneider T.D., Storz G., Rudd K.E.. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol. Microbiol. 2008; 70:1487–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andrews S.J., Rothnagel J.A.. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014; 15:193–204. [DOI] [PubMed] [Google Scholar]
- 46. Olexiouk V., Crappé J., Verbruggen S., Verhegen K., Martens L., Menschaert G.. SORFs.org: a repository of small ORFs identified by ribosome profiling. Nucleic Acids Res. 2016; 44:D324–D329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samayoa J., Yildiz F.H., Karplus K.. Identification of prokaryotic small proteins using a comparative genomic approach. Bioinformatics. 2011; 27:1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Butler J.S., Springer M., Grunberg-Manago M.. AUU-to-AUG mutation in the initiator codon of the translation initiation factor IF3 abolishes translational autocontrol of its own gene (infC) in vivo. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:4022–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Angelova M., Kalajdziski S., Kocarev L.. Computational methods for gene finding in prokaryotes. ICT Innovations 2010, Web Proceedings. 2010; 2010:11–20. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
An implementation of the REPARATION software is freely available via the GitHub development platform at https://github.com/Biobix/REPARATION.
Ribo- and RNA-seq sequencing data reported in this paper have been deposited in NCBI’s Gene Expression Omnibus with the accession number GSE91066. All MS proteomics data and search results have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005844 for the Salmonella typhimurium SL1344 datasets and PXD005901 for the E. coli K12 strain MG1655 dataset.