


Abstract
DNA methylation is an important epigenetic modification in many species that is critical for development, and implicated in ageing and many complex diseases, such as cancer. Many cost-effective genome-wide analyses of DNA modifications rely on restriction enzymes capable of digesting genomic DNA at defined sequence motifs. There are hundreds of restriction enzyme families but few are used to date, because no tool is available for the systematic evaluation of restriction enzyme combinations that can enrich for certain sites of interest in a genome. Herein, we present customised Reduced Representation Bisulfite Sequencing (cuRRBS), a novel and easy-to-use computational method that solves this problem. By computing the optimal enzymatic digestions and size selection steps required, cuRRBS generalises the traditional MspI-based Reduced Representation Bisulfite Sequencing (RRBS) protocol to all restriction enzyme combinations. In addition, cuRRBS estimates the fold-reduction in sequencing costs and provides a robustness value for the personalised RRBS protocol, allowing users to tailor the protocol to their experimental needs. Moreover, we show in silico that cuRRBS-defined restriction enzymes consistently out-perform MspI digestion in many biological systems, considering both CpG and CHG contexts. Finally, we have validated the accuracy of cuRRBS predictions for single and double enzyme digestions using two independent experimental datasets.
BACKGROUND
With the advent of next-generation sequencing (NGS) scientists are studying the biology of life at unprecedented resolution (1). Unfortunately, owing to the large size of many commonly studied genomes (human, mouse and tobacco plant for example are all >2.5 Gbp in size) (2–4), it is often still prohibitively expensive to conduct whole genome sequencing at high coverage. This creates a trade-off that negatively impacts the number of replicates that can be included and, therefore, it challenges the statistical power and the reproducibility of the studies (5,6). This is true in particular for DNA methylation, where differentially methylated regions (DMRs) are typically called by identifying changes as small as 10% and where 70–80% of the reads of Whole Genome Bisulfite Sequencing (WGBS) methods contain little to no relevant information on the DNA methylation status (7).
To address these cost inefficiencies, many methods have been developed to reduce the number of fragments that need to be sequenced for a given biological system or WGBS validation experiment (8–25). These methods can be broadly split into those that positively select for genomic fragments of interest and those that deplete for fragments that are not of interest. Positive selection-based methods involve the sites of interest being enriched from the background. This usually occurs through pull-down of these sites via an antibody (e.g. anti-5mC antibody), a recombinant binding protein (e.g. methyl-CpG-binding domains (MBD)), RNA baits for the sites of interest, array-based approaches (e.g. EPIC array in human) or PCR-based approaches (26). These methods have many limitations, including enrichment biases, complex protocols and difficulties in quantification (27).
Current evidence shows that depletion-based methods do not have enrichment biases, tend to be simpler and are more readily quantifiable (26,27). The most common depletion-based approaches use restriction enzymes to exploit the fact that the nucleotide composition in a given genome is non-random (28) and that the fragment lengths produced from a given digestion will thus reflect this. In the case of 5-methylcytosine (5mC), the most common depletion-based method is Reduced Representation Bisulfite Sequencing (RRBS) using the methylation-insensitive restriction enzyme MspI (with the recognition sequence C|CGG), although enzymes such as BglII, XmaI and TaqαI have also been used (29–31). RRBS has proven extremely useful for cost-effective, global studies of DNA methylation (10,32,33), capturing around 10% of CpG sites within mammalian genomes but with up to a 30-fold reduction in the number of fragments sequenced in comparison to WGBS (34).
Whilst restriction enzyme-based approaches are versatile, simple and cost-effective, the utility of the MspI-based RRBS approach is limited to a specific subset of CpG sites in the genome, mainly found within CpG islands and promoters (32). To allow researchers to optimise the protocol used for their specific experiment, we have developed a new computational method called customised Reduced Representation Bisulfite Sequencing (cuRRBS). cuRRBS generalises the problem of genomic enrichment with restriction enzymes by allowing the user to define both the genome and the particular sites of interest, before outputting the optimal enzyme combinations and size ranges to target these sites. In addition, cuRRBS provides the user with a variety of metrics to compare the various suggested protocols, including an estimate of the fold-reduction in sequencing costs compared to WGBS and a robustness value to assess the impact of experimental error in the size selection step.
Here, we have tested the enrichment ability of cuRRBS in several biological systems, with sites in both CpG and CHG contexts and multiple species, to showcase the generalisability and utility of the software (35–41). In addition, we take advantage of two recently published independent RRBS datasets to demonstrate the accuracy of the software predictions in both single and double enzyme experimental settings (30,31).
We hope that cuRRBS will be useful both as a tool for designing cost-effective, genome-wide studies in the future but also for validating previous results from whole genome approaches in a simple, cheap and timely fashion, something that at present is not possible.
MATERIALS AND METHODS
Restriction enzymes annotation
All the information regarding the commercially-available restriction enzymes that are used by cuRRBS was extracted from REBASE (42,43). Restriction enzymes were grouped in isoschizomer families (i.e. enzymes that recognise the same sequence and generate identical fragment length distributions) and each enzyme was manually annotated for different types of methylation-sensitivity (CpG, CHG, CHH). Only isoschizomer families that contained at least one methylation-insensitive enzyme were considered for the examples described in this manuscript.
Genome assemblies and genomic annotation
All the analyses presented here were performed in the following genome assemblies: Homo sapiens (hg38), Mus musculus (mm10) and Arabidopsis thaliana (TAIR10). Scaffolds not assembled into the main chromosomes were discarded. Genomic annotation for the human genome (hg38) was obtained from GENCODE (v25, basic gene annotation) (44), with the exception of CpG islands (CGIs), which were extracted from the UCSC Genome Browser (45). GC content and CpG content were calculated, around each restriction enzyme cleavage site, taking windows of ±25 bp and ±500 bp respectively. For each enzyme, the mean of all cleavage sites was calculated to obtain the mean GC content and the mean CpG content. Intragenic regions were defined as those regions within ±2.5 kb of a protein-coding gene, whilst the rest of the genome was considered to be intergenic. CpG shores were defined as regions 0 to 2 kb away from CGIs in both directions and CpG shelves as regions 2 to 4 kb away from CGIs in both directions (46). Promoters were defined as encompassing a 3 kb region (2.5 kb upstream and 0.5 kb downstream of the TSS) relative to the TSS of all protein-coding transcripts in GENCODE, similar to the strategy used in Taher et al. (47). Genomic annotation for the CGIs in the mouse genome (mm10) was also obtained from the UCSC Genome Browser (45). All annotations were handled using the pybedtools library (48,49).
Performing in silico digestions of a given genome
We used the Restriction package from Biopython v1.68 to digest the different genomes with the appropriate restriction enzymes in silico (50). Only the first member of a given isoschizomer family (which contained at least one methylation-insensitive enzyme) was processed to avoid redundant computations. The output of the in silico digestions was stored (pre-computed files) and subsequently read by cuRRBS when needed to reduce the computational time (see ‘cuRRBS heuristics and computational efficiency’). When assessing enzyme combinations, the information from the appropriate individual pre-computed files (i.e. the genomic coordinates where the enzyme theoretically cuts) were combined by the software to compute all the necessary variables.
cuRRBS’ enzyme flexibility
To ensure the user has full control over the enzymes that cuRRBS will use to derive the desired enrichments, one of the inputs given to cuRRBS is an enzyme annotation file. This file contains the desired isoschizomer families that the user wishes to be tested by cuRRBS. In the GitHub repository we have already defined enzyme annotation files for enzymes that are methylation-insensitive in a CG context and in CG, CHG and CHH contexts (51). However, it is also possible for the user to define a personalised set of enzymes by providing a self-generated annotation file. This can be useful, for instance, to reduce the chance of any star activity in the reported cuRRBS protocols.
In addition, the output file from cuRRBS contains, by default, 30 cuRRBS protocols that would enrich for the user's sites of interest. Therefore, the user can determine which enzyme combination and size range would be the simplest and most appropriate for the given application. This provides the user with the opportunity to consider experimental factors that may complicate the protocol, such as buffer compatibility and whether consecutive digestions would be required.
Calculating cuRRBS main variables
cuRRBS makes use of several variables in order to find the best enzyme combination and size range which enriches for a certain set of genomic sites defined by the user (see ‘cuRRBS overview’). For a given enzyme (combination) and size range, the Enrichment Value (EV) can be calculated as:
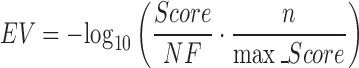 |
Here, NF is the number of genomic fragments that will theoretically be sequenced (i.e. those whose lengths after the in silico digestion are within the size range); n is the total number of sites of interest; max_Score is the Score obtained if all the sites of interest were sequenced and:
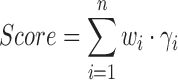 |
where wi is the weight of the ith site of interest and γi is 1 if the ith site would be theoretically sequenced (i.e. present in a size selected fragment and ≤ read length base pairs away from one of the ends of the fragment) and 0 otherwise. Since the objective of cuRRBS is to maximize the Score while minimizing the NF, the best results will be obtained when EV is minimized.
cuRRBS output also contains other variables that may be of interest to the user (see ‘cuRRBS overview’). The Cost Reduction Factor (CRF) for a given cuRRBS protocol can be calculated as:
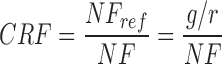 |
where NFref is the estimated number of fragments that would be sequenced in a Whole Genome Bisulfite Sequencing (WGBS) experiment, that can be roughly calculated as the genome size (g) divided by the read length (r).
Furthermore, the robustness (R) of a given enzyme (combination) is calculated as:
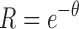 |
with
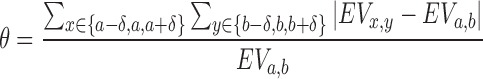 |
where EVa,b is the EV for the optimal size range (a: lower limit in size range, b: breadth) and δ is the experimental error (in bp) that is assumed during the size selection step. The robustness will take values in the interval (0,1], with higher values identifying robust cuRRBS protocols.
Flexible user-defined cuRRBS parameters
cuRRBS contains a number of user-defined parameters to ensure the greatest possible flexibility and ease of use. A table of these parameters is provided to highlight the versatility that the user has and why such versatility is useful (Table 1).
Table 1. Flexible user-defined cuRRBS parameters.
| cuRRBS parameter (abbrev.) | Significance | Default | Range |
|---|---|---|---|
| Enzymes to check (-e) | Defines the enzymes (isoschizomer families) that cuRRBS will look at | - | - |
| Annotation for the sites of interest (-a) | Allows identification and weighting of the sites of interest | - | - |
| Read length (-r) | Defines the positions in the theoretical fragments that can be ‘seen’ after sequencing | - | 30–300 |
| Adapters size (-s) | Ensures correct experimental size selection | - | - |
| C_Score constant (-c) | Sets the minimum acceptable Score | - | 0–1 |
| Genome size (-g) | Needed to calculate the CRF | - | - |
| C_NF/1000 constant (-k) | Sets the minimum acceptable CRF | 0.2 | 0–1 |
| Experimental error (-d) | Sets the assumed experimental error (δ) | 20 | 5–500 |
| Size range breadth (-b) | Constrains the breadth of the size range | 980 | - |
| Output size (-t) | Defines the number of cuRRBS protocols the user can compare | 30 | - |
| Site IDs (-i) | Enables the identification of the recovered sites of interest | No | - |
This table details the flexible user-defined parameters that cuRRBS will accept as arguments. The cuRRBS parameter full name and command-line abbreviation (in brackets) are provided alongside a simplified description of the significance of these arguments to the user. Where applicable, the defaults and ranges are also detailed.
cuRRBS heuristics and computational efficiency
cuRRBS employs several strategies to reduce the computational time needed in each run:
Restriction enzymes are grouped in isoschizomer families. Since isoschizomers generate the same genomic digestions, only one member of each family needs to be processed.
In silico digestions are read from pre-computed files. Digesting the genomes would be a limiting factor in the cuRRBS pipeline. The user can download the pre-computed files (51) and the information that they contain is read every time that an enzyme needs to be assessed.
The number of size ranges that are sampled is minimised. Since the experimental size selection step is generally imperfect, size ranges are sampled with a sliding window whose ‘resolution’ is equivalent to the experimental error specified by the user.
Parallelization. cuRRBS can use several cores to decrease the CPU time.
Moreover, we have observed that, in many enzyme combinations, one of the enzymes is providing most of the enrichment for the sites of interest, while the second one complements the targeting. Therefore, it would be possible to implement a ‘heuristic’ mode, where only those enzymes that perform well individually are used as ‘seeds’ to construct combinations (as opposed to the current implementation, where all the enzyme combinations are checked exhaustively). This could further reduce the computational time, especially if combinations of more than two enzymes were being evaluated.
The CPU time required by cuRRBS depends on several parameters, including the number of enzymes checked, the experimental error, the number of sites of interest or the genome size (Supplementary Figure S1A–D). The RAM used will be approximately equal to the size of the pre-computed files that are read by the software. A standard cuRRBS run (e.g. for a few thousand sites of interest in the human genome, checking 128 CpG methylation-insensitive isoschizomer families) takes ∼0.5–1 h and uses around 4GB RAM, which allows the user to easily run it on a dual-core laptop or desktop computer.
Obtaining the sites of interest for different biological systems
We have tested in silico the ability of cuRRBS to enrich for the sites of interest in a selection of different biological systems where DNA methylation has an important functional role. In some of these systems, described below, previous analysis was performed in order to obtain the genomic coordinates for the sites:
Exon-intron boundaries in human. Exons and introns were obtained from protein-coding genes using GENCODE annotation data. Those CpG sites that were found within ± 5 bp of a canonical splice site (5′-GT, 3′-AG) were selected.
Epigenetic clock in human. These sites were obtained from Horvath (37). Briefly, these sites make up an elastic-net regression model that is able to predict chronological and biological age in humans. These sites were lifted over to hg38 for the analysis conducted in this paper (52).
Canonical and placental imprints in human. These loci were obtained from Hanna et al., 2016 (35). These sites were lifted over to hg38 and the CpG sites were then extracted for the analysis conducted in this paper (52).
CTCF binding sites in human. We obtained the CpG sites that overlap with in vivo CTCF binding sites. Peaks from sites that seem to be affected by methylation (upregulated, reactivated) were kindly provided by Maurano (39). We scanned the peaks for high-scoring motifs according to the CTCF JASPAR model (53). Finally, we extracted those CpGs that were found in positions 5 and 15 of the motif, whose methylation status is supposed to influence the binding of the transcription factor (39).
Induced pluripotent stem cell (iPSC) demethylated and maintained sites in mouse. These were obtained as described previously (36), with an additional filter for magnitude of methylation change. In brief, a background model was obtained by calculating the global mean for the ending methylation value for each starting methylation value, comparing mouse embryonic fibroblasts (MEFs) to iPSCs. Afterwards, a binomial test was used on individual probes for their ending methylation measured against the mean for their starting methylation, to obtain p-value < 0.05 differentially methylated regions (DMRs). Adjusted p-values were calculated using a pairwise t-test with a Benjamini–Hochberg correction and only probes accounting for more than 50% loss of methylation relative to the genome average loss (from MEFs to iPSCs, commonly located at super-enhancer regions) or more than 50% gain in methylation relative to the genome average loss (from MEFs to iPSCs, commonly located at intracisternal A-particles) were used in this analysis.
NRF1 binding sites in mouse. We obtained the CpG sites that overlap with in vivo NRF1 binding sites in mouse. ChIP-seq data was processed as described in the original publication (41), where peaks were called using Peakzilla (54). We took as our final set of peaks the overlap between the two TKO replicates. Next, we scanned the peaks for high-scoring motifs according to the NRF1 JASPAR model (53). Finally, we extracted those CpGs that were found in positions 2 and 8 of the motif, whose methylation status is supposed to influence the binding of the transcription factor (41).
CHG sites in Arabidopsis thaliana. Non-CpG DMRs arising from the epigenomic diversity between Arabidopsis thaliana accessions were obtained from Kawakatsu et al. (38). The coordinates for C sites in non-CpG context were extracted.
In all the cases the sites were equally weighted (wi = 1), with the exception of the human epigenetic clock system, where the sites were assigned the absolute value of the weights in the linear model (37).
All the site annotation files are provided as Supplementary Tables S1–S9.
Running cuRRBS for different in silico systems
cuRRBS was run in the different systems described above using the default parameters (k = 0.2, d = 20, b = 980, t = 30), for a read length (r) of 75 bp and a Score threshold (c) of 0.25. In the mouse and human examples we considered 128 isoschizomer families that contained enzymes that were not sensitive to CpG methylation. In the case of Arabidopsis thaliana we used 28 isoschizomer families that contained enzymes that were not sensitive to 5mC in any context (CG, CHG, CHH).
Mapping of RRBS samples
XmaI-RRBS data generated on the Ion Torrent platform (30) and MspI&TaqαI-RRBS data generated on the Illumina HiSeq platform (31) were quality-trimmed using Trim Galore (www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and had base pairs removed from the 3′ end to avoid including filled-in nucleotides with artificial methylation states (the filled-in XmaI, MspI and TaqαI cut sites include the nucleotide sequence CCGG, CG and CG respectively). The data was then mapped to the human genome (for XmaI data, parameters: –non_directional) or the mouse genome (for MspI&TaqαI data, parameters: –directional) using Bismark (0.18.0) (55). In each of the two cases data from different experiments or replicates was merged into the same FASTQ file prior to quality trimming.
Estimating cuRRBS’ sensitivity and specificity
We assessed the performance of cuRRBS predictions in two independent experimental datasets (30,31) (see ‘Experimental validation of cuRRBS’ in Results and discussion). We ran cuRRBS fixing the theoretical size ranges tested to the ones reported in the publications (30,31) and we used as our sites of interest the CpGs that overlapped with CpG islands (CGI-CpGs) in the human (30) and the mouse genomes (31) respectively. From the cuRRBS output files, we recovered the IDs of the sites that should be theoretically sequenced. Moreover, using the experimental RRBS data (30,31), we could obtain the IDs of the sites that were actually sequenced (filtered by a given depth of coverage threshold). Afterwards, we calculated the following variables for each one of the datasets:
True positives (TP): number of CGI-CpGs that cuRRBS predicted to be sequenced and were indeed found in the RRBS data.
True negatives (TN): number of CGI-CpGs that cuRRBS predicted to be absent and were not found in the RRBS data.
False positives (FP): number of CGI-CpGs that cuRRBS predicted to be sequenced but were not found in the RRBS data.
False negatives (FN): number of CGI-CpGs that cuRRBS predicted to be absent but were found in the RRBS data.
Finally, we estimated the sensitivity and specificity, for a given dataset, as follows:
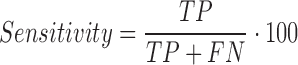 |
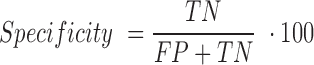 |
RESULTS AND DISCUSSION
Restriction enzyme digestion as a tool for genomic enrichment
Restriction enzymes represent an incredibly effective tool for the enrichment of certain sites of interest in a genome. This is possible due to the wide variety of motifs that commercially-available restriction enzymes can recognise (Supplementary Figure S2A and B) combined with the non-random nature of the genome composition itself. Supplementary Figure S2A and B highlight that this motif diversity is driven both by the sequence composition (GC content) and the length of the recognition sequence. Thus, different restriction enzymes will generate different fragment length distributions, dependent upon how frequently their recognition site is present in a given genome (Figure 1A, Supplementary Figure S2C).
Figure 1.

Restriction enzyme digestion as a tool for genomic enrichment. (A) Heatmap showing the fragment length distributions generated by different restriction enzymes in the human genome (hg38). Each column represents the distribution for an isoschizomer family of restriction enzymes that contains at least one member which is methylation-insensitive in a CpG context. The distributions are binned in size ranges of 200 bp, ordered as they would appear in an electrophoretic gel. Additional row annotations on top of the heatmap contain information regarding the total number of fragments (in red) and the median fragment length (in blue) produced by each in silico digestion, together with the GC content of the recognition motif in the isoschizomer family (in green). Legend is displayed on the right hand side. (B) Scatterplot showing the percentage of cleavage sites from different restriction enzymes that overlaps with CpG islands (X-axis) and promoters (Y-axis) in the human genome (hg38). The size of the circles represents the total number of cleavage sites generated by each enzyme. The enzymes MspI and BssAI are highlighted in red and blue respectively. Legend is displayed on the right hand side. (C) Scatterplot showing the percentage of cleavage sites from different restriction enzymes that overlap with intergenic regions (X-axis) and non-coding RNA genes (Y-axis) in the human genome (hg38). The size of the circles represent the total number of cleavage sites generated by each enzyme. The enzyme MspI is highlighted in red. The enzymes BsmI and MfeI are both highlighted in blue. Legend is displayed on the right hand side.
In DNA methylation studies the most common application is the use of MspI (cutting at C|CGG) in RRBS (Reduced Representation Bisulfite Sequencing), which is used to enrich for CG dinucleotides (CpGs) contained in promoters and CpG islands (32) (Figure 1B). However, in many cases, MspI is by no means the most effective restriction enzyme that could be used. For instance, MspI would be a poor restriction enzyme to choose for the enrichment of CpGs found in intergenic regions or non-coding RNA genes, which would be far better enriched for using BsmI or MfeI respectively (Figure 1C). In fact, it turns out that across many genomic features MspI is rarely the most optimal methylation-insensitive restriction enzyme (Supplementary Figure S2D).
Previous studies have tested the potential of other restriction enzymes and enzyme combinations to expand the range of CpG sites that can be targeted in a genome (8–11,28,30,56,57). However, to our knowledge, there is currently no computational method that systematically explores the capacity of all commercially-available restriction enzymes to generate ‘personalised’ reduced-representations of the genome whilst minimising the experimental cost (Supplementary Figure S2E).
cuRRBS overview
We have developed a novel computational method (cuRRBS) that determines the optimal combination of restriction enzymes and size range to enrich for any given set of sites of interest in any genome. In other words, by modifying two of the steps in the original RRBS protocol (Figure 2A), cuRRBS generalises RRBS.
Figure 2.

cuRRBS overview. (A) Outline of an RRBS protocol. Highlighted are the two steps that would be modified according to the output produced by cuRRBS (i.e. the restriction enzymes used for the genomic digestion and the size selection). Legend is displayed on the bottom left. (B) Schematic of cuRRBS. Highlighted are the two main inputs required for the software and the two thresholds that the user has to define (red and purple tags). The default output for cuRRBS is a table containing the top hits (restriction enzyme combination and size range) along with additional information that might be useful to the user (such as Cost Reduction Factor and robustness). (C) Scatterplot showing the trade-off between the number of fragments (NF) and the Score for the best enzyme combination (BsaWI & BssAI) that targets the CpGs present in the human placental-specific imprinted regions (35). NF is divided by 1000 for visualization purposes. Each point represents a different size range. Shown in dark blue and grey are the size ranges that would and would not pass filtering respectively. Shown in orange is the optimal size range in the filtered search space. The dotted lines depict the thresholds that need to be specified by the user (red: maximum NF; purple: minimum percentage of the maximum Score). In this mock example we specified an NF threshold of 150 000 fragments and a Score threshold of 25% of the maximum Score. Legend is displayed below the plot title. (D) Contour plot that depicts how the robustness (R) variable is calculated for the optimal enzyme combination (BsaWI & BssAI; size range: 60–540 bp) that targets the CpGs present in the human placental-specific imprinted regions (35). Enrichment values (EVs) are calculated for all possible size ranges in order to create an ‘EV landscape’. In this landscape, cuRRBS finds the size range with the lowest EV that still satisfies the thresholds (asterisk in green). Afterwards, cuRRBS samples EVs around the optimum (asterisks in black). The points that are sampled depend on the experimental error (in this case, δ = 20 bp). A high robustness value means that the sampled EVs do not change a lot when compared to the optimum, which implies that cuRRBS prediction will not be greatly affected by experimental errors during the size selection step.
The software takes as input the genomic coordinates that the user wants to target (Figure 2B, Supplementary Figure S3A). Afterwards, cuRRBS assesses in silico the potential of all single enzymes and double-enzyme combinations to enrich for the sites of interest using the following two key variables (see Materials and Methods):
NF, which reflects the theoretical number of fragments that will be sequenced after the size selection step. Assuming that the sequencing cost is proportional to NF, cuRRBS attempts to minimise this value.
Score, which reflects the theoretical number of sites of interest that will be sequenced after the size selection step. cuRRBS attempts to maximise this value.
The NF and Score variables are positively correlated with one another, such that the more genomic fragments sequenced, the more sites of interest are likely to be contained within the reduced representation (Figure 2C, Supplementary Figure S3B). However, this relationship disappears at higher NF values, where the Score variable becomes saturated such that any additional fragments sequenced will result in a reduction in the overall enrichment of the sites of interest. This Score saturation at high NF is mainly due to additional sites of interest being buried within long fragments that will not be sequenced due to limitations in the read length (cuRRBS parameter –r, see Table 1). For a given enzyme or enzyme combination, the NF and the Score variables depend on the size range chosen, since only the genomic fragments within the size range will be present in the reduced representation of the genome.
cuRRBS requires that the user sets thresholds for the maximum NF (i.e. minimum CRF) and minimum Score that would be acceptable for a given application (Figure 2B, Supplementary Figure S3A). These thresholds allow cuRRBS to search through all possible size ranges for a given enzyme or enzyme combination and to find the one that minimises the Enrichment Value (EV), a variable that combines both NF and Score into a single number (see Materials and Methods). cuRRBS repeats this procedure for every single enzyme and enzyme combination and reports those with the best hits (i.e. those with the lowest EVs) (Supplementary Figure S3A).
The output file contains the best scoring enzymes with their correspondent size ranges and some other useful variables for each one of the hits, such as (see Materials and Methods):
Cost Reduction Factor (CRF). This estimates the theoretical fold-reduction in sequencing costs for the cuRRBS protocol when compared to Whole Genome Bisulfite Sequencing (WGBS).
Robustness (R). This assesses how much the cuRRBS prediction varies if a slightly different size range is used (Figure 2D). The results for robust enzymes will not be greatly affected as a consequence of experimental error during the size selection step. This will help the user to make an informed decision on which enzyme combination to choose for the system of interest (Supplementary Figure S3C).
Running cuRRBS in different biological systems
cuRRBS provides a way to effectively interrogate DNA methylation in any biological system for which the reference genome is available. Besides reducing the cost for organisms currently under intensive study (e.g. human, mouse), cuRRBS opens the door to the cost-effective study of DNA methylation in species with large genomes or where DNA methylation in non-CpG contexts is common, such as plants (58), which currently lack an MspI-based RRBS protocol, owing to the enzyme's CHG methylation sensitivity (59).
We decided to test the ability of cuRRBS to enrich for genomic sites that have important functional roles in different systems. Some of the systems that we tested in silico include genomic regions whose methylation status is important during cellular reprogramming (36), an epigenetic clock (37), transcription factor binding sites that are affected by DNA methylation (39,41), imprinted loci (35), CpGs found in the exon-intron boundaries and CHG sites that are differentially methylated between different arabidopsis accessions (38) (Supplementary Figure S4). For these in silico systems we chose to run the software with the threshold set to 25% of the maximum Score.
In all cases, cuRRBS is able to dramatically reduce the cost associated with the sequencing by several orders of magnitude compared to WGBS, which is assessed using the Cost Reduction Factor (CRF) (Supplementary Figure S5). In addition, for cases where a comparison to MspI-based RRBS could be made, cuRRBS is able to improve the CRF, again, by orders of magnitude. As an example, for the placental-specific imprints, the sequencing costs are reduced by approximately 400-fold when compared to WGBS and by 12.5-fold when compared to the traditional MspI-based RRBS.
Furthermore, we have also observed that many of the top hits reported by cuRRBS are digestions of two restriction enzymes (Supplementary Figure S4), highlighting the combinatorial power of restriction enzymes to produce optimal reduced representations of the genome (28). Excitingly, we are able to show that using cuRRBS it is possible to assay a far larger number of target sites, in a far simpler experimental design than would normally be achieved using amplicon-based bisulfite sequencing.
Experimental validation of cuRRBS
To assess in an unbiased manner how well predictions from cuRRBS perform in an experimental setting, we employed two independent non-canonical RRBS datasets: one generated from a single enzyme (XmaI) and the other from a combination of two restriction enzymes (MspI and TaqαI) (30,31). By evaluating the predictive power of cuRRBS in these two datasets, we were able to observe cuRRBS’ performance in both single and double enzyme contexts and across different genomes.
To test the accuracy of cuRRBS predictions in the context of a single enzyme digestion, we utilised the non-canonical RRBS dataset generated from human DNA using the restriction enzyme XmaI (30). This dataset was previously used to show that XmaI could enrich for CpG islands (CGIs), while reducing the overall sequencing cost relative to MspI, making the protocol more cost-effective. To validate cuRRBS using this system, we therefore chose to enrich for all CpG sites that overlapped with a CGI (CGI-CpGs) in the human genome using a predetermined theoretical size range equivalent to the ‘reproducible library fragment lengths’ reported in (30) (i.e. 90–185 bp). cuRRBS predicted with high accuracy the CpG sites that were observed in the experimental XmaI-RRBS dataset (Figure 3A). In particular, only a small proportion of the total number of CGI-CpGs should be theoretically sequenced (102 253 out of 2 164 614), and this was indeed the case (Figure 3A). Furthermore, upon filtering out sites with low depth of coverage, which commonly represent noise in RRBS datasets, the sensitivity increased up to ∼80%. Importantly, the specificity remained constant at ∼100% independent of the threshold set for depth of coverage (Figure 3B). Thus, cuRRBS produces a prediction that is relatively conservative, as highlighted by the low numbers of false positives (Figure 3A), at the expense of a small decrease in sensitivity.
Figure 3.

Experimental validation of cuRRBS. (A) Barplots showing the number of true positives (TP, in green), true negatives (TN, in blue), false positives (FP, in red) and false negatives (FN, in orange) when comparing cuRRBS theoretical prediction with the actual XmaI-RRBS experimental data (30). The number of sites in each category is calculated for different thresholds in the depth of coverage (number of reads covering a CpG site as reported by Bismark). cuRRBS prediction for the CpG sites in human CpG islands was obtained enforcing a theoretical size range of 90–185 bp and running the software for XmaI with all the default parameters (with a read length of 200 bp). Legend is displayed on the right hand side. (B) Plot showing values of cuRRBS sensitivity (in light green) and specificity (in cyan) as a function of the depth of coverage threshold employed to filter the experimental data (30). The number of true positives (TP), true negatives (TN), false positives (FP) and false negatives (FN) are the same as in A. Legend is displayed below the plot curves.
Interestingly, the original theoretical size range that the study was aiming for (110–200 bp) was slightly different to the one achieved in the actual experiments (90–185 bp) (30). We ran cuRRBS using the original size range target and obtained slightly worse results for the sensitivity but not the specificity of the prediction (Supplementary Figure S6A and B). This demonstrates that the correct execution of the size selection step during the experimental protocol is key for obtaining the sites predicted by cuRRBS and highlights the importance of the robustness variable as part of the cuRRBS output in order to judge the consequences of these experimental errors.
To test the accuracy of cuRRBS predictions in the context of a double enzyme digestion, we utilised the non-canonical RRBS dataset generated from mouse DNA using the restriction enzymes MspI and TaqαI (31). To compare the accuracy of cuRRBS prediction in this double enzyme system to that of the XmaI-RRBS system, we again ran cuRRBS for CGI-CpGs, this time in the mouse genome with a theoretical size range of 80–160 bp (31). cuRRBS predicted with high accuracy the CpG sites that were observed in this double enzyme experiment (Supplementary Figure S6C). In addition, the results for sensitivity and specificity were very similar to the ones reported for the XmaI-RRBS dataset (Supplementary Figure S6D). Therefore, we conclude that cuRRBS produces robust predictions for the sites of interest that will be sequenced in RRBS protocols both for single and double enzyme combinations independent of the genome under study.
Lastly, the number of fragments that were theoretically recoverable in each of our experimental systems ranged from NF = 12 780 (for XmaI) to NF = 331 058 (for MspI and TaqαI). This represents approximately a 30-fold difference in the number of recoverable fragments and demonstrates that cuRRBS predictions, even for low NF values, are experimentally feasible. Importantly, in the nine theoretical examples that we report (Supplementary Figure S4), the number of fragments required by each cuRRBS protocol ranges from 107 248 to 974 050. Thus, the number of fragments required to achieve the stated CRF comfortably exceeds the minimum experimentally validated NF value (>8-fold).
CONCLUSIONS AND FUTURE DIRECTIONS
cuRRBS provides a new framework that allows the user to optimise RRBS for the biological system of interest by using novel combinations of restriction enzymes. Therefore, cuRRBS makes the study of DNA methylation more affordable across all species for which genomic sequences are available. Furthermore, it can open the door to the design of future studies in a clinical context (10), which require cost-effective and robust protocols.
Currently, cuRRBS only considers combinations of up to two restriction enzymes. However, in the future, it would be possible to adapt the software to explore combinations that contain higher numbers of enzymes, which could theoretically allow targeting the sites of interest even more efficiently (28). Moreover, there are several methods that are able to impute DNA methylation levels in sites that are not covered experimentally (46,60). These methods could expand the set of sites of interest that are finally measured by making use of the additional DNA methylation information that is retrieved in a cuRRBS experiment.
Finally, the potential of restriction enzymes to target different genomic coordinates is not limited to DNA methylation. As such, it would be conceivable for cuRRBS to be adapted to enrich for SNPs of interest (61,62) or to optimise chromosome conformation capture techniques (63,64). By reducing the cost associated with sequencing, we believe that cuRRBS will help to democratise high-throughput genomic studies.
DATA AVAILABILITY
cuRRBS and its documentation are freely distributed under GNU General Public License v3.0 and can be accessed via GitHub (51). The public RRBS data used in this manuscript can be found under GEO accession numbers GSE74126 (XmaI-RRBS) and GSE80961 (MspI&TaqαI-RRBS) respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the members of the Reik and Thornton laboratories for helpful discussions. Specifically, we would like to acknowledge Dr Dobril Ivanov for his very useful feedback and advice, Dr Courtney Hanna for providing the imprinted loci, Dr Ines Milagre for facilitating the iPSC sites, Dr Matthew T. Maurano for kindly providing the CTCF peaks from methylation-sensitive binding sites, Dr Oliver Stegle for his input regarding optimisation and Julia Spindel and Dr Aartjan te Velthuis for critically reviewing this manuscript.
Authors' contributions: D.E.M.H., J.M.T., W.R. and T.M.S. designed the study. D.E.M.H. and T.M.S. designed and developed cuRRBS. A.J.M.R. implemented the last version of cuRRBS. D.E.M.H. and T.M.S. conducted data analysis. F.K. processed the RRBS datasets. D.E.M.H., J.M.T., W.R. and T.M.S. interpreted data and wrote the manuscript. All authors read and approved the final manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
EMBL predoctoral fellowship [to D.E.M.H.]; EMBL postdoctoral fellowship [to A.J.M.R.]; BBSRC DTP studentship [to T.M.S.]; Biotechnology and Biological Sciences Research Council [BB/K010867/1 to W.R.]; Wellcome Trust [098565/Z/12/Z to J.M.T.; 095645/Z/11/Z to W.R.]; EU BLUEPRINT [to W.R.]; EpiGeneSys [to W.R.]. Funding for open access charge: Wellcome Trust [098565/Z/12/Z].
Conflict of interest statement. W.R. is a consultant and shareholder of Cambridge Epigenetix.
REFERENCES
- 1. Shendure J., Ji H.. Next-generation DNA sequencing. Nat. Biotechnol. 2008; 26:1135–1145. [DOI] [PubMed] [Google Scholar]
- 2. Sierro N., Battey J.N.D., Ouadi S., Bakaher N., Bovet L., Willig A., Goepfert S., Peitsch M.C., Ivanov N.V.. The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 2014; 5:3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mouse Genome Sequencing Consortium Waterston R.H., Lindblad-Toh K., Birney E., Rogers J., Abril J.F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M. et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002; 420:520–562. [DOI] [PubMed] [Google Scholar]
- 4. Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 5. Fumagalli M. Assessing the effect of sequencing depth and sample size in population genetics inferences. PLoS ONE. 2013; 8:e79667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu H., Xu T., Feng H., Chen L., Li B., Yao B., Qin Z., Jin P., Conneely K.N.. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015; doi:10.1093/nar/gkv715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ziller M.J., Gu H., Müller F., Donaghey J., Tsai L.T.Y., Kohlbacher O., De Jager P.L., Rosen E.D., Bennett D.A., Bernstein B.E. et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013; 500:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kirschner S.A., Hunewald O., Mériaux S.B., Brunnhoefer R., Muller C.P., Turner J.D.. Focussing reduced representation CpG sequencing through judicious restriction enzyme choice. Genomics. 2016; 107:109–119. [DOI] [PubMed] [Google Scholar]
- 9. Yu L., Liu C., Bennett K., Wu Y.-Z., Dai Z., Vandeusen J., Opavsky R., Raval A., Trikha P., Rodriguez B. et al. A NotI-EcoRV promoter library for studies of genetic and epigenetic alterations in mouse models of human malignancies. Genomics. 2004; 84:647–660. [DOI] [PubMed] [Google Scholar]
- 10. Lee Y.K., Jin S., Duan S., Lim Y.C., Ng D.P., Lin X.M., Yeo G.S., Ding C.. Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biol. Proced. Online. 2014; 16:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cedar H., Solage A., Glaser G., Razin A.. Direct detection of methylated cytosine in DNA by use of the restriction enzyme MspI. Nucleic Acids Res. 1979; 6:2125–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karimi M., Johansson S., Ekström T.J.. Using LUMA: a Luminometric-based assay for global DNA-methylation. Epigenetics. 2006; 1:45–48. [DOI] [PubMed] [Google Scholar]
- 13. Guo H., Zhu P., Guo F., Li X., Wu X., Fan X., Wen L., Tang F.. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat. Protoc. 2015; 10:645–659. [DOI] [PubMed] [Google Scholar]
- 14. Hahn M.A., Li A.X., Wu X., Pfeifer G.P.. Single base resolution analysis of 5-methylcytosine and 5-hydroxymethylcytosine by RRBS and TAB-RRBS. Methods Mol. Biol. 2015; 1238:273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li D., Zhang B., Xing X., Wang T.. Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods. 2015; 72:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cohen-Karni D., Xu D., Apone L., Fomenkov A., Sun Z., Davis P.J., Kinney S.R.M., Yamada-Mabuchi M., Xu S.-Y., Davis T. et al. The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:11040–11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saliba A.-E., Westermann A.J., Gorski S.A., Vogel J.. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014; 42:8845–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plongthongkum N., Diep D.H., Zhang K.. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nature Publishing Group. 2014; 15:647–661. [DOI] [PubMed] [Google Scholar]
- 19. Frankhouser D.E., Murphy M., Blachly J.S., Park J., Zoller M.W., Ganbat J.-O., Curfman J., Byrd J.C., Lin S., Marcucci G. et al. PrEMeR-CG: inferring nucleotide level DNA methylation values from MethylCap-seq data. Bioinformatics. 2014; 30:3567–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hughes S., Jones J.L.. The use of multiple displacement amplified DNA as a control for methylation specific PCR, pyrosequencing, bisulfite sequencing and methylation-sensitive restriction enzyme PCR. BMC Mol. Biol. 2007; 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Komori H.K., LaMere S.A., Torkamani A., Hart G.T., Kotsopoulos S., Warner J., Samuels M.L., Olson J., Head S.R., Ordoukhanian P. et al. Application of microdroplet PCR for large-scale targeted bisulfite sequencing. Genome Res. 2011; 21:1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyle P., Clement K., Gu H., Smith Z.D., Ziller M., Fostel J.L., Holmes L., Meldrim J., Kelley F., Gnirke A. et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 2012; 13:R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hodges E., Smith A.D., Kendall J., Xuan Z., Ravi K., Rooks M., Zhang M.Q., Ye K., Bhattacharjee A., Brizuela L. et al. High definition profiling of mammalian DNA methylation by array capture and single molecule bisulfite sequencing. Genome Res. 2009; 19:1593–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang X., Lu H., Wang J.-W., Xu L., Liu S., Sun J., Gao F.. High-throughput sequencing of methylated cytosine enriched by modification-dependent restriction endonuclease MspJI. BMC Genet. 2013; 14:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stevens M., Cheng J.B., Li D., Xie M., Hong C., Maire C.L., Ligon K.L., Hirst M., Marra M.A., Costello J.F. et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res. 2013; 23:1541–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kurdyukov S., Bullock M.. DNA methylation analysis: choosing the right method. Biology (Basel). 2016; 5:E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzuki M., Greally J.M.. Genome-wide DNA methylation analysis using massively parallel sequencing technologies. Semin. Hematol. 2013; 50:70–77. [DOI] [PubMed] [Google Scholar]
- 28. Bystrykh L.V. A combinatorial approach to the restriction of a mouse genome. BMC Res. Notes. 2013; 6:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meissner A., Gnirke A., Bell G.W., Ramsahoye B., Lander E.S., Jaenisch R.. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005; 33:5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tanas A.S., Borisova M.E., Kuznetsova E.B., Rudenko V.V., Karandasheva K.O., Nemtsova M.V., Izhevskaya V.L., Simonova O.A., Larin S.S., Zaletaev D.V. et al. Rapid and affordable genome-wide bisulfite DNA sequencing by XmaI-reduced representation bisulfite sequencing. Epigenomics. 2017; doi:10.2217/epi-2017-0031. [DOI] [PubMed] [Google Scholar]
- 31. Lim Y.C., Chia S.Y., Jin S., Han W., Ding C., Sun L.. Dynamic DNA methylation landscape defines brown and white cell specificity during adipogenesis. Mol. Metab. 2016; 5:1033–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meissner A., Mikkelsen T.S., Gu H., Wernig M., Hanna J., Sivachenko A., Zhang X., Bernstein B.E., Nusbaum C., Jaffe D.B. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008; 454:766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stubbs T.M., Bonder M.J., Stark A.-K., Krueger F. BI Ageing Clock Team . BI Ageing Clock Team von Meyenn F., Stegle O., Reik W.. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017; 18:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith Z.D., Gu H., Bock C., Gnirke A., Meissner A.. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009; 48:226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hanna C.W., Peñaherrera M.S., Saadeh H., Andrews S., McFadden D.E., Kelsey G., Robinson W.P.. Pervasive polymorphic imprinted methylation in the human placenta. Genome Res. 2016; 26:756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Milagre I., Stubbs T.M., King M.R., Spindel J., Santos F., Krueger F., Bachman M., Segonds-Pichon A., Balasubramanian S., Andrews S.R. et al. Gender differences in global but not targeted demethylation in iPSC reprogramming. Cell Rep. 2017; 18:1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kawakatsu T., Huang S.-S.C., Jupe F., Sasaki E., Schmitz R.J., Urich M.A., Castanon R., Nery J.R., Barragan C., He Y. et al. Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell. 2016; 166:492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maurano M.T., Wang H., John S., Shafer A., Canfield T., Lee K., Stamatoyannopoulos J.A.. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 2015; 12:1184–1195. [DOI] [PubMed] [Google Scholar]
- 40. Lev Maor G., Yearim A., Ast G.. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015; 31:274–280. [DOI] [PubMed] [Google Scholar]
- 41. Domcke S., Bardet A.F., Adrian Ginno P., Hartl D., Burger L., Schübeler D.. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature. 2015; 528:575–579. [DOI] [PubMed] [Google Scholar]
- 42. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015; 43:D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE–restriction enzymes and DNA methyltransferases. Nucleic Acids Res. 2005; 33:D230–D232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bock C., Walter J., Paulsen M., Lengauer T.. CpG island mapping by epigenome prediction. PLoS Comput. Biol. 2007; 3:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang W., Spector T.D., Deloukas P., Bell J.T., Engelhardt B.E.. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome Biol. 2015; 16:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taher L., Smith R.P., Kim M.J., Ahituv N., Ovcharenko I.. Sequence signatures extracted from proximal promoters can be used to predict distal enhancers. Genome Biol. 2013; 14:R117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dale R.K., Pedersen B.S., Quinlan A.R.. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics. 2011; 27:3423–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cock P.J.A., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009; 25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin-Herranz D.E., Ribeiro A.J.M., Stubbs T.M.. demh/cuRRBS: cuRRBS v1.0.4. 2017; doi:10.5281/zenodo.853452.
- 52. Kuhn R.M., Haussler D., Kent W.J.. The UCSC genome browser and associated tools. Brief. Bioinformatics. 2013; 14:144–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mathelier A., Fornes O., Arenillas D.J., Chen C.-Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. et al. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016; 44:D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bardet A.F., Steinmann J., Bafna S., Knoblich J.A., Zeitlinger J., Stark A.. Identification of transcription factor binding sites from ChIP-seq data at high resolution. Bioinformatics. 2013; 29:2705–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Krueger F., Andrews S.R.. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011; 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang J., Xia Y., Li L., Gong D., Yao Y., Luo H., Lu H., Yi N., Wu H., Zhang X. et al. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. BMC Genomics. 2013; 14:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martinez-Arguelles D.B., Lee S., Papadopoulos V.. In silico analysis identifies novel restriction enzyme combinations that expand reduced representation bisulfite sequencing CpG coverage. BMC Res. Notes. 2014; 7:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stroud H., Do T., Du J., Zhong X., Feng S., Johnson L., Patel D.J., Jacobsen S.E.. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014; 21:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun Y., Hou R., Fu X., Sun C., Wang S., Wang C., Li N., Zhang L., Bao Z.. Genome-wide analysis of DNA methylation in five tissues of Zhikong scallop, Chlamys farreri. PLoS ONE. 2014; 9:e86232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Angermueller C., Lee H.J., Reik W., Stegle O.. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017; 18:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Davey J.W., Davey J.L., Blaxter M.L., Blaxter M.W.. RADSeq: next-generation population genetics. Brief. Funct. Genomics. 2010; 9:416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davey J.W., Hohenlohe P.A., Etter P.D., Boone J.Q., Catchen J.M., Blaxter M.L.. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Publishing Group. 2011; 12:499–510. [DOI] [PubMed] [Google Scholar]
- 63. Naumova N., Smith E.M., Zhan Y., Dekker J.. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods. 2012; 58:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dekker J., Marti-Renom M.A., Mirny L.A.. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature Publishing Group. 2013; 14:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
cuRRBS and its documentation are freely distributed under GNU General Public License v3.0 and can be accessed via GitHub (51). The public RRBS data used in this manuscript can be found under GEO accession numbers GSE74126 (XmaI-RRBS) and GSE80961 (MspI&TaqαI-RRBS) respectively.