

Abstract
In recent years, a growing body of evidence has recognized the tandem repeat sequences, and specifically satellite DNA, as a functional class of sequences in the genomic “dark matter.” Using an original, complementary, and thus an eclectic experimental design, we show that the cat archetypal satellite DNA sequence, FA-SAT, is “frozen” conservatively in several Bilateria genomes. We found different genomic FA-SAT architectures, and the interspersion pattern was conserved. In Carnivora genomes, the FA-SAT-related sequences are also amplified, with the predominance of a specific FA-SAT variant, at the heterochromatic regions. We inspected the cat genome project to locate FA-SAT array flanking regions and revealed an intensive intermingling with transposable elements. Our results also show that FA-SAT-related sequences are transcribed and that the most abundant FA-SAT variant is not always the most transcribed. We thus conclude that the DNA sequences of FA-SAT and their transcripts are “frozen” in these genomes. Future work is needed to disclose any putative function that these sequences may play in these genomes.
Keywords: FA-SAT, satellite DNA, Bilateria genomes, satellite DNA transcription
Introduction
The repeated sequences named satellite DNA (satDNAs) are harbored in the heterochromatin of eukaryotic genomes, specifically at the centromeres, pericentromeres, and subtelomeric regions, but they can also be found at interstitial locations (Henikoff and Dalal 2005; Plohl et al. 2012). Traditionally, these sequences are highly abundant in genomes; the monomeric units are organized in tandem in long arrays. As a general feature, satDNAs are not well conserved, and some satDNA families are species specific. Unequal crossing-over events are responsible for differences in the monomer length, nucleotide sequence, complexity (e.g., repeats of some satDNAs are organized in high-order repeat units), and copy number. The models that best explain these hallmarks of the concerted evolution of satDNAs are the library (Fry and Salser 1977; Plohl et al. 2008) and variant library models (Cesari et al. 2003; Plohl et al. 2012), in which a specific satDNA and/or monomer variant can be shared by related species but may also experience amplification events resulting in a higher abundance for some species. Nonetheless, some satDNA sequences, designated as “frozen” satDNAs, stubbornly persist in the genomes over long evolutionary times, even in the form of low-copy number repeats, most likely because the concerted evolution of these sequences is influenced by selective constraints (Mravinac et al. 2002, 2005; Biscotti et al. 2015; Petraccioli et al. 2015).
The fact that genomes are doomed to repeat satellite DNA sequences has raised an important question that remains to be answered: what is their function? Several studies have reported important roles for satDNAs in the centromere structure, kinetochore assembly, chromosome pairing, and segregation (Biscotti et al. 2015; Ferreira et al. 2015). The evolution of satDNAs was also linked with the emergence of new species and genomes by its association with reproductive isolation and karyotype evolution, respectively (Adega et al. 2009; Chaves et al. 2012; Biscotti et al. 2015; Vieira-da-Silva et al. 2015). Finally, an increasing number of studies over the past 15 years refute the dogma that satDNAs residing in the heterochromatin are transcriptionally inert, providing evidence that satDNA transcripts or satellite noncoding RNAs (satncRNAs) can be found in different species of vertebrates, invertebrates, and plants (Ugarkovic 2005). These satDNA transcripts have been associated with heterochromatin maintenance, centromere function, kinetochore assembly, and gene expression regulation in several biological contexts (reviewed in Pezer et al. 2012; Biscotti et al. 2015) and in diseases such as cancer (Ferreira et al. 2015).
FA-SAT, the major cat satellite DNA, was first characterized by Fanning (1987) as having a monomeric unit of 483 bp and a G+C content of 64% (Fanning 1987). FA-SAT represents 2% of the cat genome, with >100,000 copies of the monomeric unit (Pontius and O’Brien 2009). According to Pontius and O’Brien (2009) the high G+C content is responsible for the low intrasequence variability (∼90% similarity among variant repeats; Tamazian et al. 2014) observed in this satDNA family. FISH mapping reveals FA-SAT localization primarily at the telomeres and also at the centromeres of the cat chromosomes; in chromosomes A1, A2, B1, B4, C1, C2, and D3, FA-SAT was not detected (Santos et al. 2004, 2006). This satDNA family can also be found in other Carnivora species (Fanning 1987; Modi et al. 1988; Zou and Li 2011) and in other mammals (i.e., mink, raccoon, rabbit, bat, African green monkey, mouse, and guinea pig) where it seems to be scattered throughout the genome and not organized in tandem arrays as in the cat (Fanning 1987).
In this work, we inspect the satDNA sequences itself, and also its physical organization in different genomes. The concept of what is a satDNA is being challenged with new discoveries, namely its localization in nonheterochromatic classic regions (e.g., Ruiz-Ruano et al. 2016, Garrido-Ramos 2017 for a review). We will consider in this work the most accepted definition that a satDNA is a tandem repeated sequence (with several units of the monomeric sequence), which in turn, can be physical organized in the genomes in a tandem of arrays (generally at heterochromatic regions, highly repeated) and/or in an interspersed fashion (these ones can be short tandem arrays). We found that FA-SAT-related sequences are present in the genomes of several Bilateria species (and not only in mammals) and have low intra- and intersequence variability, being the oldest satDNA sequence reported. The CpG island in the FA-SAT monomer has also been conserved throughout evolution in terms of sequence similarity and methylation status. We also show that FA-SAT monomer was preferentially amplified in Carnivora genomes (i.e., cat and genet), with the predominance of a specific variant, giving rise to tandem arrays of this satDNA in these genomes at the heterochromatic regions. In contrast, in non-Carnivora genomes, the FA-SAT-related tandem sequences are present with high-sequence identity but are physical organized in an interspersed fashion. The cat genome inspection at the FA-SAT array flanking regions revealed an intensive intermingling with transposable elements, which most likely explains the FA-SAT intragenomic movements and the interspersion of its sequences in this genome. Our data suggest that the evolutionary history of FA-SAT included fluctuations in copy number, particularly at the heterochromatic regions (i.e., in genet at the centromeres and in cat at the telomeres) and hypothetical independent transpositions relocating the FA-SAT in interspersion patterns. Finally, our results reveal that FA-SAT is transcribed in these Bilateria genomes. In genet, most of the FA-SAT-related sequences were naturally relocated to the centromere, and the FA-SAT transcripts do not seem to be preferentially transcribed from there. However, the widespread presence of sequences throughout the genomes most likely ensures the necessary transcription.
Materials and Methods
Cell Culture, Chromosome Preparations, Genomic DNA, and RNA Isolation
In this work, the following Bilateria species genomes were used: FCA (Felis catus), GGE (Genetta genetta), HSA (Homo sapiens), ATR (Aotus trivirgatus), MMU (Macaca mulatta), BTA (Bos taurus), SSC (Sus scrofa), RNO (Rattus norvegicus), PER (Peromyscus eremicus), DVI (Didelphis virginiana), PTR (Potorous tridactylus), PGL (Prionace glauca), DME (Drosophila melanogaster), and CGI (Crassostrea gigas). For FCA, GGE, BTA, RNO, and PER, primary cell lines were established by our group; for HSA, ATR, MMU, DVI, and PTR, commercial cell lines WS1 (ATCCCRL-1502TM), OMK (ATCCCRL-1556TM), DBS-FRhL-2 (ATCCCL-160TM), OK (ATCCCRL-1840TM), and PtK1 (ATCCCRL-6493TM) were acquired, respectively. Finally, tissue was used to obtain biological material from SSC, PGL, DME, and CGI genomes. Primary cell lines were established from samples that were collected in accordance with EU Directive 2010/63/EU and approved by the “Universidade de Trás-os-Montes e Alto Douro” (approval references POCTI/C_BIA/11285/98, POCI/CVT/62940/04, POCI/BIA_BCM/58541/04). Cell lines were maintained in DMEM supplemented with 13% AmnioMax C-100 Basal Medium, 2% AmnioMax C-100 supplement, 10% FBS, 100 U/ml and 100 µg/ml of Penicillin/Streptomycin antibiotic mixture, and 200 mM l-glutamine (all from Gibco, Thermo Fisher Scientific). Chromosome harvesting and metaphase preparations were accomplished by routine procedures. Genomic DNA isolation was performed using Quick-Gene DNA Tissue Kit S (Fujifilm Life Science) according to the manufacturer’s instructions. RNA was isolated using the mirVana Isolation Kit (Ambion, Thermo Fisher Scientific) following the manufacturer’s recommendations. RNA was purified using the TURBO DNA-free Kit (Ambion, Thermo Fisher Scientific). However, it was not possible to obtain all the biological material (i.e., RNA and chromosomes) from all species (cf. fig. results).
FA-SAT Isolation, Cloning, Sequencing, and Analysis
FA-SAT-related sequences were isolated from the genomes of the species being analysed by PCR amplification of genomic DNA previously obtained from these genomes and using specific primers (supplementary table S1, Supplementary Material online) designed using the web-based interface Primer3 (Rozen and Skaletsky 2000). Briefly, the reaction was performed as follows: one denaturation cycle at 94 °C (5 min); 30 cycles of 94 °C denaturation (1 min), 59 °C annealing (45 s), and 72 °C extension (45 s); and one final extension cycle at 72 °C (10 min). For cloning, PCR amplicons were prepared using the Fast DNA End Repair (Thermo Scientific) to blunt and phosphorylate the DNA ends, which were subsequently ligated into the SmaI site of the pUC19 vector (Thermo Scientific) with T4 DNA ligase (Thermo Scientific). Transformation was performed in DH5α competent cells (Invitrogen, Thermo Fisher Scientific). β-galactosidase blue-white alpha complementation was used to select positive clones, which were subsequently prepared for sequencing. Sequence analysis was performed using BLAST (Basic Local Alignment Search Tool from Geneious was performed in the nucleotide database provided by NCBI). Several search parameters were changed for the analysis of repetitive DNA: max_target_seqs and num_descriptions were set to 10,000, and e-value and word_size were set to 10–16 and 11, respectively. All other search parameters were set to default values. Multiple alignments were obtained using CLUSTALW cost matrix on Geneious R9 version 9.1.2 (Biomatters) with parameters set to default values.
FA-SAT In Silico Analysis in Felis catus Genome Sequencing Project
FCA chromosome sequences (felis_catus_8.0; GenBank, assembly accession: GCA_000181335.3) were obtained in FASTA format from NCBI. The FA-SAT (GenBank, sequence accession number: X06372.1) in silico search on FCA chromosomes was performed using BLAST (Basic Local Alignment Search Tool from Geneious was performed in the nucleotide database provided by NCBI). The search parameters applied were the same as above (previous section). BLAST hits with scores <90 were not considered (supplementary table S2, Supplementary Material online). BLAST hits were analysed and in silico mapped onto FCA chromosomes using Geneious R9 version 9.1.2 (Biomatters). Monomer-flanking regions were screened for the presence of repetitive elements in the Eukaryota Repbase using the CENSOR software (Kohany et al. 2006).
Bisulfite Conversion and Sequencing
For sodium bisulfite conversion, the Cells-to-CpG Bisulfite Conversion Kit (Applied Biosystems, Thermo Fisher Scientific) was used according to the manufacturer’s indications. Then, the converted DNA was used for the amplification of the converted CpG-rich region of the FA-SAT sequence by a PCR reaction using BSP primers designed using the web-based interface MethPrimer (Li and Dahiya 2002) (supplementary table S1, Supplementary Material online). Following amplification, the fragment was purified from the agarose gel using the QIAquick PCR purification kit (Qiagen) and then cloned into vector pUC19/SmaI (Thermo Scientific) for sequencing. A minimum of ten different clones per species were sequenced and analysed by MethylViewer software (Pardo et al. 2011).
Southern Blot
Genomic DNA was digested with the restriction enzymes BamHI, HpaII, and PstI and the resulting fragments were separated in a 1.2% agarose gel and blotted onto a Biodyne B Nylon Membrane (Thermo Fisher Scientific) according to the manufacturer’s indications. The membranes were probed with a FA-SAT clone previously labeled by PCR with digoxigenin-11-dUTP (Roche Biochemical reagents, Sigma–Aldrich). The procedure for nucleic acid hybridization and detection was performed using the North2South Chemiluminescent Hybridization and Detection Kit (Thermo Fisher Scientific) according to manufactures’ instructions.
Fluorescent In Situ Hybridization
Physical mapping of FA-SAT onto the chromosomes was performed by FISH using routine procedures (Heslop-Harrison and Schwarzacher 2011). The FA-SAT cloned sequence was labelled with digoxigenin-11-dUTP (Roche Biochemical reagents, Sigma–Aldrich) during PCR. The most stringent posthybridization wash was 50% formamide/2 × SSC at 42 °C. Digoxigenin-labelled probes were detected with antidigoxigenin-5′-TAMRA (Roche Biochemical reagents, Sigma–Aldrich). Chromosomes were imaged with a Zeiss Axio Imager Z2 microscope coupled to a JAI Progressive Scan (CV-M4+CL) digital camera and Cytovision software (Genus, version 4.5.2). Digitized photos were prepared using Adobe Photoshop (version 7.0). Image optimization included contrast and color adjustments and affected the whole image equally.
High-Resolution Melting Assays
Genomic DNA and DNA from FA-SAT clones, both of high purity, were analysed by High-Resolution Melting Assays (HRM) with MeltDoctor HRM Master Mix, which uses the SYTO9 dye (Applied Biosystems, Thermo Fisher Scientific), and with the 83-bp B and 83-bp C primers (supplementary table S1, Supplementary Material online). All the PCR mixtures were done according to the recommendations of the manufacturer. Experiments were performed in the StepOne real-time PCR system (Applied Biosystems, Thermo Fisher Scientific) with the next program: initial denaturation at 95 °C (10 min), 40 cycles of 95 °C for 15 s, followed by 59 °C for 1 min. Subsequently, a melt curve was performed with a denaturation step of 95 °C for 15 s, an annealing of 59 °C for 1 min and a high-resolution melting of 95 °C for 15 s with a continuous ramp rate of 0.3%, followed by a final step of annealing of 60 °C for 15 s. All reactions were performed in triplicate, and negative controls (without template) were also included on the plate. The StepOne software (version 2.2.2, Applied Biosystems, Thermo Fisher Scientific) was used for data acquisition and the High Resolution Melt software (version 3.0.1, Applied Biosystems, Thermo Fisher Scientific) for data analysis (we used the standard software parameters suggested by the manufacturer).
FA-SAT DNA Copy Number Absolute Quantification
For FA-SAT copy number absolute quantification, we used the standard curve method. The PCR primers for each FA-SAT variant are described in supplementary table S1 in the Supplementary Material online. A 10-fold serial dilution series of the different clones (plasmid DNA with the FA-SAT insert variants) ranging from 1×109 to 1×105 copies was used to obtain a standard curve (5-point serial dilutions) (supplementary table S3, Supplementary Material online). The concentration of the recombinant plasmid was obtained using the NanoDrop ND-1000 (NanoDrop Technologies), and the plasmid copy number was calculated using the following equation: DNA (copy number)=[6.022 × 1023 (copy number/mol)×DNA amount (g)]/[DNA length (bp)×660 (g/mol/bp)], which incorporates Avogadro’s number of 6.022 × 1023 (copy number)/1 mol and the average molecular weight of a double-stranded DNA molecule of 660 g/mol/bp. In this formula, the DNA length is 3,101 bp (plasmid pUC19 is 2,686 bp plus the insert of 415 bp). CT values in each dilution were measured using real-time qPCR to create the different standard curves for FA-SAT variants B and C. The standard curve contains a plot of the CT values versus the log concentration of FA-SAT. The copy number determination of the unknown total DNA sample was obtained by interpolating its CT value against the standard curve. For absolute quantification, the mass of the haploid genome species (not yet sequenced) was obtained from the Genome Size database (Gregory 2017). Real-time qPCR reactions were performed with MeltDoctor HRM Master Mix (Applied Biosystems, Thermo Fisher Scientific), according to the recommendations of the manufacturer. This experiment was performed in the StepOne real-time PCR system (Applied Biosystems, Thermo Fisher Scientific) with the PCR program: an initial denaturation at 95 °C for 10 min, 40 cycles of 95 °C for 15 s, followed by 59 °C for 1 min. Subsequently, a melt curve was performed to evaluate the primer specificity. All reactions were performed in triplicate, and negative controls (without template) were also included on the plate. The StepOne software (version 2.2.2, Applied Biosystems, Thermo Fisher Scientific) was used to create the standard curve and for data analysis. Standard curves with the following parameters were considered to be adequate: R2>0.99, slopes between −3.1 and −3.6, and reaction efficiencies between 90% and 110%. The absolute quantification was transformed into fold-change values using the standard curve equation and always compared with a reference sample.
FA-SAT Sequence Tandem Analysis
FA-SAT tandem analysis in all the genomes was proved by inverted PCR amplification of genomic DNA previously obtained from these genomes using divergent primers (supplementary table S1, Supplementary Material online). This analysis is extremely important to validate this sequence as a satDNA (cf. introduction). Briefly, the reaction was performed as follows: one denaturation cycle at 94 °C (10 min); 30 cycles of 94 °C denaturation (1 min), 59 °C annealing (45 s), and 72 °C extension (45 s); and one final extension cycle at 72 °C (10 min). The obtained amplicons were visualized in an electrophoresis agarose gel, being the 100-bp amplicons excised, reamplified, and sequenced.
FA-SAT RNA Expression Analysis by Real-Time RT-qPCR
Expression experiments with total RNA from Bilateria species were performed using the TaqMan RNA-to-CT 1-Step Kit (Applied Biosystems, Thermo Fisher Scientific). The TaqMan Gene Expression Assay Mixes (primer/probe sets) used were glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Rn01749022_g1) as reference gene and FA-SAT TaqMan assay as target (cf. supplementary table S1, Supplementary Material online, for sequences of primers and probe) (all assays were from Applied Biosystems, Thermo Fisher Scientific). The FA-SAT TaqMan assay was designed in the Primer Express software for Real-Time PCR (version 3.0, Applied Biosystems, Thermo Fisher Scientific) and ordered in the custom TaqMan assays. The PCR mixtures were prepared following the manufacturer’s recommendations and the PCR program used was an initial reverse transcriptase reaction at 48 °C for 15 min, a denaturation at 95 °C for 10 min, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. All reactions were performed in triplicate, and negative controls (without template) were also included. StepOne software (version 2.2.2, Applied Biosystems, Thermo Fisher Scientific) was applied for comparative analysis and the 2−ΔΔCT method (Livak and Schmittgen 2001) was used to calculate fold changes in the expression levels.
FA-SAT RNA Variant Analysis by Real-Time RT-qPCR
For FA-SAT RNA variant quantification, we used the standard curve method with the same dilution parameters and primers used for the standard curve described for copy number analysis. However, the Verso 1-Step RT-qPCR kit, SYBR Green, ROX (Thermo Scientific) was used (supplementary table S3, Supplementary Material online). The absolute quantification of FA-SAT RNA variants was obtained by interpolating its CT value against the standard curve. For the variant transcripts’ quantification, the Verso 1-Step RT-qPCR kit, SYBR Green, ROX (Thermo Scientific), was used with the recommendations of the manufacturer. The reactions were carried out in a 48-well optical plate (StepOne real-time PCR system, Applied Biosystems, Thermo Fisher Scientific) at 50 °C for 15 min and 95 °C for 15 min, followed by 40 cycles of 95 °C for 15 s, 59 °C for 45 s, and 72 °C for 1 min. Subsequently, a melt curve was performed to evaluate the primer specificity. All reactions were performed in triplicate, and negative controls (without DNA) were also included in the plate. The data were analysed using the same parameters and software previously described.
Statistics and Reproducibility
All data are presented as mean ± standard deviation. Statistical significance was determined using two-tailed Student’s t-test for the comparison between two independent samples and analysis of variance (ANOVA) tests when more than two groups were under analysis: ns P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Data Availability
The FA-SAT clone sequences from the different species have been deposited at the GenBank, being the all the accession information presented in supplementary table S4 in the Supplementary Material online.
Results
The FA-SAT Sequence Is Frozen in Several Bilateria Genomes
A BLAST search in NCBI revealed the FA-SAT sequence in human and Drosophila grimshawi (genomic and mRNA sequences) genomes and in uncharacterized ncRNAs (noncoding RNAs) from cat and Panthera pardus species; these sequences had high percentages of identity (supplementary fig. S1a and b, Supplementary Material online). Some of these sequences also exhibit dimeric units in tandem (supplementary fig. S1b, Supplementary Material online). Working on the premise that FA-SAT is present in nonmammalian species (Fanning already showed its presence in different mammalian genomes in 1987), we conceived an experiment to isolate this satDNA sequence by PCR using FA-SAT-specific primers (supplementary table S1, Supplementary Material online), and these amplified regions were then cloned and sequenced. We isolated FA-SAT-related sequences from 14 Bilateria species distributed in the following nine Orders (supplementary fig. S2 and table S4, Supplementary Material online): Carnivora (Felis catus, FCA; Genetta genetta, GGE), Primates (Homo sapiens, HSA; Aotus trivirgatus, ATR; Macaca mulatta, MMU), Rodentia (Rattus norvegicus, RNO; Peromyscus eremicus, PER), Cetartyodactyla (Bos taurus, BTA; Sus scrofa, SSC), Didelphimorphia (Didelphis virginiana, DVI), Diprotodontia (Potorous tridactylus, PTR), Diptera (Drosophila melanogaster, DME), Carcharhiniformes (Prionace glauca, PGL), and Ostreoida (Crassostrea gigas, CGI). This experiment revealed the presence of this FA-SAT sequence in all of these Bilateria genomes with low intra- and intersequence variability and an average 95% similarity among all FA-SAT-related sequences (see the distance matrix of pairwise alignments in fig. 1a and supplementary fig. S3, Supplementary Material online). Pontius and O’Brien (2009) infer that this low intrasequence variability among repeats in the cat genome is due to high G + C content. Our data revealed that the FA-SAT-related sequences cloned from the 14 genomes all have ∼63% G + C content. Because of this feature, it is possible to identify a CpG island in the FA-SAT monomer (fig. 1b), and thus, we decided to analyse the methylation status of these sequences in the Bilateria genomes by bisulfite sequencing (fig. 1c-d and supplementary fig. S4, Supplementary Material online). As observed in figure 1d, the methylation level of FA-SAT-related sequences for all genomes analysed is in the same range, specifically for the CpG sites in the CpG island (8 CpG sites). Furthermore, when we plot the bisulfite sequence similarities with the methylation percentages, we concluded that the similarities of all sequences are >78% (fig. 1c), which is in agreement with the low intra- and intersequence variability found in these FA-SAT-related sequences analysed. We then recognized that the CpG island in the FA-SAT monomer has been conserved throughout evolution in terms of sequence similarity and methylation status.
Fig. 1.

—FA-SAT-related sequence identity and methylation status profile (A) Distance matrix of pairwise alignments of FA-SAT clones from all of the studied species. The distances were calculated using the alignment algorithm CLUSTALW, and the matrix was generated by Geneious R9 version 9.1.2 (Biomatters) under default settings. Cells showing nucleotide identities of P98–100% (dark blue), 94–97.9% (medium blue), 90–93.9% (light blue), 88–89.9% (dark green), 84.1–87.9% (light green), 80–84% (yellow), 70–80% (orange), and <70% (red). The same matrices containing the identity values in the cells are shown in supplementary figure S3 in the Supplementary Material online. (B) CpG profile (CpG sites as red bars) of the FA-SAT monomer in red (GenBank, sequence accession number: X06372.1), highlighting the CpG island in blue. Also shown are the primers (supplementary table S1, Supplementary Material online, for sequence) used and their respective amplicon locations. These include primers for FA-SAT monomer isolation (FA-SAT, in purple), bisulfite sequence (BSP, in red), 83-bp variant B (in pink), and 83-bp variant C (in green). The scheme presented was adapted from the output of MethPrimer (Li and Dahiya 2002) for CpG island prediction (C) Graphical view showing the similarity and variation of the DNA methylation status of FA-SAT bisulfite-related sequence clones for FCA, GGE, HSA, ATR, RNO, PER, DME, and CGI genomes given by the methylation percentage of all the CpG sites in each BSP clone compared with the FA-SAT monomer BSP predicted sequence. (D) Graphical representation of the methylation percentages of the different FA-SAT-related bisulfite clones for the species indicated in (C) regarding the total 15 CpG sites analysed, the 8 CpG island sites, and the remaining 7 CpG sites.
FA-SAT-Related Sequences Are Amplified in Tandem of Arrays, in a Cluster-Type Cellular Organization, in Carnivora Genomes and Display an Interspersed Organization of Tandem Short Repeats in Other Genomes
Once the FA-SAT was identified in several genomes with extremely high-sequence conservation, we decided to investigate its genome organization to address questions about copy number variations and chromosome location with an effort to understand the evolution of this repeat.
Southern blot experiments (supplementary fig. S5, Supplementary Material online) corroborated our previous findings that all genomes analysed contain FA-SAT-related sequences. However, only FCA and GGE (Carnivore species) showed a tandem repeat organization, which is characteristic of an archetypal satellite DNA (although for the GGE species, the typical ladder pattern was only observed with the HpaII restriction enzyme) (supplementary fig. S5, Supplementary Material online). In all of the other genomes, the FA-SAT-related sequences seem to be physically organized in an interspersed fashion revealed by a smear pattern (supplementary fig. S5, Supplementary Material online). The physical location of FA-SAT in chromosomes produced specific cluster-type signals in the Carnivora chromosomes, a characteristic in situ hybridization pattern of a tandem sequence of arrays (found in satDNA) in which the FA-SAT seems to be amplified (fig. 2a). These data corroborate the Southern blot assays showing the FA-SAT tandem arrays only in these two genomes. Additionally, in these genomes, the signals are primarily located in different chromosome regions. In cat, the signal is primarily at the telomeres, as previously reported (Santos et al. 2004), and in genet, the signal resides primarily at the centromeres (but both chromosome regions are classic heterochromatic ones). This feature can be the result of the different karyotype evolution in these species, as suggested by other reports (Murphy et al. 2005; Adega et al. 2008; Paco et al. 2013; Louzada et al. 2015).
Fig. 2.

—FA-SAT DNA profile overview in different Bilateria genomes (A) Physical mapping of FA-SAT DNA by in situ hybridization (red) onto FCA and GGE chromosomes (blue). Scale bar represents 10 µm. (B) High-resolution melting (HRM) analysis of clones FCA_CL_I, FCA_CL_B, and FCA_CL_C and genomic DNA from FCA and respective FA-SAT 83-bp amplicons by agarose gel electrophoresis. (C) HRM analysis of FA-SAT 83-bp variants B and C in genomes of FCA, GGE, HSA ATR, MMU, BTA, SSC, RNO, PER, DVI, PTR, PGL, DME, and CGI. (D) FA-SAT copy number fold change of variants B and C in the different analysed genomes indicated, considering FCA as the reference genome. (E) Representativeness of each FA-SAT variant (B and C) in each of the different analysed genomes. Data are presented as mean ± SD of three replicates. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001 as determined by the Student’s t-test (two-tailed) when two samples were compared and by one-way ANOVA when more than two samples are analysed.
The information about the repetitive components of the genome, especially for satDNA, remains rather scarce, mostly because of the nature of these regions, that is, sequence complexity, the high level of polymorphisms, the repetitive essence of these sequences, the GC-rich or GC-poor content of some repeat families, the evolution of these sequences, which is usually extremely rapid for the sequence and/or the copy number (Ferreira et al. 2015), and their chromosomal locations. All of these sequence features make experimental design a major hurdle; limited tools are available to dissect genomes, and they are mainly focused on coding sequences. Moreover, because the assemblies do not cover regions of constitutive heterochromatin (where satDNA sequences traditionally reside) and the sequences contained in gene expression databases are largely purged of sequences of repetitive nature, the in silico and bioinformatics analysis of this type of sequences is quite challenging (Louzada et al. 2015; Sujiwattanarat et al. 2015; Ruiz-Ruano et al. 2016).
In an effort to develop new techniques to analyse these complex sequences, which are in fact an admixture of different sequences (i.e., variants) that coexist in a single genome and also in different genomes (in the present case, with high levels of sequence similarity, similar G + C content, and similar methylation status of the CpG island), we decided to apply high-resolution melting (HRM) dissociation profiles for the first time. Understanding the way that DNA denatures or melts is important for comprehending several DNA-dependent functions (e.g., single-stranded regions dynamically produced by its denaturation can control the binding of single-strand binding elements and thus influence functions such as transcription) (Liu et al. 2007). Our aim was to try to discriminate the different genomes in terms of their FA-SAT sequences (as a whole) and thus to understand the molecular behavior of the FA-SAT in each of these genomes. First, we considered the two most dissimilar sequence variants, one with a similarity of ∼100% and the other with ∼80% similarity with the FA-SAT monomer (GenBank sequence accession number: X06372.1), corresponding to FA-SAT FCA clones B and C and designed specific primers for these variants (supplementary table S1, Supplementary Material online). In each variant, one 83-bp fragment of the FA-SAT monomer sequence was analysed.
We conducted our first experiment with representative FA-SAT FCA clones (with a single FA-SAT molecule) and with the FCA reference genome with primers for variant B (fig. 2b and supplementary fig. S6a, Supplementary Material online, only the most distinct HRM clone curves are shown). Both the HRM dissociation profiles (fig. 2b) and the data from the melting temperatures (supplementary table S5, Supplementary Material online) showed that it is possible to discriminate FA-SAT FCA clones by their sequence variability. For example, FCA_CloneC, which presents only 82% similarity with the FA-SAT monomer, also has the lowest Tm value (80.80 ± 0.21 °C). FCA_CloneI displays the most dissimilar dissociation curve and also a higher Tm value (88.53 ± 0.06 °C). In fact, this clone is a dimer with two monomers of 97% and 96% similarity. These data are exciting, as they suggest that it is possible to detect the organization of monomer repeats in a genome by the HRM assay. In this case, the assay revealed a dimeric nature, indicative of a conspicuous tandem pattern. This feature can be corroborated by the agarose gel of the HRM amplifications (fig. 2b) in which both FCA_CloneI and the FCA samples demonstrate a tandem pattern resulting from the FA-SAT monomer amplification of ∼500 bp (FA-SAT monomer has 483 bp as specified in GenBank, sequence accession number: X06372.1).
Then, we decided to compare the FA-SAT 83-bp dissociation profiles for both variants (B and C) in all analysed genomes (fig. 2c and supplementary fig. S6b and c and table S6, Supplementary Material online, for the Tm values). This experiment clearly demonstrated that FCA shows a unique HRM profile for variant B, most likely due to its dimeric nature and tandem pattern (cf. previous paragraph, fig. 2b and supplementary table S5, Supplementary Material online). It is also clear from the shape of the HRM curves that the HRM dissociation profiles for variant C are more similar among all genomes compared with the HRM profiles for variant B (fig. 2c, supplementary table S6 and fig. S6b and c, Supplementary Material online). From the HRM data, we concluded the following: genomes are admixtures of at least two FA-SAT variants (B and C); FA-SAT variant C is more similar among all analysed genomes, but both variants still coexist in all species; and FA-SAT variant B seems to be essentially responsible for the dimeric nature in the FCA genome (suggestive of a prominent genome tandem organization; these data are also supported by Southern blot and FISH experiments).
Once in the GGE and FCA species, the FA-SAT seems to be amplified, unlike in the other genomes (cf. data from FISH analysis), we decided to evaluate the copy number of the different variants B and C (fig. 2d and supplementary tables S7 and S8, Supplementary Material online) to determine whether there is a different abundance of the two variants in each genome. As observed for the 83-bp amplicons (fig. 2e and supplementary fig. S7, Supplementary Material online), variant B is more abundant in the Carnivora genomes, where these sequences seem to be preferentially amplified during genome evolution. However, in all of the other genomes (where FA-SAT orthologous sequences do not seem to be amplified), there is not a clear variant distribution configuration that could be correlated with the sequence evolution mode. In primates, pig, cactus mouse, long-nosed potoroo, blue shark, common fruit fly, and pacific oyster species, variant B seems to be the most abundant; in all of the other genomes, FA-SAT variant C is the most represented. However, it is interesting that with genomes being admixtures of different variants (in the present work, the two most dissimilar in terms of sequence identity were analysed), in the genomes with FA-SAT amplified (Carnivora spp.), the discrepancy between the copy number values of the two variants (i.e., B and C) is exacerbated. Considering all of the data presented thus far, and by parsimony, it is possible to propose that FA-SAT is present in all of these Bilateria genomes and was preferentially amplified in FCA and GGE with a predominance of variant B giving rise to a physical organization of tandem arrays of this satDNA in these two genomes. Finally, and once PCR demonstrated to be a more sensitive method to analyse satellite DNA sequences in genomes where these sequences are represented in low copy numbers (and did not show to be amplified), we decided to use divergent primers to verify if, even in genomes that only display a FA-SAT interspersed organization, these sequences are in fact tandem repeated and hence are satDNAs. In supplementary figure S8 in the Supplementary Material online, we show a scheme that demonstrate the amplicons obtained with these primers. In all genomes analysed, at least, a dimeric nature of this sequence was evidence with the amplification of a 100-bp amplicon. However, in genomes where the sequence is amplified (i.e., Carnivora genomes), the prominent tandem genome organization, specifically observed in arrays clustered at heterochromatic regions, is easily detected by hybridization methods (i.e., Southern blot and FISH analysis). In fact, these data prove the satellite DNA nature of this sequence in all the analysed genomes.
In summary, the evolutionary history of FA-SAT appears to have occurred through fluctuations in copy number with orthologous FA-SAT sequences. These FA-SAT-related sequences are thus present in non-Carnivora genomes with a high-sequence identity and are organized in an interspersed mode. Molecular mechanisms such as unequal crossing-over and rolling circle can explain the FA-SAT amplification proposed in Carnivora genomes, where FA-SATs are organized in tandem arrays (more prominently in the cat genome); specifically the formation and migration of extrachromosomal circular DNA, can explain the expansion of these repeats to multiple chromosomes. The FISH analysis revealed that this FA-SAT amplification (with a remarkable prevalence for variant B) is most noticeable at heterochromatic locations; in cat, this is primarily at the telomeres, and in genet, it is primarily at the centromeres.
FA-SAT Sequence Can Be In Silico Mapped in Regions of the Cat Genome Other than Constitutive Heterochromatin, Revealing an Intensive Intermingling of FA-SAT with Transposable Elements
To obtain further insights into the organization of FA-SAT in the genomes, we decided to perform an in silico analysis of this sequence in the genome where this satellite DNA is highly represented and for which sufficient sequence data are available—that is, the cat genome. The cat sequencing project is currently a good representation of the genome; however, as mentioned in the previous section, and as is happening with other genomes, the heterochromatin fraction is not covered by the great majority of the assemblies (Komissarov et al. 2011; Sujiwattanarat et al. 2015; Ruiz-Ruano et al. 2016). Complementary approaches such as FISH and sequencing data can minimize these issues in mapping and annotating these repetitive sequences. FISH analysis allowed us to map FA-SAT-related sequences in the heterochromatin fraction of the genomes where FA-SAT is amplified (FCA and GGE). The in silico analysis of the sequence data from the cat genome project (felis_catus_8.0 assembly, GenBank, assembly accession: GCA_000181335.3) allowed further inspection of the FA-SAT sequence features in others regions of the genome. A BLASTN search on cat chromosomes revealed ∼3,178 BLAST hits (with score > 90), distributed on all autosomes and on the X chromosome (supplementary fig. S9a, Supplementary Material online). The F1 chromosome showed the highest percentage of this satDNA (∼35% of the BLAST hits) (supplementary fig. S9b, Supplementary Material online). The diagram presented in figure 3a is based on the karyotype scale provided by the Ensembl genome browser. Comparing the FISH data with the in silico mapping of FA-SAT on cat chromosomes, it is possible to make two main conclusions: 1) the FISH mapping essentially identifies the localization of FA-SAT at the heterochromatic regions, where this sequence is represented in a great number of copies and is organized in tandem of arrays; and 2) the in silico sequence mapping reveals other chromosome regions where FA-SAT is not so heavily repeated. Heterochromatin regions are the natural locations of satellite DNA sequences; however, these sequences can commonly be found in an interspersed fashion in the genomes, most likely as a result of intragenomic movements (Adega et al. 2009). With this premise in mind, we analysed the FA-SAT monomer/arrays-flanking regions in all chromosomes to find sequences capable of causing these genome movements. In figure 3b, it is possible to inspect a representative image of chromosome F1, which shows all possibilities regarding the sequences that flank FA-SAT monomer/arrays. In this specific chromosome, we found subtelomeric arrays of FA-SAT at the beginning of the chromosome. As observed in interstitial and subtelomeric regions, the sequences that most often flank FA-SAT monomer/arrays are mobile elements. It is also possible to find FA-SAT monomer/arrays in intergenic regions. The example from figure 3b shows two genes, a member of the RAS association domain family (RASSF5) and a kinase (IKBKE); interestingly, both genes operate in cancer pathways. Moreover, the examination of the annotation analysis in all monomers/arrays of FA-SAT and in all cat chromosomes revealed that the sequences that most often flank (in both sides) the FA-SAT monomer/arrays are NonLTR/L1, followed by NonLTR sequences and DNA transposons (fig. 3c). This analysis revealed an intensive intermingling of FA-SAT and transposable elements. This finding is in agreement with the premise that FA-SAT underwent intragenomic movements, suggesting that these sequences, specifically the L1 family, which in mammals retains the capacity to actively transpose material throughout the genome, could be responsible for genome evolution. This association pattern can also be found in other genomes (Satovic et al. 2016; Khost et al. 2017); a limited number of examples has been reported, but the topic was recently reviewed (Meštrović et al. 2015). In their article, Meštrović et al. (2015) also suggested that tandem repeat-incorporating transposable elements could represent a distinct category of multifunctional modular elements in which both types of repetitive sequences could act in synergy. In this respect, functional studies and evidence of the functionality of the sequences are mandatory to validate this hypothesis.
Fig. 3.
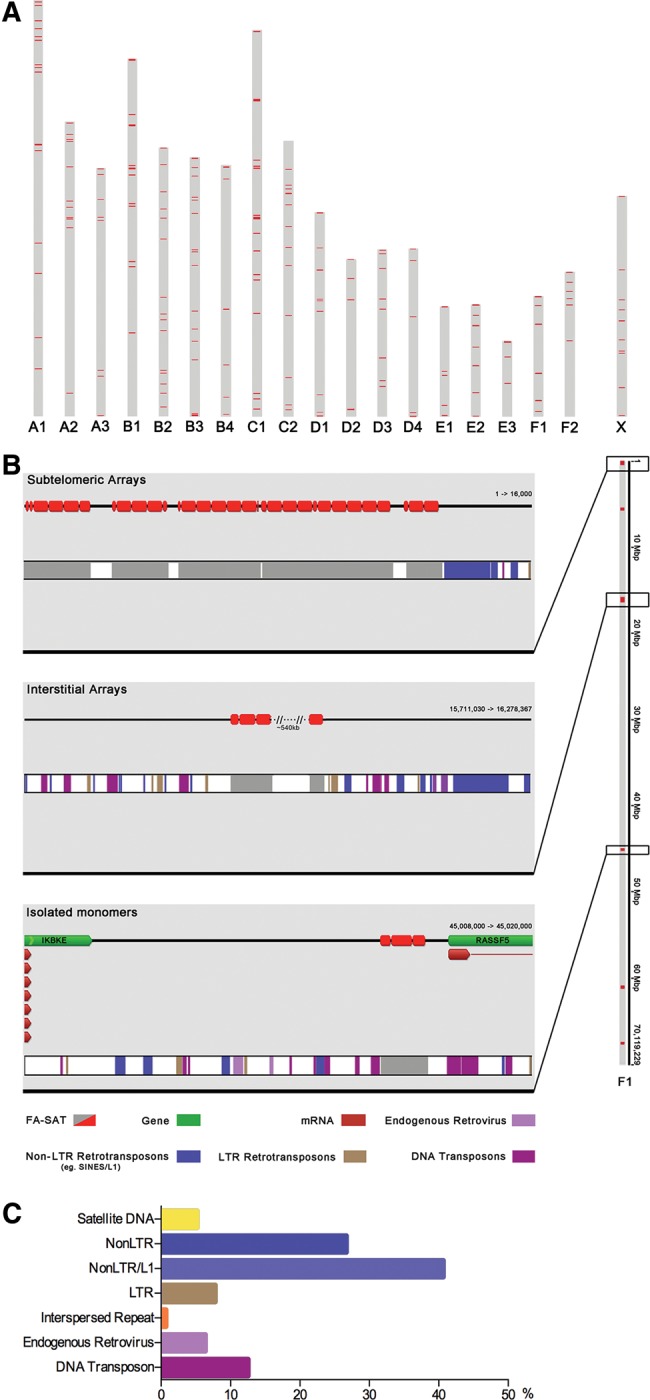
—In silico mapping of FA-SAT on the cat genome and flanking regions analysis of FA-SAT arrays (A) FA-SAT BLASTN hits in silico mapping onto FCA chromosomes based on the ideogram provided by the Ensembl genome browser. (B) Representative image of FCA chromosome F1 showing the annotation of FA-SAT BLAST hits (in red) on the top line (Geneious) and (in gray) on the bottom bar (Repbase) and flanking sequences of FA-SAT (when FA-SAT arrays are subtelomeric and interstitial or isolated monomers). (C) Quantification of the different classes of repetitive sequences flanking the FA-SAT arrays/isolated monomers along the FCA genome.
FA-SAT Is Also Transcriptionally Preserved in Bilateria Genomes
According to Palazzo and Gregory (2014), the functionality of a particular sequence in a genome can be anticipated by two main criteria: sequence conservation and sequence transcription. Although, these are far from being consensual. The conservation of the FA-SAT sequence was fully validated in the previous sections of this work. The obvious question follows: if FA-SAT is being conserved in all of these Bilateria genomes, then is it transcriptionally active? In an attempt to address this question, we first designed a specific Taqman assay (supplementary table S1, Supplementary Material online) to verify the transcription of FA-SAT in the genomes analysed. In figure 4a (and supplementary table S9, Supplementary Material online), it is possible to observe that in all genomes, the FA-SAT-related sequences are transcribed into FA-SAT ncRNA, with a higher quantity of transcripts in the genomes in which the sequence is amplified (i.e., FCA and GGE). Then, we decided for these Carnivora species to evaluate whether both variants (B and C) were transcribed. We demonstrated in a previous section that these genomes were enriched in variant B. To accomplish this goal, we used the same primers for both variants (B and C) of the 83-bp amplicon in the RNA for both species (fig. 4b and supplementary table S10, Supplementary Material online). As expected, in FCA, variant B is the most transcribed compared with variant C. However, and remarkably, in GGE, variant C seems to be preferentially transcribed, even though this variant displayed a lower number of copies within the genome. One of the putative explanations for the differential FA-SAT transcription in these two genomes could be the different FA-SAT chromosome localization. In fact, from both FISH data (fig. 2a) and copy number analysis (fig. 2d and e), the FA-SAT arrays are primarily located and amplified at the telomeres in FCA and on the centromeres in GGE. Although both chromosome regions are composed of constitutive heterochromatin with similar condensed chromatin compaction, their regulation in terms of transcription is quite different, reflecting both their function and sequence environment. For instance, in the centromere, the transcription of noncoding RNAs has been associated with certain cell stresses (Jolly et al. 2004; Valgardsdottir et al. 2008; Brown et al. 2012; Tilman et al. 2012), specific stages of the cell (e.g., senescence, heat-shock, differentiation) (Alexiadis et al. 2007; Enukashvily et al. 2007; Probst et al. 2010; Brown et al. 2012; Tilman et al. 2012; De Cecco et al. 2013; Enukashvily and Ponomartsev 2013; Plohl et al. 2014), and the cell cycle (Lu and Gilbert 2007; Bulut-Karslioglu et al. 2012). The noncoding sequences in telomeres have been associated primarily with the maintenance of telomeres (Montero et al. 2016). Until now, only one satDNA associated with telomeres was reported as being actively transcribed (PO41 in chicken) (Trofimova et al. 2015). This different location of FA-SAT arrays in the two carnivore genomes analysed linked to distinct functions and/or regulation of the telomeric and centromeric regions may explain the different expression of FA-SAT variants in both genomes; nonetheless, it seems that both variants (B and C) can perform their hypothetical tasks independently of FA-SAT amplification, that is, the number of variant copies (e.g., variant C in the GGE genome). Additionally, the FA-SAT monomer encloses telomeric motifs (complete and partial ones), which most likely reflects its primary origin in the telomeric region, where it is putatively and preferably transcribed, even when in low copy number. It is also important to note that between the cat and genet karyotypes, several chromosomal rearrangements occurred during evolution (cf. Perelman et al. 2012), explaining in part the transposition, translocation, or inversion of satDNA between the centromeric and telomeric regions. However, additional work is needed to ascertain from which genome region, or regions, this satDNA is being transcribed. Indeed, it was demonstrated from the Southern blot and the in silico analysis performed on the cat genome sequencing project that this sequence is also interspersed throughout the genome. Here, in principle, the chromatin architecture lacks the heterochromatic compaction leading to a more “open” chromatin state, which is more prone to transcription, and thus interspersed FA-SAT could be preferentially transcribed. In conclusion, this work raises several questions that need to be answered. Is FA-SAT transcribed independently of the genome region where it resides? If the sequence is transcribed from different regions of the genome, is its function the same? What is the function(s) of this satDNA and its transcripts in the genomes? In the present article, we show, for the first time, that when a satDNA sequence is naturally relocated to a new chromosome region (here, the centromere), it is not preferentially transcribed from there. However, the widespread distribution of these sequences within the genome ensures the necessary transcription, which may also suggests, according to Pallazzo and Gregory (2014) criteria referred in the beginning of this section, an important and conserved function of the FA-SAT ncRNAs.
Fig. 4.

—FA-SAT transcription in Bilateria genomes (A) Relative quantification of FA-SAT transcripts in different Bilateria genomes (FCA, GGE, HAS, ATR, BTA, PER, and DME). (B) FA-SAT transcription fold change of variants B and C in each of the Carnivora genomes, FCA and GGE, considering variant B as the reference. Data are presented as the means ± SD of three replicates. **P ≤ 0.01 ****P ≤ 0.0001 as determined by one-way ANOVA.
Discussion
In this work, we identified the presence of the major cat satellite DNA, the FA-SAT sequence, in the genomes of several Bilateria species (for a summary cf. supplementary fig. S10, Supplementary Material online), and not only in mammalian species as reported by Fanning (1987) making this the most ancient satDNA sequence ever reported. The FA-SAT-related sequences display a high level of conservation and are found in all of the analysed genomes in an interspersed genome architecture. Specifically, in the Carnivora genomes (FCA and GGE), these sequences are also amplified at heterochromatic regions, with a predominance of variant B. In FCA, FA-SAT predominates at the telomeres, whereas in GGE, it predominates at the centromeres. In fact, the relocation of these tandem repeats is responsible for these different genomic landscapes.
The next obvious question follows: How can tandem repeat sequences shuffle between different genome regions and maintain high levels of nucleotide conservation? Focusing first on the similarity of the sequences, it seems that both the high G + C content of FA-SAT and the similar methylation status of the FA-SAT CpG island can explain the low intra- and intersequence variability of FA-SAT monomers. However, it is surprising that the FA-SAT orthologous sequences, which are located at different chromosome regions of such divergent genomes, retain such high similarity. The concerted evolution of tandem repeats, such as satDNAs, can be generally explained by “burst and decay,” where there is a first-step of amplification of identical copies and a second-step where these copies can transpose and thus accumulate mutations (Maumus and Quesneville 2014). These repeat sequences do not seem to follow this general process because we could not find any informative dissimilarity of the FA-SAT-related sequences in the genomes analysed. Thus, these satDNA sequences are being preserved in these genomes with high similarity and preservation of its CpG island, despite their different chromosome environment and/or copy number. The FA-SAT seems to be, in fact, a “frozen” satellite DNA sequence, shaping differently the Bilateria genomes with its FA-SAT genome architectures (i.e., centromeric, telomeric, and interspersed). Unquestionably, we are aware that the molecular techniques used to isolate, sequence, and assemble these repeats can bias the data acquired (Louzada et al. 2015). For instance, in genome sequencing assembly, it is a common practice to exclude the satDNA domains and other highly repetitive sequences from the databases (Komissarov et al. 2011; Louzada et al. 2015; Satovic et al. 2016). These technical issues justify the following: 1) most likely, we did not analyse all the FA-SAT-related sequences; and 2) although several of the genomes analysed have already been sequenced, we could only in silico find complete FA-SAT monomers in the cat genome (in the other sequenced genomes, it is possible to find incomplete monomers not considered in this work). Regarding the first issue, the complementary techniques used to analyse the genomes as a whole, particularly the HRM analysis, corroborate that the FA-SAT variability seems to be low (fig. 2c and supplementary fig. S6b and c, Supplementary Material online), at least for variants B and C (which, respectively, represent the most similar and the most dissimilar monomeric sequences regarding the FA-SAT reference sequence). However, the copy number assay used in this work underestimates the number of copies of satDNA present in genomes because of their repetitive nature (as described by our group in Louzada et al. 2015), leaving open the hypothesis that more sequences and variants can still be discovered. Even so, the present work is certainly technically eclectic in analysing such difficult sequences in so many divergent genomes. In the future, we hope that technological advances will make the analysis of satDNAs more amenable to structural and functional studies, overcoming these limitations. We further analysed the cat genome to find molecular motifs that could explain the shuffling of FA-SAT between different genome regions and found an intensive intermingling of FA-SAT with transposable elements (TEs), which most likely are the cause of the FA-SAT intragenomic movements and the interspersion of these repeats throughout this genome. In the last few years, some studies have been published regarding the satDNA monomer-flanking regions and the sequences present at the junction ends, revealing other satDNAs, transposable elements, and other sequences as coding genes (McAllister and Werren 1999; Schueler et al. 2001; Palomeque et al. 2006; Heslop-Harrison and Schwarzacher 2011; Neumann et al. 2011; Brajkovic et al. 2012; Kuhn et al. 2012; Maumus and Quesneville 2014; Louzada et al. 2015; Satovic et al. 2016; Khost et al. 2017). Our data suggest that the evolutionary history of FA-SAT included fluctuations in copy number, especially at the heterochromatic regions (i.e., centromeres and telomeres) and hypothetical independent genome transpositions that relocated FA-SAT in interspersion patterns. Future work encompassing an extensive bulk analysis of genome sequencing data will allow us to accumulate experimental evidence on the sequences residing in these flanking regions and most likely change the way we study this class of repeats and their mobility and interspersion genome patterns. However, it seems that the presence of TEs will be a persistent finding.
Finally, there is another question that was raised by our work: if FA-SAT is responsible for shaping genomes (i.e., originating different genome organizations), how do these genomes accommodate these changes? To answer this question, it is mandatory, in the future, to determine if this DNA sequence and/or its transcripts display, in fact, a function. We showed that FA-SAT-related sequences are transcribed in the Bilateria genomes analysed. Additionally, we demonstrated that when the satDNA sequence is naturally relocated to a new chromosome region (i.e., to the centromere), it is not preferentially transcribed from there. Still, it seems that the widespread sequences guarantee the transcription needed in the genomes analysed.
It is also important to highlight that in the conserved motifs of FA-SAT, we found flanking coding genes; these conserved motifs could be linked to gene regulation and/or chromatin structure (Paar et al. 2011; Kuhn et al. 2012; Maumus and Quesneville 2014; Feliciello et al. 2015). Additionally, satncRNAs originating from sequences localized specifically at heterochromatic regions are increasingly being related to genomic instability and cancer (Lu and Gilbert 2007; Ting et al. 2011; Zhu et al. 2011; Leonova et al. 2013; Hong and Choi 2016; Kishikawa et al. 2016). In forthcoming works it will be mandatory to disclose the particular function (or functions) of this well conserved satellite DNA sequence, FA-SAT, as well as its transcripts. Finally, most reports in the literature compare different satellite DNA families that only share the same genomic region, generally the centromere. As an example, Eymery et al. (2009) reviewed the centromeric satDNAs from mouse and human, which are different in its DNA sequence and in its transcripts. In this work, the satDNA sequences found in so many different species, represent a unique opportunity to have a valuable genome’s model that allows to compare the same satDNA, with similar primary nucleotide sequence, which inhabiting different genome locations, in different species.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Science and Technology Foundation (FCT) from Portugal (PhD grants numbers SFRH/BD/80446/2011, SFRH/BD/80808/2011, SFRH/BD/41576/2007). The samples and commercial cell lines were provided in the frame of the research projects from the Science and Technology Foundation (FCT) from Portugal (grant numbers POCTI/C_BIA/11285/98, POCI/CVT/62940/04, POCI/BIA_BCM/58541/04), where the corresponding author was participant member or principal investigator. This work was funded by the BioISI project UID/Multi/04046/2013.
Author Contributions
R.C. conceived and designed the original idea, wrote the article. F.A. collaborated in the design of the experiments. S.M. conducted the experiments. A.M.-S. did the bioinformatic analysis and prepared the respective figures. F.A., D.F., and A.M.-S. collaborated in some experiments. R.C., D.F., and F.A. analyzed all data, prepared figures, tables, and graphs, and revised the article.
Literature Cited
- Adega F, Chaves R, Guedes-Pinto H.. 2008. Suiformes orthologous satellite DNAs as a hallmark of Pecari tajacu and Tayassu pecari (Tayassuidae) evolutionary rearrangements. Micron 398:1281–1287. [DOI] [PubMed] [Google Scholar]
- Adega F, Guedes-Pinto H, Chaves R.. 2009. Satellite DNA in the karyotype evolution of domestic animals–clinical considerations. Cytogenet Genome Res. 126(1–2):12–20. [DOI] [PubMed] [Google Scholar]
- Alexiadis V, Ballestas ME, Sanchez C, Winokur S, Vedanarayanan V.. 2007. RNAPol-ChIP analysis of transcription from FSHD-linked tandem repeats and satellite DNA. Biochim Biophys Acta 17691:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscotti MA, Canapa A, Forconi M, Olmo E, Barucca M.. 2015. Transcription of tandemly repetitive DNA: functional roles. Chromosome Res. 23:463–477. [DOI] [PubMed] [Google Scholar]
- Brajkovic J, Feliciello I, Bruvo-Madaric B, Ugarkovic D.. 2012. Satellite DNA-like elements associated with genes within euchromatin of the beetle Tribolium castaneum. G3 (Bethesda) 2:931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JD, Mitchell SE, O’Neill RJ.. 2012. Making a long story short: noncoding RNAs and chromosome change. Heredity (Edinb) 1081:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut-Karslioglu A, et al. 2012. A transcription factor-based mechanism for mouse heterochromatin formation. Nat Struct Mol Biol. 1910:1023–1030. [DOI] [PubMed] [Google Scholar]
- Cesari M, Luchetti A, Passamonti M, Scali V, Mantovani B.. 2003. Polymerase chain reaction amplification of the Bag320 satellite family reveals the ancestral library and past gene conversion events in Bacillus rossius (Insecta Phasmatodea). Gene 312:289–295. [DOI] [PubMed] [Google Scholar]
- Chaves R, Louzada S, Meles S, Wienberg J, Adega F.. 2012. Praomys tullbergi (Muridae, Rodentia) genome architecture decoded by comparative chromosome painting with Mus and Rattus. Chromosome Res. 206:673–683. [DOI] [PubMed] [Google Scholar]
- De Cecco M, et al. 2013. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 122:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enukashvily NI, Donev R, Waisertreiger IS, Podgornaya OI.. 2007. Human chromosome 1 satellite 3 DNA is decondensed, demethylated and transcribed in senescent cells and in A431 epithelial carcinoma cells. Cytogenet Genome Res. 1181:42–54. [DOI] [PubMed] [Google Scholar]
- Enukashvily NI, Ponomartsev NV.. 2013. Mammalian satellite DNA: a speaking dumb. Adv Protein Chem Struct Biol. 90:31–65. [DOI] [PubMed] [Google Scholar]
- Eymery A, Callanan M, Vourc’h C.. 2009. The secret message of heterochromatin: new insights into the mechanisms and function of centromeric and pericentric repeat sequence transcription. Int J Dev Biol. 53(2–3):259–268. [DOI] [PubMed] [Google Scholar]
- Fanning TG. 1987. Origin and evolution of a major feline satellite DNA. J Mol Biol. 1974:627–634. [DOI] [PubMed] [Google Scholar]
- Feliciello I, Akrap I, Ugarkovic D.. 2015. Satellite DNA modulates gene expression in the beetle Tribolium castaneum after heat stress. PLoS Genet. 118:e1005466.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira D, et al. 2015. Satellite non-coding RNAs: the emerging players in cells, cellular pathways and cancer. Chromosome Res. 233:479–493. [DOI] [PubMed] [Google Scholar]
- Fry K, Salser W.. 1977. Nucleotide sequences of HS-alpha satellite DNA from kangaroo rat Dipodomys ordii and characterization of similar sequences in other rodents. Cell 124:1069–1084. [DOI] [PubMed] [Google Scholar]
- Garrido-Ramos MA. 2017. Satellite DNA: an evolving topic. Genes 89:230.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory TR. 2017. Animal Genome Size Database. http://www.genomesize.com, last accessed January 2017.
- Henikoff S, Dalal Y.. 2005. Centromeric chromatin: what makes it unique? Curr Opin Genet Dev. 152:177–184. [DOI] [PubMed] [Google Scholar]
- Heslop-Harrison JS, Schwarzacher T.. 2011. Organisation of the plant genome in chromosomes. Plant J. 661:18–33. [DOI] [PubMed] [Google Scholar]
- Hong ST, Choi KW.. 2016. Antagonistic roles of Drosophila Tctp and Brahma in chromatin remodelling and stabilizing repeated sequences. Nat Commun. 7:12988.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, et al. 2004. Stress-induced transcription of satellite III repeats. J Cell Biol. 1641:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khost DE, Eickbush DG, Larracuente AM.. 2017. Single-molecule sequencing resolves the detailed structure of complex satellite DNA loci in Drosophila melanogaster. Genome Res. 275:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishikawa T, et al. 2016. Satellite RNAs promote pancreatic oncogenic processes via the dysfunction of YBX1. Nat Commun. 7:13006.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohany O, Gentles AJ, Hankus L, Jurka J.. 2006. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7:474.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komissarov AS, Gavrilova EV, Demin SJ, Ishov AM, Podgornaya OI.. 2011. Tandemly repeated DNA families in the mouse genome. BMC Genomics 12:531.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn GC, Kuttler H, Moreira-Filho O, Heslop-Harrison JS.. 2012. The 1.688 repetitive DNA of Drosophila: concerted evolution at different genomic scales and association with genes. Mol Biol Evol. 291:7–11. [DOI] [PubMed] [Google Scholar]
- Leonova KI, et al. 2013. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci U S A. 1101:E89–E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LC, Dahiya R.. 2002. MethPrimer: designing primers for methylation PCRs. Bioinformatics 1811:1427–1431. [DOI] [PubMed] [Google Scholar]
- Liu F, et al. 2007. The human genomic melting map. PLoS Comput Biol. 35:e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD.. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 254:402–408. [DOI] [PubMed] [Google Scholar]
- Louzada S, et al. 2015. A novel satellite DNA sequence in the Peromyscus genome (PMSat): evolution via copy number fluctuation. Mol Phylogenet Evol. 92:193–203. [DOI] [PubMed] [Google Scholar]
- Lu J, Gilbert DM.. 2007. Proliferation-dependent and cell cycle regulated transcription of mouse pericentric heterochromatin. J Cell Biol. 1793:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maumus F, Quesneville H.. 2014. Ancestral repeats have shaped epigenome and genome composition for millions of years in Arabidopsis thaliana. Nat Commun. 5:4104.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister BF, Werren JH.. 1999. Evolution of tandemly repeated sequences: what happens at the end of an array? J Mol Evol. 484:469–481. [DOI] [PubMed] [Google Scholar]
- Meštrović N, et al. 2015. Structural and functional liaisons between transposable elements and satellite DNAs. Chromosome Res. 233:583–596. [DOI] [PubMed] [Google Scholar]
- Modi WS, Fanning TG, Wayne RK, O’Brien SJ.. 1988. Chromosomal localization of satellite DNA sequences among 22 species of felids and canids (Carnivora). Cytogenet Cell Genet. 484:208–213. [DOI] [PubMed] [Google Scholar]
- Montero JJ, Lopez de Silanes I, Grana O, Blasco MA.. 2016. Telomeric RNAs are essential to maintain telomeres. Nat Commun. 7:12534.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mravinac B, Plohl M, Mestrovic N, Ugarkovic D.. 2002. Sequence of PRAT satellite DNA “frozen” in some Coleopteran species. J Mol Evol. 546:774–783. [DOI] [PubMed] [Google Scholar]
- Mravinac B, Plohl M, Ugarkovic D.. 2005. Preservation and high sequence conservation of satellite DNAs suggest functional constraints. J Mol Evol. 614:542–550. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, et al. 2005. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 3095734:613–617. [DOI] [PubMed] [Google Scholar]
- Neumann P, et al. 2011. Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mob DNA 21:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paar V, Gluncić M, Rosandić M, Basar I, Vlahović I.. 2011. Intragene higher order repeats in neuroblastoma breakpoint family genes distinguish humans from chimpanzees. Mol Biol Evol. 286:1877–1892. [DOI] [PubMed] [Google Scholar]
- Paco A, Chaves R, Vieira-da-Silva A, Adega F.. 2013. The involvement of repetitive sequences in the remodelling of karyotypes: the Phodopus genomes (Rodentia, Cricetidae). Micron 46:27–34. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Gregory TR.. 2014. The case for junk DNA. PLoS Genet. 105:e1004351.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomeque T, Antonio Carrillo J, Munoz-Lopez M, Lorite P.. 2006. Detection of a mariner-like element and a miniature inverted-repeat transposable element (MITE) associated with the heterochromatin from ants of the genus Messor and their possible involvement for satellite DNA evolution. Gene 3712:194–205. [DOI] [PubMed] [Google Scholar]
- Pardo CE, et al. 2011. MethylViewer: computational analysis and editing for bisulfite sequencing and methyltransferase accessibility protocol for individual templates (MAPit) projects. Nucleic Acids Res. 391:e5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman PL, et al. 2012. Comparative chromosome painting in Carnivora and Pholidota. Cytogenet Genome Res. 137(2–4):174–193. [DOI] [PubMed] [Google Scholar]
- Petraccioli A, et al. 2015. A novel satellite DNA isolated in Pecten jacobaeus shows high sequence similarity among molluscs. Mol Genet Genomics 2905:1717–1725. [DOI] [PubMed] [Google Scholar]
- Pezer Z, Brajkovic J, Feliciello I, Ugarkovc D.. 2012. Satellite DNA-mediated effects on genome regulation. Genome Dyn. 7:153–169. [DOI] [PubMed] [Google Scholar]
- Plohl M, Luchetti A, Mestrovic N, Mantovani B.. 2008. Satellite DNAs between selfishness and functionality: structure, genomics and evolution of tandem repeats in centromeric (hetero)chromatin. Gene 409(1–2):72–82. [DOI] [PubMed] [Google Scholar]
- Plohl M, Mestrovic N, Mravinac B.. 2012. Satellite DNA evolution. Genome Dyn. 7:126–152. [DOI] [PubMed] [Google Scholar]
- Plohl M, Mestrovic N, Mravinac B.. 2014. Centromere identity from the DNA point of view. Chromosoma 1234:313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontius JU, O’Brien SJ.. 2009. Artifacts of the 1.9x feline genome assembly derived from the feline-specific satellite sequence. J Hered. 100(Suppl 1):S14–S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst AV, et al. 2010. A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev Cell 194:625–638. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H.. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 132:365–386. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ruano FJ, Lopez-Leon MD, Cabrero J, Camacho JP.. 2016. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci Rep. 6:28333.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos S, Chaves R, Adega F, Bastos E, Guedes-Pinto H.. 2006. Amplification of the major satellite DNA family (FA-SAT) in a cat fibrosarcoma might be related to chromosomal instability. J Hered. 972:114–118. [DOI] [PubMed] [Google Scholar]
- Santos S, Chaves R, Guedes-Pinto H.. 2004. Chromosomal localization of the major satellite DNA family (FA-SAT) in the domestic cat. Cytogenet Genome Res. 107(1–2):119–122. [DOI] [PubMed] [Google Scholar]
- Satovic E, Vojvoda Zeljko T, Luchetti A, Mantovani B, Plohl M.. 2016. Adjacent sequences disclose potential for intra-genomic dispersal of satellite DNA repeats and suggest a complex network with transposable elements. BMC Genomics 171:997.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler MG, Higgins AW, Rudd MK, Gustashaw K, Willard HF.. 2001. Genomic and genetic definition of a functional human centromere. Science 2945540:109–115. [DOI] [PubMed] [Google Scholar]
- Sujiwattanarat P, et al. 2015. Higher-order repeat structure in alpha satellite DNA occurs in New World monkeys and is not confined to hominoids. Sci Rep. 51:10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamazian G, et al. 2014. Annotated features of domestic cat – Felis catus genome. Gigascience 3:13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilman G, et al. 2012. Cancer-linked satellite 2 DNA hypomethylation does not regulate Sat2 non-coding RNA expression and is initiated by heat shock pathway activation. Epigenetics 78:903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting DT, et al. 2011. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 3316017:593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofimova I, Chervyakova D, Krasikova A.. 2015. Transcription of subtelomere tandemly repetitive DNA in chicken embryogenesis. Chromosome Res. 233:495–503. [DOI] [PubMed] [Google Scholar]
- Ugarkovic D. 2005. Functional elements residing within satellite DNAs. EMBO Rep. 611:1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valgardsdottir R, et al. 2008. Transcription of Satellite III non-coding RNAs is a general stress response in human cells. Nucleic Acids Res. 362:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira-da-Silva A, Louzada S, Adega F, Chaves R.. 2015. A high-resolution comparative chromosome map of Cricetus cricetus and Peromyscus eremicus reveals the involvement of constitutive heterochromatin in breakpoint regions. Cytogenet Genome Res. 1451:59–67. [DOI] [PubMed] [Google Scholar]
- Zhu Q, et al. 2011. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 4777363:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou F, Li N.. 2011. Isolation and characterization of sixty sequences of cot-1 DNA from the Asiatic black bear, Ursus thibetanus. Afr J Biotechnol. 11:15493–15500. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The FA-SAT clone sequences from the different species have been deposited at the GenBank, being the all the accession information presented in supplementary table S4 in the Supplementary Material online.