

Abstract
Bemisia tabaci threatens production of cassava in Africa through vectoring viruses that cause cassava mosaic disease (CMD) and cassava brown streak disease (CBSD). B. tabaci sampled from cassava in eight countries in Africa were genotyped using NextRAD sequencing, and their phylogeny and population genetics were investigated using the resultant single nucleotide polymorphism (SNP) markers. SNP marker data and short sequences of mitochondrial DNA cytochrome oxidase I (mtCOI) obtained from the same insect were compared. Eight genetically distinct groups were identified based on mtCOI, whereas phylogenetic analysis using SNPs identified six major groups, which were further confirmed by PCA and multidimensional analyses. STRUCTURE analysis identified four ancestral B. tabaci populations that have contributed alleles to the six SNP-based groups. Significant gene flows were detected between several of the six SNP-based groups. Evidence of gene flow was strongest for SNP-based groups occurring in central Africa. Comparison of the mtCOI and SNP identities of sampled insects provided a strong indication that hybrid populations are emerging in parts of Africa recently affected by the severe CMD pandemic. This study reveals that mtCOI is not an effective marker at distinguishing cassava-colonizing B. tabaci haplogroups, and that more robust SNP-based multilocus markers should be developed. Significant gene flows between populations could lead to the emergence of haplogroups that might alter the dynamics of cassava virus spread and disease severity in Africa. Continuous monitoring of genetic compositions of whitefly populations should be an essential component in efforts to combat cassava viruses in Africa.
Keywords: mitochondrial DNA cytochrome oxidase I, single nucleotide polymorphism, population genetic structure, cassava mosaic disease, cassava brown streak disease
Introduction
Cassava (Manihot esculenta Crantz) is the leading crop by fresh weight produced in Africa (FAO 2014). It has potential to provide a solution to the imminent food scarcity, most notably as it is expected to be resilient to future climate change (Jarvis et al. 2012).
In Africa, the virus diseases—cassava mosaic disease (CMD) and cassava brown streak disease (CBSD)—are the most important constraints to cassava production. CMD is caused by eight cassava mosaic begomoviruses (CMBs) (family Geminiviridae: genus Begomovirus) in Africa (Bock and Woods, 1983; Hong et al. 1993; Legg and Fauquet 2004; Legg et al. 2015; ICTV 2017), and two CMBs in Asia (Alagianagalingam and Ramakrishnan 1966; Austin 1986). CBSD is caused by the ipomoviruses: Cassava brown streak virus and Ugandan cassava brown streak virus (Mbanzibwa et al. 2009; Winter et al. 2010; Patil et al. 2015; ICTV 2017).
The whitefly, Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae), is among the most damaging pests of crops worldwide, with a host range of over 1,000 plant species. The severest damage is caused by the vectoring of over 300 plant viruses (Brown and Czosnek 2002; Jones 2003; Gilbertson et al. 2015). In Africa, B. tabaci is the vector of CMBs and cassava brown streak ipomoviruses (CBSIs) (Storey and Nichols 1938; Dubern 1994; Maruthi et al. 2005; 2017). The combined damage resulting from CMD and CBSD infection is estimated to cause cassava yield losses amounting to 50% in East and Central Africa, causing annual losses equivalent to more than US$1 billion (Thresh et al. 1997; Legg et al. 2006, 2011). Virus spread in both CMD and CBSD pandemics is associated with unusually abundant populations of B. tabaci (Legg et al. 2014).
Bemisia tabaci is genetically complex (De Barro 2012), with many distinct genetic groups that have been identified based on sequences of the mitochondrial cytochrome oxidase I (mtCOI) gene (Boykin et al. 2007; Dinsdale et al. 2010; De Barro 2012). This information has been used to show that there are currently at least 34 morphologically indistinguishable species (Tay et al. 2012) under the name B. tabaci. In sub-Saharan Africa, there two major groups of B. tabaci, those that colonize cassava and are unique to this host (cassava type) and those that colonize other host plants especially vegetables such as tomato and sweet potato but do not colonize cassava (noncassava types). The noncassava types in sub-Saharan Africa mainly comprise groups designated as Indian Ocean (IO), MED, MEAM1 and Uganda (Sseruwagi et al. 2005; Delatte et al. 2006; Tocko-Marabena et al. 2017). The cassava types belong to five genetically distinct groups of B. tabaci, named sub-Saharan Africa 1 to 5 (SSA 1–5). SSA1 occurs throughout sub-Saharan Africa, SSA2 in East and West Africa, SSA3 and SSA4 in Central and West Africa, and SSA5 in South Africa (Berry et al. 2004; Esterhuizen et al. 2013; Legg et al. 2014). Based on the mtCOI sequence divergence, SSA1 has been further divided into five subgroups; SSA1 subgroup 1 (SSA1-SG1), SSA1-SG2, SSA1-SG3, SSA1-SG4 (Legg et al. 2014), and SSA1-SG5 (Ghosh et al. 2015). A genetic study of cassava-colonizing B. tabaci in the Great Lakes region of East and Central Africa over a 14-year period from 1997 to 2010 revealed that SSA1 populations went through a period of rapid expansion from 2000 to 2003 (Legg et al. 2014). During this period SSA1-SG1 became predominant and extended its geographical range within this region.
The mtCOI marker is the most commonly used for phylogenetic studies of B. tabaci due to its high degree of variability (Brown 2000; Viscarret et al. 2003; Sseruwagi et al. 2005, 2006; Boykin et al. 2007; Dinsdale et al. 2010), and it has been shown to be a useful molecular marker for phylogenetic analysis of the B. tabaci cryptic species complex (Brown 2010; Gill and Brown 2010). However, mtCOI has the drawback that it is a single locus that is also maternally inherited, hence it is strictly a marker of evolution in females. It is therefore likely to yield insufficient genetic resolution to distinguish populations, and additionally, it does not provide a full representation of phylogenetic history (Ballard and Whitlock 2004; Hurst and Jiggins 2005; Whitworth et al. 2007; Collins and Cruickshank 2013; Dupuis et al. 2012; Foster et al. 2013; Frey et al. 2013; O’Loughlin et al. 2014; Pinto et al. 2014; White et al. 2014). Furthermore, although mitochondrial DNA (mtDNA) was recognized to be a neutral marker that indicates species history, some authors have argued that it is often under strong selection (Ballard and Whitlock 2004; Bazin et al. 2006). According to species separation based on the mtCOI sequence divergence of >3.5% (Dinsdale et al. 2010), the five distinct cassava groups (SSA1, SSA2, SSA3, SSA4, and SSA5) are considered putative species meaning they are not expected to interbreed. However, mating studies conducted by Maruthi et al. (2004) demonstrated successful interbreeding between SSA2 and SSA1 individuals sourced from Uganda. Delatte et al. (2006) also reported a putative hybrid between MEAM1 and IO under field conditions. A review by Liu et al. (2012) on the species concept of B. tabaci showed that the majority of mating crosses between putative species yielded no hybrids or low proportions that were sterile or less viable, and that hybrids seldom occurred under field conditions, although there are few studies that have set out to find such hybrid populations. Accurate species identifications are often crucial for the implementation of whitefly management programmes that include the detection and prevention of spread of invasive species, (Bickford et al. 2007). Therefore, there is a need to utilize more robust markers for population genetic diversity studies (Helyar et al. 2011; Quillery et al. 2014).
Next-generation sequencing (NGS) technologies have opened up opportunities for studying genome-wide diversity of populations of target organisms (Mardis 2008; Shendure and Ji 2008). The increased speed and accuracy coupled with reduced cost and progress in bioinformatics have presented opportunities for genome-wide studies in nonmodel organisms using NGS technologies. Lately, restriction-site associated DNA (RAD) genotyping has been used for quick identification of tens to hundreds of thousands of single nucleotide polymorphisms (SNPs) distributed across whole genomes (Davey et al. 2010). RAD-based techniques have been demonstrated to be powerful in characterizing genetic diversity within groups of organisms (Etter et al. 2011; Barley et al. 2015; Szulkin et al. 2016), but the requirement for high DNA volumes has prevented their application for many insect pests with small body sizes. A new genotyping technology, NextRAD, can overcome these constraints by fragmenting and ligating adaptor sequences to genomic DNA through engineered transposomes (Nextera DNA Library Prep Reference Guide), and requires very small amounts of DNA (less than 50 ng) (Russello et al. 2015), enabling acquisition of sequence data from small organisms that could not be studied using RAD applications. This approach has been successfully utilized to study fine-scale population genetics and diversity in a mosquito species (Emerson et al. 2015) and the potato psyllid (Fu et al. 2017).
In this study, cassava whitefly Bemisia tabaci samples from eight countries in Africa were genotyped using both mtCOI and NextRAD sequencing, in an effort to infer population genetics and to describe the diversity of genetically distinct groups of cassava B. tabaci whiteflies. Sequences obtained were evaluated using phylogenetic, principal component and multidimensional scaling analyses, and population structure and gene flow were examined. Our results indicate that phylogenetic relationships inferred by mtCOI and NextRAD sequencing approaches show important differences that should have significant consequences for the taxonomic designation of the genetic groups identified. In addition, gene flow analysis suggests that there has been considerably more genetic exchange between putative species groups than has hitherto been assumed. We discuss the implications of these results both for the taxonomy of B. tabaci colonizing cassava in Africa, as well the management of this pest complex.
Materials and Methods
Whitefly Samples and DNA Extraction
Adult Bemisia tabaci used in this study were collected from eight cassava-growing countries in Africa [Burundi (BUR), Cameroon (CAM), Central African Republic (RCA), Democratic Republic of Congo (DRC), Madagascar (MAD), Nigeria (NIG), Rwanda (RWA), and Tanzania (TZ)] between 2009 and 2015. The whiteflies were sampled for at least 2 years from each country except for Rwanda and Nigeria. Detailed information on locations of sampling within countries and time of collection are reported in the Supplementary Material (supplementary table S1, Supplementary Material online). The whiteflies were aspirated alive from cassava plants, with at least ten whiteflies collected per sampling site, preserved in 95% ethanol and stored at −20 °C until DNA extraction. Bemisia tabaci whiteflies collected from cassava are mainly from cassava haplogroups but occasionally individuals from noncassava haplogroups that visit cassava are also sampled. None of the haplogroups within the B. tabaci species complex are morphologically distinguishable. Most B. tabaci colonizing cassava belong to the distinct SSA group comprising SSA1, SSA2, SSA3, SSA4, and SSA5 (Legg et al. 2014), whereas a small proportion belong to groups that have been designated as Indian Ocean, East African, MED-like, and Tanzania (a novel haplotype with 89% mtCOI similarity to GenBank accession KX397315 from a whitefly collected from gourd in India). The cassava genotypes prefer cassava though they have been reported colonizing other hosts (Sseruwagi et al. 2005) and colonies have successfully established on sweet potato and cowpea, whereas noncassava genotypes are common on vegetables and attempts to establish colonies on cassava have so far been unsuccessful as nymphs fail to reach the adult stage (Legg 1996). Bemisia afer, which also colonizes cassava, was also occasionally aspirated. Ninety-five individual whiteflies were selected for genotyping. A relatively greater number were selected from Tanzania since the north-western part of this country was a region most recently affected by spread of the CMD and CBSD pandemics associated with super-abundant whitefly populations. DNA was extracted individually from each of the 95 whiteflies. Whiteflies in groups of 5–10 from each site were removed from 95% ethanol and suspended in 0.1× TE buffer for 2 h at room temperature. The contents were emptied into a Petri dish, and with the aid of light microscope, a single adult female was picked from each group using a 10 µl pipette tip. This insect was then macerated in 20 µl lysis buffer. The lysate was incubated at −80 °C for 30 min, immediately followed by incubation at 55 °C in a water bath for 30–60 min, and then 5 µl of RNase (25 mg/ml) was added to each sample, after which samples were incubated at room temperature for 5 min. DNA extraction was completed using the Zymo gDNA miniprep kit following the manufacturer’s instructions (Zymo Research Corporation, CA, USA). DNA quality was checked on a 1.0% agarose gel (supplementary fig. S1, Supplementary Material online) and only samples with visible intact bands were selected for sequencing.
MtCOI Sequencing
DNA from the 95 whitefly samples were used for PCR and mtCOI sequencing. A partial fragment of mtCOI was amplified using three sets of newly designed primers CA, CA-1, and NSP-1 (CA: ACTCGGGCTTATTTCACTTCA-F, ACGAACCAG AAGAAAAGACT-R, 555 bp; CA-1: TTACTGTTGGGATAGAT GTGGA-F, AACCAGAAGAAAAGACTCTAAA-R, 575 bp; NSP-1: AAGAAGGAAAGATTCTAAAACAA-F, ATCATATGT TTACTGTGGGAA-R, 659 bp). Each reaction (20 µl) contained 2.5 mM MgCl2, 0.2 mM of each dNTP, 0.25 µM forward and reverse primer, 1.25 U of Taq DNA polymerase (New England Biolabs, Ipswich, MA, USA) and 1 µL extracted DNA. PCR amplification was performed at 95 °C for 2 min, followed by 35 cycles at 95 °C for 30 s, 52 °C for 30 s, and 72 °C for 1 min, and a final step at 72 °C for 10 min. The PCR amplicons were sequenced directly by Macrogen (Rockville, MD, USA), and the sequences were assembled into contigs using CLC Main Workbench 7.7.2 (Qiagen Inc. Germantown MD—USA). Multiple sequence alignment was performed using Clustal W in MEGA (version 6.06; Tamura et al. 2013). A maximum-likelihood phylogenetic tree was constructed using MEGA (version 6.06) with 1,000 bootstrap replicates. GenBank sequences were included in the phylogenetic tree to make comparisons between our mtCOI sequences and those that have previously been published. We designed the new primers because the universal primers used for mtCOI amplification (L2-N-3014 and C1-J-2195) (Frohlich et al. 1999) produced poor bands or failed to amplify some specimens. The new primers amplified some of the samples that could not be amplified using the universal primers, especially individuals in the SSA2 and SSA4 haplogroups.
NextRAD Sequencing
Genomic DNA of the same 95 whiteflies was used to construct NextRAD libraries by SNPsaurus, LLC as described in Russello et al. (2015). Genomic DNA was fragmented with Nextera reagent (Illumina Inc.), and short adapter sequences were ligated to the ends of the fragments. The Nextera reaction was scaled for fragmenting 5 ng of genomic DNA. Fragmented DNA was then amplified, with one of the primers matching the adapter and extending nine nucleotides into the genomic DNA with the selective sequence GTGTAGAGC. Thus, only fragments starting with a sequence that can be hybridized by the selective sequence of the primer was efficiently amplified. PCR amplification was done at 73 °C for 26 cycles. The NextRAD libraries were sequenced on one lane of a HiSeq 4000 system with single-end mode and read length of 150 bp, following the manufacturer’s instructions (Illumina, San Diego, CA, USA).
Raw reads from NextRAD sequencing were processed to remove adapter and low quality sequences using Trimmomatic (Bolger et al. 2014). The cleaned reads were then aligned to the MEAM1 whitefly genome (Chen et al. 2016) using BWA-MEM (Li and Durbin 2009) and only uniquely mapped reads were used for SNP calling. SNP calling was performed using TASSEL 5 (Bradbury et al. 2007). The resulting raw SNPs were filtered by the following criteria: 1) individual samples with missing data > 55% were excluded; 2) SNPs with missing data in > 20% of the samples or minor allele frequency (MAF) < 0.05 were removed; 3) SNPs with genotype quality (GQ) < 30 or SNPs with another SNP <5 bp away were excluded.
The final filtered SNPs were used for phylogenetic, population structure and principal component analyses. A Maximum-Likelihood (ML) phylogenetic tree was constructed using phyML (Guindon et al. 2010) with default parameters. STRUCTURE (v2.3.4) (Pritchard et al. 2000) was used to deduce population structure with the admixture model and to correlate allele frequencies. Twenty independent runs for each K value ranging from 1 to 10 were performed with a burn-in length of 10,000 followed by 10,000 iterations, where K is the assumed number of populations. The best K was deduced from the distribution of ΔK (Evanno et al. 2005). The derived optimal K was used in a final run with 100,000 burn-in and 100,000 iterations. Principal component analysis (PCA) was performed using PLINK (v1.9) (Chang et al. 2015). Pairwise weighted FST values (Holsinger and Weir 2009) were calculated among different groups using VCFtools (Danecek et al. 2011). To visualize the pairwise matrix, multidimensional scaling (MDS) was performed using an R function cmdscale to transfer FST values into two dimensional values for plotting.
Gene Flow Analysis
Gene flow analysis was done using the D-statistic, which is a formal test for admixture based on a four taxon statistic and can evaluate whether gene flow has occurred between populations (Patterson et al. 2012). D-statistics were calculated using AdmixTools v4.1 (Patterson et al. 2012). This programme made use of the tree structure (of out-group, x; y, SSA4), where “out-group” included Bemisia afer and B. tabaci Indian Ocean (Ind) obtained from sweet potato. Under the assumption of the model, there is no gene flow between the “out-group” and SSA4, but there is potential gene flow between either population x and y or x and SSA4, which results in negative or positive values of D, respectively. D = 0 indicates a lack of gene flow between the two populations. Significant deviations of D from 0 are estimated by the Z-score, which is considered to be significant if it is >4 or < −4.
To infer directionality of gene flow between different populations, we performed the partitioned D-statistic test, which is based on a five-taxon tree (((P1, P2), (P31, P32)), O), where P31 and P32 are two lineages within the P3 clade (Eaton and Ree 2013). In this test, three D-statistics, D1, D2, and D12, are calculated, which indicate whether a derived allele is present only in P31, only in P32, or shared by both. The test can infer directionality through its measurement of introgression of shared ancestral alleles, D12. If gene flow occurred from P31 into P2, then derived P3 alleles which arose in the ancestor of P31 and P32, and which were thus shared by both taxa, will also appear in P2. In contrast, if gene flow occurred only in the opposite direction, from P2 into P31, P2 will not contain alleles that are shared by the two P3 taxa, and thus the partitioned test would find a nonsignificant D12 (Eaton and Ree 2013). Similar to the five population tests described in Meier et al. (2017) and Razkin et al. (2016), in our tests we selected one individual with least missing data from each population. Our partitioned D-statistic tests were performed only in cases where two P3 lineages were available (SSA-CA SSA-ESA and SSA-WA), and only for populations that showed gene flows as indicated by the four taxon D-statistic tests.
GIS Mapping
Geo-referenced coordinates for samples were used to generate maps using ArcGIS 10.1 (ESRI, Redlands, California, USA). Maps were produced illustrating the geographic distributions within sampled countries of mtCOI and SNP data for B. tabaci whiteflies from cassava.
Results
MtCOI Sequencing
Out of the 95 whitefly samples, 67 produced good quality mtCOI sequences (supplementary table S1, Supplementary Material online). These sequences together with sequences available in GenBank were used to generate a Maximum Likelihood phylogenetic tree (fig. 1). Out of the 67 samples, 63 were classified as cassava haplotypes and were clustered into four major SSA groups (SSA1, SSA2, SSA3, and SSA4). The SSA1 group was further split into five subgroups, SSA1-SG1, SSA1-SG1/SG2, SSA1-SG2, SSA1-SG3, and SSA1-SG5. These groups were designated based on the topology of the phylogenetic tree which included reference sequences from GenBank, and names were used as previously (Legg et al. 2014; Ghosh et al. 2015). The SSA1 group comprised whitefly samples from all eight countries, SSA2 consisted of five samples from Cameroon, SSA3 had one sample from Cameroon, whereas SSA4 included samples from Cameroon and Central African Republic. These groupings are based solely on the phylogenetic groupings derived from the analysis of mtCOI sequences, as described by Legg et al. (2014). Individuals from these groups are not morphologically distinguishable, and there are limited studies on whether they are biological different, although SSA1-SG1 is commonly reported with high levels of abundance, and it is prevalent in the regions of East and Central Africa affected by the dual cassava virus pandemics of CMD and CBSD (Legg et al. 2014). All three sets of primers amplified fragments ranging between 555 and 659 bp within the ∼800 bp of the universal primers. The CA primer set amplified 63 out of 95 samples all of which were cassava haplotypes, whereas CA-1 and NSP-1 amplified a few cassava haplotypes, noncassava B. tabaci and B. afer. The length of fragments trimmed and aligned in this study is 535 bp. Despite these being shorter fragments the mtCOI phylogenetic tree produced population groupings identical to those previously described using universal primers (Legg et al. 2014; Ghosh et al. 2015). Our sequences were blasted using Blastn and all are within the ∼800 bp Bemisia tabaci mtCOI sequences in GenBank confirming our primers did not amplify any sequences outside those normally produced by the universal primers. A total of 25 selected sequences from this study have been submitted to GenBank under the following accession names (MF417578—MF417602). These were representatives from the four SSA groups and SSA1 subgroups as all our sequences had > 98% similarity to the numerous cassava halotype sequences that are already deposited in GenBank. The 12 cassava haplotype sequences from GenBank that were included together with our mtCOI sequences did not affect the topology of the phylogenetic tree. Their inclusion is solely to make comparison between our sequences and those that have previously been published.
Fig. 1.
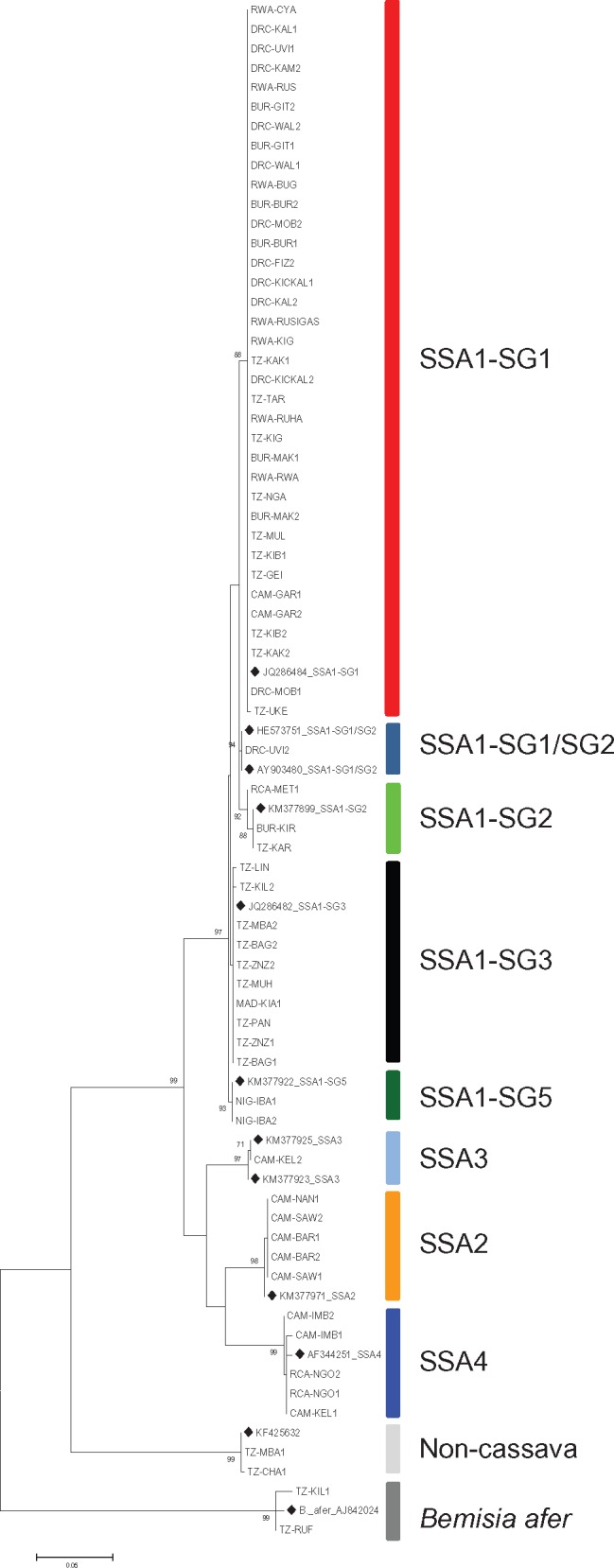
—Maximum Likelihood phylogenetic tree constructed using mtCOI sequences obtained from Bemisia tabaci (cassava and noncassava haplotypes) and B. afer adults sampled between 2009 and 2015 from eight countries in Africa, including reference sequences from GenBank (♦) for comparison.
NextRAD Sequencing and Phylogenetic Analysis
The statistics of the NextRAD sequencing reads are summarized in supplementary table S1, Supplementary Material online. After mapping the reads to the MEAM1 genome (Chen et al. 2016), a total of 7,453 SNPs were obtained. Twenty samples with missing data rates >55% were excluded from the downstream analyses (supplementary table S1, Supplementary Material online). A maximum likelihood phylogenetic tree was constructed using SNPs for all the remaining whitefly samples (72 cassava and three noncassava/B. afer as the out-group) including the reference MEAM1. The 72 cassava haplotypes included 60 out the 63 that were used to generate the mtCOI phylogenetic tree, and 12 additional ones that were unsuccessful with mtCOI. All the cassava haplotypes formed a major clade that was clearly separated from both the out-group and MEAM1 (fig. 2). This distinct separation of cassava-colonizing whiteflies and the out-group was also supported by principal component analysis (PCA) (supplementary fig. S2, Supplementary Material online).
Fig. 2.

—Maximum Likelihood phylogenetic tree constructed based on SNPs (7,453) generated by NextRAD sequencing of Bemisia tabaci (cassava and noncassava haplotypes) and B. afer adults sampled between 2009 and 2015 from eight countries in Africa. Samples are designated in different colors representing their grouping based on mtCOI sequencing, those with an NS designation at the end were not successfully sequenced using the mtCOI locus.
The Maximum Likelihood phylogenetic tree constructed using the SNPs for cassava whiteflies showed distinct genotypic groups based on individuals but not countries of collection. There were a total of six major groups, out of which four were of established clusters [sub-Saharan Africa-East and Central Africa (SSA-ECA) (closely equivalent to SSA1-SG1), sub-Saharan Africa-East and Southern Africa (SSA-ESA) (SSA1-SG3), SSA2, and SSA4] and two new groups, designated here as sub-Saharan Africa-West Africa (SSA-WA) and sub-Saharan Africa-Central Africa (SSA-CA) (fig. 2). These groups were not similar to those generated by the mtCOI locus as some haplotypes were either classified into new groups or reassigned to other groups. One major clade comprised all the sequences designated as SSA1 on the basis of mtCOI data, whilst a second clade included sequences designated as SSA2, SSA3, and SSA4 with mtCOI. The SSA1 was further divided into two major clades, each of which was further subdivided into two minor clades. The first of the major clades comprised a minor clade (SSA-ESA) which included all samples designated as SSA1-SG3 and which were from eastern and southern Tanzania as well as Madagascar, and a second minor clade (SSA-CA) including individuals from the Lake Tanganyika shore areas of eastern DRC and western Tanzania. The second major clade within SSA1 comprised two minor clades (SSA-ECA and SSA-WA). SSA-ECA included the majority of SSA1-SG1 samples as well as all the SSA1-SG2 individuals. These samples were all from CMD pandemic regions of Burundi, DRC (eastern regions), Rwanda and north-western Tanzania. The two samples from Burundi and Tanzania that were classified as SSA1-SG2 were grouped in SSA-ECA when using SNPs. The only sample from DRC that was classified as SSA1-SG1/SG2 was also part of the SSA-ECA cluster. The SSA-WA clade comprised samples from RCA and Cameroon in Central Africa, and others from Nigeria in West Africa. There was a high degree of variation within samples classified by mtCOI as SSA2, SSA3, and SSA4 and there was no clear evidence in the SNP-based tree for monophyletic groupings based on the mtCOI designations. All SSA2, SSA3, and SSA4 samples were from the central African countries of Cameroon, DRC (western regions) and RCA. Two samples from DRC (DRC-BAN1-NS and DRC-BAN2-NS) that were not successfully sequenced using mtCOI grouped closely with samples designated as SSA4 with mtCOI (fig. 2).
Population Structure Analysis Based on Bayesian Clustering
Analysis using STRUCTURE, a model-based clustering method for inferring population structure, estimated the optimal K (number of populations) to be 4, and provided evidence that the 72 cassava whitefly individuals had genetic identities derived from four ancestral populations (fig. 3). The characteristics of the STRUCTURE diagram for the K value of 4 were strongly congruent with the phylogenetic analysis of SNPs, and the six major genetic groupings were clearly distinguished. Three of the groupings (SSA2, SSA-ESA, and SSA-ECA) were largely homogenous, with little evidence of admixture. The remaining three groups were heterogeneous and constituent samples showed varying levels of population admixture. It was significant that samples where admixture between ancestral populations was most prominent (in the SSA4, SSA-CA, and SSA-WA groups) were all from countries and regions of the central parts of Africa. SSA-WA provides an example of this, as samples from Nigeria, in West Africa, were largely derived from one ancestral population, whilst other members of SSA-WA from Cameroon and Central African Republic in central Africa had a high degree of admixture with the ancestral population predominating in SSA-CA. Overall, the STRUCTURE analysis provided evidence for a much greater degree of genetic exchange between B. tabaci populations colonizing cassava in Africa than had hitherto been assumed for the putative species groups derived from mtCOI sequence analysis.
Fig. 3.

—Population structure of Bemisia tabaci (cassava haplotypes) in Africa. The estimated optimal K is 4. STRUCTURE analysis of 72 Bemisia tabaci (cassava haplotypes) adults sampled between 2009 and 2015 from eight countries in Africa. The y axis quantifies subgroup membership, and the x axis shows different accessions.
Principal Component Analysis and FST Statistics
Principal component analysis (PCA) revealed clear partitioning of populations in the first two principal components (fig. 4). The 72 cassava whitefly individuals were grouped into six distinct clusters that were similar to those generated in the SNP tree and corresponding to SSA-ECA, SSA-ESA, SSA-WA, SSA-CA, SSA2, and SSA4 (fig. 4). Clusters corresponded to the groupings generated in the STRUCTURE analysis (fig. 3). The first principal component (11.9%) clearly separated the SSA2 and SSA4 populations from the SSA-ECA, SSA-WA, SSA-CA, and SSA-ESA populations. The second principal component (7.6%) clearly separated SSA-ECA and SSA-WA from SSA-CA and SSA-ESA populations. Overall, the PCA indicated that the SSA2 cluster was the most differentiated with the largest genetic distance from the rest of the clusters. SSA-ESA was also very distinct, occupying the most distant position on the PCA axes after SSA2. SSA-ECA and SSA-WA populations grouped closely together, whereas the SSA-CA population was intermediate.
Fig. 4.
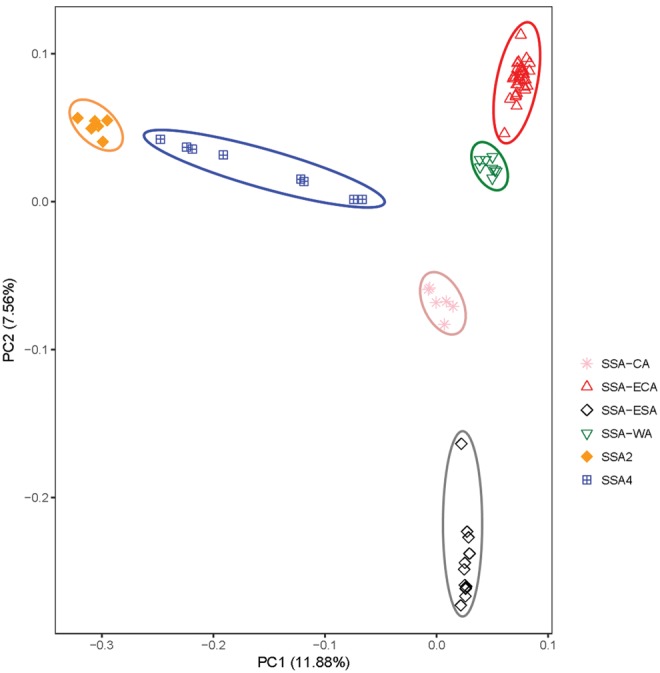
—Principal component analysis of 72 Bemisia tabaci whiteflies (cassava haplotypes) collected from eight African countries.
Pairwise weighted FST values generated among the six populations were in the range of 0.089–0.39. The lowest FST value (0.089) was between populations SSA-ECA and SSA-WA, whereas the highest (0.39) was between populations SSA2 and SSA-ESA (table 1). Interactions between SSA2 and either of the populations SSA-ECA, SSA-ESA, SSA-WA, and SSA-CA had FST values ranging between 0.31 and 0.39 (table 1). Multi-dimensional scaling to visualize the pairwise matrix of FST in two dimensions showed a clear distinction (x-axis) between SSA2 and SSA4, and between SSA-ECA, SSA-ESA, SSA-CA, and SSA-WA, with SSA2 the most distantly placed. On the Y-axis, SSA-ESA was clearly separated from SSA-ECA, SSA-CA, and SSA-WA. SSA-WA grouped closely to SSA-ECA, whereas SSA-CA was intermediate (supplementary fig. S3, Supplementary Material online). PCA analysis, FST values and multidimensional scaling all revealed the close relationship between SSA-WA and SSA-ECA, which contrasted with the relatively distant link between each of these and SSA-ESA. Consequently, SSA-ECA and SSA-ESA B. tabaci occurring in different regions of Tanzania appear to be much more distantly related than SSA-ECA B. tabaci from Tanzania and SSA-WA populations from Nigeria in West Africa.
Table 1.
Weighted Average FST between Different Populations of Bemisia tabaci (Cassava Haplogroups)
| SSA4 | SSA2 | SSA-ESA | SSA-CA | SSA-WA | SSA-ECA | |
|---|---|---|---|---|---|---|
| SSA4 | 0 | — | — | — | — | — |
| SSA2 | 0.1548 | 0 | — | — | — | — |
| SSA-ESA | 0.2540 | 0.3945 | 0 | — | — | — |
| SSA-CA | 0.1445 | 0.3362 | 0.1454 | 0 | — | — |
| SSA-WA | 0.1994 | 0.3665 | 0.2191 | 0.1335 | 0 | — |
| SSA-ECA | 0.1853 | 0.3133 | 0.1886 | 0.1116 | 0.0889 | 0 |
Gene Flow Analysis Using the D-Statistic
To test gene flow between populations using the D-statistic, we included samples of B. afer and B. tabaci Ind. as an out-group. The 7,453 SNPs were used to calculate levels of gene flow (table 2). D was significantly positive (0.41, Z-score = 18.77) under the model of [out-group, x; y, SSA4], when x was SSA2 and the other population was SSA4, representing the greatest level of gene flow between the cassava whitefly populations sampled from the eight countries in Africa. The next highest level of gene flow was between SSA-ECA and SSA-WA, with a significantly negative D (−0.22, Z-score = −12.87). Significant gene flow was also evident between SSA-CA and SSA4 (D = 0.22, Z-score = 11.50), SSA-ESA and SSA-CA (D = −0.19, Z-score = −9.34), and SSA-ECA and SSA-CA (D = −0.11, Z-score = −5.47) (table 2). No significant levels of gene flow were detected for SSA-WA versus SSA4; SSA-WA versus SSA-CA and SSA-ECA versus SSA4 (table 2). Additionally, no detectable gene flow was observed for populations SSA-ECA versus SSA-ESA, SSA-ECA versus SSA2, SSA-ESA versus SSA-WA, SSA-ESA versus SSA2, SSA-ESA versus SSA4, SSA-CA versus SSA2 and SSA-WA versus SSA2. Although D-statistic results highlight a diversity of gene exchange relationships between the major genetic groups revealed by the SNP analysis, all of the six groups were linked together in a network of gene flow (fig. 5).
Table 2.
Evidence of Gene Flow between Populations of Bemisia tabaci (Cassava Haplogroups) Determined Using D-Statistic (D [Out-Group, x; y, SSA4])
| Population x | Population y | D | Z-scorea |
|---|---|---|---|
| SSA-ECA | SSA-WA | −0.2198 | −12.872 |
| SSA-ECA | SSA-CA | −0.1046 | −5.469 |
| SSA-ECA | SSA-ESA | 0.0130 | 0.638 |
| SSA2 | SSA-ECA | 0.4083 | 18.770 |
| SSA-WA | SSA-CA | −0.0822 | −3.873 |
| SSA-WA | SSA-ESA | 0.0157 | 0.763 |
| SSA-ESA | SSA-CA | −0.1870 | −9.343 |
| SSA-CA | SSA2 | 0.2156 | 11.495 |
Note.—Significant D values are in bold.
Z-score > 4: gene flow occurs between x and SSA4; Z-score < −4: gene flow occurs between x and y.
Fig. 5.
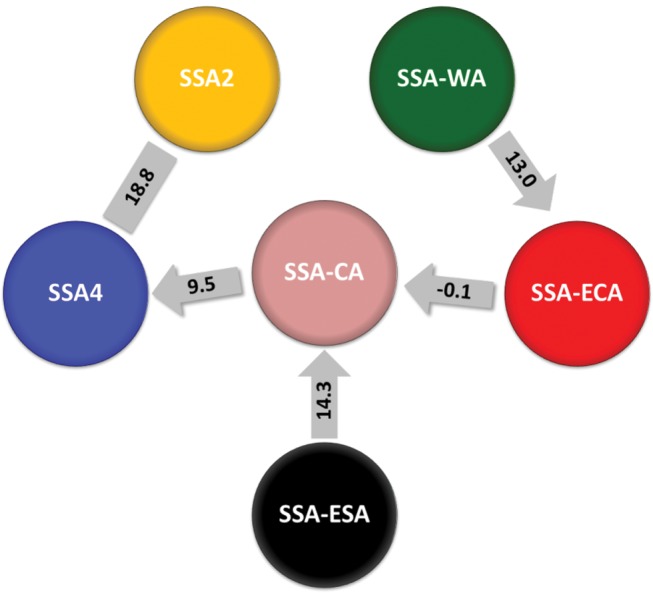
—Gene flow between the six SNP-based groups of cassava-colonizing Bemisia tabaci whiteflies sampled from eight African countries. Numbers shown in rectangle are Z-scores for D values listed in table 2 (D-statistic), whereas those in one-way arrows are Z-scores for D12 values listed in table 3 (partitioned D-statistic).
Partitioned D-statistic tests further supported gene flows between SSA-CA and SSA4, SSA-ESA and SSA-CA, and SSA-WA and SSA-ECA, as evidenced by the significant D12, whereas a weak signal of introgression was detected between SSA-CA and SSA-ECA (table 3). Based on the significance of D12, D1, and D2, the tests suggest that introgressions may have occurred predominantly from SSA-CA to SSA4, from SSA-ESA to SSA-CA, from SSA-WA to SSA-ECA, and from SSA-ECA to SSA-CA (table 3 and fig. 5).
Table 3.
Evidence of Gene Flow between Populations of Bemisia tabaci (Cassava Haplogroups) Determined Using Partitioned D-Statistic
| P1 | P2 | P31 | P32 | O | D12 (Z-score)a | D1 (Z-score) | D2 (Z-score) |
|---|---|---|---|---|---|---|---|
| SSA2 | SSA4 | SSA-CA1 | SSA-CA2 | OG | 0.7 (9.52) | 0 (0) | 0.17 (1.31) |
| SSA-WA | SSA-ECA | SSA-CA1 | SSA-CA2 | OG | −0.02 (−0.11) | −0.24 (−2.86) | 0.19 (1.15) |
| SSA2 | SSA-CA | SSA-ESA1 | SSA-ESA2 | OG | 0.63 (14.31) | 0.32 (1.58) | 0.47 (5.05) |
| SSA2 | SSA-ECA | SSA-WA1 | SSA-WA2 | OG | 0.68 (12.99) | 0.55 (4.4) | 0.56 (5.64) |
Note.—Significant D values are in bold.
Significant Z-score indicates gene introgression from P3 into P2 (P31 and P32 share derived alleles), nonsignificant Z-score indicates gene introgression from P2 into P31 or P32.
Geographical Distribution of Cassava B. tabaci
Three of the eight mtCOI haplogroups occurred in discrete geographical zones: SSA1-SG5 in south-western Nigeria, SSA2 in northern Cameroon and SSA1-SG3 was present in south-eastern Africa, including Madagascar (fig. 6A). Each of the five others shared geographical regions of occurrence with other haplogroups, although SSA4 and SSA3 were restricted to southern Cameroon/eastern RCA, whilst SSA1-SG1/2 was only reported from eastern DRC. SSA1-SG1 and SSA1-SG2 occurred in similar regions of East and Central Africa, although SSA1-SG2 was much less frequent. In addition to being the most frequently recorded haplogroup, SSA1-SG1 also occurred over the widest geographic range, as it was recorded from five of the eight countries sampled.
Fig. 6.

—Geographic distribution of cassava colonizing Bemisia tabaci in Africa based on mtCOI (A) and SNPs (B).
The most notable differences between the mtCOI and SNP maps (fig. 6A and B), were the greater uniformity in the area of occurrence of SSA-ECA in East/Central Africa, as well as the presence of SSA-CA in western Tanzania/south-eastern DRC and SSA-WA in Cameroon and RCA in addition to Nigeria. In the extremities of the geographical range of SSA1-SG1, populations had SNP identities of either SSA-WA (in Cameroon) or SSA-CA (in south-eastern DRC/western Tanzania). These results suggest that these putative virus pandemic-associated populations are hybridizing in regions into which they are expanding.
Discussion
The NextRAD sequencing approach used in this study made use of 7,453 SNPs to characterize the genetic relationships between diverse cassava-colonizing B. tabaci adult individuals collected from eight countries representing a large part of East, Central and West Africa. By comparing this SNP-based approach with mtCOI sequences for the same whitefly individuals, it was possible to make conclusions about the value of each for defining taxonomic designations. Most importantly, we have demonstrated that mtCOI sequences are an unreliable indicator of genetic identity for the set of cassava-colonizing whitefly samples considered in this study. The SNPs used here provided a detailed picture of the genomic variation amongst cassava-colonizing B. tabaci in Africa and clearly represent a much more robust means of categorizing genetic relationships than the single short sequence of mitochondrial DNA that has been used for taxonomic and molecular studies of B. tabaci (Brown 2000; Boykin et al. 2007). We also demonstrate that there is extensive genetic exchange between several of the six SNP-based groups, some of which were thought to be putative species based on mtCOI (Dinsdale et al. 2010).
MtCOI is Ineffective as a Taxonomic Marker for Cassava-Colonizing Bemisia tabaci in Africa
Accurate identification of pests and pathogens, especially cryptic complexes associated with crop plants, is critical for designing, developing, and implementing effective and sustainable control strategies. Different pest species can have contrasting degrees of invasiveness, cause different levels of damage, diverge in their ability to vector plant pathogens and have different capacities for insecticide resistance (Bickford et al. 2007). In this study, the mtCOI classification generated four major groups and five sub-groups in the major SSA1 group that are consistent with what has been reported in other studies involving cassava-colonizing B. tabaci (Legg et al. 2002, 2014; Sseruwagi et al. 2006; Mugerwa et al. 2012; Ghosh et al. 2015). However, the SNP-based classification differed from that of mtCOI.
The major mtCOI subgroup SSA1-SG1, which is the haplogroup associated with the severe CMD pandemic (Legg et al. 2014), had individuals reclassified into three SNP-based groups (SSA-ECA, SSA-CA, and SSA-WA). The SSA-CA and SSA-WA groups were previously unrecognized when using mtCOI. SSA-WA was most closely related to SSA-ECA, and was represented in this study by individuals from Nigeria (SSA1-SG5), Cameroon and RCA (SSA1-SG1 and SSA1-SG2). Those from Nigeria were initially assumed to be SSA1-SG3, since there is <0.6% divergence in their mtCOI sequences when compared with SSA1-SG3 individuals from coastal East Africa. However, these have been reported elsewhere as SSA1-SG5 (Ghosh et al. 2015).
The SNP-based phylogeny and STRUCTURE analysis revealed that there is very little shared identity between the Nigeria samples and those from coastal East Africa. SSA-CA is an intermediate population placed between SSA-WA and SSA-ESA and is the most diverse group with genetic identity derived from all the four ancestral populations detected using STRUCTURE. SSA-CA is a clear example of how mtCOI provides a misleading indication of genetic identity, since all of these individuals were identified as SSA1-SG1 with mtCOI, despite the fact that they have higher proportions of shared genetic identity with SSA4 and SSA-ESA. SSA2 individuals in this study were all from Cameroon, although SSA2 individuals identified using mtCOI have been found in countries as distantly separated as Uganda in East Africa (Legg et al. 2002; Mugerwa et al. 2012), and Spain and France in southern Europe (Hadjistylli et al. 2015). In view of the inconsistencies in the genotyping results obtained using the mtCOI and SNP methods in this study, it seems likely that SSA2 populations identified from different regions/countries using mtCOI may be genetically quite distinct. This is an important topic of future study. Individuals in the SSA4 group included one SSA3 (mtCOI) individual. This indicates that in spite of the species-level separation proposed for SSA3 and SSA4 based on mtCOI, they are likely to be closely related, although more SSA3 samples need to be examined before this conclusion can be confirmed.
The mitochondrial DNA marker mtCOI has been proposed as a means of delimiting species within the B. tabaci complex (Dinsdale et al. 2010), and has been widely used to describe genetic relationships amongst cassava-colonizing B. tabaci (Berry et al. 2004; Mugerwa et al. 2012; Legg et al. 2014; Ghosh et al. 2015; Tajebe et al. 2015). However, there are doubts about the ability of this single marker to effectively distinguish species within the complex (Legg et al. 2014; Hadjistylli et al. 2016). The findings from our study reveal that mtCOI does not provide a reliable indicator of the true genetic differences between cassava-colonizing B. tabaci haplogroups, for which the 7,453 SNPs markers produced a much more fine-scale and robust measure of genetic variability. This highlights the need to use multiple loci for species identification (Dupuis et al. 2012). Several studies have also reported conflicting patterns of species delimitation when single vs. multiple markers are used in phylogenetic studies. Attempts to delimit species/cryptic species using single locus markers, mostly mtCOI in B. tabaci (Hadjistylli et al. 2016), mosquitoes (Anopheles spp) (Foster et al. 2013), tephritid fruit flies (Frey et al. 2013), bees (Apis mellifera mellifera) (Pinto et al. 2014) and blow flies (Protocalliphora spp) (Whitworth et al. 2007), have revealed conflicting classifications compared with either multilocus genotyping or the use of morphological characteristics. There are other cases, however, where identifications of species or members of cryptic species complexes that were identified using single markers have corresponded with those where multiple markers were used. Hadjistylli et al. (2016) using microsatellites reported that most of the B. tabaci haplotypes were grouped similarly to clusters already described based on mtCOI.
Gene Flow among Cassava-Colonizing Bemisia tabaci Haplogroups
Gene flow analysis suggested that several of the SNP-based groups exchange genes, and that those groups that are geographically closer together have a higher propensity for genetic exchange. In addition, it was revealed that all groups are linked together through genetic exchange relationships. These two findings suggest the occurrence of a geographic cline in which the most distantly separated populations show no evidence of genetic exchange, but where those distant populations are linked through a set of connected intermediate populations, each of which shares genetic information with its immediate neighbors. A continuous geographic cline was observed along a south-east to north-west gradient, with extensive gene flow among populations in cassava colonizing B. tabaci. The populations at the extreme ends of the gradient (SSA2 and SSA-ESA) are the most genetically distinct and separated by the greatest distance both geographically as well as within the PCA analysis. They are connected through gene flow, however, since there is a chain of genetic exchange from SSA2 to SSA4 to SSA-CA to SSA-ESA. Communities of organisms that include humans, plants, animals, insects and microbes are known to form continuous clines connected through gene exchange under different geographical or environmental conditions (Manel et al. 2003; Vellend et al. 2014; Bhattarai et al. 2017; Stankowski et al. 2017; Classen et al. 2017; Leys et al. 2017). This is the first occasion, however, that such a pattern of continent-wide genetic exchange has been demonstrated for whiteflies.
Although this study demonstrated that geographic proximity was an important determining factor for genetic exchange, there were two significant exceptions. Firstly, no gene flow was demonstrated between SSA2 and any of the other genetic groups except SSA4, and secondly, SSA-WA, reported here from Nigeria in West Africa and from Cameroon and RCA in central Africa, showed high levels of genetic exchange with SSA-ECA, whose westernmost representative occurred in eastern DRC. It was notable, however, that SSA-WA individuals from Cameroon and RCA (geographically intermediate between Nigeria and eastern DRC) appeared from STRUCTURE to be hybrids between SSA-ECA and SSA-WA. Evidence of hybridization was strongest for the SSA-CA group for which there was evidence of gene flow with SSA-ECA, SSA-ESA, and SSA4. The SSA-ESA population appeared to be isolated from SSA-ECA in Tanzania, which is unsurprising since there is little cassava cultivated in the semi-arid central regions of the country that separate humid zones in the north-west and coastal east. By contrast, there are contiguous zones of cassava cultivation between the shores of Lake Tanganyika in both DRC and western Tanzania (from which SSA-CA was identified), and the shores of Lake Malawi, to the south of Lake Tanganyika but also part of the same western branch of the Rift Valley (from where SSA-ESA was reported). The lack of gene flow between SSA4 and SSA-WA groups despite their high proportions of shared genetic identity indicates that they share some common ancestral alleles (K = 4). However, this does not necessarily mean that there has to be gene flow (which is the transfer of alleles or genes from one population to another) between them. Further analysis of gene flow direction using the partitioned D-statistic revealed gene introgression from SSA-WA to SSA-ECA; SSA-CA to SSA4; SSA-ESA to SSA-CA, and SSA-ECA to SSA-CA. These findings indicate that most likely cause of high diversity in SSA-CA is introgession with genes from three populations: SSA-ESA, SSA-ECA, and SSA-WA.
Taken together, all of these pieces of evidence suggest that B. tabaci populations occurring on cassava in Africa are part of a single species within the B. tabaci species complex, and that these populations have strong signals of gene flow through recent history. It seems inevitable that mixing of populations over 10–100 s of km occurs, as migratory morphs of B. tabaci adults have been shown to fly considerable distances (up to 7 km) (Byrne et al. 1995), Circumstantial evidence has been presented for cassava B. tabaci population movements of up to 100 km/yr (Legg 2010), and whitefly nymphs may also be carried on leaf material inadvertently transported together with cassava stems. Given these features of whitefly population biology, it seems unsurprising that genetic evidence suggests that populations on cassava in Africa are part of an interconnected network of genetic exchange. An important practical consequence of this is that gene mutations that confer a selective advantage are likely to spread rapidly through cassava-colonizing B. tabaci populations. Although it has yet to be proven, this may well be the scenario for the super-abundance phenotype, which has spread through East and Central Africa from the late 1980s to the present day, and which continues to be the driving factor behind the expansion of the CMD and CBSD pandemics (Legg et al. 2011).
The SSA-ECA population identified in the current study maps geographically to the super-abundance phenotype of B. tabaci populations associated with the CMD pandemic, as well as recent spread through East and Central Africa of CBSD. Significant gene flow between this population and the two newly identified groups (SSA-CA and SSA-WA) is therefore a source of concern, since it raises the possibility that putative genes associated with super-abundance may be readily shared with neighboring populations. This highlights the potential threat of the continued spread of cassava virus pandemics westwards into western-central and West Africa. In addition to super-abundance, other examples of damaging traits that could potentially be exchanged through genetic transfer between cassava-colonizing whitefly populations are: broader host adaptation, enhanced migratory capability, and increased temperature tolerance. Acquiring and sharing one or more of these traits could greatly alter the dynamics of spread and severity of CMD and CBSD. This could also lead to the emergence of new virus strains adapted to transmission by new vector haplotypes.
The reported case of SSA1-SG1 colonizing non-cassava hosts in Uganda (Sseruwagi et al. 2006) is an example of how the emergence of distinct population types has the potential to impact other crops. Numerous studies of other systems have demonstrated how intraspecific genetic admixture as a result of gene flow and interbreeding can generate novel allelic combinations that can lead to increased population fitness and increased adaptive capacity with the potential for the emergence of invasive lineages capable of establishing in expanded regions (Rius and Darling 2014; Ellstrand and Rieseberg 2016; Payseur and Rieseberg, 2016). Invasive hybrid lineages produced in this way have been reported for a wide range of organisms, including: common ragweed (Ambrosia artemisiifolia) (Chun et al. 2011); fish (Cottus spp) (Nolte et al. 2005); ladybirds (Harmonia axyridis) (Turgeon et al. 2011); tamarisk shrubs (Tamarix spp) (Mayonde et al. 2016); and nematodes (Meloidogyne spp) (Lunt et al. 2014). Continuous monitoring of whitefly populations should therefore be an essential component in efforts towards combating cassava viruses in Africa.
Mating studies conducted by Maruthi et al. (2004) demonstrated successful interbreeding between mtCOI SSA2 individuals sourced from Uganda and mtCOI SSA1-SG1 individuals from the same country. By contrast, gene flow analyses from the current study indicate that there is little or no gene flow between SSA2 from Cameroon and any of the other groups, with the important exception of SSA4. Future studies should therefore use SNP-based analyses to type SSA2 individuals sampled from East Africa to determine whether their overall genetic make-up is closer to that of East or West African populations of B. tabaci. Such an effort will need to overcome the relative rarity of SSA2 in East Africa, however, as the proportion of this group declined greatly from 1997 to 2010 (Legg et al. 2014). In the study of Maruthi et al. (2004), it was reported that a cross between mtCOI SSA1 (Tanzania) and mtCOI SSA1 (Ghana) produced less progeny and a lower female to male ratio compared with mtCOI SSA2 (Uganda) × mtCOI SSA1 (Tanzania). This seems to be a contradiction, as it suggests that populations that appear to be more closely related based on their mtCOI sequences (SSA1 from Tanzania and SSA1 from Ghana) are less able to interbreed than others that look to be more distantly related (SSA1 from Tanzania and SSA2 from Uganda). The gene flow results that we have presented here highlight the point that geographical separation is more likely to be the basis for overall genetic divergence and the consequent likelihood of successful gene flow rather than mtCOI identity. Partitioned D-statistic analyses in the current study provided strong evidence for gene flow from SSA-CA to SSA4, even though the mtCOI sequence divergence between these two haplogroups was 8.6%, a figure that is much greater that the 3.5% proposed as the species boundary for haplogroups within the B. tabaci species complex (Dinsdale et al. 2010).
Conclusion
We conclude that although the current classification of cassava-colonizing B. tabaci haplogroups relies solely on the single locus mtCOI marker, this is not effective at distinguishing the major genetic groups of B. tabaci occurring on cassava in Africa, and provides a false indication of the true degree of genetic divergence between these groups. Therefore, more robust molecular markers will need to be developed for future phylogenetic studies on these whiteflies. Using more than 7,000 SNPs markers, we were able to establish a detailed picture of the genetic relatedness of cassava-colonizing B. tabaci in Africa, confirm the occurrence of six major genetic groups, and describe the relationship between these groups and former designations derived from mtCOI sequences. We also report extensive gene flow among the six SNP-based populations, some of which were presumed to be putative species based on mtCOI. Cassava whiteflies are already recognized to be the drivers of the dual pandemics of CMD and CBSD, which continue to have devastating impacts on cassava production throughout large parts of sub-Saharan Africa. Our findings demonstrate an on-going potential risk that genetic exchange may pose for the emergence of new “super-abundant” populations that may continue to expand the spread of cassava viruses to new regions or trigger the emergence of new virus strains in cassava. In addition, if new haplotypes have enhanced adaptation to other crop hosts, there is a risk that they might drive virus epidemics in these new hosts as efficient vectors. An immediate practical requirement following on from the results of our research, therefore, should be the development of a diagnostic tool based on the SNPs markers described here. This would be a valuable component of future initiatives to develop comprehensive management strategies to control whiteflies on cassava and the viruses that they transmit.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Khamis Issa for assisting with laboratory work, and Rudolph Shirima, Mathias Ndalahwa, Lava Kumar, Rachid Hanna, Simon Bigirimana, and Gervais Gashaka for assisting with field collection of whitefly specimens. Xiaowei Yang, Honghe Sun, Qiyue Ma, and Chen Jiao assisted with data analysis. This study was funded by the United States Agency for International Development Feed-the-Future programme (58-0210-3-012) to J.P.L. and Z.F. and was conducted within the framework of the Roots, Tubers, and Bananas Programme of the Consultative Group for International Agricultural Research.
Literature Cited
- Alagianagalingam MN, Ramakrishnan K.. 1966. Cassava mosaic in India. South Indian Hort. 14:71–72. [Google Scholar]
- Austin MDN. 1986. Scientists identify cassava virus. Asian Agribus. 3:10. [Google Scholar]
- Ballard JW, Whitlock MC.. 2004. The incomplete natural history of mitochondria. Mol Ecol. 13(4):729–744.http://dx.doi.org/10.1046/j.1365-294X.2003.02063.x [DOI] [PubMed] [Google Scholar]
- Barley AJ, Monnahan PJ, Thomson RC, Grismer LL, Brown RM.. 2015. Sun skink landscape genomics: assessing the roles of micro-evolutionary processes in shaping genetic and phenotypic diversity across a heterogeneous and fragmented landscape. Mol Ecol. 24(8):1696–1712. [DOI] [PubMed] [Google Scholar]
- Bazin E, Glemin S, Galtier N.. 2006. Population size does not influence mitochondrial genetic diversity in animals. Science 312(5773):570–572.http://dx.doi.org/10.1126/science.1122033 [DOI] [PubMed] [Google Scholar]
- Berry SD, et al. 2004. Molecular evidence for five distinct Bemisia tabaci (Homoptera: Aleyrodidae) geographic haplotypes associated with cassava in sub-Saharan Africa. Ann Entomol Soc Am. 97:852–859.http://dx.doi.org/10.1603/0013-8746(2004)097[0852:MEFFDB]2.0.CO;2 [Google Scholar]
- Bhattarai GP, et al. 2017. Biogeography of a plant invasion: genetic variation and plasticity in latitudinal clines for traits related to herbivory. Ecol Monogr. 87(1):57–75.http://dx.doi.org/10.1002/ecm.1233 [Google Scholar]
- Bickford D, et al. 2007. Cryptic species as a window on diversity and conservation. Trends Ecol Evol. 22(3):148–155.http://dx.doi.org/10.1016/j.tree.2006.11.004 [DOI] [PubMed] [Google Scholar]
- Bock KR, Woods RD.. 1983. The etiology of African cassava mosaic disease. Plant Dis. 67(9):994–995.http://dx.doi.org/10.1094/PD-67-994 [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120.http://dx.doi.org/10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boykin LM, et al. 2007. Global relationships of Bemisia tabaci (Hemiptera: Aleyrodidae) revealed using Bayesian analysis of mitochondrial COI DNA sequences. Mol Phylogenet Evol. 44(3):1306–1319.http://dx.doi.org/10.1016/j.ympev.2007.04.020 [DOI] [PubMed] [Google Scholar]
- Bradbury PJ, et al. 2007. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635.http://dx.doi.org/10.1093/bioinformatics/btm308 [DOI] [PubMed] [Google Scholar]
- Brown JK, Czosnek H.. 2002. Whitefly transmission of plant viruses. Adv Bot Res. 36:65–100. [Google Scholar]
- Brown JK. 2000. Molecular markers for the identification and global tracking of whitefly vector-begomovirus complexes. Virus Res. 71(1–2):233–260.http://dx.doi.org/10.1016/S0168-1702(00)00221-5 [DOI] [PubMed] [Google Scholar]
- Brown JK. 2010. Phylogenetic biology of the Bemisia tabaci sibling species group In: Stansly PA, Naranjo SE, editors. Bemisia: bionomics and management of a global pest. Dordrecht: Springer Press; p. 31–67. [Google Scholar]
- Byrne DN, Blackmer JL, Rathman RJ.. 1995. Migration and dispersal by the sweetpotato whitefly. Phytoparasitica 22:314. [Google Scholar]
- Chang CC, et al. 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4:7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, et al. 2016. The draft genome of whitefly Bemisia tabaci MEAM1, a global crop pest, provides novel insights into virus transmission, host adaptation, and insecticide resistance. BMC Biol. 14(1):110.http://dx.doi.org/10.1186/s12915-016-0321-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun YJ, Le Corre V, Bretagnolle F.. 2011. Adaptive divergence for a fitness‐related trait among invasive Ambrosia artemisiifolia populations in France. Mol Ecol. 20(7):1378–1388. [DOI] [PubMed] [Google Scholar]
- Classen A, Steffan-Dewenter I, Kindeketa WJ, Peters MK, Rezende E.. 2017. Integrating intraspecific variation in community ecology unifies theories on body size shifts along climatic gradients. Funct Ecol. 31(3):768–777. [Google Scholar]
- Collins RA, Cruickshank RH.. 2013. The seven deadly sins of DNA barcoding. Mol Ecol Resour. 13(6):969–975. [DOI] [PubMed] [Google Scholar]
- Danecek P, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158.http://dx.doi.org/10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JW, Davey JL, Blaxter ML, Blaxter MW.. 2010. RADSeq: next-generation population genetics. Brief Funct Genomics 9(5–6):416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Barro PJ. 2012. The Bemisia species complex: questions to guide future research. J Integr Agric. 11(2):187–196.http://dx.doi.org/10.1016/S2095-3119(12)60003-3 [Google Scholar]
- Delatte H, et al. 2006. Microsatellites reveal extensive geographical, ecological and genetic contacts between invasive and indigenous whitefly biotypes in an insular environment. Genet Res. 87(02):109–124.http://dx.doi.org/10.1017/S0016672306008135 [DOI] [PubMed] [Google Scholar]
- Dinsdale A, Cook L, Riginos C, Buckley YM, De Barro P.. 2010. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase I to identify species level genetic boundaries. Ann Ent Soc Am. 103(2):196–208. [Google Scholar]
- Dubern J. 1994. Transmission of African cassava mosaic geminivirus by the whitefly (Bemisia tabaci). Trop Sci. 34:82–91. [Google Scholar]
- Dupuis JR, Roe AD, Sperling FAH.. 2012. Multi-locus species delimitation in closely related animals and fungi: one marker is not enough. Mol Ecol. 21(18):4422–4436.http://dx.doi.org/10.1111/j.1365-294X.2012.05642.x [DOI] [PubMed] [Google Scholar]
- Eaton DA, Ree RH.. 2013. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst Biol. 62(5):689–706.http://dx.doi.org/10.1093/sysbio/syt032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellstrand NC, Rieseberg LH.. 2016. When gene flow really matters: gene flow in applied evolutionary biology. Evol Appl. 9(7):833–836.http://dx.doi.org/10.1111/eva.12402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson KJ, Conn JE, Bergo ES, Randel MA, Sallum MAM, Moreira LA.. 2015. Brazilian Anopheles darlingi Root (Diptera: Culicidae) clusters by major biogeographical region. PLoS One 10(7):e0130773.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterhuizen LL, et al. 2013. Genetic identification of members of the Bemisia tabaci cryptic species complex from South Africa reveals native and introduced haplotypes. J Appl Entomol. 137(1–2):122–135. [Google Scholar]
- Etter PD, Bassham S, Hohenlohe PA, Johnson EA, Cresko WA.. 2011. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol Biol. 772:157–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J.. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 14(8):2611–2620.http://dx.doi.org/10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- FAO 2014. FAOSTAT. Food and Agricultural Organization of the United Nations (FAO), Rome. Available from http://faostat.fao.org, last accessed November 1, 2017.
- Foster PG, et al. 2013. Phylogenetic analysis and DNA-based species confirmation in Anopheles (Nyssorhynchus). PLoS One 8(2):e54063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey JE, et al. 2013. Developing diagnostic SNP panels for the identification of true fruit flies (Diptera: Tephritidae) within the limits of COI-based species delimitation. BMC Evol Biol. 13:106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich DR, Torres-Jerez I, Bedford ID, Markham PG, Brown JK.. 1999. A phylogeographical analysis of Bemisia tabaci species complex based on mitochondrial DNA markers. Mol Ecol. 8(10):1683–1691. [DOI] [PubMed] [Google Scholar]
- Fu Z, et al. 2017. Using NextRAD sequencing to infer movement of herbivores among host plants. PLoS One 12(5):e0177742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Bouvaine S, Maruthi MN.. 2015. Prevalence and genetic diversity of endosymbiotic bacteria infecting cassava whiteflies in Africa. BMC Microbiol. 15:93.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RL, Batuman O, Webster CG, Adkins S.. 2015. Role of the insect supervectors Bemisia tabaci and Frankliniella occidentalis in the emergence and global spread of plant viruses. Annu Rev Virol. 2(1):67–93. [DOI] [PubMed] [Google Scholar]
- Gill RJ, Brown JK.. 2010. Systematics of Bemisia and Bemisia relatives: can molecular techniques solve the Bemisia tabaci complex conundrum—a taxonomist’s viewpoint In: Stansly PA, Naranjo SE, editors. Bemisia: bionomics and management of a global pest. Dordrecht: Springer Press; p. 5–29. [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321.http://dx.doi.org/10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Hadjistylli M, Roderick GK, Brown JK, Zhang Y.. 2016. Global population structure of a worldwide pest and virus vector: Genetic diversity and population history of the Bemisia tabaci sibling species group. PLoS One 11(11):e0165105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjistylli M, Roderick GK, Gauthier N.. 2015. First report of the Sub-Saharan Africa 2 species of the Bemisia tabaci complex in the Southern France. Phytoparasitica 43(5):679–687.http://dx.doi.org/10.1007/s12600-015-0480-3 [Google Scholar]
- Helyar SJ, et al. 2011. Application of SNPs for population genetics of nonmodel organisms: new opportunities and challenges. Mol Ecol Resour. 11:123–136.http://dx.doi.org/10.1111/j.1755-0998.2010.02943.x [DOI] [PubMed] [Google Scholar]
- Holsinger KE, Weir BS.. 2009. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat Rev Genet. 10(9):639–650.http://dx.doi.org/10.1038/nrg2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YG, Robinson DJ, Harrison BD.. 1993. Nucleotide sequence evidence for the occurrence of three distinct whitefly-transmitted geminiviruses in cassava. J Gen Virol. 74(11):2437–2443.http://dx.doi.org/10.1099/0022-1317-74-11-2437 [DOI] [PubMed] [Google Scholar]
- Hurst GD, Jiggins FM.. 2005. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc R Soc Lond B Biol Sci. 272(1572):1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICTV. 2017. Virus Taxonomy: 2016 Release. Available from https://talk.ictvonline.org/taxonomy/, last accessed November 1, 2017.
- Jarvis A, Ramirez-Villegas J, Campo BVH, Navarro-Racines C.. 2012. Is cassava the answer to African climate change adaptation?. Trop Plant Biol. 5(1):9–29. [Google Scholar]
- Jones DR. 2003. Plant viruses transmitted by whiteflies. Eur J Plant Pathol. 109(3):195–219. [Google Scholar]
- Legg JP. 1996. Host-associated strains within Ugandan populations of the whitefly Bemisia tabaci (Genn.), (Hom., Aleyrodidae). J Appl Entomol. 120(1–5):523–527. [Google Scholar]
- Legg JP, Fauquet CM.. 2004. Cassava viruses in Africa. Plant Mol Biol. 56(4):585–599.http://dx.doi.org/10.1007/s11103-004-1651-7 [DOI] [PubMed] [Google Scholar]
- Legg JP, et al. 2015. Cassava virus diseases: biology, epidemiology and management. Adv Virus Res. 91:85–142. [DOI] [PubMed] [Google Scholar]
- Legg JP, et al. 2014. Spatio-temporal patterns of genetic change amongst populations of cassava Bemisia tabaci whiteflies driving virus pandemics in East and Central Africa. Virus Res. 186:61–75.http://dx.doi.org/10.1016/j.virusres.2013.11.018 [DOI] [PubMed] [Google Scholar]
- Legg JP, et al. 2011. Comparing the regional epidemiology of the cassava mosaic and cassava brown streak pandemics in Africa. Virus Res. 159(2):161–170. [DOI] [PubMed] [Google Scholar]
- Legg JP. 2010. Epidemiology of a whitefly-transmitted cassava mosaic geminivirus pandemic in Africa In: Stansly PA, Naranjo SE, editors. Bemisia: bionomics and management of a global pest. Dordrecht: Springer Press; p. 233–257. [Google Scholar]
- Legg JP, Owor B, Sseruwagi P, Ndunguru J.. 2006. Cassava mosaic virus disease in East and Central Africa: epidemiology and management of a regional pandemic. Adv Virus Res. 67:355–418. [DOI] [PubMed] [Google Scholar]
- Legg JP, French R, Rogan D, Okao-Okuja G, Brown JK.. 2002. A distinct, invasive Bemisia tabaci (Gennadius) (Hemiptera: Sternorrhyncha: Aleyrodidae) genotype cluster is associated with the epidemic of severe cassava mosaic virus disease in Uganda. Mol Ecol. 11(7):1219–1229. [DOI] [PubMed] [Google Scholar]
- Leys M, Keller I, Robinson CT, Räsänen K.. 2017. Cryptic lineages of a common alpine mayfly show strong life‐history divergence. Mol Ecol. 26(6):1670–1686. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760.http://dx.doi.org/10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SS, Colvin J, De Barro PJ.. 2012. Species concepts as applied to the whitefly Bemisia tabaci systematics: how many species are there?. J Integr Agric. 11(2):176–186.http://dx.doi.org/10.1016/S2095-3119(12)60002-1 [Google Scholar]
- Lunt DH, Kumar S, Koutsovoulos G, Blaxter ML.. 2014. The complex hybrid origins of the root knot nematodes revealed through comparative genomics. PeerJ 2:e356.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel S, Schwartz MK, Luikart G, Taberlet P.. 2003. Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol. 18(4):189–197.http://dx.doi.org/10.1016/S0169-5347(03)00008-9 [Google Scholar]
- Mardis ER. 2008. The impact of next-generation sequencing technology on genetics. Trends Genet. 24(3):133–141.http://dx.doi.org/10.1016/j.tig.2007.12.007 [DOI] [PubMed] [Google Scholar]
- Maruthi MN, et al. 2005. Transmission of Cassava brown streak virus by Bemisia tabaci (Gennadius). J Phytopathol. 153(5):307–312. [Google Scholar]
- Maruthi MN, et al. 2004. Reproductive incompatibility and cytochrome oxidase I gene sequence variability amongst host‐adapted and geographically separate Bemisia tabaci populations (Hemiptera: Aleyrodidae). Syst Entomol. 29(4):560–568. [Google Scholar]
- Maruthi MN, et al. 2017. The role of the whitefly, Bemisia tabaci (Gennadius), and farmer practices in the spread of cassava brown streak ipomoviruses. J Phytopathol. 1–11. doi: 10.1111/jph.12609, last accessed November 1, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayonde SG, Cron GV, Gaskin JF, Byrne MJ.. 2016. Tamarix (Tamaricaceae) hybrids: the dominant invasive genotype in southern Africa. Biol Invasions 18(12):3575–3594.http://dx.doi.org/10.1007/s10530-016-1249-4 [Google Scholar]
- Mbanzibwa DR, et al. 2009. Genetically distinct strains of cassava brown streak virus in the Lake Victoria basin and the Indian Ocean coastal area of East Africa. Arch Virol. 154(2):353–359. [DOI] [PubMed] [Google Scholar]
- Meier JI, et al. 2017. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat Commun. 8:14363.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugerwa H, et al. 2012. Genetic diversity and geographic distribution of Bemisia tabaci (Gennadius)(Hemiptera: Aleyrodidae) genotypes associated with cassava in East Africa. Ecol Evol. 2(11):2749–2762.http://dx.doi.org/10.1002/ece3.379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte AW, Freyhof J, Stemshorn KC, Tautz D.. 2005. An invasive lineage of sculpins, Cottus sp.(Pisces, Teleostei) in the Rhine with new habitat adaptations has originated from hybridization between old phylogeographic groups. Proc R Soc Lond B Biol Sci. 272(1579):2379–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Loughlin SM, et al. 2014. Genomic analyses of three malaria vectors reveals extensive shared polymorphism but contrasting population histories. Mol Biol Evol. 31:889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil LB, Legg JP, Kanju E, Fauquet CM.. 2015. Cassava brown streak disease: a threat to food security in Africa. J Gen Virol. 96(Pt 5):956–968. [DOI] [PubMed] [Google Scholar]
- Patterson N, et al. 2012. Ancient admixture in human history. Genetics 192(3):1065–1093.http://dx.doi.org/10.1534/genetics.112.145037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payseur BA, Rieseberg LH.. 2016. A genomic perspective on hybridization and speciation. Mol Ecol. 25(11):2337–2360.http://dx.doi.org/10.1111/mec.13557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto MA, et al. 2014. Genetic integrity of the Dark European honey bee (Apis mellifera mellifera) from protected populations: a genome-wide assessment using SNPs and mtDNA sequence data. J Apic Res. 53(2):269–278.http://dx.doi.org/10.3896/IBRA.1.53.2.08 [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P.. 2000. Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillery E, Quenez O, Peterlongo P, Plantard O.. 2014. Development of genomic resources for the tick Ixodes ricinus: Isolation and characterization of single nucleotide polymorphisms. Mol Ecol Resour. 14(2):393–400. [DOI] [PubMed] [Google Scholar]
- Razkin O, et al. 2016. Species limits, interspecific hybridization and phylogeny in the cryptic land snail complex Pyramidula: the power of RADseq data. Mol Phylogenet Evol. 101:267–278.http://dx.doi.org/10.1016/j.ympev.2016.05.002 [DOI] [PubMed] [Google Scholar]
- Rius M, Darling JA.. 2014. How important is intraspecific genetic admixture to the success of colonizing populations? Trends Ecol Evol. 29(4):233–242. [DOI] [PubMed] [Google Scholar]
- Russello MA, Waterhouse MD, Etter PD, Johnson EA.. 2015. From promise to practice: pairing non-invasive sampling with genomics in conservation. PeerJ 3:e1106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Ji H.. 2008. Next-generation DNA sequencing. Nat Biotechnol. 26(10):1135–1145.http://dx.doi.org/10.1038/nbt1486 [DOI] [PubMed] [Google Scholar]
- Sseruwagi P, et al. 2005. Genetic diversity of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) populations and presence of the B biotype and a non-B biotype that can induce silverleaf symptoms in squash, in Uganda. Ann Appl Biol. 147(3):253–265. [Google Scholar]
- Sseruwagi P, et al. 2006. Colonisation of non-cassava plant species by cassava whiteflies (Bemisia tabaci) (Gennadius) (Hemiptera: Aleyrodidae) in Uganda. Entomol Exp Appl. 119(2):145–153. [Google Scholar]
- Stankowski S, Sobel JM, Streisfeld MA.. 2017. Geographic cline analysis as a tool for studying genome‐wide variation: a case study of pollinator‐mediated divergence in a monkeyflower. Mol Ecol. 26(1):107–122. [DOI] [PubMed] [Google Scholar]
- Storey HH, Nichols RFW.. 1938. Studies on the mosaic of cassava. Ann Appl Biol. 25(4):790–806.http://dx.doi.org/10.1111/j.1744-7348.1938.tb02354.x [Google Scholar]
- Szulkin M, Gagnaire P-A, Bierne N, Charmantier A.. 2016. Population genomic footprints of fine-scale differentiation between habitats in Mediterranean blue tits. Mol Ecol. 25(2):542–558. [DOI] [PubMed] [Google Scholar]
- Tajebe LS, et al. 2015. Abundance, diversity and geographic distribution of cassava mosaic 1 disease pandemic associated Bemisia tabaci in Tanzania. J Appl Entomol. 139(8):627–637. [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S.. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay WT, Evan GA, Boykin LM, De Barro PJ.. 2012. Will the real Bemisia tabaci please stand up?. PLoS One 7(11):e50550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thresh JM, Otim-Nape GW, Legg JP, Fargette D.. 1997. African cassava mosaic disease: the magnitude of the problem. Afr J Root Tuber Crops 2:13–19. [Google Scholar]
- Tocko-Marabena BK, et al. 2017. Genetic diversity of Bemisia tabaci species colonizing cassava in Central African Republic characterized by analysis of cytochrome c oxidase subunit I. PLoS One 12(8):e0182749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon J, et al. 2011. Experimental evidence for the phenotypic impact of admixture between wild and biocontrol Asian ladybird (Harmonia axyridis) involved in the European invasion. J Evol Biol. 24(5):1044–1052.http://dx.doi.org/10.1111/j.1420-9101.2011.02234.x [DOI] [PubMed] [Google Scholar]
- Vellend M, et al. 2014. Drawing ecological inferences from coincident patterns of population‐and community‐level biodiversity. Mol Ecol. 23(12):2890–2901. [DOI] [PubMed] [Google Scholar]
- Viscarret MM, et al. 2003. Mitochondrial DNA evidence for a distinct clade of New World Bemisia tabaci (Genn.) (Hemiptera: Aleyrodidae) from Argentina and Bolivia, and presence of the Old World B biotype in Argentina. Ann Entomol Soc Am. 96:65–72. [Google Scholar]
- White BP, Pilgrim EM, Boykin LM, Stein ED, Mazor RD.. 2014. Comparison of four species-delimitation methods applied to a DNA barcode data set of insect larvae for use in routine bioassessment. Freshw Sci. 33(1):338–348. [Google Scholar]
- Whitworth TL, Dawson RD, Magalon H, Baudry E.. 2007. DNA barcoding cannot reliably identify species of the blowfly genus Protocalliphora (Diptera: Calliphoridae). Proc R Soc Lond B Biol Sci. 274(1619):1731–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S, et al. 2010. Analysis of cassava brown streak viruses reveals the presence of distinct virus species causing cassava brown streak disease in East Africa. J Gen Virol. 91(Pt 5):1365–1372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.