

Abstract
Hybridization between closely related plant species is widespread, but the outcomes of hybridization are not fully understood. This study investigates phylogenetic relationships and the history of hybridization in the wild tomato clade (Solanum sect. Lycopersicon). We sequenced RNA from individuals of 38 different populations and, by combining this with published data, build a comprehensive genomic data set for the entire clade. The data indicate that many taxa are not monophyletic and many individuals are admixed due to repeated hybridization. The most polymorphic species, Solanum peruvianum, has two genetic and geographical subpopulations, while its sister species, Solanum chilense, has distinct coastal populations and reduced heterozygosity indicating a recent expansion south following speciation from S. peruvianum circa 1.25 Ma. Discontinuous populations west of 72° are currently described as S. chilense, but are genetically intermediate between S. chilense and S. peruvianum. Based upon molecular, morphological, and crossing data, we test the hypothesis that these discontinuous “S. chilense” populations are an example of recombinational speciation. Recombinational speciation is rarely reported, and we discuss the difficulties in identifying it and differentiating between alternative demographic scenarios. This discovery presents a new opportunity to understand the genomic outcomes of hybridization in plants.
Keywords: speciation, gene flow, hybridization, Lycopersicon, population genetics
Introduction
By some estimates, >25% of plant species hybridize in the wild, and the prevalence of hybridization has led some botanists to question if Mayr's Biological Species Concept (BSC) is appropriate for plants (Coyne and Orr 2004; Ehrlich and Raven 1969; Levin 1979; Mallet 2005; Mayr 1942). The BSC defines species as actual or potentially interbreeding populations which are reproductively isolated from other such groups. At first, the observation of interspecific hybridization does seem to be at odds with this definition, but two species can remain distinct, even if they occasionally hybridize. For example, hybrids may be produced very rarely, the hybrid embryos may abort, or the hybrids themselves may be unfit or sterile. Because of the distinction between hybridization and gene flow, most authors do not take a hard line on hybridization between otherwise “good”’ species as defined by the BSC (Coyne and Orr 2004). Furthermore, as shown by Rieseberg et al. (2006), most taxonomic plant species fit the rubric of the BSC, in that they represent reproductively independent groups.
Speciation can be a long-term process and breeding barriers will necessarily be incomplete when the taxa in question are young. In this case, hybridization can happen naturally during the divergence process and is not necessarily unexpected (Grant 1981). This idea is supported by hybridization being widespread in many taxonomic groups, including between humans and our closest relatives (Racimo et al. 2015). Hybridization is, however, more common in some groups than others. For example, nearly one in ten bird species hybridize with at least one other species (Grant and Grant 1992). Hybridization is also relatively common in flowering plants with circa 0.09 hybrids per nonhybrid species (Whitney et al. 2010).
One of the most important consequences of hybridization is the introduction of novel alleles through introgression (Anderson 1949). If first generation hybrids are not completely sterile, then they can backcross to one or both of the parental taxa, and recurrent generations of backcrossing can lead to introgression—the incorporation of one species’ alleles into the background genome of another. Introgression can transfer advantageous alleles between species, as in the case of Neanderthal introgressions which constitute 1–2% of present-day nonAfrican genomes or Denisovan introgressions into the ancestors of Tibetans (Racimo et al. 2015). Introgressions are also common in plants. For example, introgressions from oxford ragwort (Senecio squalidus) potentially increase the rate of outcrossing in the common groundsel (Strongylus vulgaris) by reintroducing a phenotype (ray flowers) into this otherwise predominately selfing species (Kim et al. 2008).
A second important outcome of hybridization is hybrid or “reticulate” speciation which combines two parental species to create a new one (Rieseberg and Willis 2007). In plants, hybrid speciation is normally associated with a doubled chromosome number in the hybrid compared with the parental taxa (Soltis and Soltis 2012). In this case, breeding barriers between the hybrid and its parents are immediate because backcrosses are sterile due to abnormal meiosis (Paun et al. 2009). However, hybrid speciation without a change in chromosome number can also occur and is known as recombinational speciation (Grant 1981). The rate of recombinational speciation is not known, but there are several well-documented examples in plants (Coyne and Orr 2004; Paun et al. 2009).
The tomato clade (Solanum sect. Lycopersicon) split from the nearest neighboring section (Juglandifolia) circa 5.8–8 Ma, but likely diversified only circa 2.5 Ma (Pease et al. 2016; Särkinen et al. 2013). Up to 13 species are recognized and these are distributed along the western coast of South America. While many species do have overlapping ranges, they have species-specific niche preferences (Nakazato et al. 2010).
Within the clade of wild tomatoes, two well-defined groups are distinguished based upon fruit color. The red-fruited clade, containing the domesticated tomato, is made up of predominantly selfing species, while the green-fruited clade contains many outcrossing species. All species are diploid (n = x = 12) and share high levels of chromosomal synteny, although cytological differences between some species are detectable (Anderson et al. 2010; Chetelat and Ji 2007; Peralta et al. 2008). Many species, including relatively distant taxa within the clade, are compatible in test crosses. Others are incompatible, normally resulting in aborted embryos and hybrid breakdown when intercrossed (postzygotic incompatibility).
Considerable intraspecific diversity in plant size, shape, habit, and other characters has made systematics following the morphological species concept difficult in wild tomato. Furthermore, incomplete lineage sorting (ILS) and introgressions continue to present a challenge for molecular taxonomic studies, even as new methods have been adopted (Breto et al. 1993; Pease et al. 2016; Zuriaga et al. 2009). Thus, although wild tomato species have been the focus of numerous morphological, phylogenetic, and biosystematic studies, the ancestry and definition of specific taxa within the clade remain unresolved.
This study is focused on sister species, Solanum chilense Dunal (S. chi) and Solanum peruvianum L. (S.per) which are the most polymorphic in wild tomato. These species are both perennial, green-fruited, and self-incompatible. They are sympatric in southern Peru, but display differences in morphology, particularly leaflet shape (fig. 1a and b), and S.chi has several adaptations for more arid habitats including grayish pubescence and deep roots (Moyle 2008). Their most recent common ancestor (MRCA) has been dated to between 0.5 and 2 Ma making them quite young (Pease et al. 2016; Stäedler et al. 2008). Hybrid seed failure is the predominant outcome when they are intercrossed in the lab, but several studies have found genetic evidence for allele sharing in the wild, including the suggestion of speciation under residual gene flow (Stäedler et al. 2005, 2008).
Fig. 1.—

Diversity of Solanum peruvianum (S.per) and Solanum chilense (S.chi). (a) Differences in flower morphology between S.per populations including one Solanum corneliomulleri (S.cor). Note that the extended stigma and large yellow petals are indicative of outcrossing. (b) Differences in leaf morphology within and between S.per, S.chi, and S.cor. (c) Collection locations of populations sampled in this study. Horizontal and vertical lines indicate the distribution of S.per and S.chi respectively.
In this study, we sequenced the RNA transcriptomes from different populations of S.chi and S.per. We aimed to determine the divergence time of the two species, test the hypothesis of speciation under residual gene flow, assemble a data set that can serve as a null model for evolutionary studies, and—by including comparable public data—tackle taxonomic problems in the entire tomato clade that have remained unresolved without a comprehensive sampling of S.chi and S.per.
By combining our data with comparable data sets, we recovered the main phylogenetic groups within the clade, but also discovered taxonomic conflicts not evident before, including even more evidence of hybridization in multiple taxa. Surprisingly, the genomic analyses reveal little allele sharing between S.chi and S.per, with the exception of populations described as S.chi near Arequipa, Peru. We tested the hypothesis that this group of populations represents a recent example of hybrid speciation, and discuss both how natural hybridization can generate new genetic entities within a clade and the difficulties in distinguishing hybrid speciation from alternate demographic scenarios.
Materials and Methods
Transcriptome Data
We sequenced 18 S.chi and 17 S.per covering the known distribution of these species (fig. 1c;supplementary table S1, Supplementary Material online). Two outgroups and one Solanum corneliomulleri J. F. Macbr. (S.cor) were also sequenced. Seeds originating from natural populations in Peru and Chile were provided by the Charles M. Rick Tomato Genetics Resource Center (TGRC), University of California, Davis (tgrc.ucdavis.edu). The seeds were germinated following TGRC guidelines and grown in a glasshouse in Düsseldorf, Germany.
We chose mRNA sequencing due to the high level (>76%) of heterochromatic repeats that would constitute a majority of the reads if genomic DNA were sequenced (Peterson et al. 1996). Leaf RNA was extracted from one individual per accession using the RNeasy Plant Mini Kit (Qiagen, Germany). The leaf mRNA was then prepared with the TruSeq RNA Library Preparation Kit v2 or the NEBNext Ultra Directional RNA Library Prep Kit for Illumina and sequenced with Illumina HiSeq2500 100-nt paired-end technology at the Max Planck Genome Center (Cologne, Germany). Final libraries had a minimum of 35 million reads with a median of 92.9 million 100-nt reads following quality control and adapter removal.
Additional Data
To systematically evaluate the wild tomato clade, 28 genomic (ENA PRJEB5235) and 14 transcriptomic (NCBI Bioproject PRJNA305880) Illumina libraries were downloaded (Aflitos et al. 2014; Pease et al. 2016) and co-analyzed. By including this data, 80 individuals from 13 species (including the two outgroups) are represented (supplementary table S1, Supplementary Material online). Additional genomic data from Lin et al. (2014) was included for one ∂a∂i analysis (supplementary table S2, Supplementary Material online).
Read Mapping to the Reference Genome and SNP Calling
Read libraries were individually mapped to the Solanum lycopersicum Heinz 1706 reference genome release SL2.50 with BWA v0.7.10 (Li and Durbin 2009; Sato et al. 2012). We allowed up to 5% divergence from the reference and disallowed insertions >25 (options: -k 1 -l 25 -n 0.05 -e 15 -i 10). For the mRNA libraries only, reads not mapped by BWA were remapped in TopHat2 (Kim et al. 2013). Alignment files were then sorted and indexed using SAMtools v0.1.19 (Li et al. 2009). All nonuniquely aligned reads and reads with mapping quality <30 were removed.
To identify polymorphisms, we used the multiallelic caller of BCFtools v1.3.1. Indels were removed, and the resulting unphased files were processed in BEAGLE 4.1 to infer haplotypes (Browning and Browning 2016). Positions with coverage <10 in any single individual were treated as missing data, and positions with >50% missing data across all individuals were excluded. Polymorphisms were categorized as 5′UTR, coding sequence (CDS), intron, 3′UTR, or intergenic using the reference GFF. Polymorphisms mapping to CDS were further characterized as synonymous, nonsynonymous, changing a start codon, changing a stop codon, or nonsense mutations.
Interspecific Relationships and Genetic Groups within Wild Tomato
We inferred the relationships of all species by maximum-likelihood (ML) using 429,881 synonymous positions. We ran 100 bootstraps under the rapid bootstrap algorithm of RAxML v8.2.9 with a GTR-GAMMA model of nucleotide substitution and an ascertainment bias for invariable sites (Stamatakis 2014). The two allied Solanum species were used to root the trees which were visualized in FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/; last accessed January 2017).
A second phylogenetic analysis was implemented with SNAPP v1.2.5 which uses a coalescent model without the need to directly infer trees (Bryant et al. 2012). To reduce computational time, we randomly selected 1,250 synonymous polymorphisms and excluded allied Solanums and the “Hirsutum” group, which is the first diverging lineage in sect. Lycopersicon. XML input files were created with the default parameters of BEAUti v2.3.1 (Drummond et al. 2012). Following 1 million MCMC iterations in BEAST v2.3.1 and examination of log files in Tracer v1.6.0 (Rambaut et al. 2014) the burn-in was set to 100,000 iterations. Coalescent trees were visualized with Densitree v2.2.2 (Bouckaert et al. 2014).
The model-based clustering software STRUCTURE v2.3.4 was used to determine the number of genetic clusters (K) in sect. Lycopersicon (Pritchard et al. 2000). To find K, we first removed the “Hirsutum” group and generated 10 independent sets of 10,000 randomly chosen synonymous polymorphisms from positions with ≥10 coverage and <10% missing data. We modeled K = 1–7 with a burn-in period of 100,000 followed by 100,000 MCMC steps under the admixture model. The greedy algorithm from CLUMPP assigned average membership coefficients from 10 independent runs (Jakobsson and Rosenberg 2007). The program STRUCTURE HARVESTER v0.6.94 was used to calculate the ad hoc statistic ΔK (Earl and Vonholdt 2012). Evanno et al. (2005) showed that ΔK, which is derived from the second order change in the log-likelihood, is accurate at finding the true K at the maximum hierarchical level.
Because of evidence for hybridization between taxa (i.e. intermediate individuals) from STRUCTURE, we built a reticulate network using SplitsTree4 (Huson and Bryant 2006). Reticulate networks do not require a tree-like model which allows more complicated evolutionary histories to be represented. Similarly, a principle component analysis (PCA) on synonymous polymorphisms was calculated using the R package APE. The PCA was run using the prcomp function (Paradis et al. 2004; R Core Team 2014).
Within-Species Nucleotide Diversity and Individual Heterozygosity
To estimate pairwise nucleotide diversity (π) within species, we derived accession-specific genomes for all individuals using the reference genome and called SNPs. Sites were called for all positions with coverage ≥10 reads. CDSs based on the ITAG2.4 genome annotation were then extracted and π at synonymous (πsyn) and at nonsynonymous (πnonsyn) sites were calculated following Nei and Gojobori (1986). Heterozygosity was calculated for all individuals by dividing the total number of heterozygous positions with ≥10 coverage by all positions with ≥10 coverage in that individual. FST was calculated between different species and subgroups with VCFtools v0.1.13 (Danecek et al. 2011).
Modeling the Joint Demography of S.chi and S.per
We estimated the joint demography of S.chi and S.per using ∂a∂i (Gutenkunst et al. 2009). Demographic inference in ∂a∂i uses a diffusion-based approach to model the distribution of multi-population allele frequency spectra. The joint site frequency spectrum (JSFS) was derived from 289,563 synonymous polymorphisms that had a nonzero allele frequency in S.chi or S.per. Individuals of Solanum huaylasense Peralta (S.hua) and S.cor were included as S.per by default, but any individual with >10% mixed ancestry in the STRUCTURE analysis was excluded.
Demographic parameters were estimated in a simple model that had an ancestral speciation event at time tau (T1) followed by the potential for population size changes and migration between species (Supplementary Information, Supplementary Material online). We did 100 independent 10,000-iteration runs from randomized starting parameter values to find the optimum global parameter values and 100 conventional bootstraps to determine confidence intervals. To normalize for sequence length, we divided theta by the total number of potentially synonymous positions in all individuals (Supplementary Information, Supplementary Material online).
Test Crosses
Test crosses were made to determine if reproductive barriers were present between two cryptic hybrid populations (see Results) and their parental species. Multiple individuals from the following accessions were grown: Hybrid-LA1930, Hybrid-LA1932, S.chi-LA2930, S.chi-LA1960, S.per-LA1954, S.per-LA2732, S.per-LA0153, S.per-LA2964, and S.cor-LA1274. Test crosses were done for all combinations with the exception of S.chi-LA1960 and Hybrid-LA1930, which failed to flower. From the remaining 7 accessions, 815 flowers were bagged and hand-pollinated. Following a minimum of 50 days, fruit diameter, the number of seeds per fruit, and the number of seed-like structures (SLS; i.e. ovules not completely developed into seeds) were counted. The germination of seeds and some larger SLSs were determined following TGRC germination protocols (tgrc.ucdavis.edu; last accessed August 2017).
Results
We analyzed 80 individuals from 11 wild tomato species and two outgroups. On average 78% of the mRNA reads were uniquely aligned to the Heinz 1706 reference genome with a mapping quality ≥30 (supplementary fig. S1a and table S1, Supplementary Material online). In contrast, likely due to heterochromatic repeats, only 56% of the reads from the genomic data (ENA PRJEB5235) aligned uniquely. A mean of 63 million positions per individual had ≥10 mapping coverage for mRNA data (mean of 285 million positions for genomic data), but only 8.04 million positions had a ≥10 coverage in all individuals (supplementary figs. S1b and S2, Supplementary Material online). By allowing an intermediate amount of missing data (50%) as recommended by Streicher et al. (2016), the number of positions increases to 35.64 million. This represents 4.2% of the total genome, but contains >66% of coding positions. In total, 4,866,729 bi-allelic polymorphisms with ≥10 coverage and ≤50% missing data were identified. This included 1,385,292 synonymous, 1,243,177 nonsynonymous, 903 start codon change, 2,410 stop codon change, and 62,000 nonsense mutations. The remaining polymorphisms were intergenic, 5′UTR, intronic, or 3′UTR (supplementary fig. S3, Supplementary Material online).
Distribution of Genetic Variation across Groups
Three major genetic groups were consistently identified (figs. 2b and 3, supplementary figs. S4 and S5, Supplementary Material online). One group contained only individuals of S.chi, one group contained individuals of S.per sensu lato (including S.cor and S.hua) and the third group contained all of the mostly autogamous taxa in the Esculentum and Arcanum species groups (named here Arc + Esc, fig. 2). Sequence polymorphism was the greatest for the S.per group (πsyn = 1.69%) followed by S.chi (1.27%). The autogamous group (Arc + Esc) had the lowest diversity (πsyn = 1.04%). The level of nonsynonymous polymorphism was similar, but considerably lower with πnon = 0.22% (S.per), πnon = 0.18% (S.chi), and πnon = 0.16% (Arc + Esc). Average individual heterozygosity (proportion of heterozygous positions per individual) was greatest in the allogamous-SI S.per (0.51%) and S.chi (0.43%; supplementary fig. S6 and table S3, Supplementary Material online).
Fig. 2.—

Phylogeny of sect. Lycopersicon. (a) A maximum-likelihood phylogeny from all accessions excluding those with >10% admixture according to STRUCTURE. The species groups are delineated by black lines and labeled. Node labels give bootstrap support. (b) Coalescent phylogeny with all accessions excluding the early diverging “Hirsutum” group. Taxon abbreviations: Solanum arcanum (S.arc), Solanum chmielewskii (S.chm), Solanum corneliomulleri (S.cor), Solanum chilense (S.chi), Solanum galapagense (S.gal), Solanum huaylasense (S.hua), Solanum habrochaites (S.hab), Solanum lycopersicoides (S.lyc), Solanum neorickii (S.neo), Solanum ochranthum (S.och), Solanum pennellii (S.pen), Solanum peruvianum (S.per), Solanum pimpinellifolium (S.pim).
Solanum peruvianum showed strong population subdivision, evident in nearly all analyses (figs. 2 and 3). One subpopulation contains low-elevation collections from the sandy coast and/or Lomas formations of the Peruvian desert (fig. 4). These populations (LA1951, LA1954, LA1336, LA1333, LA1474, LA2964, and LA3218) are located between 14° and 17° S, and are <5 km from the coast and collected at sites at <600 m in elevation (with the exception of LA1474 which is located at 1,300 m, 14 km from the coast). The second subpopulation has collections distributed across many different watersheds, mainly in central Peru, but also includes three S.per from northern Chile, S.cor-LA0118, S.cor-LA1274, and all of S.hua. This noncoastal subpopulation has higher nucleotide diversity (πsyn = 1.57%) than the coastal one (πsyn = 1.07%). Individuals from the noncoastal subpopulation also have significantly more unique SNPs per individual (65,127 ± 15,531, SD) compared with the coastal ones (30,205 ± 7,025 SD, t-test P < 10−5).
Fig. 4.—
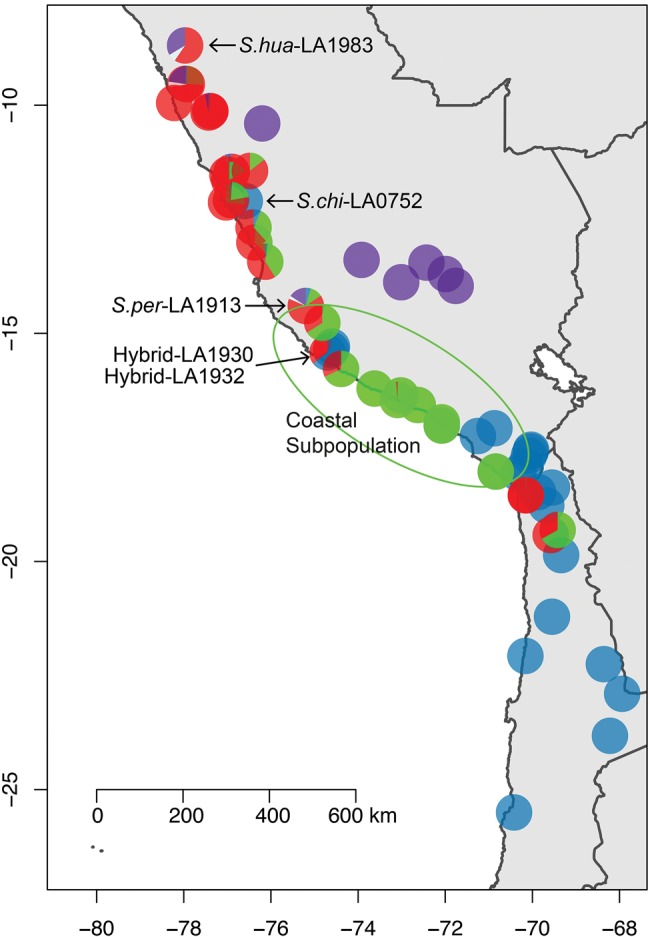
A pie chart of STRUCTURE groups based on K = 5 for all individuals shown in figure 3 at their collection location. The coastal subpopulation of Solanum peruvianum is circled. Two highly admixed accessions, S.per-LA1913 and Solanum huaylasense (S.hua) LA1983 as well as the anomalous Solanum chilense (S.chi) accession LA0752 and the cryptic hybrid populations LA1930 and LA1932 are marked.
Mean FST between S.chi and S.per was 0.070 which was less than the mean FST between the two subpopulations of S.per (FST = 0.074; supplementary fig. S7, Supplementary Material online). The coastal subpopulation of S.per has a higher mean FST to S.chi (0.14) and Esculentum (0.33) than the noncoastal deme does (0.09 and 0.13).
Solanum chilense shows relatively little population structure compared with S.per. Individual heterozygosity decreases in the more southern populations of S.chi (r = 0.72, P < 1.1 × 10−5, supplementary fig. S8, Supplementary Material online). Furthermore, three accessions (LA2750, LA2930, and LA0752) are always distinguishable as a clade (figs. 2 and 5). Two of these accessions, LA2750 and LA2930, are from low-elevation coastal regions of Chile, and LA0752 is a northern S.chi population described in the final results section.
Recent Speciation of S.chi and S.per
We used ∂a∂i to model the ancestry of the sister species S.chi and S.per because ∂a∂i is appropriate for detecting recent demographic events (Gutenkunst et al. 2009). We fit the synonymous JSFS to a simple model and this returned an estimate for the speciation time (tau) at 1.46–1.56 times the size of the MRCA (supplementary table S4, Supplementary Material online). If we assume a per site mutation rate of 5.1 × 10−9 (Roselius et al. 2005; Stäedler et al. 2008), then the speciation time is estimated to be between 1.2 and 1.4 million generations ago. We found evidence of population expansion in both species relative to their MRCA. S.per has an estimated population size of 1.54–1.70 million individuals which is nearly three times larger than the estimate for S.chi (0.52–0.58 million). We detect low levels of reciprocal gene flow between the two species when compared with previous reports; gene flow from S.chi into S.per was 0.27 individuals per generation and the reciprocal was 0.12 individuals per generation. All ML parameter values are provided in supplementary table S4, Supplementary Material online and the model fit is visualized in supplementary figure S9, Supplementary Material online.
Evidence of Natural Hybrid Populations between S.chi × S.per in Southern Peru
Three accessions of S.chi (LA1782, LA1930, and LA1932) were all collected in the Acari river drainage near Arequipa, in southern Peru. These populations were considered to be among the northernmost of S.chi (fig. 1b), but the following observations indicate that these populations are genetically not individuals of S.chi:
1.The genomes of all individuals show circa 35% corresponding to S.per and the remainder corresponding to S.chi in the STRUCTURE analysis (fig. 3).
Fig. 3.—

STRUCTURE analysis of all sect. Lycopersicon accessions excluding the “Hirsutum” group. The most likely number of clusters (K) was 3, but data for K = 4 and K = 5 is also shown. The subdivision of Solanum peruvianum is noticeable at K > 4. Taxon abbreviations are given in figure 2.
2.These populations are intermediate between the S.chi and S.per in the network and PCA analyses (fig. 5, supplementary fig. S4, Supplementary Material online).
Fig. 5.—

Reticulate network based on SplitsTree4. The reticulate network shows reticulation within the “Peruvianum” group species and between Solanum peruvianum and Solanum chilense, including reticulation of the hybrid populations. Species abbreviations are given in figure 2.
3. They form a monophyletic clade located between S.chi and S.per in all phylogenetic analyses (fig. 2).
According to collection records from the TGRC (tgrc.ucdavis.edu), wild individuals from these populations are described as vigorous, stress-tolerant, and long-lived (>10-years-old). To characterize them morphologically and to exclude the possibility of seed or sample contamination, we regrew individuals from accessions LA1930, LA1932, S.chi-LA2930, S.chi-LA0752, S.per-LA0153, S.per-LA1954, S.per-LA2732, and S.cor-LA1274. Individuals from LA1930 and LA1932 were fast-growing, large, and could be distinguished from both S.chi and S.per although they were similar in leaf shape to northern S.chi populations (figs. 1 and 6). Three-month-old plants had significantly longer leaves, thicker stems, and reduced lateral branching in comparison to the tested S.chi, S.per, and S.cor (t-test, P < 0.01; supplementary fig. S10, Supplementary Material online). They also had a very high density of type I trichomes on the lower stem, although this character was not quantified (fig. 6i). These populations flowered reluctantly and later than the other species in our hands. The corollas were frequently recurved and 2–3 cm in diameter. The style was straight and extended circa 2 mm beyond the anther tube. This and an absence of fruits on unpollinated flowers are consistent with outcrossing. Pollen fertility was normal (data not shown).
Fig. 6.—

Phenotype of the hybrid accessions. (a–c) Leaflet diversity across individuals from a single population of (a) Solanum chilense LA2930, (b) Hybrid-LA1930, and (c) Solanum peruvianum LA1954. (d–f) Typical 3-week-old (d) S. chilense LA2930, (e) Hybrid-LA1932, and (f) S. peruvianum LA1954. (g) 10-Week S. chilense LA2930 (left), Hybrid-LA1932 (center), and S. peruvianum LA1954 (right). All plants were germinated and grown together. (h) Typical leaf of Hybrid-LA1932 at 10 weeks. (i) Typical Hybrid-LA1932 stem at 10 weeks showing the thickness and high density of type I trichomes. In all cases, the phenotype of Hybrid-LA1930 was indistinguishable from Hybrid-LA1932.
Because of their genotype, phenotype, and the fact that two tetraploid populations of S.chi have been reported from southern Peru (Chetelat and Ji 2007; Rick 1990), we considered the possibility that these populations represent allotetraploids. However, flow cytometry indicated that they have a diploid C content (supplementary Supporting Information and fig. S11, Supplementary Material online).
These individuals appeared to be hybrids between S.chi and S.per, but it is difficult to distinguish hybridization from ILS or population subdivision using methods such as STRUCTURE, PCA, and phylogenetic reconstruction. The three-population test of (Patterson et al. 2012) is a formal test for admixture and results in a negative f3 if the tested population is admixed between parental populations by essentially looking for intermediate allele frequencies in the tested population with respect to the parents. This test did not result in a negative f3 and therefore does not provide evidence for or against a recent history of admixture.
A second method for potentially differentiating hybridization from other scenarios is to look for chromosomal blocks from the parental species in the hybrids (Ungerer et al. 1998). This was done with the program HAPMIX (Price et al. 2009; supplementary fig. S12, Supplementary Material online). We ran HAPMIX with a uniform recombination rate using all S.chi and S.per individuals to identify parental haplotypes. The admixed individuals were indeed composed of S.chi and S.per haplotypes. The mean haplotype was 112 kb long. Consistent with our previous analyses, the hybrid individuals appeared more S.chi-like than S.per-like, and the mean S.chi haplotype (218 kb) was longer than the mean S.per haplotype (55 kb). This analysis indicated that these individuals did indeed have a history of hybridization.
Test crosses with Hybrid-LA1932 largely failed to produce viable seeds with the four tested S.per populations and with S.chi-LA2930 (summary by species pair in table 1, summary by accession pair in supplementary fig. S13, Supplementary Material online). Fruits of these crosses instead contained a large number of SLS. The total number of SLS for crosses to a hybrid parent were 1,892 and only a small number of seeds were recovered (total seeds across all crosses with hybrid parent = 47; supplementary fig. S13, Supplementary Material online). In contrast, seed number from fruits of conspecific crosses always substantially outnumbered SLSs (total seeds of conspecific crosses = 2,950 seeds; total SLS of conspecific crosses = 520). This indicates a high degree of incompatibility between the hybrid and the putative parental species.
Table 1.
The Mean Number of Seeds and Seed-like Structures (SLS) per Fruit for All Cross Combinations. Germination Tests on Seeds Were Carried Out Following TGRC Guidelines (http://tgrc.ucdavis.edu/seed_germ.aspx)
| Cross | Mean Seed Number per Fruit | Mean SLS Number per Fruit | Germination Rate | Offspring Relative to within Deme S.per × S.per Crosses |
|---|---|---|---|---|
| S.per × S.per—within deme | 44.6 | 7.5 | 72.4% | 100% |
| S.per × S.per—between deme | 33.2 | 2.9 | 74.6% | 77% |
| S.cor × S.per | 16.7 | 8 | 35% | 18% |
| S.cor × S.chi | 0 | 10.9 | n/a | 0% |
| S.chi × S.per | 0.82 | 17.7 | 100% | 3%a |
| S.chi × Hybrid | 0.92 | 17.2 | 0% | 0% |
| S.per × Hybrid | 0.49 | 23.5 | 50% | 1% |
All of these individuals died before reaching maturity.
Furthermore, the small seeds from the LA1932 (hybrid) × S.chi cross all failed to germinate (N = 12; table 1). These hybrid populations are known to set seed in test crosses with other S.chi populations, but in reduced numbers (Chetelat R, personal communication). Crosses between the hybrid and two S.per accessions (LA0153 and LA2964) resulted in a total of 35 seeds (average of two seeds per fruit for these two crosses). These seeds had a 50% germination frequency, indicating some potential for backcrossing to S.per, but all of the interspecific F1 individuals eventually died while conspecific seedlings did not. No other S.per × LA1932 crosses produced seeds (supplementary fig. S13, Supplementary Material online).
Interspecific crosses between individuals of S.chi and S.per recapitulated the outcome of crosses between the hybrid and each of these species (table 1, supplementary fig. S13, Supplementary Material online). Fruits had an excess of SLS and few viable seeds (S.chi × S.per crosses resulted in 55 fruits, containing in total 971 SLS and 45 seeds). These interspecific seeds germinated, but all individuals died before reaching maturity.
Some intraspecific reproductive incompatibility was detected within S.per between the coastal and noncoastal demes (supplementary fig. S13, Supplementary Material online). In one case, 56% of the ovules were aborted in the 14 fruits from the S.per-LA1954 × S.per-LA0153 crosses (but not the reciprocal). In a second example, seeds from S.per-LA2964 × S.per-LA2732 and the reciprocal cross had only 25% germination rate despite normal seed set. Overall, crosses were between the two genetic groups identified within S.per had significantly lower number of seeds per fruit compared with within-deme crosses (Wilcoxon test, P < 0.05). Some incompatibility was also detected between S.cor-LA1274 and S.per-LA1954 (mean of 5 seeds and 12 SLS per fruit). However, the S.cor-LA1274 × S.per-LA1954 cross was not different than within-deme S.per crosses. The entire outcome of all crosses is reported in supplementary table S5, Supplementary Material online.
Broader Phylogenetic Implications
This large genetic data set allowed us to test and validate some earlier phylogenetic observations within the Lycopersicon clade. Three of four well-established species groups within sect. Lycopersicon were monophyletic, independent of the phylogenetic method (fig. 2). In contrast, the Peruvianum group was not monophyletic in either the ML or coalescent phylogenies. In the ML analysis, Arc + Esc was initially derived from within S.per making S.per paraphyletic (supplementary fig. S14, Supplementary Material online). To test if this was due to the inclusion of potentially admixed individuals (eight individuals had mixed membership between S.per and Arc + Esc in the STRUCTURE analysis), accessions with >10% mixed membership were removed and the ML analysis was redone. This resulted in Arc + Esc as a relative outgroup to S.chi and S.per as expected and restored monophyly to the Peruvianum group (fig. 2a). However, S.per itself remained paraphyletic due to inclusion of S.hua and S.cor, both of which themselves are polyphyletic.
In contrast to the ML analysis, Arc + Esc is not derived from within S.per in the coalescent analysis. The Peruvianum group was, however, polyphyletic due to S.per-LA1913 and S.hua-LA1983 which are in a clade with Arc + Esc. Furthermore, in contrast to the ML analysis excluding admixed accessions, Arc + Esc and S.per are sister taxa with S.chi as a relative outgroup (fig. 2b). These differences appear to be dependent on the inclusion of certain admixed accessions, giving strong indication that reticulate events are influencing phylogenetic relationships within the clade.
The species Solanum arcanum Peralta (S.arc) was polyphyletic in the ML and coalescent phylogenies. This is due to S.arc-LA2157 which is always an outgroup to all other species of the Arcanum group (fig. 2). Evidence for a close relationship of S.arc-LA2157 and S.per is present in the STRUCTURE analysis (fig. 3) and in the reticulate network (fig. 5) suggesting that the polyphyly S.arc may be the result of past hybridization between an individual of Arc + Esc and S.per.
One S.hua accession, S.hua-LA1358, is indistinguishable from S.per while the five others appear to have mixed ancestry between Arc + Lyc and S.per. One of these accessions, S.hua-LA1360 was shown to have introgressions from the Esculentum group by Pease et al. (2016) using the ABBA-BABA test. This accession and S.hua-LA1983 are nearly intermediate between S.per and Arc + Lyc in the STRUCTURE and network analyses (figs. 3 and 5). Like S.hua, S.cor is also polyphyletic in all phylogenetic analyses. Two S.cor individuals are indistinguishable from S.per while S.cor-LA0118 has circa 1% and S.cor-LA1274 circa 10% Arc + Esc component. The S.cor individuals never group together in any phylogenetic analyses.
We detect a minor signature of S.chi component in S.per-LA3636, S.per-LA1616, S.cor-LA0444, S.cor-LA0107, and S.hua-LA1358. This is also seen in the association of these accessions with S.chi along the first principle component (supplementary fig. S4, Supplementary Material online). S.per-LA1913 was unique in having mixture from all three STRUCTURE groups. Interestingly, as the number of clusters inferred by STRUCTURE increases, S.per-LA1913 continues to have genetic material from all of them (fig. 3).
Solanum chilense was always monophyletic, and there was no evidence of mixed ancestry in any S.chi accessions with the exception of three samples that appear nearly intermediate between S.chi and S.per, as described above.
One “S.per” accession, LA0752, was collected in central Peru, yet is closest genetically to the southern coastal populations of S.chi. This anomalous accession was described as S.chi-like when collected, and we confirmed this by growing multiple individuals (supplementary fig. S15, Supplementary Material online). On the basis of the genetic and phenotypic evidence, LA0752 has been re-annotated as S.chi. To our knowledge, this is the most northerly accession of S.chi described. Furthermore, S.chi-LA0752 has the lowest heterozygosity of any S.chi individual (supplementary fig. S8, Supplementary Material online), and plant growth was weak. Its disjunct location, weak growth and low heterozygosity may indicate a long-distance dispersal and subsequent founder effect in the history of this population. However, collection error (i.e. mislabeling) cannot be excluded.
Finally, in the independent data sets used to build phylogenies of sect. Lycopersicon, individuals of six accessions from four species were duplicated: S.arc-LA2172, S.hua-LA1364, S.neo-LA2133, S.per-LA1954, S.per-LA2744, and S.per-LA2964. These duplicates are always sister taxa in the ML, coalescent, and network analyses (figs. 2 and5). This concordance demonstrates the feasibility and consistency of combining sequence data from many independent studies (and labs) to address problematic questions in evolutionary biology.
Discussion
Our comprehensive population genomic analysis provides an in-depth view of population and lineage divergence among a group of closely related plant species. While many of our analyses confirmed the existence of three previously well-established groups within the section including 1) the monophyletic Hirsutum group, 2) the Esculentum clade, and 3) the Arcanum group, the novelty of our study is the extensive sampling and analysis of the two most polymorphic species in the clade: S.chi and S.per. In fact, the focus on these taxa leads to the surprising discovery of a new entity of hybrid origin.
Demography and Speciation of S.chi and S.per
Prior estimates of the divergence time between S.chi and S.per indicate a very recent speciation time of 0.18 × 2Ne, where Ne is the estimated size of S.chi (Naduvilezhath et al. 2011). Assuming one generation per year and a mutation rate of 5.1 × 10−9 site per year, this corresponds to a split 730,000 years ago. This estimate was based upon a modest data set of seven nuclear genes containing 954 polymorphic positions. However, the sample unknowingly included up to seven Quicacha individuals that we now know to represent S.chi × S.per hybrids (discussed below). The inclusion of these individuals not only contributed to the signature of on-going gene flow between species, but likely caused the speciation time to be underestimated. Grounded upon a much larger data set and following the identification and exclusion of admixed individuals, our analysis indicates that the species split was 1.51 × 2Ne generations before present. Assuming the same generation time and mutation rate, this corresponds to ∼1.25 Ma, which is consistent with a previous family-wide dated phylogeny (Särkinen et al. 2013). However, age estimates based on the molecular clock need to be approached cautiously given disagreement between molecular data and new fossil evidence (Wilf et al. 2017). The recency is, however, consistent with the observed low FST and few fixed differences between these species. In our analyses, migration rates of 0.12 and 0.27 individuals per generation were estimated—in contrast to higher estimates from data which included cryptic hybrid individuals (e.g. Stäedler et al. 2005).
We detect a lower amount of synonymous nucleotide diversity in S.per (1.7%) compared with previous estimates of 2.1%, 2.5%, and 3.1% (Arunyawat et al. 2007; Stäedler et al. 2005, 2012). This could be in part due to method-specific differences (i.e. next generation versus Sanger sequencing). However, our numbers may reflect a more accurate estimate of nucleotide diversity since it is based upon orders of magnitude larger number of nucleotide positions, more populations from both species, and did not include interspecific hybrid individuals.
We detected two genetic groups within S.per, similar to those described by Rick (1963) based upon morphology and by Nakazato et al. (2012) based upon AFLPs. These two groups appear to represent distinct geographic demes and/or subspecies occupying different ecological niches. One contained seven low-elevation populations restricted to the coast and/or lomas formations of southern Peru. The second deme contained noncoastal central Peruvian populations, including most individuals of S.cor and S.hua.
Rick (1963) described the coastal S.per group as having less variation in shape, size, and habit between populations, but greater variation within any single population. Conversely, the noncoastal Peru populations were described as more restricted and more idiosyncratic (Rick 1963). This morphological observation is reinforced by our genetic data showing higher diversity and more private polymorphism in the noncoastal Central populations: An observation which could be explained by limited dispersal between different Andean river drainages.
In contrast, there are few geographical barriers to inhibit gene flow between coastal populations. The coastal deme also has higher mean FST with both S.chi and Esculentum than the noncoastal deme. We interpret this difference due to recent gene flow between the noncoastal deme and both S.chi and the Esculentum group: The S.chi × S.per hybrids have noncoastal S.per component and the admixed Peruvianum group accessions (including S.cor and S.hua) are from the noncoastal deme whereas the coastal deme shows no admixture. Alternatively, the greater mean FST could reflect differentiation of the coastal deme, perhaps from ecological adaptation.
Climatic conditions differ between the two S.per subpopulations: Fog is abundant along the coast in the Lomas formation from May to October, while rainfall is abundant in the central river drainages November to May (Taylor 1986). These climatic differences may influence flowering time resulting in a prezygotic barrier that could account for the population subdivision. The subpopulations also show reduced interfertility (i.e. reduced seed number in LA0153 × LA1954 crosses). Recognizing these geographic races/subspecies of S.per as distinct species is not warranted due the low amount of genetic differentiation and the absence of pronounced incompatibility. Overall, these geographic races are a good opportunity for the “magnifying glass” approach to study speciation-in-action (Via 2009).
The five S.per populations showing a low amount (<10%) of genetic similarity to S.chi according to STRUCTURE are all from the noncoastal deme near Lima, Peru. We do not detect admixture between sympatric populations, consistent with previous crossing studies (Rick and Lamm 1955). Interestingly, the five S.per populations with S.chi admixture are physically near the S. chi-LA0752 population. Thus, if S.chi-LA0752 is not a collection error and represents a long-distance dispersal event, this could explain how genes from S.chi could be introduced into noncoastal S.per populations.
Solanum corneliomulleri and S. huaylasense
Relationships in the Peruvianum group remain challenging, even in the face of such an extensive data set. Although many different species concepts exist, there is general consensus that species form discrete, evolutionarily independent lineages (de Queiroz 2005). Our data show that neither S.cor nor S.hua form discrete genetic clades as currently circumscribed. C.H. Muller (1940) first described S.cor as Lycopersicon glandulosum, but Macarthur and Chiasson (1947) and later Rick (1963) demonstrated the compatibility of L. glandulosum with other S.per accessions. Therefore, L. glandulosum was renamed L. peruvianum var. glandulosum and later designated as a race of S.per (Warnock 1988). In fact, Rick (1963) noted at least five additional races, some currently included within S.cor, that were equally distinct from S.per. Our data are in agreement with other studies reporting the lack of genetic or ecological differentiation between S.cor and S.per (Labate et al. 2014; Nakazato et al. 2010; Pease et al. 2016; Rodriguez et al. 2009; Zuriaga et al. 2009).
Solanum huaylasense was delineated from S.per using morphologically by Peralta et al. (2005). Our data included six individuals of this species from five accessions. One accession, LA1958, is indistinguishable from S.per, but the remaining five show some admixture with Arc + Esc. For example, LA1364, described to be admixed by Labate et al. (2014) and again by Pease et al. (2016), has circa 7% mixed ancestry. Other S.hua accessions show even more admixture, and the species is consistently polyphyletic. Thus, as currently defined, S.hua does not appear to be a natural group and some, maybe most, of the individuals described as S.hua could be of hybrid origin.
Given the increasing evidence, recognizing S.cor and S.hua as distinct species seems untenable. Instead, we have identified two well-differentiated demes in S.per: The coastal and noncoastal demes. The currently recognized representatives of S.cor and S.hua belong to the noncoastal deme. This deme is highly idiosyncratic and there has undoubtedly also been admixture, which has further contributed to the taxonomic difficulties and confusion.
Solanum arcanum
A different variety of S.per called humifusum was first described by Muller (1940) and largely corresponds to the species now recognized as S.arc. Morphologically S.arc can be distinguished from other taxa by its unbranched inflorescences, straight anther tubes, and short styles (Müller 1940; Rick 1986). Although Rick and colleagues detected a reduction of cross-compatibility between the typical S.per and variety humifusum (aka S.arc), Rick, following the BSC, did not feel justified in recognizing humifusum as a distinct species from S.per because gene flow was theoretically possible through several intermediate populations (Rick 1979; Rick and Lamm 1955), Instead, Rick (1986) delineated four more-or-less reproductively isolated assemblages based on extensive reciprocal test-crosses: Chotano-humifusum, Chamaya-Cuvita, Marañón, and typical S.per. Today’s S.arc is the sum of the first three assemblages (Peralta et al. 2008). But gene flow is possible between these assemblages and the typical S.per, through, for example, Chotano-humifisum populations (Rick 1979). In our data, S.arc-LA2172 (Marañón) shared a more recent ancestor with Solanum neorickii while S.arc-LA2157 (Chotano) is an outgroup to the Arcanum group species and appears to have S.per ancestry. Interestingly, these two S.arc accessions have actually been shown to be incompatible with one another by Rick (1986). Thus, while evidence supports the Arcanum group as biologically meaningful, the number of species within the group and the species-level assignment of individual accessions (particularly of individuals of S.arc) deserves further study and clarification.
Evidence for Widespread Cryptic Hybrid Populations
Herbarium records and TGRC collection data indicate that there are several other collections of S.chi from Arequipa near the interspecific hybrids identified in this study, and that all of these collections are geographically discontinuous from the rest of S.chi (supplementary fig. S16a, Supplementary Material online). This does not appear to be a sampling artifact because many other wild tomatoes have been collected from this area (supplementary fig. S16b, Supplementary Material online). The reported collections of S.chi west of 72° are LA0869, LA1782, LA1917, LA1930, LA1931, LA1932, LA1934, LA1938, LA1939, LA3780, LA3784, LA3785, and LA3786. Most of these were collected in 1979 or 1996 and field notes indicated that they included many “tall upright plants” with “long peduncles” and “very large growth and very heavy fruit set,” resembling our morphological observations. The genetic evidence for hybridization in three of these populations, the distinct and consistent morphological differences of these populations, and their geographic discontinuity with other S.chi led to the hypothesis that all of these discontinuous northern S.chi populations are of hybrid origin.
This hypothesis is supported by the following observations from previously published work. First, LA1782 and LA4117A were chosen to represent S.chi in a study of wild tomato evolution by Pease et al. (2016). LA1782 was collected independently and circa 9 km from LA1930 and LA1932 in 1977, and is genetically indistinguishable from these populations (figs. 2 and 3). The finding is consistent with the relatively long coalescent of the two sampled S.chi individuals in figures 2a and b of Pease et al. (2016).
Second, Boendel et al. (2015) sequenced 30 genes from 23 S.chi populations, including Hybrid-LA1930 and another putative hybrid, LA3784. The similarity between these two accessions and the separation of these two accessions from other S.chi in their analyses is consistent with LA3784 being genetically comparable to Hybrid-LA1930. In fact, Boendel et al. (2015) explored the idea of hybridization in these populations in their discussion, but were not able to make conclusions because they had data only from S.chi and not from S.per.
Third, Stäedler and colleagues collected plants and seeds from S.chi and S.per in Peru and Chile in 2004 (supplementary fig. S16a, Supplementary Material online; Roselius et al. 2005). We examined the voucher specimens from this 2004 trip, including two S.chi collections from Arequipa, Peru near Acari: Quicacha (QUI) and Nazca (NAZ; supplementary fig. S17, Supplementary Material online). The NAZ collection includes a specimen of S.chi that is phenotypically very similar to the S.chi QUI population. The leaf morphology of both QUI and NAZ specimens is more similar to our sampled hybrids than to typical S.chi or S.per, indicating that the QUI and NAZ S.chi samples are also hybrids. Furthermore, genetic studies using this material came to conclusions consistent with our phenotypic observations. On the basis of this material, Stäedler and colleagues estimated a very recent split time between the two species (<0.55 Ma), found an absence of fixed differences, and concluded that speciation occurred under residual gene flow (Stäedler et al. 2005, 2008). Subsequent studies based on this material describe trans-specific allele sharing and selection (due to the presence of S.per alleles in the QUI population; Mboup et al. 2012; Xia et al. 2010). While there is no reason to question the data, we argue that the allele sharing resulted from the inclusion of cryptic hybrids in their sample rather than natural selection as they hypothesize.
Together, these observations support the conclusion that all of the populations described as S.chi west of 72° are genetically equivalent and therefore of hybrid origin. Given that their genomic constitution is composed of S.chi and S.per haplotype blocks and the hybrid accessions are diploid, these data are consistent with this being an example of recombinational speciation in wild tomato. However, because they are not shown to be definitively admixed when formally tested, other hypotheses must also be considered, including, for example, recent introgressions of S.per haplotypes into distinct populations of S.chi. Note that the lack evidence for admixture according to the f3 test could be due to the small number of hybrid individuals tested or drift within the hybrid populations following their creation.
A Potential Example of Recombinational Speciation
Recombinational speciation is the rapid formation of a new species resulting from a cross between two closely related species, without a change in chromosome number. It is rare, with only circa 20 examples in plants, and many of these examples are unconvincing (Rieseberg 1997; Rieseberg and Willis 2007; Stuessy et al. 2014). Rarity may be due to poor documentation because there is no cytotaxonomic evidence and because hybrid species may be difficult to recognize and distinguish morphologically (Rieseberg 1997). However, this form of speciation may simply be less common because barriers to gene flow, such as those introduced by a change in ploidy, are not present at the outset. Without such barriers, hybrids inevitably backcross to the more abundant parental species, leading to their eventual disappearance (Baack et al. 2005).
Theoretical studies on hybrid speciation have therefore emphasized the role of ecology and the necessity of an open habitat for the hybrids to separate them from their parental taxa (Anderson 1949; Buerkle 2000; Buerkle and Rieseberg 2008; Gross and Rieseberg 2005). Simulations also show that this type of rapid speciation is more likely in perennial species (reviewed in Stuessy et al. 2014), a condition met by S.chi and S.per. While recombinational speciation is theoretically more likely in self-compatible species, it can also occur in outcrossing taxa, and, interestingly, most of the convincing examples are outcrossers such as S.chi and S.per (McCarthy et al. 1995; Rieseberg 1997).
The hybrid populations show strong reproductive barriers to all tested S.per populations. While the cross-compatibility of the hybrid populations to northern S.chi (R. Chetelat, pers. comm.) would allow backcrossing, their nonoverlapping distribution would generally shield them from gene swamping by S.chi. However, their morphological similarity to the northern Chilean S.chi populations and their genomic composition seems to indicate historical backcrossing to S.chi. Alternatively, their similarity to S.chi could be accounted for by differential segregation in the F2 or later generation hybrids, or these populations could be a distinct subpopulation of S.chi with introgressions from S.per. It is difficult to distinguish between these scenarios, but the consistent phenotype and genotype of the hybrids from independent collections from the 1970s to 2004 and the small haplotype size make it clear that they are stabilized derivatives and not first or early generation hybrids. Because they are older, the different scenarios of hybridization, introgression, and population subdivision are especially difficult to distinguish.
Schumer et al. (2014) give three criteria that need to be met in a definitive example of recombinational speciation. Most purported examples fail to meet all of these criteria. The first criterion is reproductive isolation of the hybrid species from its parents. The second is genetic evidence of hybridization. These two criteria are fulfilled here, but the third criterion—showing that hybridization resulted in reproductive barriers and speciation—is more challenging. This has only been demonstrated once in plants by Rieseberg et al. (2003) who recreated the extreme phenotypes of hybrid sunflower species. These tomato populations are a good starting point for tests of reproductive barriers, and further mapping and cytological work employing them could narrow down the incompatibility loci as done for other species pairs in the clade (Moyle and Nakazato 2008). Such work is also ultimately needed to demonstrate recombinational speciation in wild tomato. Further studies can also help clarify the date of admixture and the exact compatibilities of these populations.
In conclusion, section Lycopersicon provides a window into the speciation continuum, from population subdivision to speciation, and includes one and possibly more hybrid taxa. Knowing the ancestry of these populations and species is fundamental for addressing future questions about the genomics of ecological adaptation and the development of breeding barriers in the clade.
Data Accessibility
Nucleotide sequence data generated in this study have been deposited in NCBI under Bioproject PRJNA329478.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors thank the TGRC for seed and Roger Chetelat for comments, Sandra Knapp for herbarium records, and Hajo Esser for herbarium specimens. This research was supported by the Research Training Group GRK1525 and Deutsche Forschungsgemeinschaft grants: Ro 2491/5-2 and Ro 2491/6-1. I.B. was supported by a fellowship from the International Graduate School for Plant Science program (Deutsche Forschungsgemeinschaft GRK1525).
Author Contributions
L.R. and I.B. conceived the study. I.B. and A.R. grew the samples and A.R. extracted RNA. I.B. and T.K. analyzed data. I.B. and L.R. wrote the manuscript. All authors approved the final version of the manuscript.
Literature Cited
- Aflitos S, et al. 2014. Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. Plant J. 80(1):136–148.http://dx.doi.org/10.1111/tpj.12616 [DOI] [PubMed] [Google Scholar]
- Anderson E. 1949. Introgressive hybridization. New York: John Wiley and Sons. [Google Scholar]
- Anderson LK, et al. 2010. Structural differences in chromosomes distinguish species in the tomato clade. Cytogenet Genome Res. 129(1–3):24–34. [DOI] [PubMed] [Google Scholar]
- Arunyawat U, et al. 2007. Using multilocus sequence data to assess population structure, natural selection, and linkage disequilibrium in wild tomatoes. Molec Biol Evol. 24(10):2310–2322.http://dx.doi.org/10.1093/molbev/msm162 [DOI] [PubMed] [Google Scholar]
- Baack EJ, et al. 2005. Hybridization and genome size evolution: timing and magnitude of nuclear DNA content increases in Helianthus homoploid hybrid species. New Phytol. 167(2):623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boendel KB, et al. 2015. North-south colonization associated with local adaptation of the wild tomato species Solanum chilense. Molec Biol Evol. 32:2932–2943.http://dx.doi.org/10.1093/molbev/msv166 [DOI] [PubMed] [Google Scholar]
- Bouckaert R, et al. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 10(4):e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breto MP, et al. 1993. Genetic variability in Lycopersicon species and their genetic relationships. Theor Appl Genet. 86:113–120. [DOI] [PubMed] [Google Scholar]
- Browning BL, Browning SR.. 2016. Genotype imputation with millions of reference samples. Am J Hum Genet. 98(1):116–126.http://dx.doi.org/10.1016/j.ajhg.2015.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D, et al. 2012. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Molec Biol Evol. 29(8):1917–1932.http://dx.doi.org/10.1093/molbev/mss086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerkle CA. 2000. The likelihood of homoploid hybrid speciation. Heredity 84(4):441–451.http://dx.doi.org/10.1046/j.1365-2540.2000.00680.x [DOI] [PubMed] [Google Scholar]
- Buerkle CA, Rieseberg LH.. 2008. The rate of genome stabilization in homoploid hybrid species. Evolution 62(2):266–275.http://dx.doi.org/10.1111/j.1558-5646.2007.00267.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat RT, Ji YF.. 2007. Cytogenetics and evolution In: Razdan MK, Mattoo AK, editors. Genetic improvement of Solanaceous crops. Enfield (NH: ): Science Publishers; p. 77–112. [Google Scholar]
- Coyne JA, Orr HA.. 2004. Speciation. Sunderland (MA: ): Sinauer Associates. [Google Scholar]
- Danecek P, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158.http://dx.doi.org/10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Queiroz K, 2005. Ernst Mayr and the modern concept of species In: Hey J, et al. editors. Systematics and the origin of species. Washington (DC: ): National Academy of Sciences; p. 243–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, et al. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molec Biol Evol. 29(8):1969–1973.http://dx.doi.org/10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl DA, Vonholdt BM.. 2012. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 4(2):359–361.http://dx.doi.org/10.1007/s12686-011-9548-7 [Google Scholar]
- Ehrlich PR, Raven PH.. 1969. Differentiation of populations. Science 165(3899):1228–1232.http://dx.doi.org/10.1126/science.165.3899.1228 [DOI] [PubMed] [Google Scholar]
- Evanno G, et al. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molec Ecol. 14(8):2611–2620.http://dx.doi.org/10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Grant PR, Grant BR.. 1992. Hybridization of bird species. Science 256(5054):193–197.http://dx.doi.org/10.1126/science.256.5054.193 [DOI] [PubMed] [Google Scholar]
- Grant V. 1981. Plant speciation. New York: Columbia University Press. [Google Scholar]
- Gross BL, Rieseberg LH.. 2005. The ecological genetics of homoploid hybrid speciation. J Hered. 96(3):241–252.http://dx.doi.org/10.1093/jhered/esi026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, et al. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5(10):e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Molec Biol Evol. 23(2):254–267.http://dx.doi.org/10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA.. 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23(14):1801–1806.http://dx.doi.org/10.1093/bioinformatics/btm233 [DOI] [PubMed] [Google Scholar]
- Kim D, et al. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, et al. 2008. Regulatory genes control a key morphological and ecological trait transferred between species. Science 322(5904):1116–1119.http://dx.doi.org/10.1126/science.1164371 [DOI] [PubMed] [Google Scholar]
- Labate JA, et al. 2014. Genetic structure of the four wild tomato species in the Solanum peruvianum s.l. species complex. Genome 57(3):169–180.http://dx.doi.org/10.1139/gen-2014-0003 [DOI] [PubMed] [Google Scholar]
- Levin DA. 1979. The nature of plant species. Science 204(4391):381–384.http://dx.doi.org/10.1126/science.204.4391.381 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760.http://dx.doi.org/10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079.http://dx.doi.org/10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, et al. 2014. Genomic analyses provide insights into the history of tomato breeding. Nat Genet. 46(11):1220–1226.http://dx.doi.org/10.1038/ng.3117 [DOI] [PubMed] [Google Scholar]
- Macarthur JW, Chiasson LP.. 1947. Cytogenetic notes on tomato species and hybrids. Genetics 32(2):165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J. 2005. Hybridization as an invasion of the genome. Trends Ecol Evol. 20(5):229–237.http://dx.doi.org/10.1016/j.tree.2005.02.010 [DOI] [PubMed] [Google Scholar]
- Mayr E. 1942. Systematics and the origin of species. New York: Columbia University Press. [Google Scholar]
- Mboup M, et al. 2012. Trans-species polymorphism and allele-specific expression in the CBF gene family of wild tomatoes. Molec Biol Evol. 29(12):3641–3652.http://dx.doi.org/10.1093/molbev/mss176 [DOI] [PubMed] [Google Scholar]
- McCarthy EM, et al. 1995. A theoretical assessment of recombinational speciation. Heredity 74(5):502–509.http://dx.doi.org/10.1038/hdy.1995.71 [Google Scholar]
- Moyle LC. 2008. Ecological and evolutionary genomics in the wild tomatoes (Solanum sect. Lycopersicon). Evolution 62(12):2995–3013.http://dx.doi.org/10.1111/j.1558-5646.2008.00487.x [DOI] [PubMed] [Google Scholar]
- Moyle LC, Nakazato T.. 2008. Comparative genetics of hybrid incompatibility: sterility in two Solanum species crosses. Genetics 179(3):1437–1453.http://dx.doi.org/10.1534/genetics.107.083618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CH. 1940. A revision of the genus Lycopersicon. Vol. 382. Washington (DC): U.S. Department of Agriculture. p. 1–28. [Google Scholar]
- Naduvilezhath L, et al. 2011. Jaatha: a fast composite-likelihood approach to estimate demographic parameters. Molec Ecol. 20(13):2709–2723.http://dx.doi.org/10.1111/j.1365-294X.2011.05131.x [DOI] [PubMed] [Google Scholar]
- Nakazato T, et al. 2012. Population structure, demographic history, and evolutionary patterns of a green-fruited Tomato, Solanum peruvianum (Solanaceae), revealed by spatial genetics analysis. Am J Bot. 99(7):1207–1216.http://dx.doi.org/10.3732/ajb.1100210 [DOI] [PubMed] [Google Scholar]
- Nakazato T, et al. 2010. Ecological and geographic modes of species divergence in wild tomatoes. Am J Bot. 97(4):680–693.http://dx.doi.org/10.3732/ajb.0900216 [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T.. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molec Biol Evol. 3(5):418–426. [DOI] [PubMed] [Google Scholar]
- Paradis E, et al. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289–290.http://dx.doi.org/10.1093/bioinformatics/btg412 [DOI] [PubMed] [Google Scholar]
- Patterson N, et al. 2012. Ancient admixture in human history. Genetics 192(3):1065–1093.http://dx.doi.org/10.1534/genetics.112.145037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paun O, et al. 2009. Hybrid speciation in angiosperms: parental divergence drives ploidy. New Phytol. 182(2):507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease JB, et al. 2016. Phylogenomics reveals three sources of adaptive variation during a rapid radiation. PLoS Biol. 14(2):e1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta IE, et al. 2005. New species of wild tomatoes (Solanum section Lycopersicon: Solanaceae) from northern Peru. Syst Bot. 30(2):424–434.http://dx.doi.org/10.1600/0363644054223657 [Google Scholar]
- Peralta IE, et al. 2008. Taxonomy of wild tomatoes and their relatives. Syst Bot Monogr. 84. [Google Scholar]
- Peterson DG, et al. 1996. DNA content of heterochromatin and euchromatin in tomato (Lycopersicon esculentum) pachytene chromosomes. Genome 39(1):77–82.http://dx.doi.org/10.1139/g96-011 [DOI] [PubMed] [Google Scholar]
- Price AL, et al. 2009. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 5(6):e1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, et al. 2000. Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2012. ISBN:3-900051-07-0. [Google Scholar]
- Racimo F, et al. 2015. Evidence for archaic adaptive introgression in humans. Nat Rev Genet. 16(6):359–371.http://dx.doi.org/10.1038/nrg3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, et al. 2014. Tracer v1.6, Available from: http://beast.bio.ed.ac.uk/Tracer.
- Rick CM. 1963. Barriers to interbreeding in Lycopersicon peruvianum. Evolution 17(2):216–232.http://dx.doi.org/10.1111/j.1558-5646.1963.tb03272.x [Google Scholar]
- Rick CM, 1979. Biosystematic studies in Lycopersicon and closely related species of Solanum In: Hawkes JG, et al. editors. The biology and taxonomy of the Solanaceae. London: Academic Press; p. 667–678. [Google Scholar]
- Rick CM. 1990. New or otherwise noteworthy accessions of wild tomato species. Tomato Genet Coop Rep. 40:30. [Google Scholar]
- Rick CM, 1986. Reproductive isolation in the Lycopersicon peruvianum complex In: D'Arcy WG, editor. Solanaceae biology and systematics. New York: Columbia University Press; p. 477–495. [Google Scholar]
- Rick CM, Lamm R.. 1955. Biosystematic studies on the status of Lycopersicon chilense. Am J Bot. 42(7):663–675.http://dx.doi.org/10.2307/2485327 [Google Scholar]
- Rieseberg LH. 1997. Hybrid origins of plant species. Ann Rev Ecol Syst. 28(1):359–389.http://dx.doi.org/10.1146/annurev.ecolsys.28.1.359 [Google Scholar]
- Rieseberg LH, et al. 2003. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301(5637):1211–1216.http://dx.doi.org/10.1126/science.1086949 [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Willis JH.. 2007. Plant speciation. Science 317(5840):910–914.http://dx.doi.org/10.1126/science.1137729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH, et al. 2006. The nature of plant species. Nature 440(7083):524–527.http://dx.doi.org/10.1038/nature04402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez F, et al. 2009. Do potatoes and tomatoes have a single evolutionary history, and what proportion of the genome supports this history? BMC Evol Biol. 9:191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselius K, et al. 2005. The relationship of nucleotide polymorphism, recombination rate and selection in wild tomato species. Genetics 171(2):753–763.http://dx.doi.org/10.1534/genetics.105.043877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Särkinen T, et al. 2013. A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol Biol. 13:214.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, et al. 2012. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485(7400):635–641.http://dx.doi.org/10.1038/nature11119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, et al. 2014. How common is homoploid hybrid speciation? Evolution 68(6):1553–1560. [DOI] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE.. 2012. Polyploidy and genome evolution. Berlin: Springer. [Google Scholar]
- Stäedler T, et al. 2005. Genealogical footprints of speciation processes in wild tomatoes: demography and evidence for historical gene flow. Evolution 59:1268–1279. [PubMed] [Google Scholar]
- Städler T, et al. 2008. Population genetics of speciation in two closely related wild tomatoes (Solanum section Lycopersicon). Genetics 178(1):339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Städler T, et al. 2012. Testing for “snowballing” hybrid incompatibilities in Solanum: impact of ancestral polymorphism and divergence estimates. Molec Biol Evol. 29(1):31–34. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313.http://dx.doi.org/10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicher JW, et al. 2016. How should genes and taxa be sampled for phylogenomic analyses with missing data? An empirical study in iguanian lizards. Syst Biol. 65(1):128–145.http://dx.doi.org/10.1093/sysbio/syv058 [DOI] [PubMed] [Google Scholar]
- Stuessy TF, et al. 2014. Plant systematics: the origin, interpretation, and ordering of plant biodiversity. Königstein: Koeltz Scientific Books. [Google Scholar]
- Taylor IB. 1986. Biosystematics of the tomato In: Atherton JG, Rudich J, editors. The tomato crop: a scientific basis for improvement. London: Chapman and Hall; p. 1–34. [Google Scholar]
- Ungerer MC, et al. 1998. Rapid hybrid speciation in wild sunflowers. Proc Natl Acad Sci U S A. 95(20):11757–11762.http://dx.doi.org/10.1073/pnas.95.20.11757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via S. 2009. Natural selection in action during speciation. Proc Natl Acad Sci U S A. 106(Suppl. 1):9939–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock SJ. 1988. A review of taxonomy and phylogeny of the genus Lycopersicon. HortScience 23:669–673. [Google Scholar]
- Whitney KD, et al. 2010. Patterns of hybridization in plants. Perspect Plant Ecol Evol Syst. 12(3):175–182.http://dx.doi.org/10.1016/j.ppees.2010.02.002 [Google Scholar]
- Wilf P, et al. 2017. Eocene lantern fruits from Gondwanan Patagonia and the early origins of Solanaceae. Science 355(6320):71–74.http://dx.doi.org/10.1126/science.aag2737 [DOI] [PubMed] [Google Scholar]
- Xia H, et al. 2010. Nucleotide diversity patterns of local adaptation at drought-related candidate genes in wild tomatoes. Molec Ecol. 19(19):4144–4154.http://dx.doi.org/10.1111/j.1365-294X.2010.04762.x [DOI] [PubMed] [Google Scholar]
- Zuriaga E, et al. 2009. Classification and phylogenetic relationships in Solanum section Lycopersicon based on AFLP and two nuclear gene sequences. Genet Resour Crop Evol. 56(5):663–678.http://dx.doi.org/10.1007/s10722-008-9392-0 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Nucleotide sequence data generated in this study have been deposited in NCBI under Bioproject PRJNA329478.