


Abstract
Predicting pre-disease state or tipping point just before irreversible deterioration of health is a difficult task. Edge-network analysis (ENA) with dynamic network biomarker (DNB) theory opens a new way to study this problem by exploring rich dynamical and high-dimensional information of omics data. Although theoretically ENA has the ability to identify the pre-disease state during the disease progression, it requires multiple samples for such prediction on each individual, which are generally not available in clinical practice, thus limiting its applications in personalized medicine. In this work to overcome this problem, we propose the individual-specific ENA (iENA) with DNB to identify the pre-disease state of each individual in a single-sample manner. In particular, iENA can identify individual-specific biomarkers for the disease prediction, in addition to the traditional disease diagnosis. To demonstrate the effectiveness, iENA was applied to the analysis on omics data of H3N2 cohorts and successfully detected early-warning signals of the influenza infection for each individual both on the occurred time and event in an accurate manner, which actually achieves the AUC larger than 0.9. iENA not only found the new individual-specific biomarkers but also recovered the common biomarkers of influenza infection reported from previous works. In addition, iENA also detected the critical stages of multiple cancers with significant edge-biomarkers, which were further validated by survival analysis on both TCGA data and other independent data.
INTRODUCTION
Disease progression can be generally divided into three stages or states, i.e. normal state, pre-disease state (or tipping point) and disease state. Traditional molecular biomarkers are designed to diagnose disease states based on the molecular data, rather than the prediction for pre-disease state. Recent advance on the high-throughput technologies provides an unprecedented opportunity to study the occurrence and progression of a disease in a person (or patient) (1,2), and pave a new way to make accurate and earlier disease diagnosis on individuals, which is a key concept and action for precision medicine (3).
One representative example is the prediction of acute respiratory diseases. The analysis of temporal transcriptome data from individual suspects has revealed many biological and biomedical insights (4–6). The traditional differential gene expression analysis, i.e. traditional molecular biomarker, is expected to capture the responsive genes to virus infection (7); then the gene module analysis tends to find interactive genes with particular biological functions associated with virus infection (6,8); and further the gene network analysis tries to extract the systematical features of virus infection by inferring the biological pathway between up-stream and down-stream genes (9). All of those works mainly focus on the diagnosis of disease state by exploiting information of the first-order statistics (e.g. ‘mean value’ for differential expressions of genes or proteins) from the observed data. In contrast to such traditional node-network analysis, recently, edge-network analysis (ENA) (10) (Figure 1A) combined with dynamic network biomarker (DNB) (11) was proposed to detect the early-warning signals or the pre-disease state (or tipping point) before the serious disease deterioration by considering the second-order statistics (e.g. ‘covariance’ for differential expressions among genes or proteins) from the observed data. Different from general anomaly detection (12–15), ENA can detect the tipping point or pre-disease state just before the critical transition to the disease state. Actually, ENA in our earlier work has shown the ability to identify the pre-disease or pre-symptom states (10), but it requires multiple samples for such prediction, which is, however, not generally available for each individual in clinical practice (10,16,17). To overcome this problem, we aim to extend the ENA from multiple-samples to one-sample in this work so as to achieve personalized diagnosis, prognosis and prediction on an individual basis.
Figure 1.

Concept of edge-network and iENA. (A) The comparison between new edge-network and conventional node-network. (B) The computational framework of iENA.
Specifically, we propose the individual-specific ENA (iENA) with DNB to identify the individual-specific biomarkers (Figure 1B), which are applied to disease prediction of individual subjects (or patients) on the basis of single samples. According to the three conditions of the tipping point from DNB theory (10), iENA can capture the high-order statistical information during dynamical disease progression for detecting the pre-disease state. On the other hand, ENA is based on a well-founded stochastic dynamics, and thus it can predict pre-disease states for individuals by reducing the false-positive in the discovery of individual-specific edge-biomarkers.
Based on the differential expression mean, differential expression variance and differential expression co-variance (18,19), we extend the ENA framework to iENA, which has been implemented for the analysis on personalized disease prediction, e.g. influenza infection and cancer deteriorations. From the data of H3N2 cohorts on individual subjects, iENA shows its ability on detecting early-warning signals or pre-disease state as the tipping point of influenza infection for each individual, in contrast to the disease state diagnosed by traditional molecular biomarkers. In particular, (i) iENA recovered the common biomarkers of influenza infection reported in the previous work (7), which is also more effective than common modules derived from WGCNA approach (20); (ii) iENA detected individual specific biomarkers, depending on the temporal (or spatial) transcriptome data from each subject and the reference data of multiple subjects; (iii) different from previous static prediction of disease states, a dynamical prediction model of disease, i.e. DNB, was adopted in iENA, and successfully predicted both the disease event and time in an accurate manner. For TCGA cancer data, iENA also demonstrates its power to detect the critical cancer stages with significant edge-biomarkers. We show that iENA can identify the critical stages on both breast cancer and liver cancer, and the results were further validated by survival analysis on both TCGA data and other independent data.
In a brief summary, iENA provides a powerful network-analysis tool for the study of complex diseases based on edge induced data transformation, and also opens a new way to predict the pre-disease states during disease progression (e.g. hunt for cancer tipping point) on the basis of individual samples for the personalized or precision medicine (21). The matlab code of iENA is available on http://sysbio.sibcb.ac.cn/cb/chenlab/software.htm.
MATERIALS AND METHODS
Node-network of multiple samples
Biochemical master equation is generally used to simulate the stochastic dynamics of a biological system at a molecular level (see Supplementary Information A1). Given the linearization with Gaussian distribution assumption for such a master equation, the biological system is usually approximated by a group of equations on the mean vector of molecules in previous study (10), i.e. node-network dynamics:
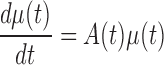 |
(1) |
where, μ is the mean vector of variables as network nodes; A is the adjacent matrix of the underlying network and a node represents a molecule (i.e. μi) in terms of concentration or number. Obviously, this group of linear differential equations represents the first-order statistic information of the molecular network, i.e. mean values. It can construct the conventional network, called as a node-network in this paper, e.g. using each molecule as a node in a molecular network.
In practice, the node-network (e.g. a molecular network) includes node set and edge set, where the node set is a list of molecules and the edge set is a list of molecule-pairs. A molecule-pair is determined by the correlation (or association) between two molecules on the observed data, e.g. Pearson correlation coefficient (PCC) between two genes based on gene expression profiles. As well known to us, such correlation for a node-network with a group of samples can be calculated as:
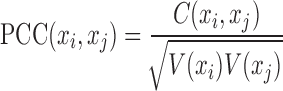 |
(2) |
where, xi and xj represent two molecules' expression profiles; their expression covariance for a group of samples is 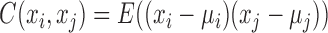 ; and their expression variance is
; and their expression variance is 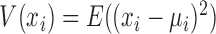 . Here, E(x) is the operation of expectation for variable x over a group of samples. When the absolute PCC is significantly large, it is to say there is an edge or association between xi and xj, and all edges connect nodes into a node-network (Figure 1A). Note that, PCC includes both direct and indirect associations between two variables, and thus to better represent the topological structure of a node-network, direct association measurement, such as partial correlation (22,23) or part mutual information (24) can be adopted instead of Equation (2).
. Here, E(x) is the operation of expectation for variable x over a group of samples. When the absolute PCC is significantly large, it is to say there is an edge or association between xi and xj, and all edges connect nodes into a node-network (Figure 1A). Note that, PCC includes both direct and indirect associations between two variables, and thus to better represent the topological structure of a node-network, direct association measurement, such as partial correlation (22,23) or part mutual information (24) can be adopted instead of Equation (2).
Edge-network of multiple samples
In addition to the node-network dynamics of Equation (1), to exactly and completely characterize the stochastic biological system, it is also required the other group of equations on the covariance matrix of molecules according to the Lyapunov differential equation (see Supplementary Information A1), i.e. edge-network dynamics:
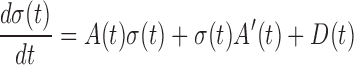 |
(3) |
Where, σ is the covariance matrix of variables; A is the adjacent matrix of the original node-network; A’ is the transpose of A; and D is a state-independent (e.g. constant) cyclic matrix. Obviously, this is a group of Lyapunov differential equations characterizing the second-order statistic information of a biological system, i.e. covariance. It can construct the new edge-network of molecules, by using each pair of molecules (i.e. σij) or each molecule interaction as a node.
A link in an edge-network is a fourth-order statistic due to its relationship between two molecule-pairs, which could be approximately inferred by using the corresponding correlations between two molecule-pairs. Given, for four molecules, i.e. two molecule-pairs, the correlation measurement is calculated as a high-order PCC (hPCC) of a group of samples (10):
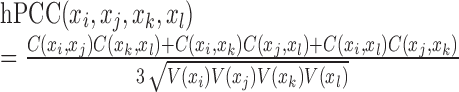 |
(4) |
where, covariance and variance over a group of samples are respective 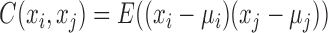 and
and 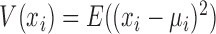 . Provided that there are time-course data or multi-sample data, such correlation (hPCC) between two molecule-pairs can be calculated, and two molecule-pairs with significant hPCC can be connected as a high-order edge. All high-order edges connect original edges of the node network into a new edge-network in a usual co-expression form (Figure 1A) (10). Clearly, an edge-network can characterize the high-order moment information of a biological system. The dynamics of a biological system with stochastic fluctuations can be fully recovered by its node-network and edge-network dynamics, given the Gaussian distribution assumption of all molecules.
. Provided that there are time-course data or multi-sample data, such correlation (hPCC) between two molecule-pairs can be calculated, and two molecule-pairs with significant hPCC can be connected as a high-order edge. All high-order edges connect original edges of the node network into a new edge-network in a usual co-expression form (Figure 1A) (10). Clearly, an edge-network can characterize the high-order moment information of a biological system. The dynamics of a biological system with stochastic fluctuations can be fully recovered by its node-network and edge-network dynamics, given the Gaussian distribution assumption of all molecules.
Edge-network of a single sample
For both node-network or edge-network, their constructions require multiple samples, which, however, are generally unavailable in clinical practice (25). It is strongly demanded to characterize or infer edge-network (also node-network) on the basis of single sample (25), which is expect to obtain the individual-specific or sample-specific edge-network.
Previously, the quantification of PCC in one sample was proposed to be calculated by correlation-like vectors (or delta PCC) (26,27). In fact, when the number of the reference samples is sufficiently large, these measurements can be reduced to calculate single-sample PCC (sPCC, see Supplementary Information A1 and A2):
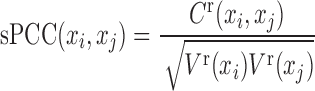 |
(5) |
where, the variance of the reference samples is 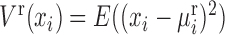 , and the covariance of a single sample is
, and the covariance of a single sample is 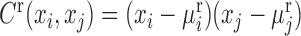 . For a single sample, the expression mean and variance are unknown, and thus
. For a single sample, the expression mean and variance are unknown, and thus 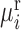 is the expression mean of gene i on the reference group, and the
is the expression mean of gene i on the reference group, and the 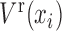 is the expression variance of gene i on the same reference group. Actually, Equation (5) can be viewed as a data transform from individual variables xi or xj to variable-pairs xi-xj against a given group of reference samples, e.g., from molecular concentrations to correlations. Thus, instead of the original data of a single sample, we can use the transformed data of this single sample for further analysis (e.g., differential analysis or clustering analysis), which may reveal different features or “Dark Matter" of the molecular expressions on the basis of a single sample.
is the expression variance of gene i on the same reference group. Actually, Equation (5) can be viewed as a data transform from individual variables xi or xj to variable-pairs xi-xj against a given group of reference samples, e.g., from molecular concentrations to correlations. Thus, instead of the original data of a single sample, we can use the transformed data of this single sample for further analysis (e.g., differential analysis or clustering analysis), which may reveal different features or “Dark Matter" of the molecular expressions on the basis of a single sample.
In a similar way to PCC and sPCC, we extend hPCC with multiple samples to shPCC with only one sample based on a number of the reference samples (see Supplementary Information A1), i.e. single-sample high-order PCC (shPCC) is computed as:
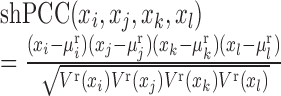 |
(6) |
where the high-order variance of the reference samples is 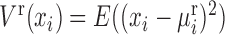 , and the high-order covariance of a single sample is
, and the high-order covariance of a single sample is 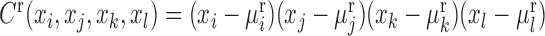 . Others are similarly defined. Hence, similar to the reconstruction of edge-network, we can obtain individual-specific edge-network based on shPCC from the observed single-sample data. Note that, to further eliminate the indirect association between variables in Equations (5)–(6), a sophisticated measurement such as partial correlation (22,23) or part mutual information (24) can be similarly adopted.
. Others are similarly defined. Hence, similar to the reconstruction of edge-network, we can obtain individual-specific edge-network based on shPCC from the observed single-sample data. Note that, to further eliminate the indirect association between variables in Equations (5)–(6), a sophisticated measurement such as partial correlation (22,23) or part mutual information (24) can be similarly adopted.
Individual-specific edge-network analysis (iENA)
Originally, the ENA was proposed to study molecular associations based on multiple samples or population features (10), e.g. cohorts’ risk estimation. However, it is increasingly demanded to analyse the molecular mechanisms of diseases on the basis of individuals. Thus, to address this general problem of network analysis (25), in this study we propose an advanced framework, i.e. iENA, based on our proposed measurement of shPCC on one-sample omics data. The details of this new approach will be described in a step-by-step manner (Figure 1B).
Collecting data: to evaluate the performance of iENA, we downloaded several gene expression datasets from NCBI GEO and TCGA to mainly predict the live influenza infection and cancer deterioration on individual subjects.
Selecting reference samples: in order to solve the calculation of mean and variance against a single-sample (i.e. for each sample of one subject at one time point), we need a group of reference samples (i.e. control samples, or normal samples). Here, we set the samples from baseline date to latter a few points as a reference group of each individual for influenza infection, and normal (or early-stage) samples as a reference group for cancers. Actually, any samples with the similar property can serve as a reference group (at least, five samples are required for the reference group). Once the reference samples are determined, they should be kept unchanged for whole studies.
Constructing node-network by sPCC calculation: when we have reference samples, we can construct the co-expression network of one sample by our single-sample measurement of PCC (sPCC, Equation (5)) consistent with previous studies (19,26,28). Note that a direct cut-off for edge correlations is difficult to decide because the distribution of the new sPCC values is not the normal distribution, and our experiments also suggest that the general threshold for PCC (e.g. 0.8 or else) seems not work well in this situation. Hence, we only focused on the edges from STRING database (29) to reduce computational complexity in biological context. Note that theoretically and computationally, instead of normal distribution, we can also construct the distribution of sPCC from the reference samples without the approximation for such a purpose (statistical test for the cut-off), but a large number of reference samples are required to construct this distribution. Furthermore, for each sample at a time point, the candidate edges/relations are required to have big changes on associations comparing to the same relations observed in the reference samples, i.e. the edges/relations have larger standard deviations than other edges. Then, the top-ranked edges with strong relations and significant changes will be finally selected. These candidate edges consist of conventional node-network, and will be used as the background ‘nodes’ for constructing the edge-network.
Constructing edge-network by shPCC calculation: for an edge-network, we use a background set of edges (gene-pairs) from the above steps as new nodes. Between two gene-pairs, we can carry on the estimation of the fourth-order single-sample correlation coefficient for each edge-pair (i.e. two gene-pairs) by shPCC (Equation (6)) for each single-sample (e.g. for each sample of one subject at one time point). Note that, in this step, we actually only compute the correlations between the pre-selected edges from above steps, and thus we can reduce the unnecessary computations drastically. Finally, we will get the edge-network corresponding to each sample at a particular time point, and each subject has its personalized/individual features on the observed samples or time series in the edge-networks.
Recognizing individual edge-biomarkers: similar to the edge selection, we select top-ranked edge-pairs as edge-biomarkers, which have strong relations with each other in terms of the high-order correlations. Those strong correlated gene-pairs are considered as DNB candidates, represented as a set called ‘Marker’. Then, for each individual, the gene-pairs involved in the edge-network (i.e. Marker) are used as individual edge-biomarkers and these genes (i.e. marker genes) are applied in the disease prediction.
-
Detecting the tipping point and DNB members by sCI: the DNB has been developed to identify the pre-disease state or the tipping point just before a sudden deterioration during disease progression (16,30) as general early-warning signals based on three statistical conditions (10,16,17). Recently, DNB model with its criterion (i.e. CI: composite index) based on multiple samples (11,31–34) has been adopted to successfully identify the tipping points of cell fate decision (35,36), to study immune checkpoint blockade (37,38), and also to quantify edge-biomarkers (10):
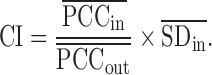 In this work, DNB criterion is further re-defined by the above second-order moment measurements on the basis of single-sample, i.e. single-sample composite index (sCI) is defined as:
In this work, DNB criterion is further re-defined by the above second-order moment measurements on the basis of single-sample, i.e. single-sample composite index (sCI) is defined as: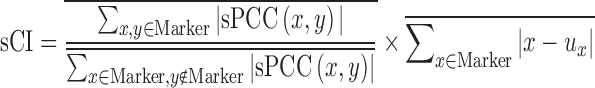
(7) where, the numerator is
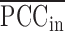 , which is the average sPCC of the expressions of genes in the dominant group or DNB (e.g. a group of marker genes or molecules) in absolute value in one sample; the denominator is
, which is the average sPCC of the expressions of genes in the dominant group or DNB (e.g. a group of marker genes or molecules) in absolute value in one sample; the denominator is 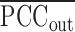 , which is the average sPCC of the expressions of genes between the dominant group and other in absolute value in one sample; the multiplier is
, which is the average sPCC of the expressions of genes between the dominant group and other in absolute value in one sample; the multiplier is 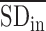 , which is the average standard deviation of the expressions of genes in the dominant group or DNB. ‘Marker’ is the set of DNB members. The superscript line means the average value. Thus, the sCI of individual edge-biomarkers will indicate the early-warning signals when its value is larger than a threshold. Note that we can evaluate sCI of Equation (7) by sPCC of Equation (5) without performing shPCC of Equation (6).
, which is the average standard deviation of the expressions of genes in the dominant group or DNB. ‘Marker’ is the set of DNB members. The superscript line means the average value. Thus, the sCI of individual edge-biomarkers will indicate the early-warning signals when its value is larger than a threshold. Note that we can evaluate sCI of Equation (7) by sPCC of Equation (5) without performing shPCC of Equation (6). Comparing edge-biomarkers: for each individual, we can use the differential gene-pairs in each single-sample (i.e. the edge associations in each sample or time point) as novel edge-biomarkers to indicate the early-warning signals or the tipping point. We will obtain sCI value with edge biomarkers for each subject or sample, and we can observe different sCI scores at consecutive time points or stages. Thus, we can set a threshold to indicate the criticality, i.e. warning or not for a subject. In addition, for the influenza infection data, we also examine the edge-biomarkers induced from each subject, and compare them with previously reported 50-gene markers and 22-gene markers from population focused studies (6–8,10).
RESULT AND DISCUSSION
Datasets for influenza infection
To evaluate the applicability of iENA, we downloaded two datasets GSE30550 and GSE52428 (7,8) from NCBI GEO to predict the live influenza infection on subjects in a temporal model. The analysis settings on dataset GSE52428 are described next mainly for convenience.
The transcriptome datasets contain 17 subjects (or adults) challenged with influenza H3N2/Wisconsin. In such challenge, nine subjects have been actually infected (i.e. appearance of the clinical symptom) but other eight subjects still stay healthy (without the appearance of clinical symptom). The gene expression profiles were obtained and measured on whole peripheral blood drawn from all subjects at an interval of 8 h post-inoculation (hpi) through 108 hpi. Totally, 268 gene micro-arrays were obtained for all subjects at 16 time points including baseline (24 h before subjects were injected with influenza virus, e.g. −24 hpi) (7), the pre-processions on which are similar to previous study (10).
As shown in Figure 2, according to iENA, we divided subjects into two groups according to the clinical symptom chart based on the standardized symptom scoring (7): symptomatic (Sx) group with nine subjects (subjects 1,5,6,7,8,10,12,13,15) and asymptomatic (Asx) group with eight subjects (subjects 2,3,4,9,11,14,16,17). And the samples from baseline date (as green in Figure 2) to latter a few points will be used as the reference group of samples; the non-symptom samples (as grey in Figure 2) which have no significant clinical symptom, may have obvious changes in molecular level and we will identify edge-biomarkers for detecting early-warning signals before the time point of symptom samples (as red in Figure 2). In calculation, we set five samples including baseline data as a reference group, i.e. there are five reference samples available for each subject (except the data of the 13th subject missed at a time point in dataset GSE52428). After that, we can calculate sPCC (with mean and variance from reference group) for each sample. Experimentally, we focused on the edges with great changes comparing to the reference group (with log2(fold change) > 0.8) and finally determined the top-ranked 1000 strong relations at each time point. Then, these pre-selected edges are used as the background ‘nodes’ for constructing the edge-network, and capturing the significant signal peaks of edge-biomarkers across multiple time points, from which 59 genes are finally obtained as related edge-biomarkers for Sx group (called Sx gene) compared to 215 candidates for Asx group. Those genes are candidate DNB members.
Figure 2.

The sample organization of dataset about influenza infection. The subjects are divided into two groups according to the clinical symptom chart based on the standardized symptom scoring: symptomatic (Sx) group with nine subjects (subjects S1,S5,S6,S7,S8,S10,S12,S13,S15) and asymptomatic (Asx) group with eight subjects (subjects S2,S3,S4,S9,S11,S14,S16,S17). The non-symptom samples (in grey) which have no significant clinical symptom, may have obvious changes in a network level; and we will identify edge-biomarkers for detecting early-warning signals before the time point of symptom samples (in red). The samples labeled in yellow colour indicates the time points predicted by our iENA based on dataset GSE52428, which is clearly earlier than the clinical symptom except one subject (S5). Actually, for the subject S5, the tipping point predicted by iENA coincides with the first symptom point at 45 h, but in this paper, we count it as an incorrect prediction.
Edge-biomarkers of Influenza infection identified by iENA are consistent with literature reports
Based on each sample from dataset GSE52428, iENA predicted the tipping points of each subject, shown in Figure 2 (i.e. samples labeled in yellow colour), which are clearly earlier than the clinical symptom except one subject (S5). Actually, for the subject S5, the tipping point predicted by iENA coincides with the first symptom point at 45 h, but in this paper, we count it as an incorrect prediction. iENA recovered the common biomarkers of influenza infection reported in the previous works (10), which demonstrates the effectiveness of iENA on biomarker discovery for diseases (see Supplementary Information A3). As shown in Figure 3A, the marker genes detected in Sx individuals are very different from those identified in Asx individuals; that is why the Sx-specific edge markers can be used to predict the symptom appearance (or disease occurrence). As shown in Figure 3B, our final edge-biomarkers have significant overlap with 22-gene markers reported in our previous work (10) and the 50-gene markers reported in the original work (6–8,10). That means our new method can efficiently find the key genes involved in influenza infection, and also discover many new potential disease-associated genes.
Figure 3.

The consistence between our findings and previously reported markers in the study of influenza infection from dataset GSE52428. (A) The overlap between marker genes identified from Sx individuals and Asx individuals. (B) The overlap among iENA identified disease marker genes (i.e. Sx genes) and previously reported 22 marker genes and 50 marker genes. (C) The overlap ratio as the percentage of individual edge-biomarkers recovered from prior-known genes in each sample. (D) The overlap ratio as the percentage of individual node-biomarkers recovered from prior-known genes in each sample.
Particularly, we evaluated the efficiency of edge-network for recovering previously reported markers on individuals, and used the overlap ratio (i.e. the ratio between the overlapping genes and the whole detected marker genes on all samples for each individual) to evaluate such efficiency. As shown in Figure 3C, almost 10% genes have been reported for Sx individuals, meanwhile <5% genes for Asx individuals. This fact means that it is more possible to observe the alteration of Influenza infection on Sx individuals than Asx ones, and edge-network is effective to predict diseases. In addition, we also evaluated the similar efficiency of node-network (i.e. constructed by the background edges). As shown in Figure 3D, the overlap ratio is <1% for both Sx and Asx individuals, and there is no significant difference between them, which supports again that edge-network is more effective than node-network in iENA.
Next, our edge-biomarkers (i.e. Sx-specific edge biomarkers) have many dense associations on STRING network (Figure 4A), which indicate that edge-network can actually detect interactive gene-pairs. These marker genes have significant enrichments on many virus-related KEGG pathways, such as NOD-like receptor signalling pathway, Herpes simplex infection, Influenza A, Hepatitis C, Cytosolic DNA-sensing pathway and Measles. For Influenza A pathway, our marker genes (as red in Figure 4B) are widely distributed on the IPA annotated network as ‘Antimicrobial Response, Inflammatory Response’, which has been reported to be related with early immune response to influenza A virus infection (39). That strongly supports that our biomarkers can detect the early-warning signals of infection before the appearance of symptom (i.e. the prediction of pre-symptom state or the tipping point).
Figure 4.

The edge-biomarkers identified by iENA for dataset GSE52428. (A) Protein–protein associations on STRING network. (B) Regulatory associations on IPA network annotated as ‘Antimicrobial Response, Inflammatory Response’.
Detecting early-warning signals of Influenza symptom by iENA
iENA with DNB can detect biomarkers and predict pre-symptom state in a single-sample manner, in contrast to the traditional methods (7,8). In other words, due to the nature of single-sample, we can also analyze the data without the temporal information. In this study, the dominant group or DNB is the molecules or genes of edge-biomarkers from each Sx or Asx sample, and others (all genes except those in the dominant group) are the selected edges' genes for the same sample. When calculating the single-sample DNB score, i.e. sCI index, there is no need to pre-define the time window for expression correlation calculation. Thus, each Sx or Asx subject can have 15 checkpoints and the corresponding index values (sCI) can represent his/her diagnostic score over time (Figure 5A). When the predicted score becomes significantly larger than a given threshold, this time point will be regarded as the early-warning point/tipping point of the disease occurrence. By using such model to predict influenza infection in practice, e.g. on dataset GSE52428, the ROC can be drawn along with the change of the threshold, then the corresponding AUC is used to evaluate the accuracy of such prediction. The results show that our edge-biomarkers achieve about 0.9 accuracy (Figure 5B).
Figure 5.

Prediction performance of iENA for dataset GSE52428. (A) The predicted curve of early-warning signals or tipping points for all subjects by our edge-biomarkers. (B) The prediction accuracy as ROC curve. (C) The individual prediction curves. The red circle points the clinically diagnosed infection-time (symptom) for the corresponding subject, and the green star mark indicates the predicted infection-time (tipping point) by our prediction cut-off. Clearly, we correctly predicted all of the symptom cases except S5 before their clinical symptom. Actually, for the subject S5, the tipping point predicted by iENA coincides with the first clinical symptom point at 45 h, but in this paper, we count S5 as an incorrect prediction.
An advantage of our model is to predict both tipping point and key molecules of diseases, rather than only disease state. Especially, iENA can make the prediction based on each single sample on any time point rather than multiple samples on a time window. As shown in Figure 5C each sub-figure displays the early-warning signal for each Sx subject during the progression of the influenza infection, where the green star mark indicates the predicted tipping point or pre-symptom state by our prediction cut-off and the red circle points the clinically diagnosed infection-time or symptom state for the corresponding subject. According to the cut-off for judging the pre-disease state during the progression of influenza infection, all nine Sx subjects can be correctly predicted the final disease occurrence. Even more, we can also correctly predict the symptom for each subject (green label in Figure 5C) before the influenza infection diagnosed by the standardized symptom scoring record (red label in Figure 5C), although the predicted time for subject 5 is a little delayed. All results imply that edge-biomarker by our iENA can predict the time of onset influenza infection effectively and accurately.
Robustness and reproducibility of iENA
On purpose of practical prediction on both occurred event and time, different markers have been compared. For instance, the consistent module genes significantly associated with phenotype by WGCNA (20), which has AUC <0.5. Thus, the gene module from WGCNA would have more false positives, and have less power on disease prediction. By contrast, iENA achieves the higher performance than WGCNA, because it not only considers the prediction of disease occurrence but also captures the occurred time point. On the other hand, some of the genes enriched in known KEGG influenza pathways can be potential biomarkers. But, the KEGG influenza pathway is still incomplete, and thus, there is need to find additional marker genes (40). Different from conventional common markers (e.g. KEGG influenza pathway genes), iENA can investigate the individual-specific makers and therefore provides a new way for such a purpose.
In addition, we have also analyzed another dataset of influenza infection, i.e. GSE30550 dataset, and obtained significant reproducibility, i.e. re-discovered markers (Figure 6A), high precision on disease occurrence and time (Figure 6B), and superior performance on individual prediction (Figure 6C). The results validated the effectiveness of our method.
Figure 6.

Prediction results on another dataset of influenza symptom from GSE30550 for validation. (A) The detected marker genes (recovered genes) from GSE30550 analysis, and the Common Sx gene from GSE52428 analysis, and the 22-gene marker from our previous study. (B) The ROC of prediction. (C) The prediction curves of individuals. The red circle points the clinically diagnosed infection-time for the corresponding subject, and the green star mark indicates the predicted infection-time by our prediction cut-off. Clearly, we correctly predicted all of the symptom cases except S5 before the clinical symptom, and the accuracy of the prediction is about 90%. Although the tipping point of subject S5 predicted by iENA coincides with the first clinical symptom point at 45 h, we count S5 as an incorrect prediction in this paper.
Detecting the tipping points of cancer deteriorations by iENA on TCGA data
Not limited to time course data, iENA can also analyse the phased or sample-based data, e.g. the TCGA data about cancer progression. In clinic, the cancer progression is measured with different stages, and some stage would be critical one or tipping point, after which the disease drastically or seriously deteriorates. Before and after the tipping point, the patient survival times will be significantly changed. A few works have studied the gene expressions during cancer occurrence and development (41), but, the protein expressions (i.e. reverse phase protein array in TCGA) still remain less focused on a systematical level.
In this study, for the first time, we applied iENA to systematically investigate the contribution of proteins in cancer progression, especially in the critical stage of cancer progression. We downloaded the TCGA RPPA data for breast cancer (BRCA with 226 proteins on 929 samples) and liver cancer (LIHC with 219 proteins on 172 samples) respectively (42), and used the samples known with clinical stage information. We took four sample groups labelled with sequentially clinical stages as stage i, stage ii, stage iii and stage iv. Using the samples in stage i as references, each sample can be analyzed by iENA to construct an edge-network and further identify its edge-biomarkers. In such a way, each sample can be quantified by sCI score. Note that even if a sample is in the reference group, it can also be analysed for iENA as the same as other single samples but the group of the reference samples cannot be changed for the whole study. The detail procedure and results for the analysis is described as follows.
For each stage, the sCI scores of its all samples are averaged as this stage’s early-warning signals. The average sCI score with re-sampling achieved the highest value at stage ii for both BRCA and LIHC, which suggests that the second clinical stage would be the critical stage during cancer progression. And in stage ii, according to the sample number of a protein selected as edge-biomarkers, the proteins with marker frequency no <0.1 are considered as the protein signatures or DNB members of the corresponding critical stage (Figure 7A and E).
To validate the tipping point, we also analyzed the distribution of survival days for samples in critical stage ii and the periods before and after it. We can see that the average survival days significantly decrease at stage ii or later (Figure 7B and F), which provide the clinical evidence for stage ii as a tipping point before patients suffer from serious deteriorations on their disease risks.
In addition, we used the coding-genes of protein signatures (see Supplementary Information A3) to infer their expression associations with patient survival time by survival analysis (43), i.e. to test the effectiveness of DNB members. Still on TCGA expression data, these coding-genes can actually divide all patients (disregarding specific stages) into discriminative high-risk and low-risk groups, for BRCA and LIHC respectively (Figure 7C and G). To avoid platform dependence, we also tested these coding-genes on the third-party data from NCBI GEO with large sample sizes (43), and again they indeed have significant survival analysis results on those independent breast cancer or liver cancer cohorts (Figure 7D and H).
Figure 7.

The results on TCGA cancer data. (A) The iENA analysis on breast cancer (BRCA), where stage ii is identified as the tipping point. (B) The survival-day comparison for different breast cancer stages. (C) The survival analysis by coding-genes of edge-biomarkers on TCGA BRCA data. (D) The survival analysis by coding-genes of edge-biomarkers on GEO independent data. (E) The iENA analysis on liver cancer (LIHC), where stage ii is identified as the tipping point. (F) The survival-day comparison for different liver cancer stages. (G) The survival analysis by coding-genes of edge-biomarkers on TCGA LIHC data. (H) The survival analysis by coding-genes of edge-biomarkers on GEO independent data. Here, the edge-biomarkers are the DNB members.
All these results illustrate that iENA is a general method to study complex diseases including cancers, and provide effective edge-biomarkers for disease prediction or tipping point detection.
CONCLUSION
We have presented a new framework, i.e. iENA, based on DNB to identify the pre-disease state or the tipping point during disease progression, by extracting the high-order statistics and dynamical information from biological data in a one-sample manner. In this paper, as demonstration examples of iENA, we have mainly analyzed two time-course datasets of 17 subjects (healthy adults) with risk of influenza infection, and two cancer datasets of TCGA. The results of the influenza infection data from the iENA analysis indicate: (i) for nine Sx subjects, the early-warning signals of the symptom were correctly found before clinical diagnosis except one case, and their critical time points (i.e. tipping points) with edge-biomarkers (i.e. DNBs) were detected; (ii) the edge-biomarkers are significantly related to the disease progression and development (e.g. virus infection); and (iii) the edge-biomarkers are able to predict the infection occurrence and time simultaneously. In addition, we applied iENA to the analysis of TCGA cancer proteomic data (breast cancer and liver cancer), which also revealed critical stages during cancer progression and provided survival-relevant protein signatures. These results demonstrate the effectiveness of iENA analysis with DNB on disease study, which makes it practical in clinic applications due to no requirement on multiple samples, which is a useful tool for personalized medicine on different types of omics data (25).
Indeed, the iENA is the further development of the ENA from the prediction of common risk factors over multiple samples to the prediction of individual-specific biomarkers on a single sample. This new method actually utilizes individual samples for the prediction of disease states by exploring differential edge-network based on differential expression, variance and covariance analysis. Although our evaluations have shown that iENA would be robust on the parameter setting (see Supplementary Information A3), it requires careful quality control to reduce experimental errors for the case of the limited reference samples. In this work, we focused on omics data rather than low-throughput data, and thus it is still unclear if or not iENA can be directly applied to the clinical panel assay. How to optimally choose edge-biomarkers as DNB members is also a future topic. iENA will be an important step towards precision medicine, and especially its ability to predict the tipping points or critical transitions of diseases including cancer is also an important topic in the field of translational medicine. In addition to medicine, iENA can also be directly applied to the study for the tipping points and key regulators of many biological processes, e.g., cell differentiation or molecular evolution.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2017YFA0505500]; Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) [XDB13040700]; National Natural Science Foundation of China [61403363, 11401222, 31200987, 91529303, 91439103, 81471047, 31771476]; National Key R&D Program (Special Project on Precision Medicine) [2016YFC0903400]; Natural Science Foundation of Shanghai [17ZR1446100]. Funding for open access charge: Strategic Priority Research Program of the CAS [XDB13040700].
Conflict of interest statement. None declared.
REFERENCES
- 1. Zeng T., Wang D.C., Wang X., Xu F., Chen L.. Prediction of dynamical drug sensitivity and resistance by module network rewiring-analysis based on transcriptional profiling. Drug Resist. Updat. 2014; 17:64–76. [DOI] [PubMed] [Google Scholar]
- 2. Cohen A., Bont L., Engelhard D., Moore E., Fernandez D., Kreisberg-Greenblatt R., Oved K., Eden E., Hays J.P.. A multifaceted ‘omics’ approach for addressing the challenge of antimicrobial resistance. Future Microbiol. 2015; 10:365–376. [DOI] [PubMed] [Google Scholar]
- 3. Hood L., Friend S.H.. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat. Rev. Clin. Oncol. 2011; 8:184–187. [DOI] [PubMed] [Google Scholar]
- 4. Banchereau R., Baldwin N., Cepika A.M., Athale S., Xue Y., Yu C.I., Metang P., Cheruku A., Berthier I., Gayet I. et al. Transcriptional specialization of human dendritic cell subsets in response to microbial vaccines. Nat. Commun. 2014; 5:5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jain S., Gitter A., Bar-Joseph Z.. Multitask learning of signaling and regulatory networks with application to studying human response to flu. PLoS Comput. Biol. 2014; 10:e1003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsalik E.L., Henao R., Nichols M., Burke T., Ko E.R., McClain M.T., Hudson L.L., Mazur A., Freeman D.H., Veldman T. et al. Host gene expression classifiers diagnose acute respiratory illness etiology. Sci. Transl. Med. 2016; 8:322ra311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang Y., Zaas A.K., Rao A., Dobigeon N., Woolf P.J., Veldman T., Oien N.C., McClain M.T., Varkey J.B., Nicholson B. et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet. 2011; 7:e1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woods C.W., McClain M.T., Chen M., Zaas A.K., Nicholson B.P., Varkey J., Veldman T., Kingsmore S.F., Huang Y., Lambkin-Williams R. et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PloS One. 2013; 8:e52198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y., Jin S., Lei L., Pan Z., Zou X.. Deciphering deterioration mechanisms of complex diseases based on the construction of dynamic networks and systems analysis. Sci. Rep. 2015; 5:9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu X., Li G., Chen L.. Prediction and early diagnosis of complex diseases by edge-network. Bioinformatics. 2014; 30:852–859. [DOI] [PubMed] [Google Scholar]
- 11. Liu R., Wang X., Aihara K., Chen L.. Early diagnosis of complex diseases by molecular biomarkers, network biomarkers, and dynamical network biomarkers. Med. Res. Rev. 2014; 34:455–478. [DOI] [PubMed] [Google Scholar]
- 12. Kim H., Gelenbe E.. Anomaly detection in gene expression via stochastic models of gene regulatory networks. BMC Genomics. 2009; 10(Suppl. 3):S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cao Y., Li Y., Coleman S., Belatreche A., McGinnity T.M.. Adaptive hidden Markov model with anomaly States for price manipulation detection. IEEE transactions on neural networks and learning systems. 2015; 26:318–330. [DOI] [PubMed] [Google Scholar]
- 14. Silva J., Willett R.. Hypergraph-based anomaly detection of high-dimensional co-occurrences. IEEE Trans. Pattern Anal. Mach. Intell. 2009; 31:563–569. [DOI] [PubMed] [Google Scholar]
- 15. Wang K., Langevin S., O’Hern C.S., Shattuck M.D., Ogle S., Forero A., Morrison J., Slayden R., Katze M.G., Kirby M.. Anomaly detection in host signaling pathways for the early prognosis of acute infection. PLoS One. 2016; 11:e0160919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen L., Liu R., Liu Z.P., Li M., Aihara K.. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep. 2012; 2:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zeng T., Zhang C.C., Zhang W., Liu R., Liu J., Chen L.. Deciphering early development of complex diseases by progressive module network. Methods. 2014; 67:334–343. [DOI] [PubMed] [Google Scholar]
- 18. Yu X., Zeng T., Li G.. Integrative enrichment analysis: a new computational method to detect dysregulated pathways in heterogeneous samples. BMC Genomics. 2015; 16:918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu X., Zeng T., Wang X., Li G., Chen L.. Unravelling personalized dysfunctional gene network of complex diseases based on differential network model. J. Transl. Med. 2015; 13:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Langfelder P., Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sankar P.L., Parker L.S.. The Precision Medicine Initiative's All of Us Research Program: an agenda for research on its ethical, legal, and social issues. Genet. Med. 2016; 19:743–750. [DOI] [PubMed] [Google Scholar]
- 22. Zhang X., Zhao J., Hao J.K., Zhao X.M., Chen L.. Conditional mutual inclusive information enables accurate quantification of associations in gene regulatory networks. Nucleic Acids Res. 2015; 43:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zeng T., Chen L.. Tracing dynamic biological processes during phase transition. BMC Syst. Biol. 2012; 6(Suppl. 1):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao J., Zhou Y., Zhang X., Chen L.. Part mutual information for quantifying direct associations in networks. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:5130–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zeng T., Zhang W., Yu X., Liu X., Li M., Chen L.. Big-data-based edge biomarkers: study on dynamical drug sensitivity and resistance in individuals. Brief. Bioinform. 2016; 17:576–592. [DOI] [PubMed] [Google Scholar]
- 26. Zhang W., Zeng T., Chen L.. EdgeMarker: Identifying differentially correlated molecule pairs as edge-biomarkers. J. Theor. Biol. 2014; 362:35–43. [DOI] [PubMed] [Google Scholar]
- 27. Liu X., Wang Y., Ji H., Aihara K., Chen L.. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 2016; 44:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang W., Zeng T., Liu X., Chen L.. Diagnosing phenotypes of single-sample individuals by edge biomarkers. J. Mol. Cell Biol. 2015; 7:231–241. [DOI] [PubMed] [Google Scholar]
- 29. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K.P. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang Y., Lin X., Lu X., Luo G., Zeng T., Tang J., Jiang F., Li L., Cui X., Huang W. et al. Interferon-microRNA signalling drives liver precancerous lesion formation and hepatocarcinogenesis. Gut. 2016; 65:1186–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu R., Li M., Liu Z.P., Wu J., Chen L., Aihara K.. Identifying critical transitions and their leading biomolecular networks in complex diseases. Sci. Rep. 2012; 2:813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu R., Yu X., Liu X., Xu D., Aihara K., Chen L.. Identifying critical transitions of complex diseases based on a single sample. Bioinformatics. 2014; 30:1579–1586. [DOI] [PubMed] [Google Scholar]
- 33. Liu R., Chen P., Aihara K., Chen L.. Identifying early-warning signals of critical transitions with strong noise by dynamical network markers. Sci. Rep. 2015; 5:17501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen P., Liu R., Li Y., Chen L.. Detecting critical state before phase transition of complex biological systems by hidden Markov model. Bioinformatics. 2016; 32:2143–2150. [DOI] [PubMed] [Google Scholar]
- 35. Richard A., Boullu L., Herbach U., Bonnafoux A., Morin V., Vallin E., Guillemin A., Papili Gao N., Gunawan R., Cosette J. et al. Single-cell-based analysis highlights a surge in cell-to-cell molecular variability preceding irreversible commitment in a differentiation process. PLoS Biol. 2016; 14:e1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mojtahedi M., Skupin A., Zhou J., Castano I.G., Leong-Quong R.Y., Chang H., Trachana K., Giuliani A., Huang S.. Cell fate decision as high-dimensional critical state transition. PLoS Biol. 2016; 14:e2000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lesterhuis W.J., Bosco A., Millward M.J., Small M., Nowak A.K., Lake R.A.. Dynamic versus static biomarkers in cancer immune checkpoint blockade: unravelling complexity. Nat. Rev. Drug Discov. 2017; 16:264–272. [DOI] [PubMed] [Google Scholar]
- 38. Li M., Li C., Liu W.X., Liu C., Cui J., Li Q., Ni H., Yang Y., Wu C., Chen C. et al. Dysfunction of PLA2G6 and CYP2C44-associated network signals imminent carcinogenesis from chronic inflammation to hepatocellular carcinoma. J. Mol. Cell Biol. 2017; doi:10.1109/CCA.2009.5281071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. LeMessurier K.S., Lin Y., McCullers J.A., Samarasinghe A.E.. Antimicrobial peptides alter early immune response to influenza A virus infection in C57BL/6 mice. Antiviral Res. 2016; 133:208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang C., Liu J., Shi Q., Zeng T., Chen L.. Comparative network stratification analysis for identifying functional interpretable network biomarkers. BMC Bioinformatics. 2017; 18:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zeng T., Sun S.Y., Wang Y., Zhu H., Chen L.. Network biomarkers reveal dysfunctional gene regulations during disease progression. FEBS J. 2013; 280:5682–5695. [DOI] [PubMed] [Google Scholar]
- 42. Akbani R., Ng P.K., Werner H.M., Shahmoradgoli M., Zhang F., Ju Z., Liu W., Yang J.Y., Yoshihara K., Li J. et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat. Commun. 2014; 5:3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aguirre-Gamboa R., Gomez-Rueda H., Martinez-Ledesma E., Martinez-Torteya A., Chacolla-Huaringa R., Rodriguez-Barrientos A., Tamez-Pena J.G., Trevino V.. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 2013; 8:e74250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.