

Abstract
Prostate cancer is the most frequently diagnosed malignancy and the leading cause of cancer related death in men. First line therapy for disseminated disease relies on androgen deprivation, leveraging the addiction of these tumors on androgens for both growth and survival. Treatment typically involves antagonizing the androgen receptor (AR) or blocking the synthesis of androgens. Recurrence is common and within 2–3 years patients develop castration resistant tumors that become unresponsive to AR-axis targeted therapies. In order to provide a more effective treatment, we are utilizing an approach that targets a key scaffolding protein, Sigma1 (also known as sigma-1 receptor), a unique 26-kilodalton integral membrane protein that is critical in stabilizing the AR. Herein we report on a new series of Sigma1 compounds for lead optimization derived from a hybrid pharmacophore approach.
Keywords: Sigma1, Sigma-1 receptor, Sigma-1 chaperone, Prostate cancer, Guanidine, Small molecule inhibitor
Prostate cancer is the most frequently diagnosed malignancy and the leading cause of cancer related death in men.1 It is estimated that 217,000 men in the US will be diagnosed with prostate cancer and up to 32,000 men will die of advanced disease. Early detection of the primary tumor and treatment of localized disease by radical prostatectomy and/or radiation therapy generally leads to favorable prognostic outcomes.2 However once the disease disseminates it becomes much more difficult to treat and unlike other solid tumors, prostatic adenocarcinoma responds poorly to standard chemotherapeutic intervention.3
Thus first line therapy for disseminated disease relies on androgen deprivation, leveraging the addiction of these tumors on androgens for both growth and survival.3 Treatment typically includes the androgen receptor (AR) antagonist enzalutamide (Xtandi®)4,5 that directly antagonizes the AR or abiraterone acetate (Zytiga®), a molecule that blocks the synthesis of androgens.6,7 Despite this chemical castration approach, recurrence is common and within 2–3 years patients develop castration resistant tumors that become unresponsive to AR-axis targeted therapies such as enzalutamide and abiraterone.3 At this stage of the disease, advanced castration resistant prostate cancer (CRPC), the AR in these tumors has adapted to the stress of treatment resulting in alternative splicing, AR mutation, or over expression of the AR to overcome treatment regimes.3 In order to provide a more effective treatment for CRPC patients we are utilizing an approach that targets a key scaffolding protein that is critical in stabilizing the AR.
We are targeting Sigma1 (also known as sigma-1 receptor), a unique 26-kilodalton integral membrane protein8,9 primarily located in the ER in cancer cells. Sigma1 acts as a chaperone or scaffolding protein that coordinates the maturation and transport of client proteins critical to the AR axis. Sigma1 is highly expressed in prostate cancer cells and RNAi-mediated knockdown of Sigma1 results in tumor growth inhibition.11 Upon profiling known Sigma1 inhibitors for their ability to affect Sigma1 mediated translational repression10 and ER homeostasis pathways,11 we identified IPAG (1-adamantan-1-yl)-3-(4-iodophenyl)guanidine) and the anti-psychotic drug, haloperidol, as potent inhibitors of Sigma1-mediated AR signaling, protein homostasis, as well as inhibitors of cancer cell proliferation and growth.10,11 In an effort to create new composition of matter for intellectual property protection, and to provide a new series of Sigma1 compounds for lead optimization, we utilized a hybrid pharmacophore approach to identify novel Sigma1 compounds.
IPAG12 is a high affinity Sigma1 ligand (Ki = 2.7 nM; [3H]-(+)-pentazocine) based on the 1,3-di-o-tolylguanidine (DTG) template13,14 and is a valuable probe for studying Sigma1 function. IPAG shows antiproliferative activity in AR-positive prostate cancer cell lines and cellular activity in the androgen reporter assay15–17 (EC50 AR = 9.7 μM) in a comparable range to the indirect androgen synthesis blocker, abiraterone (EC50 AR = 17.1 μM), and about 20-fold less potent than the direct androgen receptor antagonist, enzalutamide (EC50 AR = 0.42 μM).17 However, it is unstable when in solution, and particularly unstable when exposed to normal fluorescent light due to the aryl iodide functionality. It is also slightly more lipophilic than desired for optimal drug-like qualities (CLogP = 4.46). Haloperidol, an antipsychotic neuroleptic drug widely used in the treatment of schizophrenia. It shows high affinity to Sigma1 (Ki = 0.21 nM; [3H]-(+)-pentazocine).18 Cellular activity in the androgen reporter assay was about 2-fold greater than abiraterone. It has potent affinity for D2 dopamine receptors, as well as affinity at several other receptors. However, CYP3A4 oxidation of the 4-hydroxyphenyl piperidine moiety of haloperidol results in the formation of pyridinium metabolites, such as, 4-(4-chlorophenyl)-1-[4-(4-fluorophenyl)-4-oxobutyl]pyridinium (HPP+), that have been shown to bind irreversibly to Sigma1 protein,19 and display neurotoxic properties resembling those of their structural analog MPP+ thus making this functionality undesirable in a compound moving forward due to its potential toxic liability. For these reasons, and the desire for novelty we envisioned a hybrid approach to combine the best features of the two potent Sigma1 ligands and create a new series (Fig. 1).
Fig. 1.
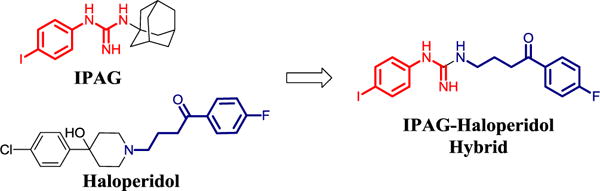
Conceptually, combining the aryl guanidine of IPAG and the side chain of haloperidol provides a novel series of hybrid analogs with potential to improve on drug-like properties, potency, and selectivity.
Weber et al.20–22 have identified symmetrically substituted guanidines (i.e. DTG), and further developed the SAR13 to demonstrate the importance of the aryl guanidine in DTG analogs for Sigma1 activity. We envisioned elaborating the right hand side chain of a symmetrical di-substituted aryl guanidine by replacing it with side chains of various Sigma1 ligands, and then using this mix and match approach we sought to identify new Sigma1 ligands suitable for lead optimization. We also noted that the molecular volume of the aryl piperidine motif in haloperidol is approximately the same size as the aryl guanidine motif in IPAG suggesting a hybrid. To synthesize these hybrid analogs we started with the same chemistry used to expand the aryl guanidine SAR study by Scherz, et al.13 This method (Scheme 1) provided the desired product, 1, but also a significant amount of the symmetric di-substituted guanidine, 2, that required reverse phase HPLC purification due to the polar nature of the molecules. We used the equivalent and more metabolically stable aryl ether bioisosteric side chain22 instead of the aryl ketone side chain in haloperidol for ease of synthesis. Using a similar approach we also made the symmetric di-substituted guanidine, 2, to confirm the structure and to provide enough material to test both analogs side-by-side (Scheme 1). In order to improve the yield and ease of synthesis, we attempted the synthesis via the side chain amino nitrile, unfortunately this only lead to poor yields of the side chain dimer, 3, which we purified by reverse phase chromatography and tested as an additional compound for potency (Scheme 2).
Scheme 1.
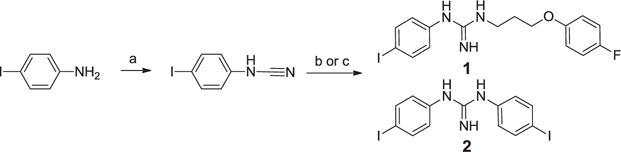
Reagents and conditions: (a) Cyanogen bromide, THF, DIEA, rt, 48 h, 20% (b) 3-(4-fluorophenoxy)propan-1-amine, chlorobenzene, TsOH, 200 °C (microwave), 40 min., 32% (c) 4-iodoaniline, chlorobenzene, TsOH, 200 °C (microwave), 40 min., 30%.
Scheme 2.

Reagents and conditions: (a) Cyanogen bromide, DCM, H2O, NaHCO3, 0 °C to rt, 1 h., 10%.
We also utilized the activated thiourea synthesis of guanidines to make the first batch of the 1-(4-chlorophenyl)-3-(3-(4-fluorophenoxy)propyl)guanidine analog, 4 (Scheme 3). This route provided product in good yield, however purification by chromatography was still challenging due to the polar nature of the guanidine product. Seeking a general method and an improved synthetic route for the unsymmetrical guanidines we evaluated the “pyrazole guanidine” reagent reported by Bernatowicz24 that was developed for the synthesis of mono-substituted guanidines or arginine containing peptides. Important advantages of this method were that the intermediates are N-Boc protected and relatively non-polar so that they can be more readily purified by normal phase flash chromatography. Using the commercially available bis-boc-pyrazolocarboxamidine (AstaTech, Inc.; CAS No.: 862686-58-6) we synthesized the alcohol side chain,23 and then installed it via a Mitsunobu reaction27 to provide the di-N-boc pyrazole guanidine reagent (Scheme 4; R = H) as a suitable advanced intermediate envisioned for the synthesis of numerous analogs derived from anilines.
Scheme 3.
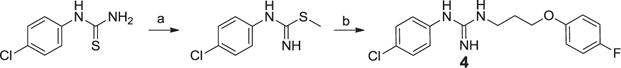
Reagents and conditions: (a) Methyl iodide, acetone, reflux, 1 h., 83% (b) 3-(4-fluorophenoxy)propan-1-amine, ethanol, reflux, 12 h., 78%.
Scheme 4.
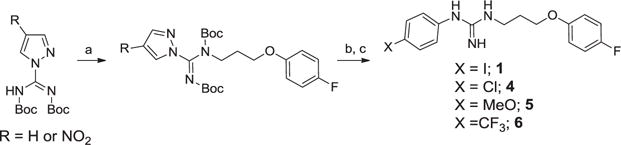
Reagents and conditions: (a) DIAD, Ph3P, THF, 3-(4-fluorophenoxy)propan-1-ol, 0 °C to rt, 18 h, 61%. (b) The corresponding aniline, anhyd. MeOH, Et3N, 24–36 h., 86%. (c) DCM, 4N HCl in dioxane, 0 °C to rt, 18–24 h., 92%.
This methodology also provided a facile means to incorporate various alcohol side chains in high yield. Although this worked satisfactorily on a small scale, scale up to multigram level was still difficult because of the poor nucleophilicity of the anilines used to generate the aryl guanidine analogs. In addition, when heating was used in order to drive the reactions to completion typically one of the N-Boc protecting groups was lost, making purification difficult. Thus a further improvement of the synthesis was found using the more electrophilic variant,25,26 4-nitro-1H-pyrazole-1-N,N′-bis(tert-butoxycarbonyl)carboxamidine (Scheme 4; R = NO2). This more reactive guanidylating reagent provided a facile method for analog synthesis. Importantly the reactions proceeded to completion at ambient temperature without loss of the N-Boc protection. This method was then successfully utilized to synthesize key analogs for SAR purposes and for multigram scale synthesis for further profiling and in vivo efficacy studies.
In vitro radio-ligand binding assays using membranes from MDA-MB-468 cancer cells, which express Sigma111,28,29 provided confirmation of Sigma1 binding affinity for our new hybrid analogs compared to IPAG and haloperidol (Table 1). The hybrids 1, 4, 5, 6 all demonstrated reasonable affinity to Sigma1 with about a 5–10-fold decrease in binding affinity compared to IPAG. Notably the dimer 3 had no affinity for Sigma1. The di-aryl dimer 2 maintained binding affinity similar to IPAG.
Table 1.
Sigma affinity of haloperidol, IPAG, and new hybrid sigma ligands.
| Structure | ID | Ki (nM)a | CLogP |
|---|---|---|---|
|
|
Haloperidol | 0.21 | 3.85 |

|
IPAG | 2.76 | 4.46 |
|
|
1 | 46.08 | 4.50 |

|
2 | 3.0b | 4.25 |
|
|
3 | >1000b | 4.55 |
|
|
4 | 38.00 | 4.09 |
|
|
5 | 13.36 | 3.30 |
|
|
6 | 45.32 | 4.26 |
Ki values represent duplicate or triplicate determinations against [3H] – (+) – pentazocine.
Ki determined against [125I]-IPAG.
We also noted that the aryl iodides 1 and 2 showed a photo-stability issue similar to IPAG. Evaluation of IPAG, 1, 4, and 5 in a cell viability assay (Cell Titer Glo; MDA-MB-468 cells) demonstrated that the new analogs were not acutely cytotoxic, and showed antiproliferative activity with potencies in the 3–20 μM range. We further profiled analogs 1, 4, and 5, for plasma protein binding, plasma stability, liver microsome stability, and water solubility (Table 2).
Table 2.
In vitro ADME properties of new hybrid analogs.
| ID | % Plasma protein binding
|
Plasma Stability
|
Liver microsome stability (min)
|
Water solubility
|
|||
|---|---|---|---|---|---|---|---|
| Mouse | Human | Mouse | Human | Mouse | Human | PBS buffer pH 7.4 (mg/mL) | |
| 1 | 97.9 | 96.6 | >6 h | >6 h | 24.6 | >90 | 5.37 |
| 4 | 88.8 | 93.4 | >6 h | >6 h | 24.8 | >90 | 4.58 |
| 5 | 82.1 | 75.1 | >6 h | >6 h | 7.0 | 23.2 | 8.45 |
In vitro profiling suggests that the new hybrid series has good drug-like properties with regards to in vitro ADME. The compounds show modest plasma protein binding with compound 4 and 5 below 95%. The compounds are stable in mouse and human plasma. Mouse liver microsome stability for compounds 1 and 4 is acceptable in the mouse and very good in human. Compound 5, the 4-methoxy aryl analog, is most likely being degraded due to dealkylation of the methoxy group, or oxidative metabolism of the electron rich aromatic ring. Water solubility for these analogs is very good. We further evaluated compound 4 for plasma and brain exposure in the mouse, as well as oral bioavailability at two different oral doses for an initial evaluation of escalating dose linearity (Table 3).
Table 3.
Mouse pharmacokinetic exposure of compound 4.
| A. Levels of compound 4 in Male Swiss Mice
| ||
|---|---|---|
| 10 mg/kg, 5 mL/kg, IP | Plasma (ng/mL) | Brain (ng/g) |
| Cmax, ng/mL | 141 (438.18 nM) | 691 (2,147.4 nM) |
| Tmax, h | 0.25 | 0.25 |
| AUC0−t, ng*h/mL | 161 | 829 |
| AUC0−¥, ng*h/mL | 166 | 856 |
| t1/2, h | 1.1 | 1.1 |
| Mean, n = 3 per time point | ||
| B. Plasma levels of compound 4 in Male Swiss Mice
| |||||
|---|---|---|---|---|---|
| 2 mg/kg IV | 2 mg/kg IV | 10 mg/kg Oral | 30 mg/kg Oral | ||
| t1/2, h | 1.4 | 1.8 | Cmax, ng/mL | 98 (304.5 nM) | 217 (674.3 nM) |
| AUC0−t, ng*h/mL | 67 | 53 | Tmax, h | 0.25 | 1.00 |
| AUC0−¥, ng*h/mL | 71 | 57 | AUC0−t, ng*h/mL | 119 | 414 |
| V, L/kg | 57.6 | 91.0 | AUC0−¥, ng*h/mL | 141 | 480 |
| CL, mL/min/kg | 469 | 586 | t1/2, h | >3 | >3 |
| Mean, n = 3 per time point | Data set 2 | Data set 1 | Bioavailability, % | 40 | 56 |
The mouse PK study shows that compound 4 has good plasma exposure in the 400 nM range by the IP route of administration at this 10 mg/kg dose and has a brain: plasma ratio of about 5 showing this compound has good brain exposure of about 2 μM at this dose (Table 3A). The dose escalation by the oral route of administration (Table 3B) from a 10 mg/kg dose to the 30 mg/kg dose is showing less than a 3-fold escalation. This could be due to the relatively high volume of distribution, suggestive of tissue accumulation. The fivefold increase in brain levels versus plasma levels (Table 3A) also suggests this tendency for high tissue distribution. Compound 4 is also showing a high clearance from plasma, that could be due to the compound’s tendency to accumulate in tissue rather than poor stability, because of the relatively good mouse liver microsome stability. The compound is showing good oral bioavailability, and thus overall, this appears like a reasonable pre-clinical lead for further optimization.
Finally we evaluated compound 4 in the CEREP safety screen 44 panel.30 This panel is a good cost effective compilation of targets for off-target profiling to identify undesirable off-target activities that may hinder or halt development of a compound. In summary we observed only six off target activities out of the 44 that were greater than 50% at a 1 μM concentration, suggestive of a highly selective series. We then evaluated IPAG and compounds 1, 4,and 5 for hERG binding (performed at Reaction Biology Corporation, Malvern, PA) and hERG functional Patch clamp inhibition (QPatch; performed at Eurofins Panlabs, Inc., St. Charles, MO).
We found modest hERG binding, and modest inhibition of hERG functional activity, which will be addressed in lead optimization (Table 4). In conclusion, compound 4 provides an interesting new chemotype for lead optimization based on its modest off-target activity, affinity at Sigma1, high plasma exposure in the mouse. Due to its relatively clean off target profile and good in vivo exposure, compound 4 is a useful tool compound for use in future studies to validate sigma for in vivo efficacy in mouse models of CRPC. We are actively improving this compound series to improve potency, off-target selectivity, and pharmacokinetic exposure and will report on these activities in the future.
Table 4.
hERG binding and QPatch functional inhibition.
Not Available.
Supplementary Material
Acknowledgments
This work was funded by an American Cancer Society Institutional Research Grant, Drexel University Clinical and Translational Research Institute Grant, Drexel University College of Medicine Professional Enrichment Grant, Sidney Kimmel Cancer Center Consortium Pilot Study Award to FJK, and a Coulter-Drexel Translational Research Partnership Program Award (FJK/JMS). JMS would like to thank Mr. Martin Lehr and Professor James E. Barrett for resources and helpful discussions.
A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2017.03.030. These data include MOL files and InChiKeys of the most important compounds described in this article.
References
- 1.Jermal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer J Clin. 2009;59:225. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Klein EA, Ciezki J, Kupelian PA, Mahadevan A. Oncology. 2009;27:67. doi: 10.1016/j.urolonc.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Augello MA, Den RB, Knudsen KE. Cancer Metastasis Rev. 2014;33:399. doi: 10.1007/s10555-013-9471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher HI, Fizazi K, Saad F, et al. N Engl J Med. 2012;367:1187. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 5.Dhingra R, Sharma T, Singh S, et al. Mini Rev Med Chem. 2013;13:1475. doi: 10.2174/13895575113139990003. [DOI] [PubMed] [Google Scholar]
- 6.Bonomo S, Hansen CH, Petrunak EM, et al. Sci Rep. 2016;6:29468. doi: 10.1038/srep29468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin L, Hu Q. Nat Rev Urol. 2014;11:32. doi: 10.1038/nrurol.2013.274. [DOI] [PubMed] [Google Scholar]
- 8.Hanner M, Moebius FF, Flandorfer A, et al. Glossmann. Proc Natl Acad Sci. 1996;93:8072. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kekuda R, Prasad PD, Fei YJ, Leibach FH, Ganapathy V. Biochem Biophys Res Commun. 1996;229:553. doi: 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- 10.Kim FJ, Schrock JM, Spino CM, Marino JC, Pasternak GW. Biochem Biophys Res Commun. 2012;426:177. doi: 10.1016/j.bbrc.2012.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schrock JM, Spino CM, Longen CG, et al. Mol Pharmacol. 2013;84:751. doi: 10.1124/mol.113.087809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whittemore ER, Ilyin VI, Woodward RM. J Pharmacol Exp Ther. 1997;282:326. [PubMed] [Google Scholar]
- 13.Scherz MW, Fialeix M, Fischer James B, et al. J Med Chem. 1990;33:2421. doi: 10.1021/jm00171a016. [DOI] [PubMed] [Google Scholar]
- 14.Kimes AS, Wilson AA, Scheffel U, Campbel BG, London ED. J Med Chem. 1992;35:4683. doi: 10.1021/jm00103a005. [DOI] [PubMed] [Google Scholar]
- 15.Araki N, Ohno K, Takeyoshi M, Iida M. Toxicol In Vitro. 2005;19:335. doi: 10.1016/j.tiv.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Luccio-Camelo DC, Prins GS. J Ster Biochem Mol Biol. 2011;127:74. doi: 10.1016/j.jsbmb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Human Androgen Receptor Reporter Assay System Product # IB03001-32 Indigo biosciences.
- 18.Vilner BJ, Bowen WD. JPET. 2000;292:900. [PubMed] [Google Scholar]
- 19.Cobos E, del Pozo E, Baeyens JM. J Neurochem. 2007;102:812. doi: 10.1111/j.1471-4159.2007.04533.x. [DOI] [PubMed] [Google Scholar]
- 20.Weber E, Sonders M, Quarum M, McLean S, Pou S, Keana JFW. Proc Natl Acad Sci USA. 1986;83:8784. doi: 10.1073/pnas.83.22.8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams JT, Teal PM, Sonders MS, et al. Eur J Pharmacol. 1987;142:61. doi: 10.1016/0014-2999(87)90654-6. [DOI] [PubMed] [Google Scholar]
- 22.Kavanaugh MP, Tester BC, Scherz MW, Keana JFW, Weber E. Proc Natl Acad Sci USA. 1988;85:2844. doi: 10.1073/pnas.85.8.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peprah K, Zhua XY, Eyunnia SVK, Setolab V, Roth BL, Ablordeppey SY. Bioorg Med Chem. 2012;20:1291. doi: 10.1016/j.bmc.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernatowicz MS, Wu Y, Matsueda GR. Tetrahedron Lett. 1993;34:3389. [Google Scholar]
- 25.Yong YF, Kowalski JA, Lipton MA. J Org Chem. 1997;62:1540. [Google Scholar]
- 26.Yong YF, Kowalski JA, Thoen JC, Lipton MA. Tetrahedron Lett. 1999;40:53. [Google Scholar]
- 27.Kim H-O, Mathew F, Ogbu C. Synlett. 1999;193 [Google Scholar]
- 28.Spruce BA, Campbell LA, McTavish N, et al. Cancer Res. 2004;64:4875. doi: 10.1158/0008-5472.CAN-03-3180. [DOI] [PubMed] [Google Scholar]
- 29.Aydar E, Stratton D, Fraser SP, Djamgoz MBA, Palmer C. Eur Biophys J. 2016;45:671. doi: 10.1007/s00249-016-1135-0. [DOI] [PubMed] [Google Scholar]
- 30.Bowes J, Brown AJ, Hamon J, et al. Nat Rev Drug Dis. 2012;11:909. doi: 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.