

Summary
Delivery of neurotrophins and neuropeptides via long-range trafficking of dense core vesicles (DCVs) from the cell soma to nerve terminals is essential for synapse modulation and circuit function. But the mechanism by which transiting DCVs are captured at specific sites is unknown. Here, we discovered that Synaptotagmin-4 (Syt4) regulates the capture and spatial distribution of DCVs in hippocampal neurons. We found that DCVs are highly mobile and undergo long-range translocation but switch directions only at the distal ends of axons, revealing a circular trafficking pattern. Phosphorylation of serine 135 of Syt4 by JNK steers DCV trafficking by destabilizing Syt4-Kif1A interaction, leading to a transition from microtubule-dependent DCV trafficking to capture at en passant presynaptic boutons by actin. Furthermore, neuronal activity increased DCV capture via JNK-dependent phosphorylation of the S135 site of Syt4. Our data reveal a mechanism that ensures rapid, site-specific delivery of DCVs to synapses.
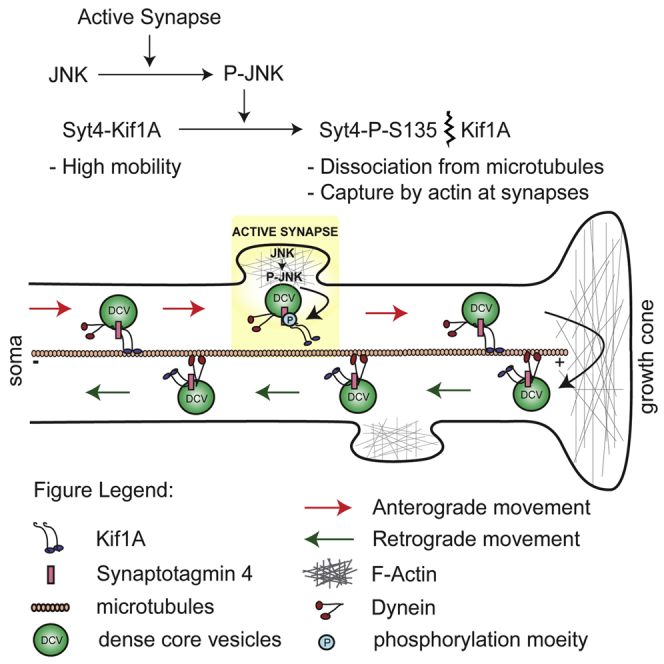
Graphical Abstract
Highlights
-
•
Syt4-bearing dense core vesicles in axons traffic continually in a circular pattern
-
•
Phosphorylation of S135 of Syt4 by JNK destabilizes Syt4-Kif1A binding
-
•
Destabilized Syt4-Kif1A binding promotes capture of vesicles at synapses by actin
-
•
Neuronal activity increases vesicle capture via S135-dependent JNK phosphorylation
Bharat et al. show that Synaptotagmin-4 (Syt4) on highly mobile dense core vesicles (DCVs) in axons binds the motor protein Kif1A. Phosphorylation of the S135 site of Syt4 by JNK destabilizes Syt4-Kif1A binding, leading to capture of DCVs at synapses by actin. Neuronal activity increases capture via this mechanism.
Introduction
Neurotrophins and neuropeptides are essential for a wide range of brain functions, including metabolism, pain perception, and cognition. Dense core vesicles (DCVs) in neurons facilitate these crucial functions by transporting and releasing neurotrophins and neuropeptides (Scalettar, 2006, Wong et al., 2012). Unlike synaptic vesicles (SVs), which are locally refilled and recycled at synaptic terminals, DCVs are filled with cargo at the Golgi and must then be trafficked from the cell soma to distal sites in neuronal processes (Burgoyne and Morgan, 2003, Kim et al., 2006, Wong et al., 2012), where they accumulate in presynaptic boutons (Bulgari et al., 2014, Shakiryanova et al., 2006, Wong et al., 2012). The majority of axonal processes in the CNS specify multiple sequential en passant boutons along their length to which DCVs must be distributed. In addition, neuropeptides are released during neuronal activity to modulate synapse and circuit function; under conditions of high neuronal activity, the demand for DCVs at synapses is necessarily greater, but how DCVs are captured to sustain the constant demand of neurotrophins and neuropeptides at active synapses remains unknown.
A possible mechanism is suggested by reports that phosphorylation of motor proteins can disrupt vesicle-motor protein interaction, leading to detachment from microtubules and deposit of cargo (Gibbs et al., 2015), but no such mechanism of cargo delivery has been reported for DCVs; neither a kinase nor a kinase substrate are known. One candidate kinase is JNK, which has been reported to reduce cargo mobility under pathological conditions in hippocampal neurons (Stagi et al., 2006), and to suppress aberrant clustering of synaptic proteins in C. elegans mutants (Wu et al., 2013). However, DCV cargos were not tested in these studies, and whether JNK itself directly promotes cargo-microtubule dissociation and which substrates it might act upon to do so is unknown.
We previously identified Syt4 as an integral vesicle protein present on brain-derived neurotrophic factor (BDNF)-harboring DCVs in neurons (Dean et al., 2009). Interestingly, in non-neuronal PC12 cells, JNK can phosphorylate Syt4 and enhance exocytosis of secretory vesicles (Mori et al., 2008). This may be due to an increase in fusion efficacy, given that JNK caused Syt4 to translocate from immature to mature fusion-competent vesicles in this study, but it could also be caused by a change in trafficking of vesicles. We hypothesized that, in neurons, which have long axonal processes and numerous en passant synaptic contact sites, phosphorylation of Syt4 might affect long-range DCV trafficking and vesicle capture. To test this, we used a series of live imaging experiments to investigate the trafficking dynamics of Syt4 phosphomutants in hippocampal neurons.
We found that Syt4 itself is a target of activated JNK in hippocampal neurons, which steers the activity-dependent capture of DCVs at synapses. Phosphorylation of the S135 site of Syt4 by JNK at active synapses destabilizes a direct interaction of Syt4 with the motor protein Kif1A, leading to capture of DCVs at synaptic sites by actin. This mechanism allows fast recruitment of DCVs to active synapses on a timescale of seconds to minutes, bypassing delays associated with signaling between synapses and the soma, and may promote competition and sharing of DCVs between synapses in axons based on synaptic activity.
Results
DCVs Traffic in a Circular Pattern between the Soma and Distal Processes
We first verified the localization of Syt4 to DCVs marked with chromogranin A (CgA), a cargo present in the majority of DCVs (Adams et al., 1993, Machado et al., 2010), by immunostaining hippocampal neuron cultures. Syt4 was highly co-localized with CgA; 82.4% ± 1.1% of Syt4 signal co-localized with CgA, and 68.3% ± 1.5% of CgA signal co-localized with Syt4 (Figure 1A). As a control, we immunostained for Syt4 and synaptophysin (syp), a synaptic vesicle marker; only 29.8% ± 3.7% of syp signal co-localized with Syt4, and 24.1% ± 2.6% of Syt4 signal co-localized with syp. We further confirmed, by immuno-organelle isolation of synaptic vesicles with anti-Syt1 and anti-Syb2 antibodies, that Syt4 was not present on synaptic vesicles (Figure S1A). Because we planned to use mCherry-tagged Syt4 in trafficking experiments, we also tested co-localization of mCherry-Syt4 and CgA-GFP and found a similar amount of co-localization in co-transfected neurons; 82.2% ± 2.2% of mCherry-Syt4 puncta were CgA-GFP+, and 77.3% ± 3.4% of CgA-GFP puncta were mCherry-Syt4+ (Figure 1B). We then examined the trafficking pattern of mCherry-Syt4-labeled DCVs in neurons. Syt4-harboring DCVs exhibited high mobility that was microtubule-dependent (Figures S1B and S1C). Although the long length of mammalian hippocampal axons precludes imaging an entire axon to track single vesicle trajectories in mature neurons, we tracked the movement of 992 vesicles in 4,948 μm of mid-axonal regions and 200 vesicles in 1,008 μm of distal axons near growth cones in hippocampal neurons. In mid-axonal regions, 99.2% of mobile vesicles moved unidirectionally (either in the anterograde or retrograde direction), and only 0.8% of vesicles switched directions (Figures 1C and 1D). Conversely, in distal axons, within approximately 200 μm of growth cones, a much larger fraction of mobile vesicles, 54.8%, switched directions, compared with 45.2% that were unidirectional (Figures 1C and 1D). This suggests that DCVs in hippocampal neurons are routed in a circular trafficking pattern, from the soma to distal ends of axons and back again.
Figure 1.

Syt4 Phosphomutant-Harboring DCVs Have Altered Trafficking after Synapse Formation
(A) Hippocampal cultures immunostained for Syt4 and CgA (left; n = 10 images, 3 cultures) or for Syt4 and syp (right; n = 8 images, 3 cultures). Scale bar, 5 μm.
(B) Hippocampal neurons co-transfected with mCherry-Syt4 and CgA-GFP (n = 11 images, 3 transfections). Scale bar, 5 μm.
(C) Kymographs of Syt4 vesicles in mid-axon and distal regions. Select colored vesicle trajectories indicate unidirectional or switching vesicles. Raw underlying kymographs are deposited at Mendeley Data (https://doi.org/10.17632/5b9zw3tm4f.1). Scale bar, 10 μm.
(D) Quantitation of vesicle trajectories in mid-axon and distal regions (mid-axonal regions = 4948.16 μm, vesicles = 992; distal regions = 1,008 μm, vesicles = 200).
(E) Hippocampal neurons at DIV6 and DIV13 immunostained with MAP2, PSD-95, and syp to mark dendrites and postsynaptic and presynaptic sites, respectively. Right: quantitation of number of synapses identified by puncta positive for both syp and PSD95.
(F and G) Color-coded vesicle tracks (top; scale bars, 10 μm) with vesicles shown in red and kymographs (bottom; scale bars, 5 μm) of Syt4 WT, S135A, and S135E vesicles over 5 min at DIV6 (F) and DIV13 (G).
(H and I) Quantitation of velocity (H) and mobile vesicle percentage (I) (n = 3,703, 4,619, and 3,937 vesicles for velocity and n = 11, 13, and 12 movies for mobile vesicle percentage for DIV6 neurons; n = 2,686, 2,965 and 3,189 vesicles for velocity and n = 12, 11, and 14 movies for mobile vesicle percentage, for DIV13 neurons and for WT, S135A, and S135E Syt4 vesicles, respectively, from 3 cultures.)
Significance was determined by Student’s t test with Bonferroni correction; error is SEM; ∗∗p < 0.01, ∗∗∗p < 0.001.
Phosphorylation of Syt4 at S135 Reduces DCV Mobility after Synapse Formation
We next examined the trafficking of mCherry-tagged wild-type (WT), phosphodeficient (S135A), and phosphomimetic (S135E) Syt4 mutant-harboring DCVs in hippocampal neurons at two developmental stages: before the majority of synapses have formed, at day in vitro (DIV) 6, and after, at DIV13 (Figure 1E). mCherry-tagged WT, S135A, and S135E Syt4 were overexpressed at similar levels; 2.5 ± 0.3-, 2.4 ± 0.1-, and 2.3 ± 0.2-fold, respectively, as assessed by comparison of anti-Syt4 fluorescence intensity in cell bodies. Vesicle movement over 5 min was visualized by color-coded tracks over time (Figures 1F and 1G, top) and kymographs (Figures 1F and 1G, bottom) of trafficking vesicles (Movies S1 and S2). The average intrinsic speed of vesicles while in motion (not including pauses) was not significantly different between WT, S135A, and S135E vesicles (0.76 ± 0.10 μm/s, 0.78 ± 0.08 μm/s, and 0.76 ± 0.09 μm/s, respectively) and was similar to previously reported DCV trafficking speeds (Adachi et al., 2005, de Wit et al., 2006). Interestingly, however, S135A vesicle velocity remained high at DIV13 and was unchanged compared with DIV6, while WT and S135E vesicle velocity was reduced at DIV13 compared with DIV6 (Figure 1H). The percentage of mobile vesicles (moving for at least 10 s over a distance of at least 1 μm over 5 min) was increased for S135A, unchanged for WT, and decreased for S135E mutants at DIV13 compared with DIV6 (Figure 1I). Thus phosphorylation of S135 of Syt4 reduces vesicle mobility in DIV13 cultures.
DCVs Are Highly Mobile in Axons, and Phosphorylation of Syt4 Reduces Mobility
To examine Syt4 vesicle mobility specifically in axons, compared with dendrites, at DIV13, we co-transfected hippocampal neurons with EGFP to distinguish axons, which have a small diameter and are the only process that projects far away from the cell body, from dendrites, which have a larger diameter, are decorated by dendritic spines, and branch at angles of less than 90° within approximately 100 μm of the cell soma. In axons (Figure 2A), S135A vesicles had the highest velocity and covered the most distance, whereas S135E vesicles had the lowest velocity and covered the least distance compared with WT (Figure 2B). The S135A mutant also had a higher percentage of mobile vesicles, and S135E had a lower percentage of mobile vesicles, compared with WT (Figure 2C).
Figure 2.

Phosphorylation of the S135 Site of Syt4 Reduces DCV Mobility in Axons
(A) Axons marked with EGFP (top) co-transfected with mCherry-tagged WT, S135A, or S135E Syt4; vesicle trafficking is indicated by color-coded vesicle tracks (center) and kymographs (bottom).
(B–E) Quantitation of velocity (B), mobile vesicle percentage (C), pause frequency (D), and pause time (E) of S135A and S135E Syt4 vesicles in axons normalized to WT (n = 4,476, 6,502, and 4,228 vesicles for velocity, pause frequency, and pause time and n = 27, 31, and 32 movies for mobile percentage of WT, S135A, and S135E vesicles, respectively, from 4 cultures).
(F) Trafficking of WT, S135A, and S135E vesicles in axons of transfected Syt4 knockout neurons indicated by color-coded vesicle tracks (center) and kymographs (bottom).
(G–J) Quantitation of velocity (G), mobile vesicle percentage (H), pause frequency (I), and pause time (J) of S135A and S135E Syt4-harboring vesicles in axons normalized to WT in a Syt4 knockout background (n = 12,549, 18,991, and 9,880 vesicles for velocity, pause frequency, and pause time and n = 28, 34, and 31 movies for mobile percentage of WT, S135A, and S135E vesicles, respectively, from 4 cultures). Scale bars, 5 μm.
Significance was determined by Student’s t test with Bonferroni correction; error indicates SEM; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Velocity, mobile vesicle percentage, and pause time of S135A and S135E vesicles relative to WT were not significantly different between WT and Syt4 knockout backgrounds (2-way ANOVA). The pause frequency of S135A vesicles was slightly higher in a WT background (p < 0.05, 2-way ANOVA, Tukey’s multiple comparisons test).
The decreased mobility of S135E vesicles and increased mobility of S135A vesicles in axons could be caused by a change in pause frequency or duration. Surprisingly, the pause frequency was highest in the most mobile vesicle population, S135A vesicles, compared with S135E and WT vesicles (Figure 2D). The average pause time of S135A vesicles, however, was the shortest (Figure 2E). Because S135A vesicles travel the farthest distance, they may scan a larger number of putative capture/release sites (possibly corresponding to en passant synapses), resulting in an increased number of pauses at these putative sites. However, an inability to be captured at these sites would result in shorter pauses, causing the observed overall increase in S135A vesicle mobility. S135E and WT vesicles, on the other hand, have longer pause durations (Figure 2E) and may therefore have the potential to be captured at release sites. Syt4 DCVs were much less mobile in dendrites (Figure S2A) compared with axons for all parameters tested (p < 0.001), consistent with previous studies (Adachi et al., 2005, de Wit et al., 2006). We observed similar (although less pronounced) trafficking effects of the phosphomutants in dendrites: S135A vesicles had a higher velocity than S135E and WT vesicles (Figure S2B), but the percentage of mobile vesicles was unchanged (Figure S2C).
Because, in these experiments, endogenous Syt4 remained in which S135 could be phosphorylated or non-phosphorylated, we also examined trafficking in a Syt4 knockout background (Figure 2F). Interestingly, we observed similar trafficking effects: S135A vesicles were the most mobile and S135E vesicles were the least mobile, in terms of vesicle velocity (Figure 2G) and mobile vesicle percentage (Figure 2H), which were not significantly different from a WT background. Likewise, pause analysis revealed that highly mobile S135A vesicles had a higher pause frequency (Figure 2I) and shorter pause time (Figure 2J) compared with WT and S135E vesicles, with no significant differences in trafficking compared with a WT background, except for a slight increase in pause frequency of S135A vesicles in a WT background (p < 0.05, 2-way ANOVA, Tukey’s test). Thus, Syt4 phosphomutants expressed in WT neurons appear to act in a dominant fashion to supplant the function of endogenous Syt4.
Phosphomimetic Syt4 Has Reduced Direct Interaction with Kif1A
To determine why phosphomutant vesicles have altered trafficking, we tested whether phosphorylation of Syt4 affects its interaction with motor proteins. Knockdown of Kif1A, a motor protein involved in fast axonal transport of DCVs (Lo et al., 2011), significantly reduced the number of Syt4 DCVs in axons, and this reduction was rescued by re-introducing knockdown-resistant Kif1A (vesicles/20 μm axon = 4.2 ± 0.4 [WT], 2.5 ± 0.3 [Kif1A knockdown], and 4.7 ± 0.4 [Kif1A rescue]; p = 0.003 for WT versus Kif1A knockdown, p = 0.0004 for Kif1A knockdown versus Kif1A rescue). The vesicles that remained in Kif1A knockdown neurons had severely reduced mobility with virtually no translocation of vesicles in kymographs (Figure 3A), reduced velocity (Figure 3B), and a reduced percentage of mobile vesicles (Figure 3C). Co-expression of knockdown-insensitive Kif1A fully restored vesicle trafficking, indicating that Kif1A is the motor protein that transports Syt4-harboring DCVs (Figures 3A–3C).
Figure 3.

Kif1A Transports Syt4-Harboring DCVs, and S135E Syt4 Has Reduced Binding to Kif1A
(A) Trafficking of mCherry-Syt4 vesicles in control, Kif1A knockdown, or Kif1A rescue axons indicated by color-coded vesicle tracks (top) and kymographs (bottom).
(B and C) Normalized velocity (B) and mobile vesicle percentage (C) under control, Kif1A knockdown, and Kif1A rescue conditions (n = 9,319, 3,625, and 7,493 vesicles for velocity and n = 14, 15, and 15 movies for mobile vesicle percentage of control, Kif1A knockdown, and Kif1A rescue, respectively, from 4 cultures). Scale bars, 5 μm. Significance was determined by Student’s t test with Bonferroni correction; error indicates SEM; ∗∗∗p < 0.001.
(D) Western blot of Kif1A-GFP (left) and mCherry-tagged WT, S135A, or S135E Syt4 co-immunoprecipitation using GFP-Trap beads (+). Agarose beads (−) controlled for binding specificity. The uppermost band (right) corresponds to full-length mCherry-Syt4 (n = 6).
(E) Western blot of mCherry-tagged WT, S135A, or S135E Syt4 (left) and Kif1A-GFP (right) co-immunoprecipitation using an RFP antibody (n = 6).
(F) Pull-down of Kif1A from brain lysates using His-tagged WT, S135A, or S135E Syt4 bound to nickel beads; input, pellet (P) and supernatant (S), and bead-only negative controls are indicated (n = 6).
(G) Direct binding of GST-tagged Kif1A and His-tagged WT, S135A, or S135E Syt4; bead-only negative controls are indicated (n = 6).
(H and I) Color-coded vesicle tracks (top; scale bar, 5 μm) and kymographs (bottom; scale bar, 10 μm) of WT, S135A, and S135E Syt4 vesicle trafficking in a WT background (H) and of S135E Syt4 vesicle trafficking in a Syt4 knockout background (I) in neurons co-transfected with Kif1A-GFP. Kymographs show WT, S135A, or S135E mCherry-Syt4 (red) and co-transfected Kif1A-GFP (green).
(J–M) Quantitation of velocity (J), mobile vesicle percentage (K), pause frequency (L), and pause time (M) of WT, S135A, and S135E Syt4-harboring vesicles in axons co-transfected with Kif1A-GFP, normalized to WT (without Kif1A-GFP). Control conditions without Kif1A-GFP are shown for comparison (n = 4,278, 6,325, 9,507, and 6,839 vesicles for average velocity, pause frequency, and pause time and n = 7, 9, 12, and 23 movies for mobile percentage of WT, S135A, S135E, and S135E/ Syt4 knockout background, respectively, from 4 cultures). Scale bars, 5 μm.
Significance was determined by Student’s t test with Bonferroni correction; error indicates SEM; ∗p < 0.05, ∗∗∗p < 0.001.
Although most motor proteins bind their cargo via adaptor proteins (Hirokawa et al., 2010), it is possible that Syt4 and Kif1A bind directly to transport DCVs and that phosphorylation of S135 of Syt4 destabilizes this interaction. To test this idea, we first used structural modeling. The S135 site of Syt4 (Figure S3A) is in a largely exposed and intrinsically disordered linker (Figure S3B) between the transmembrane and C2A domains of Syt4, at the N-terminal cap of a region with transient helical structure (Figure S3C). Although it is intrinsically disordered, several hydrophobic residues within the linker argue for a preferentially compact structure (Figure S3D). Interestingly, we identified, with 98% confidence, a C2-like domain and a DEP-like domain similar to that in pleckstrin in the vesicle membrane-proximal undefined region (UDR) of Kif1A (Figure S3E). Structural models indicate that Syt4 may interact with this C2 domain of Kif1A when Syt4 is in a compact non-phosphorylated state (Figure S3F). Thus, Syt4 and Kif1A may bind directly.
We first examined an interaction between Syt4 and Kif1A in co-immunoprecipitation (coIP) experiments in HEK293 cells co-transfected with Kif1A-GFP and mCherry-tagged WT, S135A, or S135E Syt4. WT Syt4 co-immunoprecipitated with Kif1A, but the amount of S135E Syt4 co-immunoprecipitated by Kif1A was reduced (42.9% ± 10.8% of WT), whereas the amount of S135A Syt4 co-immunoprecipitated by Kif1A was increased (134.4% ± 11.1%) compared with the WT (p = 0.04 for WT versus S135E, and p = 0.02 for S135A versus S135E) (Figure 3D). Reverse IP using a red fluorescent protein (RFP) antibody revealed the same trends: the amount of Kif1A-GFP co-immunoprecipitated by S135E Syt4 was reduced (87.0% ± 22.9% of WT), and the amount of Kif1A-GFP co-immunoprecipitated by S135A Syt4 was increased (158.19% ± 24.49% of WT) (Figure 3E). We additionally tested binding of endogenous Kif1A to WT and phosphomutant Syt4 in pull-down experiments from brain lysates. We again found reduced Kif1A binding to S135E Syt4 (43.6% ± 10.7% of WT, p = 0.0017), with no significant change in Kif1A binding to S135A Syt4 in this case (Figure 3F). Finally, we purified recombinant WT and phosphomutant Syt4 and Kif1A and examined direct binding. We found that there is indeed a direct interaction between Kif1A and WT Syt4. In addition, Kif1A binding to S135E Syt4 was reduced (36.7% ± 5.5% of control, p = 0.0003), whereas S135A Syt4 showed similar binding to WT Syt4 (100.4% ± 16.8% of control) (Figure 3G).
The reduced interaction of S135E Syt4 with Kif1A may destabilize cargo-motor trafficking complexes and immobilize phosphorylated DCVs. If this is the case, then overexpression of Kif1A, providing an excess of motor proteins, might “rescue” the immobility of S135E Syt4 in WT neurons (where remaining copies of WT Syt4 can bind Kif1A normally) but not in Syt4 knockout neurons. To test this, we co-expressed mCherry-tagged WT, S135A, or S135E Syt4 with Kif1A-GFP. The ratio of mCherry-Syt4 to Kif1A-GFP expression, assayed by fluorescence intensity, was similar under all conditions (p > 0.15). Overexpression of Kif1A-GFP (Figures 3H and 3I; Movie S3) did indeed increase the vesicle velocity (Figure 3J), mobile vesicle percentage (Figure 3K), and pause frequency (Figure 3L) of S135E and S135A vesicles to similar levels in WT neurons (where the S135E vesicle pause frequency was even higher than that of S135A) but not in Syt4 knockout neurons. Similarly, overexpression of Kif1A-GFP decreased the vesicle pause time of S135E and S135A vesicles to similar levels in WT neurons but not in Syt4 knockout neurons (Figure 3M). This further confirms that the decreased mobility of S135E vesicles is due to decreased binding of phosphorylated Syt4 to Kif1A.
Syt4 Vesicles Accumulate at Presynaptic Sites in an Actin- and S135 Phosphorylation-Dependent Manner
Capture of DCVs at specific sites likely requires that they detach from microtubules and interact with a new substrate. In mature neurons, presynaptic sites are enriched in actin filaments (Cingolani and Goda, 2008, Hirokawa et al., 1989; Figure S4A), which may promote vesicle capture. To test this, we examined localization of WT and phosphomutant Syt4 to presynaptic sites marked with syp-GFP in control cultures and in cultures treated with 10 μM latrunculin for 30 min to disrupt actin. Under control conditions, there was no difference in the proportion of WT, S135A, or S135E vesicles localized to axons (Figures 4A and 4B) or dendrites (Figures 4A and 4C) or in the fluorescence intensity of Syt4 puncta in axons (WT = 3.5 ± 1.0, S135A = 2.5 ± 0.6, S135E = 2.8 ± 0.4 [a.u.]) or dendrites (WT = 4.6 ± 0.6, S135A = 5.3 ± 0.8, S135E = 5.4 ± 0.7 [a.u.]). However, S135E vesicles were increased and S135A vesicles were decreased at presynaptic sites (marked with co-expressed syp-GFP) compared with WT vesicles (Figure 4D). Latrunculin treatment decreased synaptic localization of S135E and WT vesicles to levels similar to S135A vesicles, whose localization was unchanged by latrunculin (Figures 4E and 4F). Thus, actin recruits Syt4 vesicles that are phosphorylated at the S135 site to presynaptic boutons. WT, S135A, and S135E vesicles were less prominent at post-synaptic sites marked with PSD95-GFP, with no significant difference in localization of phosphomutants compared with the WT (Figures S4B and S4C).
Figure 4.

Actin is required for S135 phosphorylation-dependent capture of Syt4 DCVs at presynaptic sites
(A) GFP-tagged WT, S135A and S135E Syt4 transfected cells immunostained with MAP2.
(B and C) (B) Quantitation of WT, S135A, and S135E GFP puncta in axons (MAP2-negative) and dendrites (C; MAP-2-positive).
(D and E) Cropped axonal regions of hippocampal neurons co-transfected with syp-GFP and mCherry-tagged WT, S135A or S135E Syt4 in control (D) and latrunculin-treated (E) conditions.
(F) Quantitation of percentage of mCherry-tagged WT, S135A or S135E Syt4 colocalizing with syp-GFP, in control and latrunculin-treated conditions (n=37, 33 and 33 cells in control conditions, and n=31, 25 and 28 cells in latrunculin-treated conditions for WT, S135A and S135E, respectively, from 3 cultures).
(G and H) (G) Quantitation of velocity and mobile vesicle percentage (H) of WT, S135A and S135E Syt4 vesicles in control and and latrunculin-treated conditions, normalized to WT control (n=5109, 5609 and 2317 vesicles for velocity and n=22, 21 and 15 videos for mobile percentage for WT, S135A and S135E vesicles, respectively, from 4 cultures).
(I and J) (I) Quantitation of velocity and mobile vesicle percentage (J) of WT, S135A and S135E Syt4 vesicles in control and jasplakinolide-treated conditions, normalized to WT control (n=5625, 5977 and 8556 vesicles for velocity, and n=13, 13 and 15 videos for mobile percentage for WT, S135A and S135E vesicles, respectively, from 3 cultures). Scale bars = 5 μm. Significance determined by Student’s t-test with Bonferroni correction; error indicates s.e.m.; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
To test whether actin depolymerization affects the mobility of DCVs, we examined trafficking of mCherry-tagged WT, S135A, or S135E vesicles under control conditions and following treatment with 10 μM latrunculin for 30 min. Although the relatively high velocity of S135A vesicles was unchanged, the velocity of S135E and WT vesicles significantly increased following latrunculin treatment (Figure 4G). The percentage of mobile S135A vesicles was also unchanged, whereas that of S135E and WT vesicles trended toward an increase upon latrunculin treatment (Figure 4H). Latrunculin also slightly increased the pause frequency (108.3% ± 2.4%, p = 0.01) and decreased the pause time (71.1% ± 6.5%, p = 0.002) of S135E vesicles and WT vesicles (68.5% ± 3.2%, p = 0.0007) without affecting these parameters for S135A vesicles. Thus, disrupting actin specifically increased the mobility of S135E vesicles to (or above) S135A levels but had no effect on S135A vesicle mobility. This suggests that actin is specifically involved in a Syt4 S135 phosphorylation-dependent mechanism of vesicle capture and not a more general unrelated effect, which would result in a uniform increase in the mobility of both S135A and S135E vesicles.
To further test the role of actin, we examined the effects of the actin polymerizing agent jasplakinolide. We again found phosphomutant-specific effects: the S135A vesicle velocity (Figure 4I) was dramatically reduced by 1 μM jasplakinolide treatment for 30 min prior to imaging, below S135E levels (which were reduced more slightly). Similarly, the mobile percentage of S135A vesicles was significantly reduced, whereas that of S135E vesicles was unchanged by jasplakinolide (Figure 4J). Pause analysis revealed similar trends. Jasplakinolide significantly decreased the pause frequency of S135A vesicles (89.2% ± 1.4%, p = 0.001) and WT vesicles (90.3% ± 1.4%, p = 0.001) but had no effect on S135E vesicles, whereas the pause duration of S135A and S135E vesicles was increased by jasplakinolide to the same level (121.4% ± 7.1%, 122.5% ± 6.0%, and 119.4% ± 5.5% for WT, S135A, and S135E vesicles, respectively).
JNK Binds to and Phosphorylates Syt4 at S135
JNK is reported to bind to and phosphorylate Syt4 at S135 in PC12 cells (Mori et al., 2008) and, therefore, may phosphorylate Syt4 in neurons to promote the capture of DCVs at synapses. In HEK293 cells co-transfected with FLAG-MKK7-JNK1 and GFP-Syt, we were able to co-immunoprecipitate GFP-Syt4 with a FLAG antibody (Figure 5A) and FLAG-MKK7-JNK1 with a GFP antibody in a reverse approach (Figure 5B), confirming an interaction between Syt4 and JNK. In addition, using an in silico approach (with c-Jun, a known substrate of JNK1, as a positive control), we found that JNK1 is predicted to phosphorylate Syt4 at S135 (Figure 5C). Finally, we used an in vitro kinase assay to test whether JNK1 specifically phosphorylates the S135 site of Syt4. As a control, we used a c-Jun peptide containing S63, which is phosphorylated by JNK1. Replacing S135 of Syt4 with alanine reduced phosphorylation by approximately 90%, and replacing S63 of c-Jun with alanine reduced phosphorylation by approximately 80% (Figure 5D). The 10% kinase activity remaining for the S135A peptide likely reflects autophosphorylation of JNK1, although we cannot exclude phosphorylation of other serine or threonine residues in the peptide.
Figure 5.

JNK Phosphorylates Syt4 at S135 to Promote DCV Capture
(A and B) Western blot of co-immunoprecipitation of FLAG-JNK and GFP-Syt4 co-expressed in HEK cells, using anti-FLAG beads (A, +), anti-GFP beads (B, +), or control beads without antibody (−). Input, IP, and S are indicated.
(C) GPS kinase prediction, where a score/cutoff > 2 is positive.
(D) In vitro kinase assay quantitation of percent JNK1-dependent phosphorylation of WT and phosphodeficient peptides of Syt4 and c-Jun.
(E) Color-coded vesicle tracks (top; scale bar, 10 μm) and kymographs (bottom; scale bar, 5 μm) from hippocampal neurons transfected with mCherry-tagged Syt4 alone (control) or co-expressed with dominant-negative JNK1 (JNK1(APF)) or overexpressed active JNK1 (MKK7-JNK1) and S135A Syt4 with overexpressed JNK1 (S135A/MKK7-JNK1). Scale bar, 10 μm.
(F–I) Quantitation of velocity (F), mobile percentage (G), pause frequency (H), and pause time (I) of Syt4 vesicles under JNK1(APF), MKK7-JNK1, and S135A/MKK7-JNK1 conditions, normalized to WT control (n = 5,216, 7,478, 3,172, and 4,232, vesicles for velocity, pause frequency, and pause time and n = 22, 27, 27, and 18 movies for mobile vesicle percentage for control, JNK1(APF), MKK7-JNK1, and S135A/MKK7-JNK1, respectively, from 4 cultures).
(J) Axons of neurons co-transfected with syp-GFP and mCherry-Syt4, mCherry-Syt4-P2A-JNK1(APF), mCherry-Syt4-P2A-MKK7-JNK1, or mCherry-Syt4(S135A)-P2A-MKK7-JNK1. Scale bar, 10 μm.
(K) Quantitation of the percentage of mCherry-Syt4 vesicles co-localized with syp-GFP (n = 12, 17, and 21 cells for control, JNK1(APF), and MKK7-JNK1, respectively, from 3 cultures).
Significance was determined by Student’s t test with Bonferroni correction; error is SEM; ∗∗p < 0.01, ∗∗∗p < 0.001.
JNK Phosphorylation of the S135 Site of Syt4 Captures DCVs at Synapses
To test whether JNK phosphorylation of the S135 site of Syt4 immobilizes vesicles, we first tried manipulating JNK pharmacologically with anisomycin (a JNK activator) and SP600125 (a JNK inhibitor) (Bennett et al., 2001, Curtin and Cotter, 2002) and examined mCherry-Syt4 trafficking. We observed a significant increase in p-JNK levels and a decrease in the mobility of Syt4 vesicles following anisomycin treatment but little to no change in vesicle trafficking following application of SP600125, possibly because of similar p-JNK levels in control and SP600125-treated conditions (Figure S5). Because anisomycin has additional effects (most notably in blocking protein synthesis), we tested the effect of JNK phosphorylation using a more specific approach. We transfected hippocampal neurons with mCherry-Syt4-P2A-JNK1(APF), where JNK1(APF) corresponds to a dominant-negative form of JNK1 (Dérijard et al., 1994), or with mCherry-Syt4-P2A-MKK7-JNK1, where MKK7-JNK1 increases activate JNK1 (Lei et al., 2002). We hypothesized that increased active JNK1 would phosphorylate Syt4 and reduce vesicle mobility by capturing vesicles, whereas dominant-negative JNK1 would prevent phosphorylation of Syt4 and increase vesicle mobility by preventing capture. Consistent with this hypothesis, Syt4 vesicles had increased velocity in the presence of JNK1(APF) and decreased velocity in the presence of increased active JNK1 (MKK7-JNK1) compared with the control (Figures 5E and 5F). The percentage of mobile vesicles was also decreased by active JNK1 (Figure 5G). Pause analysis corroborated these changes in mobility: highly mobile vesicles under JNK1(APF) conditions paused more frequently but for shorter durations (Figure 5H), whereas vesicles in MKK7-JNK1-expressing neurons showed the opposite effect and paused less frequently but for longer durations (Figure 5I). JNK1 could affect DCV capture by phosphorylating other sites on Syt4 or a site on a different protein, which might then indirectly immobilize DCVs via another mechanism. To test whether the S135 site of Syt4 is specifically phosphorylated by JNK1 to capture DCVs, we transfected neurons with mCherry-Syt4(S135A)-P2A-MKK7-JNK1, where S135A vesicles are co-expressed with excess active JNK1. If the S135 site of Syt4 is specifically required for JNK1-dependent phosphorylation and capture of vesicles, S135A vesicles would remain mobile even in the presence of increased active JNK1. We found that this was indeed the case. S135A vesicles remained highly mobile in neurons with increased active JNK1 (Figure 5E) in terms of velocity (Figure 5F), mobile percentage (Figure 5G), pause frequency (Figure 5H), and pause duration (Figure 5I). We also found that Syt4 vesicle localization to presynaptic sites increased in the presence of excess MKK7-JNK1 and decreased in the presence of JNK1(APF) compared with the control (Figures 5J and 5K), indicating that JNK1 phosphorylation of Syt4 captures DCVs at presynaptic sites.
Activity-Dependent Capture of DCVs Requires Phosphorylation of S135 of Syt4
To determine whether capture of DCVs at presynaptic sites is increased by neuronal activity, we examined the trafficking of Syt4 vesicles during treatment of cultures with 70 mM KCl for 3 min to depolarize neurons in the middle of 10 min time-lapse recordings. We defined capture events as immobility of a transiting vesicle at a site for greater than 120 s (Wong et al., 2012). We found a significant increase in capture of transiting Syt4-harboring vesicles during depolarization (Figures 6A and 6D). These vesicles then either resumed movement following stimulation or remained immobilized following washout of KCl. In contrast to WT Syt4 vesicles, S135A vesicles remained highly mobile during increases in neuronal activity, similar to non-depolarized conditions (Figures 6B and 6D), indicating that phosphorylation of the S135 site is important for activity-dependent capture. Syt4 vesicles also remained highly mobile during increased neuronal activity in the presence of dominant-negative JNK1 in mCherry-Syt4-P2A-JNK1(APF) transfected neurons, similar to non-depolarizing conditions, further confirming that JNK1 phosphorylation is necessary for activity-dependent capture of transiting DCVs (Figures 6C and 6D).
Figure 6.

Capture of DCVs by Increased Activity Requires Phosphorylation of S135 of Syt4
(A–C) Kymographs of mCherry-Syt4 WT (A), mCherry-Syt4 S135A (B), and mCherry-Syt4/JNK1(APF) (C) vesicle trafficking during stimulation. Equal lengths of processes are shown for comparison, which may include more than one example. Neurons were depolarized with 70 mM KCl for 3 min (indicated by white dashed lines). Select representative vesicle trajectories used for analysis are indicated in color. Raw underlying kymographs were deposited at Mendeley Data (https://doi.org/10.17632/5b9zw3tm4f.1).
(D) Quantitation of the percentage of vesicles captured (pausing for > 2 min) during and in the absence of stimulation under the indicated conditions (n = 17, 15, and 9 movies of WT Syt4, S135A Syt4, and WT Syt4/JNK1(APF), respectively, from 4 cultures). Scale bars, 5 μm.
(E) GFP-Syt4/ NPY-mCherry co-localization in a co-transfected axons (n = 23 images from 3 cultures).
(F) Kymographs of NPY-mCherry vesicle trafficking in WT and Syt4 knockout neurons during stimulation (white dashed lines).
(G) Quantitation of the percentage of NPY-mCherry vesicles captured (pausing for > 2 min) in response to stimulation in WT and Syt4 knockout neurons (n = 12 and 15 movies from 3 cultures, respectively).
(H) Kymographs of Kif1A-GFP trafficking during stimulation.
(I) Quantitation of the percentage of Kif1A-GFP pauses > 2 min in response to stimulation and under unstimulated conditions (n = 19 movies from 3 cultures).
Significance was determined by Student’s t test with Bonferroni correction; error is SEM; ∗p < 0.05, ∗∗∗p < 0.001.
If Syt4 is necessary for activity-dependent DCV capture at synapses, we would also expect that, in the absence of Syt4, DCVs cannot be captured by increased neuronal activity. We used NPY-mCherry as a DCV marker to compare vesicle trafficking in response to stimulation in WT and Syt4 knockout neurons. GFP-Syt4 and the DCV cargo NPY-mCherry are highly co-localized on DCVs (91.3% ± 1.9% of NPY-mCherry vesicles co-localized with GFP-Syt4, and 88.5% ± 2.5% of GFP-Syt4 vesicles co-localized with NPY-mCherry; Figure 6E). We found that capture of NPY-mCherry vesicles was significantly increased during stimulation in WT neurons but abolished in Syt4 knockout neurons (Figures 6F and 6G). If captures correspond to dissociation of DCVs from Kif1A motors, then Kif1A-GFP should continue to traffic along microtubules under conditions of increased neuronal activity without pausing. We tested this and found that increasing neuronal activity did not affect Kif1A movement (Figures 6H and 6I), indicating that Syt4 vesicles dissociate from Kif1A during activity-dependent capture events.
Neuronal Activity- and JNK-Dependent Capture of DCVs Occurs at Synapses
To test whether Syt4 vesicle capture occurs at synaptic sites, we co-transfected neurons with mCherry-Syt4 and syp-GFP to mark presynaptic boutons. We found significantly more (twice as many) pause events (immobility for longer than 2 min) at synaptic sites identified by syp-GFP fluorescence compared with non-synaptic sites (Figures 7A and 7B). We further examined whether endogenous Syt4 is captured at synaptic sites under conditions of increased neuronal activity. Because 70 mM KCl causes a transient depolarization and transient capture of mCherry-Syt4 vesicles in some cases (Figure 6A), we tested the effects of longer-term increases in neuronal activity by treating hippocampal neurons with 40 μM bicuculline for 1 h First, we tested the effects of bicuculline on overall levels of Syt4 and p-JNK by western blot. Endogenous Syt4 expression in neurons increased with bicuculline treatment (Figure 7C), consistent with previously reported activity-dependent expression of Syt4 (Dean et al., 2012, Vician et al., 1995). Bicuculline treatment also increased p-JNK levels (Figure 7C). We further tested the efficacy of the JNK inhibitor SP600125 in hippocampal neurons by western blot. Treatment of hippocampal neurons with the JNK activator anisomycin increased phosphorylation of c-Jun, a substrate of JNK, whereas addition of SP600125 decreased anisomycin-induced JNK activation back to control levels, showing that SP600125 inhibits JNK activity (Figure 7D). We then examined endogenous Syt4 and p-JNK localization at synapses. Interestingly, endogenous p-JNK was more concentrated at synapses compared with total JNK (Figure S6A), and the level of p-JNK at synapses was higher at DIV13 compared with DIV6 (Figures S6B and S6C). Increasing neuronal activity with 40 μM bicuculline treatment for 1 h increased p-JNK (Figures 7E and 7F) and Syt4 (Figures 7G and 7H) at synapses, and this increase was blocked by inhibition of JNK with SP600125. Thus, increased neuronal activity increases p-JNK at synapses, which captures Syt4 vesicles.
Figure 7.

Neuronal Activity Increases p-JNK at Synapses and Recruits Syt4 Vesicles
(A) Kymographs of mCherry-Syt4 vesicle trafficking during stimulation with 70 mM KCl for 3 min (white dashed lines) in axons co-transfected with syp-GFP to mark presynaptic sites.
(B) Quantitation of mCherry-Syt4 vesicle capture events (pauses > 2 min) at synaptic (syp-GFP+) and non-synaptic sites.
(C) Western blots of p-JNK, total JNK, and Syt4 in hippocampal neurons treated with bicuculline (bic) for 1 hr compared with the control. Tubulin was a loading control. Quantitation of p-JNK and Syt4 levels normalized to control is shown at the center and right, respectively.
(D) Western blot of phosphorylated c-Jun in hippocampal neurons treated with SP600125 (a JNK inhibitor), anisomycin (a JNK activator), and anisomycin + SP600125 compared with the control. Tubulin was a loading control.
(E) Hippocampal cultures immunostained for p-JNK, syp, and MAP2 under control, bic, and bicuculline + SP600125 (bic + SP6) treatment conditions (n = 33, 34, and 17 images for control, bic, and bic + SP6 conditions, respectively, from 4 cultures).
(F) Quantitation of p-JNK at synapses, normalized to control.
(G) Hippocampal cultures immunostained for Syt4, syp, and MAP2 under control, bic, and bic + SP6 conditions.
(H) Quantitation of Syt4 at synapses, normalized to control (n = 46, 49, and 17 images under control, bic, and bic + SP6 conditions, respectively, from 4 cultures).
Scale bars, 5 μm. Significance was determined by Student’s t test with Bonferroni correction; error is SEM; ∗p < 0.05, ∗∗∗p < 0.001.
Discussion
Our results shed light on a fundamental question regarding the distribution of essential modulatory components—neuropeptides and neurotrophins—at synapses and reveals a capture mechanism of DCVs. We found that, prior to capture, DCVs in hippocampal neurons traffic in a circular route from the soma to distal processes and back again, as previously reported at the Drosophila NMJ (Wong et al., 2012), ensuring an even distribution of DCVs at all synapses. Capture of Syt4-harboring DCVs occurs through a single phosphorylation site, S135, on Syt4 that steers DCVs to active synapses upon phosphorylation by JNK. Using biochemical and structural modeling approaches, we showed that phosphorylation of Syt4 at S135 decreases its direct interaction with Kif1A, which leads to destabilization of a Syt4-Kif1A trafficking complex and DCV capture at synapses.
Capture of phosphorylated Syt4 vesicles is activity-dependent and requires JNK phosphorylation of the S135 site of Syt4. Depolarization of neurons increased capture of WT Syt4-harboring DCVs but had no effect on S135A phosphodeficient Syt4 vesicles or on WT Syt4 vesicles in neurons in which JNK1 was inactivated. Importantly, S135A vesicles remained highly mobile both during stimulation and in the presence of increased active JNK1, demonstrating that Syt4 vesicles are captured by JNK-dependent phosphorylation of the S135 site of Syt4 specifically. Syt4 is necessary for DCV capture because, in the absence of Syt4 in Syt4 knockout neurons, activity-dependent capture of DCVs was abolished.
Although the precise mechanism by which neuronal activity leads to JNK activation is not yet known, calcium influx is likely to be important; JNK activation in hippocampal neurons depends on calcium influx (Ko et al., 1998), and calcium influx has been reported to be necessary for longer pauses of transport vesicles at synapses (Sabo et al., 2006). Syt4 is upregulated by neuronal activity (Vician et al., 1995), but JNK-dependent capture of WT Syt4 vesicles occurs within minutes of increased activity, before Syt4 protein levels begin to increase (Dean et al., 2012). Upregulation of Syt4 over longer time periods of increased activity, however, would provide more Syt4 on vesicles that can potentially be phosphorylated and recruited to synaptic sites. This would allow an additional level of control to provide sufficient DCVs to active sites under conditions of high neuronal activity.
Capture could involve a single step, where phosphorylated DCVs detach from microtubules and are unable to get back on as long as they are phosphorylated, or it could be a multi-step process where phosphorylated vesicles pause at synapses and are then sequestered by a separate mechanism. In either case, capture would require the detachment of Syt4 vesicles from microtubules. We found that Syt4 vesicles are transported by Kif1A and that depolarization immobilizes Syt4 vesicles but does not affect trafficking of Kif1A, which remains mobile. This suggests that Syt4 vesicles do indeed dissociate from Kif1A upon activity-dependent capture. A two-step mechanism of vesicle capture is supported by our finding that actin is necessary for S135 phosphorylation-dependent capture of Syt4-harboring vesicles. Disrupting actin specifically increased the mobility of S135E vesicles, which bind less tightly to Kif1A and are therefore more susceptible to capture by actin, but had no effect on the mobility of S135A vesicles. Conversely, polymerizing actin dramatically decreased the mobility of S135A vesicles but had less effect on S135E vesicles. This suggests that actin is specifically involved in a Syt4 S135 phosphorylation-dependent mechanism of vesicle capture and not a more general unrelated effect, which would result in a uniform increase or decrease in mobility of both S135A and S135E vesicles following perturbation of actin.
Interestingly, vesicles that are more mobile (i.e., S135A vesicles or vesicles under conditions where Kif1A is overexpressed or actin is disrupted) pause more frequently but for a short duration. Conversely, vesicles that are less mobile (harboring the S135E mutation or in the presence of increased JNK or increased polymerized actin) pause less frequently but for a longer duration. This suggests that pauses do not necessarily always lead to capture and that the duration of vesicle pausing determines whether a vesicle is captured by actin.
Our data are consistent with a “tug of war” model in which vesicle mobility versus capture is determined by relative binding to microtubules via Kif1A or to actin. This mechanism has been described for transition of cargo from microtubules to actin via kinesin/dynein and myosin motors, where the number of motors (i.e., the amount of force provided by each) controls cargo switching (Schroeder et al., 2010). KIF1a dimerizes in a conformation that promotes more force and, thus, faster processivity only when bound to cargo (Soppina et al., 2014, Tomishige et al., 2002). Reduced interaction of phosphorylated Syt4 with Kif1A could lead to reduced processivity, resulting in less force exerted by Kif1A on microtubules, where actin-based transport then “wins” and captures Syt4-harboring vesicles in actin-rich presynaptic sites.
The S135 site of Syt4 is located within a largely exposed and intrinsically disordered 114-amino acid linker connecting the transmembrane and C2A domains of Syt4 at a site 20 amino acids upstream of the C2A domain. Because S135 is located at the N-terminal cap of a region with transient helical structure, its phosphorylation might stabilize an α helix (Andrew et al., 2002, Schwalbe et al., 2013). This could interfere/compete with structural changes the linker is required to undergo upon binding to Kif1A and, therefore, attenuate interaction with Kif1A; for example, via leucine zipper interactions (Perin et al., 1991).
Interestingly, we identified, with 98% confidence, a C2-like domain and a DEP-like domain similar to that in pleckstrin in the Kif1A UDR. In our Kif1A dimer structural model, a central symmetric PH dimer is flanked by DEP and C2 domains. C2 domains can oligomerize (Wang et al., 2015, Wu et al., 2003, Zhou et al., 2015). Thus, the C2 domains of Syt4 might interact with the C2 domain of Kif1A when Syt4 is in the compact non-phosphorylated state. In addition, Syts and Kif1A can both bind lipids via their C2 and PH domains, respectively (Klopfenstein et al., 2002, Klopfenstein and Vale, 2004, Lin et al., 2014), where binding of the PH domain of Kif1A to vesicle membranes is important for cargo transport (Klopfenstein et al., 2002, Klopfenstein and Vale, 2004). We propose a model (Figure S7) in which the C2A-C2B domains of two Syt4 molecules in the compact non-phosphorylated state interact with the C2 domains of Kif1A dimers in a 1:1 ratio to “clamp” and stabilize Kif1A dimers. This would also displace the negatively charged DEP domain from the positively charged PH domain of Kif1A (Civera et al., 2005, Consonni et al., 2014), allowing the PH domain of Kif1A to interact with vesicle membranes. This trafficking complex could be further stabilized by interaction of Syt4 and Kif1A C2 domains with vesicle membranes. In this model, the S135 site of Syt4 is surface-accessible by JNK. Upon phosphorylation of S135 by JNK, a change in linker conformation loosens the clamp between C2 domains, resulting in decreased interaction of C2 and PH domains with vesicle membranes and release of vesicles.
This model is consistent with the idea that binding of the PH domain of Kif1A to cargo vesicle membranes is important for their transport (Klopfenstein et al., 2002, Klopfenstein and Vale, 2004), but other adaptor proteins that bind in parallel to Kif1A and proteins in cargo membranes provide specificity/stability (Hirokawa et al., 2010). For example, binding of Kif1A to GTP-bound Rab3 on vesicles via DENN/MADD (Niwa et al., 2008) and to the adaptor protein liprin-alpha (Shin et al., 2003) is necessary for synaptic vesicle transport (Miller et al., 2005). Syt4 is unique in that it does not require an adaptor protein and binds directly to Kif1A.
Alterations in axonal transport, capture, and release of DCVs have been implicated in neurodegenerative diseases, including Huntington’s and Alzheimer’s diseases, and autism (Chevalier-Larsen and Holzbaur, 2006, Dubey et al., 2015, Sadakata and Furuichi, 2009). Pathogenic huntingtin (polyQ-htt), for example, activates axonal JNK3, which phosphorylates kinesin-1 and reduces its binding to microtubules (Morfini et al., 2009). Thus, other JNKs or additional kinases could phosphorylate additional sites on DCV proteins or other organelles, such as mitochondria, to target them to active synapses. Increasing evidence suggests synaptic dysfunction as a hallmark of pathophysiology (Lu et al., 2013), where activity-dependent capture of DCVs is an important mechanism to modulate synaptic function. For example, BDNF, a known cargo of Syt4-harboring DCVs, is neuroprotective against synaptic deficits in animal models of neurodegeneration (Lu et al., 2013, Nagahara et al., 2009). Our study contributes to an understanding of the basic mechanisms of trafficking, capture, and fusion of DCVs, which may uncover strategies for combating neurodegenerative diseases.
Experimental Procedures
All research involving animals was approved by the Institutional Animal Care and Ethics Committees of Göttingen University (T10.31) and was done in accordance with German animal welfare laws.
Hippocampal Neuron Culture and Transfection
Hippocampal neurons were isolated from embryonic day 18 (E18)–E19 Wistar rats or post-natal day 0 (P0) Syt4 knockout mice (Jackson Laboratory; Ferguson et al., 2000)) as described previously (Banker and Cowan, 1977) and plated at a density of 80,000 cells/well for rat neurons or 120,000 cells/well for mouse neurons in 24-well plates. Neurons were transfected at DIV3 or DIV10 using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
Live-Cell Imaging
For trafficking experiments, cells were imaged on DIV13–DIV15 in a live imaging chamber (Warner Instruments) containing 150 μL of Tyrode’s solution (140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 5.5 mM glucose, and 20 mM HEPES [pH 7.3]) on a Zeiss AxioObserver inverted microscope. For activity-dependent pause experiments, high-KCl solution (100 mM NaCl, 70 mM KCl, 10 mM CaCl2, 2 mM MgCl2, 5.5 mM glucose, and 20 mM HEPES [pH 7.3]) was applied by pipette for 3 min during time-lapse recording, followed by perfusion with base Tyrode’s solution. Vesicle trafficking was analyzed using Imaris 7.6.4 (Bitplane) and MATLAB (MathWorks, Natick, MA, USA). Kymographs were generated using Metamorph software. Statistical significance was determined by Student’s t test with Bonferroni correction for multiple comparisons or as otherwise indicated.
Immunocytochemistry and Fixed-Cell Imaging and Analysis
For immunocytochemistry, cultures were fixed at DIV14–DIV16, immunostained with antibodies in buffer D (2% donkey serum, 0.1% Triton X-100, and 0.05% sodium azide in 2× PBS), imaged with a 40× 1.3 numerical aperture (NA) oil Zeiss Plan-Apochromat differential interference contrast (DIC) objective on a Zeiss LSM 710 confocal microscope and analyzed with ImageJ and Metamorph software.
HEK Cell Transfection and CoIP
HEK cells were transfected using calcium phosphate. For coIP experiments, cell lysates were incubated with 30 μL of antibody-coupled protein A/G Dynabeads (Invitrogen) or GFP-Trap beads (Chromotech) for 2 hr at 4°C on a rotator. Bound proteins were eluted, and samples were analyzed by SDS-PAGE and immunoblotting.
In Silico Kinase Prediction and In Vitro Kinase Assay
GPS 3.0 software (Zhou et al., 2004) was used as a tool to predict kinases capable of phosphorylating Syt4 at S135 and c-Jun (as a control) at S63. Peptides for WT and phosphodeficient Syt4 (S135A) and WT and phosphodeficient c-Jun (S63A) were synthesized by GenScript (Piscataway, NJ, USA). The ADP-Glo kinase assay kit (Promega) was used with JNK1 (R&D Systems) according to the manufacturer’s instructions.
Pull-Downs from Brain Lysates and Direct Binding Assays
Recombinant His-tagged WT, S135A, or S135E Syt4 was coupled to nickel beads and incubated with mouse brain homogenate or with purified GST-tagged full-length Kif1A for 2 hr at 4°C. The beads were then loaded onto SDS-PAGE gels and analyzed by western blot.
Sequence and Analysis and Structural Modeling
Swiss-Prot entries Syt4_Rat (P50232) and Kif1A_Rat (F1M4A4) were used to generate structural models. Coordinates of human C2A (PDB: 1UGK; 90% identity to rat) and rat C2B (PDB: 1W15) from Syt4 were arranged to generate a C2A-C2B tandem. The compact N-terminal domain of Syt4 was modeled ab initio by threading using PHYRE2 (Kelley et al., 2015). For Kif1A, PHYRE2 generated a PH domain with 99% confidence (PDB: 2COA) and a C2 domain (residues 1,007–1,154) with 98% confidence, and a positional psi/phi blast identified a DEP-like fold (residues 1,165–1,265, 23% ID, 40% similarity; PDB: 1UHW) in the Kif1A UDR (residues 849–1592). For complex presentation, Syt4 was manually docked to the Kif1A structure. Missing loops in structures were refined using FOLDIT (Kleffner et al., 2017). All structures were rendered and visualized using PyMOL.
Statistical Analysis
Statistical methods and sample size are described in each figure legend. Statistical analyses were performed using Prism 6 (GraphPad) and plotted as mean ± SEM. Because vesicles exhibited the highest variance and SD (compared with neuronal processes, movies, coverslips, or cultures), vesicles were used as sample size for all trafficking parameters except mobile percentage, for which movies were used. Statistical significance was determined by Student’s two-tailed unpaired t test with Bonferroni correction for multiple comparisons or 2-way ANOVA with Tukey’s multiple comparison test, as indicated in the figure legends. Significance values (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) are indicated in plots and in the text for analyses not represented graphically.
Author Contributions
V.B. designed and performed experiments, analyzed data, and wrote the paper. M.S. designed MATLAB code for time-lapse imaging analysis and analyzed data. K.B., S.A., and M.K.-N. designed and performed experiments. C.R. performed structural modeling. V.S. performed experiments. M.Z. performed sequence analysis and prediction. J.T.T. designed experiments and contributed reagents. C.D. designed experiments, analyzed data, and wrote the paper.
Acknowledgments
This work was funded by a Dorothea Schloezer fellowship (to K.B.), a Sofja Kovalevskaja grant from the Alexander von Humboldt Foundation, European Research Council Starting Grant SytActivity FP7 260916, Deutsche Forschungsgemeinschaft grants DE1951/1-1 and DE1951/2-1 (to C.D.), and the Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB).
Published: November 21, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three movies and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.10.084.
Data and Software Availability
Raw underlying kymographs are available at Mendeley (https://doi.org/10.17632/5b9zw3tm4f.1).
Supplemental Information
Five minute timelapse movies of vesicle movement in mCherry-Syt4 (left), mCherry-Syt4 S135A (middle), and mCherry-Syt4 S135E (right) transfected hippocampal neurons at DIV6 imaged at 1 frame/sec (frame rate = 20 fps, scale bar = 10 μm).
Five minute timelapse movies of vesicle movement in mCherry-Syt4 (left), mCherry-Syt4 S135A (middle), and mCherry-Syt4 S135E (right) transfected hippocampal neurons at DIV6 imaged at 1 frame/sec (frame rate = 20 fps, scale bar = 10 μm).
Five minute timelapse movies of mCherry-Syt4 S135E vesicle movement in neurons with (right panel) and without (left panel) co-transfection of Kif1A-GFP (left), imaged at 1 frame/sec (frame rate = 20 fps, scale bar = 10 μm).
References
- Adachi N., Kohara K., Tsumoto T. Difference in trafficking of brain-derived neurotrophic factor between axons and dendrites of cortical neurons, revealed by live-cell imaging. BMC Neurosci. 2005;6:42. doi: 10.1186/1471-2202-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams L.A., Ang L.C., Munoz D.G. Chromogranin A, a soluble synaptic vesicle protein, is found in cortical neurons other than previously defined peptidergic neurons in the human neocortex. Brain Res. 1993;602:336–341. doi: 10.1016/0006-8993(93)90699-n. [DOI] [PubMed] [Google Scholar]
- Andrew C.D., Warwicker J., Jones G.R., Doig A.J. Effect of phosphorylation on alpha-helix stability as a function of position. Biochemistry. 2002;41:1897–1905. doi: 10.1021/bi0113216. [DOI] [PubMed] [Google Scholar]
- Banker G.A., Cowan W.M. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–442. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Bennett B.L., Sasaki D.T., Murray B.W., O’Leary E.C., Sakata S.T., Xu W., Leisten J.C., Motiwala A., Pierce S., Satoh Y. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgari D., Zhou C., Hewes R.S., Deitcher D.L., Levitan E.S. Vesicle capture, not delivery, scales up neuropeptide storage in neuroendocrine terminals. Proc. Natl. Acad. Sci. USA. 2014;111:3597–3601. doi: 10.1073/pnas.1322170111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne R.D., Morgan A. Secretory granule exocytosis. Physiol. Rev. 2003;83:581–632. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen E., Holzbaur E.L. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta. 2006;1762:1094–1108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Cingolani L.A., Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Civera C., Simon B., Stier G., Sattler M., Macias M.J. Structure and dynamics of the human pleckstrin DEP domain: distinct molecular features of a novel DEP domain subfamily. Proteins. 2005;58:354–366. doi: 10.1002/prot.20320. [DOI] [PubMed] [Google Scholar]
- Consonni S.V., Maurice M.M., Bos J.L. DEP domains: structurally similar but functionally different. Nat. Rev. Mol. Cell Biol. 2014;15:357–362. doi: 10.1038/nrm3791. [DOI] [PubMed] [Google Scholar]
- Curtin J.F., Cotter T.G. Anisomycin activates JNK and sensitises DU 145 prostate carcinoma cells to Fas mediated apoptosis. Br. J. Cancer. 2002;87:1188–1194. doi: 10.1038/sj.bjc.6600612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J., Toonen R.F., Verhaagen J., Verhage M. Vesicular trafficking of semaphorin 3A is activity-dependent and differs between axons and dendrites. Traffic. 2006;7:1060–1077. doi: 10.1111/j.1600-0854.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- Dean C., Liu H., Dunning F.M., Chang P.Y., Jackson M.B., Chapman E.R. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat. Neurosci. 2009;12:767–776. doi: 10.1038/nn.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C., Liu H., Staudt T., Stahlberg M.A., Vingill S., Bückers J., Kamin D., Engelhardt J., Jackson M.B., Hell S.W., Chapman E.R. Distinct subsets of Syt-IV/BDNF vesicles are sorted to axons versus dendrites and recruited to synapses by activity. J. Neurosci. 2012;32:5398–5413. doi: 10.1523/JNEUROSCI.4515-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dérijard B., Hibi M., Wu I.H., Barrett T., Su B., Deng T., Karin M., Davis R.J. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Dubey J., Ratnakaran N., Koushika S.P. Neurodegeneration and microtubule dynamics: death by a thousand cuts. Front. Cell. Neurosci. 2015;9:343. doi: 10.3389/fncel.2015.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson G.D., Anagnostaras S.G., Silva A.J., Herschman H.R. Deficits in memory and motor performance in synaptotagmin IV mutant mice. Proc. Natl. Acad. Sci. USA. 2000;97:5598–5603. doi: 10.1073/pnas.100104597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs K.L., Greensmith L., Schiavo G. Regulation of Axonal Transport by Protein Kinases. Trends Biochem. Sci. 2015;40:597–610. doi: 10.1016/j.tibs.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Hirokawa N., Sobue K., Kanda K., Harada A., Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J. Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N., Niwa S., Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T., Gondré-Lewis M.C., Arnaoutova I., Loh Y.P. Dense-core secretory granule biogenesis. Physiology (Bethesda) 2006;21:124–133. doi: 10.1152/physiol.00043.2005. [DOI] [PubMed] [Google Scholar]
- Kleffner R., Flatten J., Leaver-Fay A., Baker D., Siegel J.B., Khatib F., Cooper S. Foldit Standalone: a video game-derived protein structure manipulation interface using Rosetta. Bioinformatics. 2017;33:2765–2767. doi: 10.1093/bioinformatics/btx283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein D.R., Vale R.D. The lipid binding pleckstrin homology domain in UNC-104 kinesin is necessary for synaptic vesicle transport in Caenorhabditis elegans. Mol. Biol. Cell. 2004;15:3729–3739. doi: 10.1091/mbc.E04-04-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein D.R., Tomishige M., Stuurman N., Vale R.D. Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell. 2002;109:347–358. doi: 10.1016/s0092-8674(02)00708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko H.W., Park K.Y., Kim H., Han P.L., Kim Y.U., Gwag B.J., Choi E.J. Ca2+-mediated activation of c-Jun N-terminal kinase and nuclear factor kappa B by NMDA in cortical cell cultures. J. Neurochem. 1998;71:1390–1395. doi: 10.1046/j.1471-4159.1998.71041390.x. [DOI] [PubMed] [Google Scholar]
- Lei K., Nimnual A., Zong W.X., Kennedy N.J., Flavell R.A., Thompson C.B., Bar-Sagi D., Davis R.J. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell. Biol. 2002;22:4929–4942. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.C., Seikowski J., Pérez-Lara A., Jahn R., Höbartner C., Walla P.J. Control of membrane gaps by synaptotagmin-Ca2+ measured with a novel membrane distance ruler. Nat. Commun. 2014;5:5859. doi: 10.1038/ncomms6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo K.Y., Kuzmin A., Unger S.M., Petersen J.D., Silverman M.A. KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci. Lett. 2011;491:168–173. doi: 10.1016/j.neulet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Lu B., Nagappan G., Guan X., Nathan P.J., Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013;14:401–416. doi: 10.1038/nrn3505. [DOI] [PubMed] [Google Scholar]
- Machado J.D., Díaz-Vera J., Domínguez N., Alvarez C.M., Pardo M.R., Borges R. Chromogranins A and B as regulators of vesicle cargo and exocytosis. Cell. Mol. Neurobiol. 2010;30:1181–1187. doi: 10.1007/s10571-010-9584-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller K.E., DeProto J., Kaufmann N., Patel B.N., Duckworth A., Van Vactor D. Direct observation demonstrates that Liprin-alpha is required for trafficking of synaptic vesicles. Curr. Biol. 2005;15:684–689. doi: 10.1016/j.cub.2005.02.061. [DOI] [PubMed] [Google Scholar]
- Morfini G.A., You Y.M., Pollema S.L., Kaminska A., Liu K., Yoshioka K., Björkblom B., Coffey E.T., Bagnato C., Han D. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009;12:864–871. doi: 10.1038/nn.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y., Higuchi M., Hirabayashi Y., Fukuda M., Gotoh Y. JNK phosphorylates synaptotagmin-4 and enhances Ca2+-evoked release. EMBO J. 2008;27:76–87. doi: 10.1038/sj.emboj.7601935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara A.H., Merrill D.A., Coppola G., Tsukada S., Schroeder B.E., Shaked G.M., Wang L., Blesch A., Kim A., Conner J.M. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa S., Tanaka Y., Hirokawa N. KIF1Bbeta- and KIF1A-mediated axonal transport of presynaptic regulator Rab3 occurs in a GTP-dependent manner through DENN/MADD. Nat. Cell Biol. 2008;10:1269–1279. doi: 10.1038/ncb1785. [DOI] [PubMed] [Google Scholar]
- Perin M.S., Brose N., Jahn R., Südhof T.C. Domain structure of synaptotagmin (p65) J. Biol. Chem. 1991;266:623–629. [PubMed] [Google Scholar]
- Sabo S.L., Gomes R.A., McAllister A.K. Formation of presynaptic terminals at predefined sites along axons. J. Neurosci. 2006;26:10813–10825. doi: 10.1523/JNEUROSCI.2052-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakata T., Furuichi T. Developmentally regulated Ca2+-dependent activator protein for secretion 2 (CAPS2) is involved in BDNF secretion and is associated with autism susceptibility. Cerebellum. 2009;8:312–322. doi: 10.1007/s12311-009-0097-5. [DOI] [PubMed] [Google Scholar]
- Scalettar B.A. How neurosecretory vesicles release their cargo. Neuroscientist. 2006;12:164–176. doi: 10.1177/1073858405284258. [DOI] [PubMed] [Google Scholar]
- Schroeder H.W., 3rd, Mitchell C., Shuman H., Holzbaur E.L., Goldman Y.E. Motor number controls cargo switching at actin-microtubule intersections in vitro. Curr. Biol. 2010;20:687–696. doi: 10.1016/j.cub.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalbe M., Biernat J., Bibow S., Ozenne V., Jensen M.R., Kadavath H., Blackledge M., Mandelkow E., Zweckstetter M. Phosphorylation of human Tau protein by microtubule affinity-regulating kinase 2. Biochemistry. 2013;52:9068–9079. doi: 10.1021/bi401266n. [DOI] [PubMed] [Google Scholar]
- Shakiryanova D., Tully A., Levitan E.S. Activity-dependent synaptic capture of transiting peptidergic vesicles. Nat. Neurosci. 2006;9:896–900. doi: 10.1038/nn1719. [DOI] [PubMed] [Google Scholar]
- Shin H., Wyszynski M., Huh K.H., Valtschanoff J.G., Lee J.R., Ko J., Streuli M., Weinberg R.J., Sheng M., Kim E. Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J. Biol. Chem. 2003;278:11393–11401. doi: 10.1074/jbc.M211874200. [DOI] [PubMed] [Google Scholar]
- Soppina V., Norris S.R., Dizaji A.S., Kortus M., Veatch S., Peckham M., Verhey K.J. Dimerization of mammalian kinesin-3 motors results in superprocessive motion. Proc. Natl. Acad. Sci. USA. 2014;111:5562–5567. doi: 10.1073/pnas.1400759111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagi M., Gorlovoy P., Larionov S., Takahashi K., Neumann H. Unloading kinesin transported cargoes from the tubulin track via the inflammatory c-Jun N-terminal kinase pathway. FASEB J. 2006;20:2573–2575. doi: 10.1096/fj.06-6679fje. [DOI] [PubMed] [Google Scholar]
- Tomishige M., Klopfenstein D.R., Vale R.D. Conversion of Unc104/KIF1A kinesin into a processive motor after dimerization. Science. 2002;297:2263–2267. doi: 10.1126/science.1073386. [DOI] [PubMed] [Google Scholar]
- Vician L., Lim I.K., Ferguson G., Tocco G., Baudry M., Herschman H.R. Synaptotagmin IV is an immediate early gene induced by depolarization in PC12 cells and in brain. Proc. Natl. Acad. Sci. USA. 1995;92:2164–2168. doi: 10.1073/pnas.92.6.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Takeuchi H., Gao J., Zhang Z., Hirata M. Hetero-oligomerization of C2 domains of phospholipase C-related but catalytically inactive protein and synaptotagmin-1. Adv. Biol. Regul. 2015;57:120–129. doi: 10.1016/j.jbior.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Wong M.Y., Zhou C., Shakiryanova D., Lloyd T.E., Deitcher D.L., Levitan E.S. Neuropeptide delivery to synapses by long-range vesicle circulation and sporadic capture. Cell. 2012;148:1029–1038. doi: 10.1016/j.cell.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., He Y., Bai J., Ji S.R., Tucker W.C., Chapman E.R., Sui S.F. Visualization of synaptotagmin I oligomers assembled onto lipid monolayers. Proc. Natl. Acad. Sci. USA. 2003;100:2082–2087. doi: 10.1073/pnas.0435872100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.E., Huo L., Maeder C.I., Feng W., Shen K. The balance between capture and dissociation of presynaptic proteins controls the spatial distribution of synapses. Neuron. 2013;78:994–1011. doi: 10.1016/j.neuron.2013.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F.F., Xue Y., Chen G.L., Yao X. GPS: a novel group-based phosphorylation predicting and scoring method. Biochem. Biophys. Res. Commun. 2004;325:1443–1448. doi: 10.1016/j.bbrc.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Zhou Q., Lai Y., Bacaj T., Zhao M., Lyubimov A.Y., Uervirojnangkoorn M., Zeldin O.B., Brewster A.S., Sauter N.K., Cohen A.E. Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature. 2015;525:62–67. doi: 10.1038/nature14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Five minute timelapse movies of vesicle movement in mCherry-Syt4 (left), mCherry-Syt4 S135A (middle), and mCherry-Syt4 S135E (right) transfected hippocampal neurons at DIV6 imaged at 1 frame/sec (frame rate = 20 fps, scale bar = 10 μm).
Five minute timelapse movies of vesicle movement in mCherry-Syt4 (left), mCherry-Syt4 S135A (middle), and mCherry-Syt4 S135E (right) transfected hippocampal neurons at DIV6 imaged at 1 frame/sec (frame rate = 20 fps, scale bar = 10 μm).
Five minute timelapse movies of mCherry-Syt4 S135E vesicle movement in neurons with (right panel) and without (left panel) co-transfection of Kif1A-GFP (left), imaged at 1 frame/sec (frame rate = 20 fps, scale bar = 10 μm).