

Abstract
Non-small-cell lung cancer (NSCLC) is one of the leading death-related malignancies worldwide with elusive molecular mechanisms. A-kinase interacting protein 1 (AKIP1) is an important regulator controlling metastasis, lymphangiogenesis and angiogenesis. However, the role of AKIP1 in NSCLC progression is still little known. Here, we found that AKIP1 was overexpressed in NSCLC specimens as well as cell lines. Overexpression of AKIP1 in NSLCC tissues was positively correlated with TNM stage, lymph node metastasis and poor prognosis. Knockdown of AKIP1 inhibited NSCLC cell migration, invasion and epithelial-mesenchymal transition (EMT), as indicated by the up-regulation of mesenchymal markers (fibronectin and vimentin) and down-regulation of epithelial marker E-cadherin, whereas overexpression of AKIP1 showed the opposite effects. Moreover, AKIP1 transactivated Zinc Finger E-Box Binding Homeobox 1 (ZEB1) expression via directly binding to ZEB1 promoter, thereby leading to E-cadherin transcriptional repression. Additionally, we observed that the binding efficiency of AKIP1 within ZEB1 promotor was determined by the interaction between AKIP1 and SP1. In conclusion, AKIP1 promoted EMT of NSCLC via transactivating ZEB1, suggesting AKIP1 as a potential therapeutic target.
Keywords: Non-small-cell lung cancer, A-kinase interacting protein 1, epithelial-mesenchymal transition, Zinc Finger E-Box Binding Homeobox 1
Introduction
Non-small-cell lung cancer (NSCLC), which accounts for more than 80% of lung cancer cases, is an extremely lethal disease worldwide [1-3]. The prognosis of NSCLC patients continues to be poor, despite significant improvements in surgical and adjuvant treatment strategies in recent years, with a 5-year survival rate of about 16% [4]. Metastasis is the major reason for the high mortality of NSCLC patients after surgical resection [5,6]. However, the molecular characteristics and related biological mechanisms underlying NSCLC metastasis are still elusive and a better understanding in it might contribute to the development of therapeutic targets.
A-kinase interacting protein 1 (AKIP1) was initially reported as a novel human breast cancer associated gene 3 (BCA3) which encoded an alternatively spliced proline-rich protein [7]. Accumulated evidences indicated that AKIP1 dysregulation reflected physiological and pathological alterations in many human malignancies, and might be involved in tumor metastasis. In esophageal cancer, AKIP1 was found to be overexpressed in esophageal squamous cell carcinoma (ESCC) cell lines and clinical samples, and predicted poorer outcomes of ESCC patients [8]. Mechanistic investigations by Lin et al. revealed that AKIP1 was a downstream target of NK2 homeobox 8 (Nkx2-8) and a transcription factor of VEGF-C, which induced lymphangiogenesis and lymphatic metastasis in ESCC [8,9]. Moreover, another study in breast carcinoma showed that AKIP1 promoted metastasis through Akt/GSK-3β/Snail signaling pathway [10]. Although AKIP1 has been implicated as a potential mediator of metastasis in several human tumors, a consensus knowledge of its molecular functionality, clinical significance and novel mechanisms involved in promoting distant metastasis especially in NSCLC has not yet been reached.
In the current study, we first focused on the clinical significance of AKIP1 by analysing its expression in a series of NSCLC cases, followed by analysing the relationship between the expression level of AKIP1 and the clinico-pathologic parameters. To determine the biological role of AKIP1 in NSCLC, loss-of-function and overexpression studies were conducted, which showed critical effect of AKIP1 on cellular behaviors, including cell proliferation, colony formation, migration and invasion of NSCLC cells. In addition, we addressed potential mechanism for metastasis of NSCLC in a series of in vitro and in vivo experiments. We demonstrated that AKIP1 was involved in epithelial-mesenchymal transition (EMT) process through transcriptionally activating Zinc Finger E-Box Binding Homeobox 1 (ZEB1). These results provide evidences that AKIP1 is a promising regulator of tumor metastasis, and may serve as a therapeutic target for NSCLC.
Materials and methods
Patient samples and immunohistochemistry (IHC) staining
From 2008 to 2011, a total of 139 adult patients with NSCLC who underwent curative resection at Henan Provincial People’s Hospital (Henan, China) were enrolled in this study. The study protocol was approved by the research ethics committee of Henan Provincial People’s Hospital, and written informed consent was obtained from each patient. None of these patients had received neoadjuvant therapy. The clinico-pathologic information was shown in Table 1. IHC staining of tissue microarray was conducted according to the manufacturer’s protocol (Immunostain SP kit, DakoCytomation, USA). Antibodies used for IHC analysis were anti-C11orf17/AKIP1 antibody (Abcam) and anti-AREB6/ZEB1 antibody (Abcam). Quantification of the IHC scores of paraffin-embedded NSCLC specimens was performed by two independent pathologists who were blinded to the histopathological features of the enrolled patients. The final IHC scores were determined by multiplying the proportion of positively stained cells and the staining intensity as previously reported [11].
Table 1.
Correlation between AKIP1 expression and clinico-pathological characteristics of NSCLC patients
| AKIP1 expression | ||||
|---|---|---|---|---|
|
|
||||
| Parameters | n | Low | High | p |
| Total | 139 | 58 | 81 | |
| Age (years) | ||||
| <55 | 51 | 19 | 32 | 0.416 |
| ≥55 | 88 | 39 | 49 | |
| Gender | ||||
| Male | 94 | 37 | 57 | 0.414 |
| Female | 45 | 21 | 24 | |
| Smoking | ||||
| No smoking | 55 | 19 | 36 | 0.165 |
| Smoking | 84 | 39 | 45 | |
| Histopathology type | ||||
| Adenocarcinoma | 71 | 33 | 38 | 0.246 |
| Squamous cell carcinoma | 68 | 25 | 43 | |
| Lymph node metastasis | ||||
| No | 73 | 39 | 34 | 0.003 |
| Yes | 66 | 19 | 47 | |
| TNM stage | ||||
| Stage I | 38 | 23 | 15 | 0.011 |
| Stage II | 55 | 22 | 33 | |
| Stage III-IV | 46 | 13 | 33 | |
| Differentiation | ||||
| Low grade | 21 | 11 | 10 | 0.221 |
| Middle grade | 86 | 31 | 55 | |
| High grade | 32 | 16 | 16 | |
Cell culture and generation of stable cell lines
Human NSCLC cell lines A549, NCI-H441 and the normal immortalized bronchial epithelial cell line HBE were purchased from American Type Culture Collection (Manassas, VA, USA). NCI-H292, NCI-H358, NCI-H460, NCI-H1299 and NCI-H1650 were purchased from Shanghai Cell Bank (Chinese Academy of Sciences, Shanghai, China). Cells were maintained at 37°C/5% CO2 in a humidified incubator with recommended medium containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA). Stable cell lines expressing AKIP1 or shAKIP1 were generated by transfection of pCMV-AKIP1 or pRS-shAKIP1 into A549 and NCI-H1299 cells and cultured for 10 days with 400 μg/ml G418 or 1 μg/ml puromycin 48 h after infection. Positive clones were then selected, and transfection was verified by immunoblotting assay.
Quantitative real time polymerase chain reaction (qRT-PCR)
Total RNA was isolated using TRIzol Reagent (Invitrogen). Complementary DNA (cDNA) was obtained by using the High Capacity cDNA Reverse Transcription Kit (Promega), and quantification of gene expression was conducted as previously described [11]. The primers were listed in Supplementary Table 1.
Cell proliferation and colony formation assay
Cell proliferation was assessed using cell counting kit 8 (CCK-8, Dojindo, Japan). Cells were plated into 96-well plates at 1000 cells per well, and cell viability (absorbance 450 nm) was measured every 24 h for 6 days. For colony formation assay, A549 or NCI-H1299 cells were plated into 6-well plates at 1000 cells per well and incubated for 10 days. The number of colonies was counted after stained with 0.1% crystal violet solution.
Cell migration and invasion assay
For migration assay, cells were seeded into the upper chamber of a transwell (8 μm, 24-well format, Corning). DMEM medium containing 10% fetal bovine serum (FBS) was added to the lower chamber. After incubation for 48 h at 37°C, the upper chambers were washed with phosphate-buffered saline (PBS), and cells on the top surface were softly wiped by a cotton swab. For invasion assay, the insert chambers were coated with diluted Matrigel (BD Biosciences), and other procedures were similar to the cell migration assay. The migrated/invased cells were stained with 0.1% crystal and counted in three random fields at × 200 magnification.
Co-Immunoprecipitation (Co-IP) and immunoblotting analysis
For Co-IP assays, cells were washed with cold PBS and lysed with cold lysis buffer at 4°C for 30 min. Cell lysates were then incubated with 1 μg primary antibodies or normal IgG on a rotator at 4°C overnight, followed by the addition of protein A/G sepharose beads for 2 h at 4°C. Immunocomplexes were then washed with lysis buffer three times and separated by SDS-PAGE. Immunoblotting was performed following standard procedures [11].
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed according to the EZ ChIP™ Chromatin Immunoprecipitation Kit (Millipore). In brief, Cells (2 × 106) in a 10-cm culture dish were treated with 1% formaldehyde to cross-link proteins to DNA. Cell lysates were sonicated to shear DNA to sizes of 200-1000 bp. Equal aliquots of chromatin supernatants were incubated with 1 μg of an anti-C11 or f17/AKIP1, or an anti-RNA polymerase II, or an anti-IgG antibodies (Millipore) overnight at 4°C with rotation. PCR was performed after reverse cross-link of protein/DNA complexes.
Transient transfection and luciferase assay
Cells at 60% confluence were transfected with 100 ng luciferase reporter gene construct containing different fragments of ZEB1 promoter, plus 1 ng of pRL-TK renilla plasmid (Promega) using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Luciferase activity was measured by Dual Luciferase Assay (Promega) according to the manufacturer’s recommendations. The final transfection efficiencies were normalized according to the Renilla activity.
Animal model of tumor xenograft
Animal experiments was approved by the Institutional Animal Care and Use Committee of Henan Provincial People’s Hospital. A549, A549/shNC and A549/shAKIP1 cells were injected into nude mice subcutaneously (2 × 106 in 100 μl PBS, 5 mice per group). Tumor length (L) and width (W) were measured every 5 days and tumor volumes were calculated using the following formula: volume = (W+L)/2 × W × L × 0.5236 [12]. Animals were sacrificed at the 30th day and the tumor specimens were collected, weighed and fixed by formalin.
Statistical analysis
Statistical analyses were performed using the SPSS software package (V. 16.0, SPSS, Inc., Chicago, IL). Differences between two groups were compared using the paired Student t test. Correlation between AKIP1 and clinico-pathologic parameters was evaluated by nonparametric chi-square test. Kaplan-Meier’s method was used to analysing the survival rates for AKIP1 expression. P < 0.05 was considered statistically significant.
Results
AKIP1 is overexpressed and associated with advanced clinical stage and metastasis in NSCLC
To study the clinical significance of AKIP1 in NSCLC, we examined the expression levels of AKIP1 in 20 paired NSCLC tumor and adjacent normal tissues. The result showed that AKIP1 expression was significantly increased in tumor tissues compared with normal tissues, both in mRNA (P < 0.01, Figure 1A, 1B) and protein (P < 0.01, Figure 1C, 1D) levels. The clinical significance of AKIP1 was further investigated by IHC staining of 139 paired NSCLC specimens. Representative pictures were shown in Figure 1E, and the final scoring result was presented in Figure 1F. We observed a significant relationship between AKIP1 expression and TNM stage as well as lymph node metastasis (Table 1; TNM stage, P = 0.011; lymph node metastasis, P = 0.003). Kaplan-Meier analysis revealed that patients in the AKIP1 high expression group had poorer 5-year overall survival (OS) (P = 0.007, Figure 1G) and shorter disease-free survival (DFS) time (P = 0.006, Figure 1H) than those in the AKIP1 negative expression group. We also detected higher mRNA and protein level of AKIP1 in eight NSCLC cell lines than the normal immortalized human bronchial epithelial cell line HBE, as indicated in Figure 1I, 1J. These results suggested that AKIP1 was overexpressed in NSCLC and might be involved in tumor progression.
Figure 1.
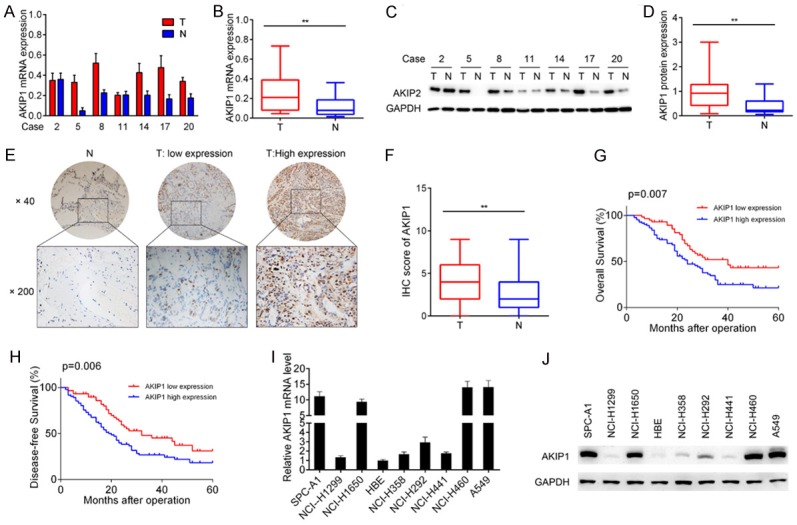
Overexpression of AKIP1 indicated poor prognosis of NSCLC patients. A, B. qRT-PCR analysis of AKIP1 expression in 20 pairs of NSCLC tissues and non-tumor tissues. C, D. Immunoblotting analysis of AKIP1 in 20 pairs of NSCLC tissues and non-tumor tissues. E. Representative images of AKIP1 staining in NSCLC tissues and paired non-tumor tissues. F. The IHC scores of AKIP1 in NSCLC tissues and matched non-tumor tissues. G. Kaplan-Meier analysis of the correlation between AKIP1 expression and the overall survival of NSCLC patients. H. Kaplan-Meier analysis of the correlation between AKIP1 expression and the disease free survival of NSCLC patients. I, J. The mRNA and protein level of AKIP1 expression in NSCLC cell lines was evaluated by qRT-PCR and immunoblotting, respectively. **P < 0.01.
AKIP1 is required for lung cancer cell growth
To test whether AKIP1 was required for NSCLC cell growth, we first carried out AKIP1 loss-of-function study in A549 cells, which showed a relatively higher level of AKIP1. The knockdown efficiency of AKIP1 was confirmed by immunoblotting (Figure 2A), and the result of CCK-8 assay showed that AKIP1 silencing significantly reduced cell viability as shown by the growth curve (P < 0.05, Figure 2B). We next performed colony formation assays and showed that AKIP1 silencing markedly inhibited the clonogenicity of A549 cells (P < 0.01, Figure 2C, 2D), which was commonly considered as indicators of tumorigenesis. In contrast, overexpression of AKIP1 promoted the cell proliferation (P < 0.05) and clonogenicity (P < 0.01) of NCI-H1299 cells (Figure 2E-H).
Figure 2.
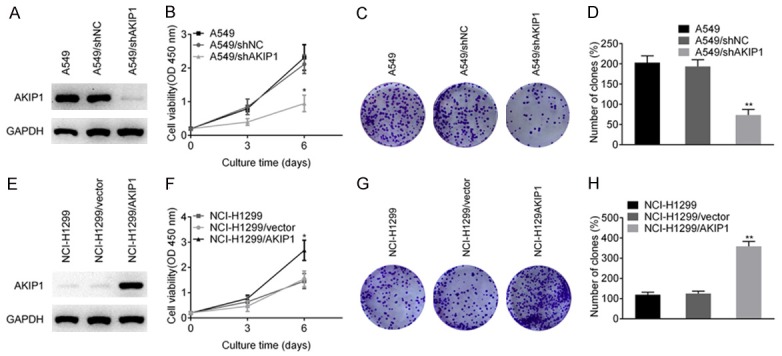
The effect of AKIP1 on NSCLC cell proliferation and colony formation. A. Knockdown efficiency of AKIP1 was confirmed by immunoblotting. B. AKIP1 silencing inhibited the proliferation of A549 cells. C, D. AKIP1 silencing inhibited the colony formation of A549 cells. E. Overexpression of AKIP1 was confirmed by immunoblotting. F. Overexpression of AKIP1 in NCI-H1299 cells promoted the proliferation ability. G, H. Overexpression of AKIP1 in NCI-H1299 cells promoted the colony formation ability. All experiments were performed at least three times. *P < 0.05, **P < 0.01.
AKIP1 promotes migration, invasion and EMT of NSCLC cells
Transwell assays were then applied to further assess other biological function of AKIP1 include cell migration and invasion ability. The result indicated that AKIP1 silencing significantly inhibited the migration and invasion ability of A549 cells (P < 0.01, Figure 3A, 3B), while overexpression of AKIP1 promoted the migration and invasion ability of NCI-H1299 cells (P < 0.01, Figure 3E, 3F). Because EMT was a well-reported mechanism for tumor cell migration and invasion, we hypothesized that AKIP1 could regulate EMT of NSCLC cells, resulting in aggressive effects. As indicated in Figure 3C, 3D, AKIP1 silencing promoted the level of epithelial marker (E-cadherin), and decreased the expression levels of mesenchymal markers (fibronectin and vimentin), (P < 0.01). In contrast, overexpression of AKIP1 promoted EMT of NCI-H1299 cells as shown in Figure 3G, 3H (P < 0.01). Several transcription factors, including Slug, Snail, Twist, ZEB1 and ZEB2, was responsible for the transcriptional repression of E-cadherin [13,14]. Thus, we evaluated whether AKIP1 inhibited E-cadherin expression through these repressors. The result of qRT-PCR showed that AKIP1 silencing inhibited the mRNA expression of ZEB1, while overexpression of AKIP1 promoted the level of ZEB1 (P < 0.01, Figure 3I). These results were further confirmed by immunoblotting assay (Figure 3J).
Figure 3.
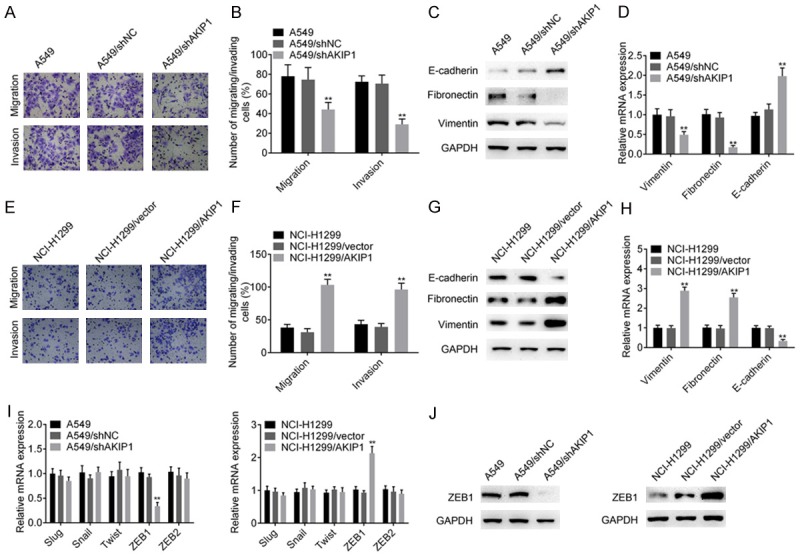
AKIP1 promoted the migration, invasion and epithelial-mesenchymal transition of NSCLC cells. A, B. AKIP1 silencing inhibited the migratory and invasive ability of A549 cells. C, D. The effect of knockdown of AKIP1 on epithelial and mesenchymal markers was evaluated by immunoblotting and qRT-PCR analysis, respectively. E, F. Overexpression of AKIP1 promoted the migratory and invasive ability of NCI-H1299 cells. G, H. Immunoblotting and qRT-PCR analysis showed that overexpression of AKIP1 promoted the expression of mesenchymal markers, while inhibited the expression of epithelial marker E-cadherin. I, J. The effect of AKIP1 on several transcriptional repressors of E-cadherin was evaluated by qRT-PCR assay and confirmed by immunoblotting assay. All experiments were performed at least three times. **P < 0.01.
ZEB1 acts as a downstream effector in AKIP1-induced invasion and migration
To gain more insight into AKIP1 mediated EMT of NSCLC cells, a luciferase reporter assay was conducted to determine whether AKIP1 regulates ZEB1 promoter activity. As indicated in Figure 4A, 4B, overexpression of AKIP1 promoted, whereas knockdown of AKIP1 attenuated, the luciferase activity of ZEB1 promoter in a dose-dependent manner in NSCLC cells (P < 0.01). Additionally, the luciferase reporter assay indicated that a deletion from nt -1250 to nt -499 had no effect on AKIP1-induced ZEB1 promoter activity, whereas further deletion from nt -499 to nt -350 markedly decreased AKIP1-induced ZEB1 promoter activity (P < 0.01, Figure 4C). The result of ChIP assay showed that AKIP1 could bind to nt -499 to nt -350 within the ZEB1 promoter (Figure 4D and 4F). Using the JASPAR database (http://jaspar.genereg.net/), we got a potential SP1 binding site within nt -499 to nt -350. Immunoprecipitation (IP) assay showed that AKIP1 could interact with SP1 in A549 cells (Figure 4E), which was coincided with previous reports [8]. Importantly, knockdown of SP1 in A549 cells significantly attenuated the binding efficiency of AKIP1 within ZEB1 promotor, which suggested SP1 was involved in AKIP1 mediated transactivation of ZEB1 (Figure 4G).
Figure 4.
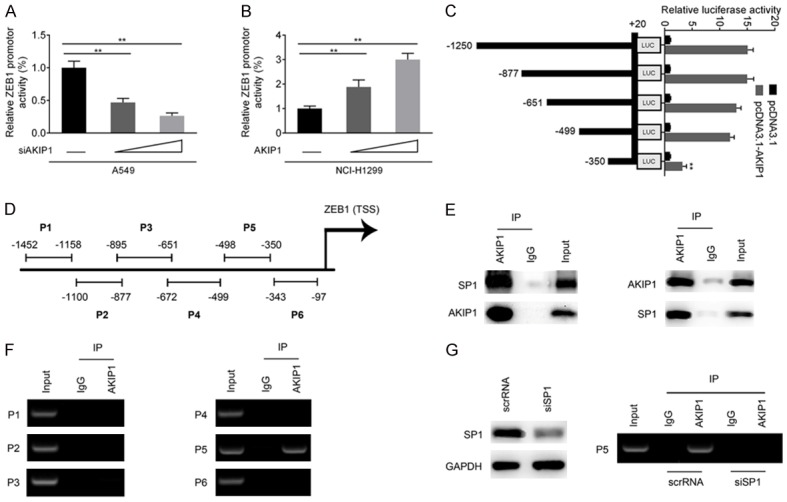
AKIP1 regulated ZEB1 promotor activity through a SP1 dependent manner. Transcriptional repression (A) and activation (B) of ZEB1 promotor by AKIP1, as indicated by dual luciferase reporter assay system. (C) Serially deletion analysis in A549 cells identified an AKIP1-responsive region within the ZEB1 promoter. (D, F) ChIP assay showed a physical association between AKIP1 and ZEB1 promotor. (E) The interaction between AKIP1 and SP1 was confirmed by IP assay. Left: Cell lysates of A549 cells were immunoprecipitated with anti-AKIP1 and immunoblotted with anti-SP1 (first line)/anti-AKIP (second line); right: Cell lysates of A549 cells were immunoprecipitated with anti-SP1 and immunoblotted with anti-AKIP1 (first line)/anti-SP1 (second line). (G) Knockdown of SP1 in A549 cells inhibited the binding efficiency of AKIP1 within ZEB1 promotor. **P < 0.01.
ZEB1 was responsible for AKIP1 induced effects
Given that ZEB1 was a direct downstream target of AKIP1, we next investigated whether ZEB1 was responsible for AKIP1 induced effects. We first evaluated the effects of AKIP1 following knockdown of ZEB1 in NCI-H1299/AKIP1 cells. NCI-H1299/AKIP1 were transiently transfected with scrRNA or siRNA targeting ZEB1 (Figure 5A). The result of CCK8, colony formation and transwell assay showed that knockdown of ZEB1 in NCI-H1299/AKIP1 cells inhibited the proliferation (Figure 5B), colony formation (Figure 5C), migration and invasion ability (Figure 5D). Furthermore, we transiently transfected ZEB1 overexpressing plasmid to A549/shAKIP1 cells, to explore whether ZEB1 could restore the effects induced by AKIP1 depletion. The restoration of ZEB1 was verified by western blot in Figure 5E. Functional studies revealed that ectopic expression of ZEB1 significantly promoted the cell proliferation, colony formation, migration and invasion of A549/shAKIP1 cells (Figure 5F-H), suggesting ZEB1 was responsible for AKIP1 mediated effects.
Figure 5.
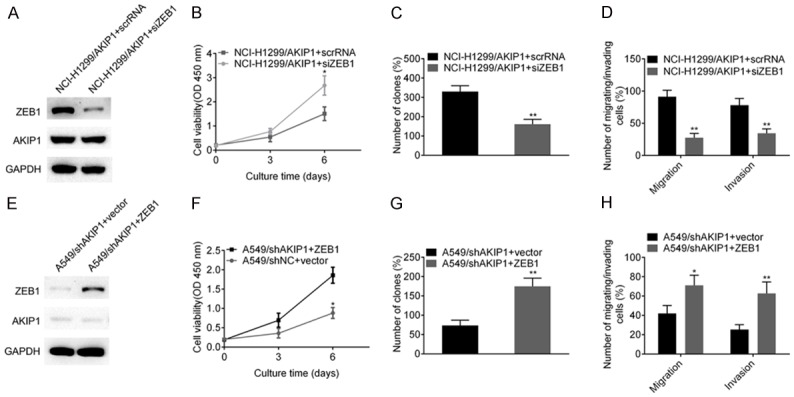
ZEB1 was responsible for AKIP1 induced effects. A. NCI-H1299/AKIP1 cells were transiently transfected with scrRNA or siAKIP1, and the knockdown efficiency of AKIP1 was confirmed by western blot assay. B. Depletion of ZEB1 in NCI-H1299/AKIP1 cells inhibited the cell proliferation by CCK8 assay. C. Depletion of ZEB1 in NCI-H1299/AKIP1 cells inhibited the colony formation assay. D. Depletion of ZEB1 in NCI-H1299/AKIP1 cells inhibited cell migration/invasion ability. E. A549/shAKIP1 cells were transiently transfected with ZEB1 expressing plasmid or empty vector, and ZEB1 restoration was examined by western blot assay. F-H. Forced expression of ZEB1 in A549/shAKIP1 cells restored the proliferation, colony formation, migration and invasion ability. *P < 0.05, **P < 0.01.
AKIP1 could regulate NSCLC cell growth and ZEB1 expression in vivo
The role of AKIP1 in regulating tumor growth was confirmed by in vivo assays. A549, A549/shNC and A549/shAKIP1 cells were subcutaneously implanted into nude mice. As shown in Figure 6A, the progressive growth of A549/shAKIP1 cells was slower compared with A549 or A549/shNC cells. Additionally, average weight of subcutaneously in A549/shAKIP1 group was lighter than the other two groups (Figure 6B). To further assess whether AKIP1 could regulate ZEB1 expression in vivo, we determined the expression of AKIP1 and ZEB1 in subcutaneous tumors. Histological analysis of the subcutaneous tumors showed fewer AKIP1 and ZEB1 antigen-positive cells in A549/shAKIP1 group, compared with A549 or A549/shNC group (Figure 6C). These results suggested that AKIP1 could regulate NSCLC cell growth and ZEB1 expression in vivo.
Figure 6.
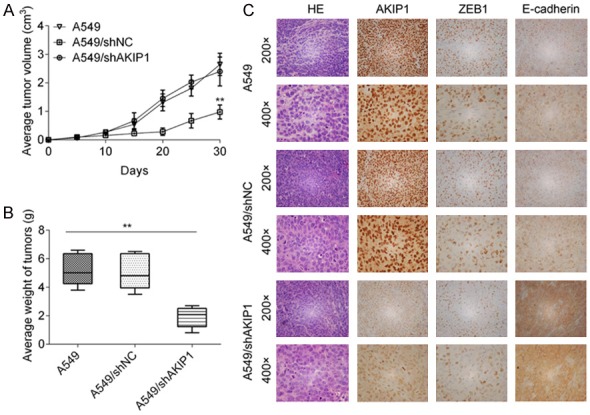
AKIP1 regulated NSCLC cell growth and ZEB1 expression in vivo. A. Tumor growth curves of A549, A549/shNC and A549/shAKIP1 group. B. Average tumor weight of each group. C. IHC assay of AKIP1 and ZEB1 expression in xenograft tumours. **P < 0.01.
The prognostic value of combination of AKIP1 and ZEB1
Given that AKIP1 promoted EMT of NSCLC through transactivating ZEB1, we then asked whether the combination of AKIP1 and ZEB1 could better predict patient prognosis than AKIP1/ZEB1 alone. The expression patterns of AKIP1 and ZEB1 in primary tumor tissues from the same patient were evaluated by IHC assay. The representative images for ZEB1 staining were shown in Figure 7A, and a scatter plot of AKIP1 and ZEB1 expression in NSCLC specimens revealed a positive correlation between AKIP1 and ZEB1 at the protein level (R = 0.629, P < 0.001, Figure 7B). Additionally, the level of AKIP1 and ZEB1 also presented a positive correlation in NSCLC cell lines (R = 0.914, P = 0.001, Figure 7C). The result of Kaplan-Meier survival analysis indicated that ZEB1 overexpression was strongly correlated with poor survival (both OS and DFS) of NSCLC patients (OS: P = 0.026, DFS: P = 0.032, Figure 7D). Moreover, NSCLC patients bearing both high levels of AKIP1 and ZEB1 exhibited worst prognoses (OS: P < 0.001, DFS: P = 0.004, Figure 7E).
Figure 7.
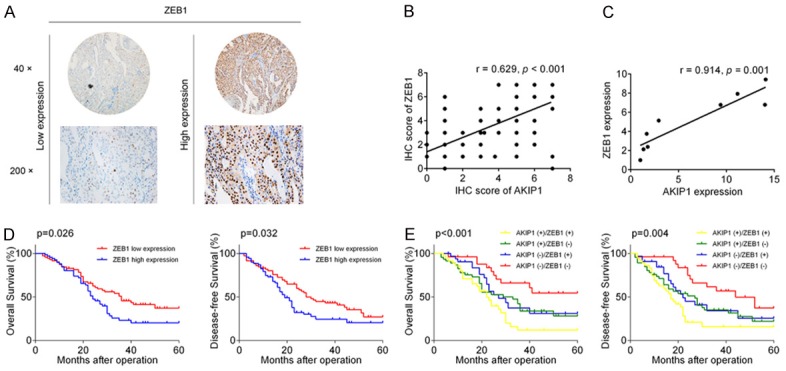
Expression of AKIP1-ZEB1 axis and prognostic significance in NSCLC patients. A. Representative IHC images of ZEB1 in NSCLC specimens. B. Scatter plot of AKIP1 and ZEB1 expression in NSCLC tissues. C. Correlation between AKIP1 expression and ZEB1 expression in NSCLC cell lines. D, E. A Kaplan-Meier analysis of ZEB1 or AKIP1/ZEB1 different combination in NSCLC tissues.
Discussion
The development of clinically useful prognostic markers is crucial for predicting risk of recurrence and/or outcomes of cancer patients [15-17]. In this study, we investigated the clinical significance of AKIP1 expression in a series of NSCLC samples. We found that AKIP1 expression was significantly up-regulated in NSCLC tumors and NSCLC cell lines. Additionally, the IHC staining of tissue microarray identified that overexpression of AKIP1 was significantly associated with TNM stage and lymph node metastasis. Further clinical analysis suggested that NSCLC patients carrying tumors with high-expression of AKIP1 had a markedly reduced 5-year OS and DFS. These evidences indicated that AKIP1 might be a novel oncogene contributing to the carcinogenesis of NSCLC.
AKIP1 has been shown to modulate NF-ĸB and protein kinase A (PKA) activity, which was involved in immunoinflammatory responses, tumorigenesis and metastasis [18-20]. Gain- and loss-of-function experiments characterized that AKIP1 induced not only cell proliferation and colony formation, but also invasive and migratory capacity of NSCLC cell lines, as evidenced from a series of in vitro and in vivo studies. As EMT could link to invasive and metastatic behavior of various human tumors [21,22], including NSCLC [23,24]. We further evaluated whether AKIP1 could regulate the expression of EMT-related markers. We provided evidence that AKIP1 silencing promoted the expression of epithelial marker E-cadherin, whereas AKIP1 overexpression could decrease the level of E-cadherin. Disruption of the E-cadherin-mediated adhesion system is a well-known mechanism for invasion [25,26]. Thus, we further focused our study on investigating the mechanisms underlying AKIP1 mediated E-cadherin down-regulation. Several transcription factors, including Snail, Slug, ZEB1, ZEB2 and Twist, could bind to E-box elements within E-cadherin promotor and induce transcriptional inactivation of E-cadherin [13,27]. In this study, we systematically investigated potential role of AKIP1 and reported a novel function for AKIP1 in modifying ZEB1-E-cadherin axis in NSCLC (Figure 8). Using serial deletion and ChIP assays, we identified ZEB1 as a direct transcriptional target of AKIP1. Clinically, AKIP1 expression was positively correlated with ZEB1 expression. NSCLC patients whose tumors expressing high levels of AKIP1 and ZEB1 exhibited worse 5-year OS and DFS, which suggested that the combination of AKIP1 and ZEB1 could better predict prognosis than either protein. Thus, AKIP1-mediated ZEB1 transactivation might play important roles in promoting NSCLC metastasis.
Figure 8.
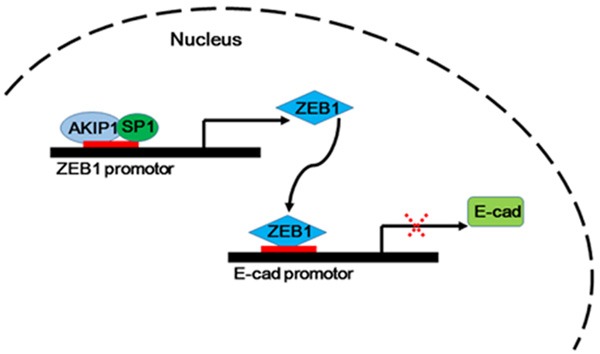
Schematic algorithm of AKIP1-ZEB1-E-cadherin axis demonstrated a critical transcriptional network in NSCLC cells.
Another important finding of our study was that we identified SP1 as a binding partner of AKIP1 in NSCLC cells. SP1 was identified as an interacting protein of AKIP1 in esophageal cancer cells [8]. Interestingly, a potential SP1 binding site was predicted within the AKIP1 binding region (nt -499 to nt -350). Therefore, we hypothesized that the function of AKIP1 in NSCLC cells might be associated with SP1. We first confirmed the interaction between AKIP1 and SP1 by IP assay. Moreover, knockdown of SP1 significantly attenuated the recruitment of AKIP1 within ZEB1 promotor. Given these evidences, we concluded that AKIP1 and SP1 constituted a transcriptional complex within ZEB1 promotor.
In conclusion, our investigation from clinical, functional and mechanistic studies indicated that AKIP1 regulated EMT of NSCLC cells through transactivating ZEB1 and might be served as a prognostic marker as well as therapeutic target in NSCLC.
Acknowledgements
We thank the patients and their families who generously donated valuable tissue samples.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Lei Z, Liu RY, Zhao J, Liu Z, Jiang X, You W, Chen XF, Liu X, Zhang K, Pasche B, Zhang HT. TGFBR1 haplotypes and risk of non-small-cell lung cancer. Cancer Res. 2009;69:7046–7052. doi: 10.1158/0008-5472.CAN-08-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Siegel RL, Jemal A. Lung Cancer Statistics. Adv Exp Med Biol. 2016;893:1–19. doi: 10.1007/978-3-319-24223-1_1. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Popper HH. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016;35:75–91. doi: 10.1007/s10555-016-9618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mirrielees JA, Kapur JH, Szalkucki LM, Harter JM, Salkowski LR, Strigel RM, Traynor AM, Wilke LG. Metastasis of primary lung carcinoma to the breast: a systematic review of the literature. J Surg Res. 2014;188:419–431. doi: 10.1016/j.jss.2014.01.024. [DOI] [PubMed] [Google Scholar]
- 7.Kitching R, Li H, Wong MJ, Kanaganayakam S, Kahn H, Seth A. Characterization of a novel human breast cancer associated gene (BCA3) encoding an alternatively spliced proline-rich protein. Biochim Biophys Acta. 2003;1625:116–121. doi: 10.1016/s0167-4781(02)00562-6. [DOI] [PubMed] [Google Scholar]
- 8.Lin C, Song L, Liu A, Gong H, Lin X, Wu J, Li M, Li J. Overexpression of AKIP1 promotes angiogenesis and lymphangiogenesis in human esophageal squamous cell carcinoma. Oncogene. 2015;34:384–393. doi: 10.1038/onc.2013.559. [DOI] [PubMed] [Google Scholar]
- 9.Lin C, Song L, Gong H, Liu A, Lin X, Wu J, Li M, Li J. Nkx2-8 downregulation promotes angiogenesis and activates NF-kappaB in esophageal cancer. Cancer Res. 2013;73:3638–3648. doi: 10.1158/0008-5472.CAN-12-4028. [DOI] [PubMed] [Google Scholar]
- 10.Mo D, Li X, Li C, Liang J, Zeng T, Su N, Jiang Q, Huang J. Overexpression of AKIP1 predicts poor prognosis of patients with breast carcinoma and promotes cancer metastasis through Akt/GSK-3beta/Snail pathway. Am J Transl Res. 2016;8:4951–4959. [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu X, Guo X, Wu S, Wei L. ANGPTL4 correlates with NSCLC progression and regulates epithelial-mesenchymal transition via ERK pathway. Lung. 2016;194:637–646. doi: 10.1007/s00408-016-9895-y. [DOI] [PubMed] [Google Scholar]
- 12.Bandyopadhyay S, Zhan R, Chaudhuri A, Watabe M, Pai SK, Hirota S, Hosobe S, Tsukada T, Miura K, Takano Y, Saito K, Pauza ME, Hayashi S, Wang Y, Mohinta S, Mashimo T, Iiizumi M, Furuta E, Watabe K. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat Med. 2006;12:933–938. doi: 10.1038/nm1444. [DOI] [PubMed] [Google Scholar]
- 13.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488–494. doi: 10.1038/ncb2976. [DOI] [PubMed] [Google Scholar]
- 15.Soland TM, Brusevold IJ. Prognostic molecular markers in cancer-quo vadis? Histopathology. 2013;63:297–308. doi: 10.1111/his.12184. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki K, Kachala SS, Kadota K, Shen R, Mo Q, Beer DG, Rusch VW, Travis WD, Adusumilli PS. Prognostic immune markers in non-small cell lung cancer. Clin Cancer Res. 2011;17:5247–5256. doi: 10.1158/1078-0432.CCR-10-2805. [DOI] [PubMed] [Google Scholar]
- 17.Woodard GA, Jones KD, Jablons DM. Lung cancer staging and prognosis. Cancer Treat Res. 2016;170:47–75. doi: 10.1007/978-3-319-40389-2_3. [DOI] [PubMed] [Google Scholar]
- 18.Gao N, Asamitsu K, Hibi Y, Ueno T, Okamoto T. AKIP1 enhances NF-kappaB-dependent gene expression by promoting the nuclear retention and phosphorylation of p65. J Biol Chem. 2008;283:7834–7843. doi: 10.1074/jbc.M710285200. [DOI] [PubMed] [Google Scholar]
- 19.Gao N, Hibi Y, Cueno M, Asamitsu K, Okamoto T. A-kinase-interacting protein 1 (AKIP1) acts as a molecular determinant of PKA in NF-kappaB signaling. J Biol Chem. 2010;285:28097–28104. doi: 10.1074/jbc.M110.116566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sastri M, Barraclough DM, Carmichael PT, Taylor SS. A-kinase-interacting protein localizes protein kinase A in the nucleus. Proc Natl Acad Sci U S A. 2005;102:349–354. doi: 10.1073/pnas.0408608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 22.Katsuno Y, Lamouille S, Derynck R. TGFbeta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 23.Datar I, Schalper KA. Epithelialmesenchymal transition and immune evasion during lung cancer progression: The Chicken or the Egg? Clin Cancer Res. 2016;22:3422–3424. doi: 10.1158/1078-0432.CCR-16-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li A, Gao HF, Wu YL. Targeting the MET pathway for potential treatment of NSCLC. Expert Opin Ther Targets. 2015;19:663–674. doi: 10.1517/14728222.2014.995093. [DOI] [PubMed] [Google Scholar]
- 25.Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126:393–401. doi: 10.1242/jcs.100115. [DOI] [PubMed] [Google Scholar]
- 26.Labernadie A, Kato T, Brugues A, Serra-Picamal X, Derzsi S, Arwert E, Weston A, Gonzalez-Tarrago V, Elosegui-Artola A, Albertazzi L, Alcaraz J, Roca-Cusachs P. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19:224–237. doi: 10.1038/ncb3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giroldi LA, Bringuier PP, de Weijert M, Jansen C, van Bokhoven A, Schalken JA. Role of E boxes in the repression of E-cadherin expression. Biochem Biophys Res Commun. 1997;241:453–458. doi: 10.1006/bbrc.1997.7831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.