

Abstract
Esophageal squamous cell carcinoma (ESCC) is one of the most common malignant tumors with poor survival and limited therapeutic options. The aim of this study is to identify novel anticancer strategies from existing Food and Drug Administration (FDA)-approved drugs that have been used to clinically treat other diseases. Here, propafenone, an antiarrhythmic medication, was found to induce apoptosis and exert a significantly inhibitory effect on the proliferation and colony-forming ability of ESCC cells in a dose-dependent manner without observed cytotoxicity on normal esophageal epithelial cells. Furthermore, propafenone markedly suppressed growth of tumor xenografts in nude mice by reducing the Ki-67 proliferation index and angiogenesis but did not damage the vital organs of the animals. Mechanistically, our data from the proteomics, Western blot and flow cytometry analyses demonstrated that propafenone caused mitochondrial dysfunction as indicated by a decreased mitochondrial membrane potential and reduced expression of Bcl-xL and Bcl-2. In summary, this study provides the first evidence that propafenone, an FDA-approved drug to treat arrhythmias, could be a novel therapeutic strategy for treating ESCC without obvious side effects.
Keywords: Drug repositioning, propafenone, apoptosis, mitochondria dysfunction, ESCC
Introduction
Esophageal squamous cell carcinoma (ESCC), a tumor of the digestive tract, has been ranked as the eighth most common cancer and the fifth leading cause of cancer-related deaths worldwide with poor overall survival [1]. Despite the great development of therapeutic regimens, the resistance of tumors to chemo- and radiotherapies leads to recurrence and poor prognosis of this lethal disease [2,3]. Many the chemotherapeutic agents were found to possess serious adverse effects in addition to limited efficacy [4]. Therefore, there is an urgent need to invent novel therapeutic strategy.
In the field of drug discovery, the concept of “old drugs for new applications” is gaining increasing traction [5]. When a new function of a clinical drug is found, the drug could have much more opportunities for clinical use. Propafenone HCL, a Vaughan-Williams class IC antiarrhythmia medication [6], has been extensively applied to treat paroxysmal supraventricular tachycardia, paroxysmal atrial fibrillation, and ventricular arrhythmia. The functions of propafenone in the treatment of arrhythmias could be explained by its activities in blocking beta-receptors and calcium channels [7]. However, the effects of propafenone in cancer were rarely reported, and the underlying mechanisms are largely unknown. In this study, in vitro and in vivo experiments were performed to provide evidence on the role of propafenone in cancer treatment. We found that propafenone elicited a strong ability to suppress the growth of ESCC cells in vitro and in vivo.
Quantitative proteomics is a systemic tool to uncover the mechanisms of anticancer compounds [8,9]. Data-independent acquisition (DIA) mass spectrometry (MS) is a label-free quantification method that offers more consistent peptide detection and accurate proteome quantification [10]. A recent study used DIA-MS to screen and verify protein biomarkers in ESCC [11]. In the present study, DIA-MS coupled with bioinformatics was performed to elucidate the molecular mechanisms involved in the anticancer effect of propafenone in ESCC cells. The proteomics data suggested that propafenone may induce apoptosis and inhibit cancer cell proliferation by causing mitochondrial dysfunction. Mitochondria, a dynamic cellular network of organelles, harbor the tricarboxylic acid (TCA) cycle and oxidative phosphorylation machinery to produce energy in the form of ATP [12]. Recent studies showed that mitochondria act as an important regulator of tumorigenesis, and its activity is strongly related to cancer development and progression [13]. Interestingly, mitochondria-mediated apoptosis, an intrinsic pathway of apoptosis, is activated when cancer cells are exposed to cell stress or damage, and several key molecules, including anti-apoptotic proteins Bcl-2 and Bcl-xL, are involved in this process [14,15]. Thus, we aim to investigate the anticancer property of propafenone and to determine the significance of the mitochondrial pathway in this effect. The outcome of this study could prove the potential of propafenone, an FDA-approved antiarrhythmic medication, as a therapeutic strategy in the treatment of cancer.
Materials and methods
Cell lines and culture
The human ESCC cell lines KYSE30, KYSE150 and KYSE270 (obtained from DSMZ, Braunschweig, Germany) were cultured in RPMI 1640 medium (Life Technologies, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS; Life Technologies) at 37°C in 5% CO2.
Cell viability assay
Cell viability was measured by the WST-1 assay (Beyotime, Jiangsu, China) as described previously [8]. ESCC cells were seeded into 96-well plates and treated with propafenone at various concentrations for different duration. WST-1 was added and incubated at 37°C for 2 h, and then the absorbance was measured on an automated microplate spectrophotometer (BioTek Instruments, Vermont, USA) at a wavelength of 450 nm.
Colony formation assay
A colony formation assay was performed as described previously [16]. In brief, ESCC cells were seeded into 6-well plates at a density of 3000 cells per well and treated with different concentrations of propafenone for 14 days. After washing twice with PBS, the plates were fixed with methanol for 15 min at room temperature and then stained with 1% crystal violet for 10 min. The number of colonies was counted, and all statistical measurements were obtained from three independent experiments.
Annexin V-FITC/PI staining assay
Cell apoptosis was determined by using an Annexin V-FITC/PI Apoptosis Detection kit (KeyGen, Jiangsu, China) [9]. Cells were suspended in binding buffer, stained with annexin V-FITC and propidium iodide (PI) for 15 min at room temperature in the dark. Apoptosis was assessed and analyzed by using a C6 flow cytometer (BD Biosciences, San Diego, CA, USA).
Measurement of the mitochondrial membrane potential
A JC-1 assay kit (Beyotime) was used to measure the mitochondrial membrane potential as described previously [17]. In brief, cells were collected after exposure to propafenone at various concentrations for 72 h, incubated with JC-1 (5 μg/ml) for 20 min at 37°C, and then rinsed twice with PBS. The cell resuspension was assayed by using a C6 flow cytometer to measure the JC-1 fluorescence intensities of red aggregates (hyperpolarization) and green fluorescence monomers (depolarization). The change in mitochondrial depolarization was calculated as the green/red fluorescence intensity ratio.
Western blot assay
Whole-cell lysates were prepared in lysis buffer (Cell Signaling Technology, Beverly, MA, USA) [18]. A BCA kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to determine the protein concentration. The samples were loaded onto a sodium dodecyl sulfate (SDS)-PAGE gel and subsequently electrotransferred to a PVDF membrane (Millipore, Bedford, MA, USA). After blocking with 5% nonfat milk for 1 h, the membrane was incubated with primary antibodies for 2 h at room temperature and washed three times for 10 min per wash with 1x Tris-buffered saline with Tween (TBST). Next, the membrane was incubated with corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. The reaction was visualized using Clarity Western ECL substrate (Bio-Rad, Hercules, CA, USA) and detected by exposure to autoradiographic film [19]. The antibodies used targeted caspase-3, cleaved caspase-3, Bcl-2, Bax, ERK, p-ERK, and survivin (Cell Signaling Technology); Bcl-xL (Abcam, Cambridge, UK), and actin (from Santa Cruz Biotechnology, Santa Cruz, CA, USA).
MS and bioinformatics analyses
KYSE270 cells were treated for 10 μM propafenone for 72 h, and whole-cell lysates were homogenized in RIPA lysis buffer. After further trypsin digestion through the method of filter-aided sample preparation (FASP), the peptide samples were vacuum-freeze-dried and resuspended in anhydrous acetonitrile solution for further desalination with a MonoTIPTM C18 Pipette Tip (GL Sciences, Tokyo, Japan). Next, the peptides were analyzed with an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). The raw data were searched using Proteome Discoverer (Thermo Fisher Scientific) and Spectronaut (Omicsolution Co., Ltd., Shanghai, China) software. An FDR of 1% was set to identify proteins. The differently expressed proteins were analyzed by Ingenuity Pathway Analysis (IPA, Ingenuity Systems, Redwood City, CA, USA) as described previously [8].
Tumor xenograft experiments
Female BALB/c nude mice aged 6-8 weeks were maintained under standard conditions and cared for according to the institutional guidelines for animal care. All the animal experiments were approved by the Ethics Committee for Animal Experiments of Jinan University. KYSE270 cells (5 × 105 cells in equal volumes of PBS and Matrigel) were subcutaneously implanted into the flanks of mice to establish tumor xenografts [20]. When the tumor xenografts reached 5 mm in diameter, the mice were randomly divided into treatment and control groups. The treatment groups received propafenone (10 mg/kg or 20 mg/kg) through intraperitoneal injection every other day, whereas the control group received vehicle only. The body weight of the mice was monitored weekly during the experiments to evaluate overall health. Tumor size was measured every three days, and the tumor volume was calculated using the following equation: V = (length × width2)/2. All animals were killed at the end of the study, and their tumors, lungs, liver, and kidneys were collected for further analysis. Immunohistochemical analyses of the proliferative index and microvessel density (MVD) were performed using antibodies against Ki-67 (Dako, Mississauga, ON, Canada) and CD31 (Santa Cruz Biotechnology), respectively [21].
Statistical analysis
All in vitro experiments were performed in triplicate. GraphPad Prism software (San Diego, CA, USA) was used to calculate statistically significant differences with a Student’s t-test method. All values were expressed as the means ± SD from three independent experiments. All statistical tests were two-sided, and P < 0.05 was considered statistically significant.
Results
Propafenone inhibits ESCC cell proliferation
To evaluate the inhibitory effect of propafenone on cancer cell proliferation, ESCC cells were exposed to various concentrations of propafenone for 24 h, 48 h and 72 h, and the results showed that propafenone gradually decreased cell proliferation over time and potently inhibited cell proliferation with increasing concentrations in KYSE30, KYSE150 and KYSE270 cells (Figure 1A). The cell viability decreased by half when ESCC cells were treated with 10 μM propafenone for 72 h. Of note, treatment with propafenone did not exert any toxic effects in normal esophageal epithelial cells (Figure 1B). In addition, we determined the effect of propafenone on the colony-forming ability of ESCC cells. As shown in Figure 1C, the number of colonies formed by ESCC cells was significantly reduced by propafenone in a dose-dependent manner. Taken together, these results suggest that propafenone has a tumor suppressive effect in ESCC.
Figure 1.
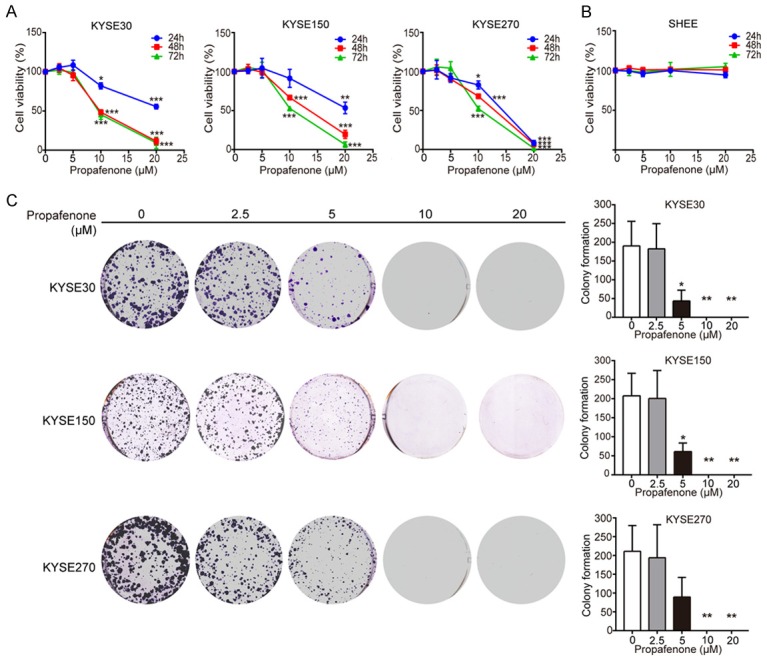
Propafenone inhibits ESCC cell proliferation. A. A WST-1 assay was performed to determine the cell viability of KYSE30, KYSE150, and KYSE270 cells treated with the indicated concentrations of propafenone for 24, 48 and 72 h. B. Propafenone did not affect the proliferation of the normal esophageal epithelial cell line SHEE. C. Propafenone inhibited the colony-forming ability of ESCC cells. Bars, SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with control cells.
Propafenone induces apoptosis in ESCC cells
Next, we studied the effect of propafenone on apoptosis in ESCC cells. ESCC cells were treated with different concentrations of propafenone (up to 20 μM), and an annexin V-FITC/PI double staining assay was performed using flow cytometric analysis. As shown in Figure 2A, a dose-dependent induction of apoptosis was observed in KYSE30, KYSE150 and KYSE270 cells treated with propafenone. The effect of propafenone on apoptosis was also evidenced by the increased expression levels of cleaved caspase-3 in propafenone-treated ESCC cells (Figure 2B). Collectively, these results indicated that propafenone can trigger apoptosis to inhibit cell proliferation in ESCC cells.
Figure 2.
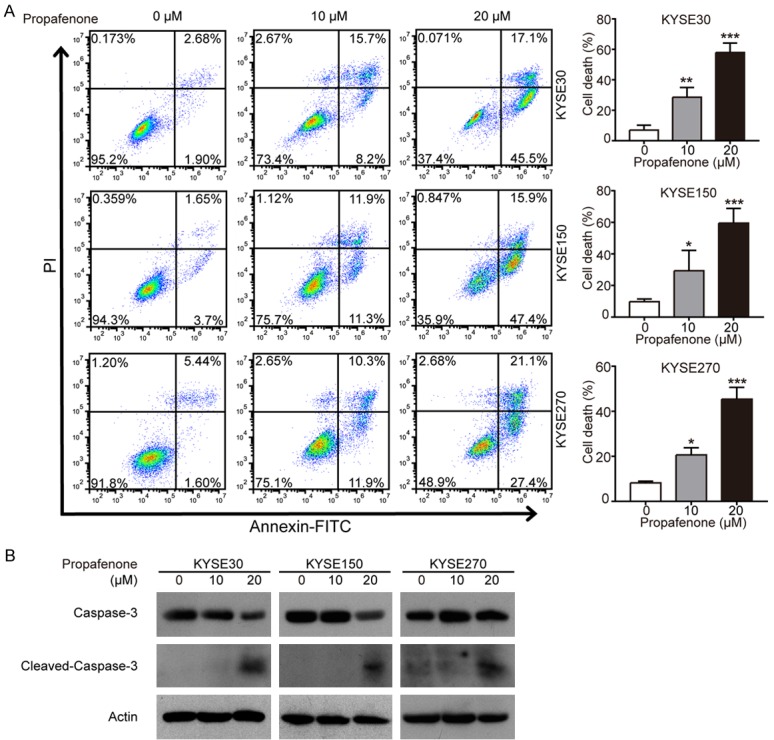
Propafenone induces apoptosis in ESCC cells. A, B. KYSE30, KYSE150, and KYSE270 cells were exposed to the indicated concentrations of propafenone (up to 20 μM) for 72 h, and apoptotic cells were determined with annexin V-FITC/PI double staining by flow cytometry (A), and Western blotting was performed to compare the expression levels of caspase-3 and cleaved caspase-3. Bars, SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with control cells.
Proteomics analysis suggests the involvement of mitochondrial signaling in antitumor activity of propafenone in cancer cells
To quantify the proteomic alterations and elucidate the potential molecular mechanism of propafenone treatment on ESCC cells, we used DIA-MS, which has been justified as a good tool for protein quantitation, especially for low abundance proteins. Here, a total of 2799 proteins were identified and quantified from the triplicate samples after propafenone treatment (10 μM for 72 h) against samples with DMSO treatment. Meanwhile, a total of 428 differentially expressed proteins (Supplementary Table 1) was identified through the power law global error model (PLGEM) algorithm [22], a statistical test for analyzing protein abundance, with a slope of 0.777 and an adjusted r2 of 0.986 (Pearson r = 0.748) (Figure 3A). The quantile-quantile (Q-Q) plot presented that the data had a fitted normal distribution of the residual standard deviations between the modeled and the actual values (Figure 3B). Next, to explore the molecular mechanism involved in the growth inhibition of propafenone in ESCC cells, we used IPA software to characterize the canonical pathways in which the differently expressed proteins participated. As shown in Figure 3C, we noticed that the signaling associated with mitochondrial dysfunction, one of the top five canonical pathways with P value of 5.92E-7, was significantly affected by propafenone treatment.
Figure 3.
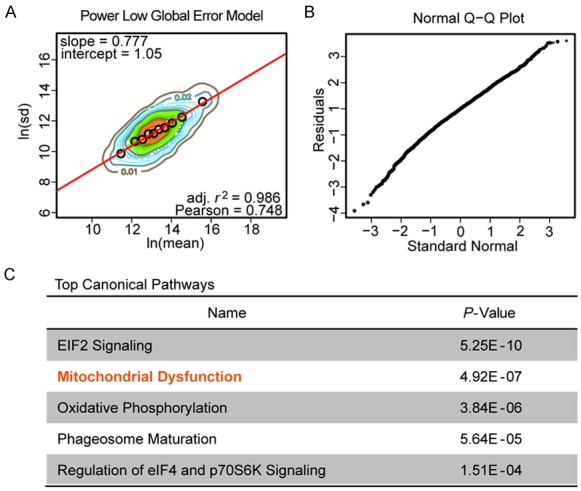
Proteomics analysis for the identification of the potential mechanism(s) of action of propafenone. A. PLGEM model using a regression fitting analysis with a contour plot; black circles showed a good fitting of the DIA-MS data by the PLGEM model. B. A Q-Q plot was used to detect a normal distribution of the residual standard deviations between the modeled and the determined data. C. The top five canonical pathways from the IPA analysis of differently expressed proteins in propafenone-treated cells.
Propafenone causes dysregulation of mitochondria function
Based on the proteomics data above, we investigated effect of propafenone on mitochondria function. The integrity of mitochondrial membrane permeability was determined with the fluorescent dye JC-1, which fluoresces a red color in healthy mitochondria but a green color when mitochondria are damaged. KYSE30, KYSE150 and KYSE270 cells were treated with increasing concentrations of propafenone for 72 h and stained with JC-1. A dose-dependent decrease of the red/green fluorescence ratio was observed (Figure 4A), suggesting that propafenone may induce apoptosis through decreasing the mitochondrial membrane potential. Simultaneously, we detected the expression levels of proteins involved in the mitochondrial pathway. Western blot data showed that propafenone treatment significantly downregulated the expression levels of the anti-apoptotic proteins Bcl-xL and Bcl-2 in ESCC cells (Figure 4B). In addition, reduced expression of p-ERK was observed in propafenone-treated cells (Figure 4B), indicating the inactivation of the ERK signaling pathway, which has been reported to be an upstream regulator of Bcl-2 and Bcl-xL [23,24]. Taken together, these results demonstrated that propafenone may induce apoptosis through the intrinsic (i.e., mitochondrial) pathway.
Figure 4.
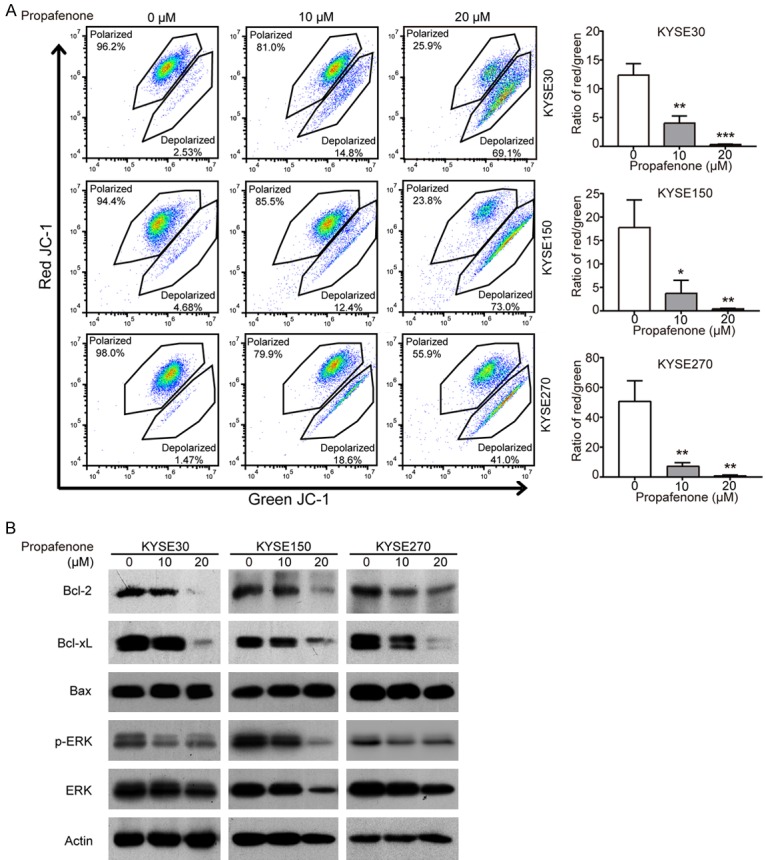
Propafenone causes dysregulation of mitochondria function. A. A JC-1 assay was performed to measure the mitochondrial membrane potential in ESCC cells treated with propafenone for 72 h. B. Western blot comparison of the protein levels of ERK, p-ERK, Bax, Bcl-xL and Bcl-2 in ESCC cells treated with various concentrations of propafenone. Bars, SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with control cells.
Propafenone suppresses tumor growth in vivo
Given that propafenone exerted a significant inhibitory effect on the proliferation of cancer cells, we next evaluated the therapeutic potential of propafenone using a mouse model. Propafenone was intraperitoneally injected into nude mice bearing established subcutaneous tumor xenografts, and its effect on tumor growth was monitored. The results showed that the tumor burden was markedly suppressed by propafenone treatment with a decrease of 69.2% in the group receiving 20 mg/kg propafenone (Figure 5A). As shown in Figure 5B, the immunohistochemical analysis of the Ki67 proliferation index also provides evidence that tumor cell proliferation was significantly inhibited by propafenone (mean index decreased from 56.3 ± 6.7% in the DMSO-treated group to 20.7 ± 5.1% in the 10 mg/kg propafenone-treated group and 11.3 ± 4.0% in the 20 mg/kg propafenone-treated group). Furthermore, we investigated the effect of propafenone on tumor angiogenesis, and an analysis of microvessel density (indicated by CD31 staining) showed that treatment of propafenone resulted in a decrease in tumor angiogenesis (Figure 5B). Mechanistically, same with the in vitro data (Figure 4B), significant downregulation of Bcl-xL and Bcl-2 expression levels was observed in propafenone-treated tumors compared with those in the control group by Western blot (Figure 5C). No significant difference between the treated and control groups in terms of body weight was observed (Figure 5D). Histological examination of vital organs, including the lung, liver and kidneys, did not reveal any overt changes in morphology (Figure 5E), suggesting that propafenone treatment had no toxic effects on animals.
Figure 5.
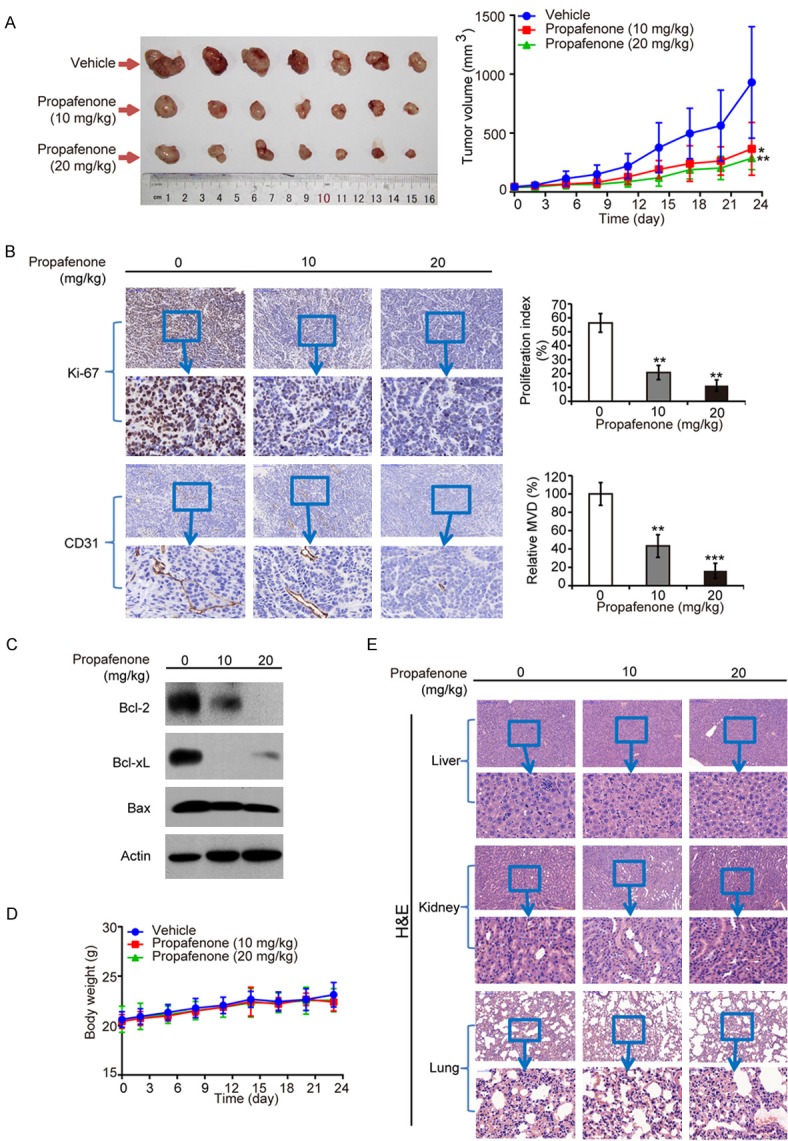
Propafenone suppresses tumor growth in vivo. Nude mice bearing KYSE270-derived xenografts were intraperitoneally injected with either propafenone (10 mg/kg or 20 mg/kg) or vehicle every other day (n = 7 per group). A. Left, images of tumors; Right, tumor curves showed that propafenone exerted a significantly inhibitory effect on the growth of tumor xenografts. B. Immunohistochemical analysis of the Ki-67 proliferative index and CD31-based MVD. C. Expression levels of Bcl-xL, Bcl-2 and Bax in tumors from mice treated with propafenone or vehicle were detected by Western blot. D. Comparison of the body weight of nude mice during the experimental period. E. Representative images of liver, kidney and lungs stained with hematoxylin and eosin (H&E). Bars, SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with the control group.
Discussion
Finding new functions for existing drugs, also known as “drug repositioning”, is a promising approach for identifying more therapeutic strategies in translational and clinical studies [5]. Since FDA-approved drugs have clear descriptions of their physicochemical properties and pharmacokinetics as well as stronger evidence of drug safety, it may be a faster and more effective strategy to repurpose existing drugs for cancer treatment. Propafenone, a Vaughan-Williams class IC antiarrhythmic medication, is often used to manage atrial fibrillation and other supraventricular arrhythmias and has no severe side effects and a favorable drug tolerance [25,26]. Several studies revealed that propafenone can block the activities of potent sodium channel and calcium channel signaling [27-30]. However, there are few reports on the implication of propafenone in the treatment of cancer. In our study, we found that propafenone triggered apoptosis and displayed a significantly inhibitory effect on the proliferation and colony-forming ability of ESCC cells in a dose-dependent manner (Figure 1). More importantly, the animal experiment demonstrated that propafenone markedly suppressed the growth of tumor xenografts in nude mice (Figure 5). It is worth noting that the dose used in our in vivo study, 120 mg every other day, is much lower than the recommended dose of propafenone (from 450 mg to 900 mg each day) for the clinical treatment of arrhythmias according to current guidelines [31] and clinical studies [32,33]. In addition to the data showing that propafenone did not exert any obvious toxic effects on normal esophageal epithelial cells or the vital organs of animals, we believe that propafenone could be a potential therapeutic drug for the management of ESCC.
With the rapid development of high-throughput technologies and computational frameworks, genetic alterations in human cancers and the mechanisms of action of anticancer drugs can be discovered more accurately and systematically. Mass spectrometry-based shotgun proteomics has been used to perform the data-dependent acquisition (DDA) mode of analysis, in which there is a priority of isolating and fragmenting more abundant precursor peptides to generate tandem mass (MS/MS) spectra [34]. However, the DIA mode has an advantage in the detection and quantification of both high and low abundance peptides, which leads to more complete quantitative coverage [10,35]. In the DIA analysis mode, a set of wide windows is used to cover the entire mass range relevant to the researcher and to trigger the acquisition of fragment ion spectra for an unbiased set of precursors [36]. Recently, DIA-MS has been used to determine the biological mechanisms of human trabecular meshwork cells in response to dexamethasone and prednisolone, and the results showed that integrin cell surface interactions and other additional pathways were involved in corticosteroid-induced ocular hypertension [37]. In our study, we carried out DIA-MS to determine the potential mechanisms of how propafenone exerts an inhibitory effect on the proliferation of ESCC cells. As indicated by the proteomics data and IPA pathway analysis, a cluster of differentially expressed proteins was enriched in pathways related to mitochondrial dysfunction (Figure 3).
Mitochondria not only contain the components for energy production, including the TCA cycle and the core of the cellular respiratory chain and oxidative phosphorylation, but also play an important role in regulating cell apoptosis. In response to cellular and environmental alterations, the mitochondrial membrane equilibrium will be disturbed, and the permeabilization of outer membrane will increase, which leads to pro-apoptotic factors infiltrating the cytoplasm and inducing apoptosis. The anti-apoptotic proteins Bcl-2 and Bcl-xL engage the outer membrane to control mitochondrial permeability and suppress the release of apoptotic factors such as cytochrome C [38,39]. One study showed that MT3-037, a novel microtubule inhibitor, could inactivate the pro-survival proteins Bcl-2 and Bcl-xL and activate the cleavage of caspases, resulting in the apoptosis of cancer cells [40]. It has reported that ABT-199 (navitoclax), a Bcl-2/Bcl-xL inhibitor, can cooperate with doxorubicin or dinaciclib to reduce Bcl-2 and Bcl-xL expression levels and activate mitochondria-mediated cell death in small cell lung cancer cells [41]. In the present study, our results indicated that propafenone downregulated the expression levels of Bcl-2 and Bcl-xL as well as disordered the equilibrium and damaged the polarity of mitochondria (Figure 5). Taken together, our data is the first evidence that propafenone may cause mitochondrial dysfunction by regulating Bcl-2 and Bcl-xL to therefore induce apoptosis and inhibit the proliferation of ESCC cells.
More than 50% of ESCC cases worldwide occur in China [42]. The incidence is still on the rise, and its overall survival rate remains low. Traditional chemotherapeutic drugs have many toxic side effects, such as toxic disturbance of the immune system, toxicity against human organs and myelosuppression [43]. Unfortunately, to date, there is no ideal targeted therapeutic drug for adjuvant treatment. All these obstacles compel us to develop new drugs or find new therapeutic regimens for cancer treatment. Nevertheless, the development of a completely new anticancer drug not only needs extensive time and cost but also elicits a higher treatment failure rate. Here, we demonstrated for the first time that propafenone, an FDA-approved antiarrhythmic medication, not only triggered the intrinsic pathway of apoptosis mediated by mitochondria but also suppressed the growth of ESCC cells in vitro and in vivo accompanied by an inhibited Ki-67 proliferation index and reduced angiogenesis. Our data suggest that propafenone could be a novel treatment option for ESCC.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2017YFA0505100), the National Natural Science Foundation of China (31570828, 31770888, 81773085), the Guangzhou Science and Technology Project (201707010260), and a Guangdong Natural Science Research Grant (S2013030013315, 2016A030313838). We thank Mr. Xing-Feng Yin and Mr. Zheng-Hua Sun for their technical assistance on mass spectrometry.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Wheeler JB, Reed CE. Epidemiology of esophageal cancer. Surg Clin North Am. 2012;92:1077–1087. doi: 10.1016/j.suc.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Khaider NG, Lane D, Matte I, Rancourt C, Piche A. Targeted ovarian cancer treatment: the TRAILs of resistance. Am J Cancer Res. 2012;2:75–92. [PMC free article] [PubMed] [Google Scholar]
- 4.Liu H, Lv L, Yang K. Chemotherapy targeting cancer stem cells. Am J Cancer Res. 2015;5:880–893. [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Q. Editorial: old drugs learn new tricks: Aadvances and applications for drug repurposing. Curr Top Med Chem. 2016;16:3627–3628. doi: 10.2174/1568026616999160812144924. [DOI] [PubMed] [Google Scholar]
- 6.Stoschitzky K, Klein W, Stark G, Stark U, Zernig G, Graziadei I, Lindner W. Different stereoselective effects of (R)- and (S)-propafenone: clinical pharmacologic, electrophysiologic, and radioligand binding studies. Clin Pharmacol Ther. 1990;47:740–746. doi: 10.1038/clpt.1990.102. [DOI] [PubMed] [Google Scholar]
- 7.Alsaad AA, Ortiz Gonzalez Y, Austin CO, Kusumoto F. Revisiting propafenone toxicity. BMJ Case Rep. 2017:2017. doi: 10.1136/bcr-2017-219270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Yu RY, Zhang J, Zhang WX, Huang ZH, Hu HF, Li YL, Li B, He QY. Inhibition of Nrf2 enhances the anticancer effect of 6-O-angeloylenolin in lung adenocarcinoma. Biochem Pharmacol. 2017;129:43–53. doi: 10.1016/j.bcp.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Zhang J, Huang ZH, Huang XH, Zheng WB, Yin XF, Li YL, Li B, He QY. Isodeoxyelephantopin induces protective autophagy in lung cancer cells via Nrf2-p62-keap1 feedback loop. Cell Death Dis. 2017;8:e2876. doi: 10.1038/cddis.2017.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11:O111.016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou G, Lou X, Sun Y, Xu S, Zi J, Wang Q, Zhou B, Han B, Wu L, Zhao X, Lin L, Liu S. Biomarker discovery and verification of esophageal squamous cell carcinoma using integration of SWATH/MRM. J Proteome Res. 2015;14:3793–3803. doi: 10.1021/acs.jproteome.5b00438. [DOI] [PubMed] [Google Scholar]
- 12.Ernster L, Schatz G. Mitochondria: a historical review. J Cell Biol. 1981;91:227s–255s. doi: 10.1083/jcb.91.3.227s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trotta AP, Chipuk JE. Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci. 2017;74:1999–2017. doi: 10.1007/s00018-016-2451-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011;18:1414–1424. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem. 2010;285:6053–6062. doi: 10.1074/jbc.M109.082990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong Y, Yang J, Xu WW, Wang Y, Zheng CC, Li B, He QY. KCTD12 promotes tumorigenesis by facilitating CDC25B/CDK1/Aurora A-dependent G2/M transition. Oncogene. 2017;36:6177–6189. doi: 10.1038/onc.2017.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Huang ZH, Li YJ, He GW, Yu RY, Yang J, Liu WT, Li B, He QY. Dynamic quantitative proteomics characterization of TNF-alpha-induced necroptosis. Apoptosis. 2016;21:1438–1446. doi: 10.1007/s10495-016-1300-z. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Tsao SW, Chan KW, Ludwig DL, Novosyadlyy R, Li YY, He QY, Cheung AL. Id1-induced IGF-II and its autocrine/endocrine promotion of esophageal cancer progression and chemoresistance--implications for IGF-II and IGF-IRtargeted therapy. Clin Cancer Res. 2014;20:2651–2662. doi: 10.1158/1078-0432.CCR-13-2735. [DOI] [PubMed] [Google Scholar]
- 19.Li B, Xu WW, Guan XY, Qin YR, Law S, Lee NP, Chan KT, Tam PY, Li YY, Chan KW, Yuen HF, Tsao SW, He QY, Cheung AL. Competitive binding between Id1 and E2F1 to Cdc20 regulates E2F1 degradation and thymidylate synthase expression to promote esophageal cancer chemoresistance. Clin Cancer Res. 2016;22:1243–1255. doi: 10.1158/1078-0432.CCR-15-1196. [DOI] [PubMed] [Google Scholar]
- 20.Li B, Xu WW, Han L, Chan KT, Tsao SW, Lee NPY, Law S, Xu LY, Li EM, Chan KW, Qin YR, Guan XY, He QY, Cheung ALM. MicroRNA-377 suppresses initiation and progression of esophageal cancer by inhibiting CD133 and VEGF. Oncogene. 2017;36:3986–4000. doi: 10.1038/onc.2017.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu WW, Li B, Guan XY, Chung SK, Wang Y, Yip YL, Law SY, Chan KT, Lee NP, Chan KW, Xu LY, Li EM, Tsao SW, He QY, Cheung AL. Cancer cell-secreted IGF2 instigates fibroblasts and bone marrow-derived vascular progenitor cells to promote cancer progression. Nat Commun. 2017;8:14399. doi: 10.1038/ncomms14399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavelka N, Pelizzola M, Vizzardelli C, Capozzoli M, Splendiani A, Granucci F, Ricciardi-Castagnoli P. A power law global error model for the identification of differentially expressed genes in microarray data. BMC Bioinformatics. 2004;5:203. doi: 10.1186/1471-2105-5-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Zheng Y, Zhu R, Zhu Y, Yao W, Liu W, Gao X. The ERK/eIF4F/Bcl-XL pathway mediates SGP-2 induced osteosarcoma cells apoptosis in vitro and in vivo. Cancer Lett. 2014;352:203–213. doi: 10.1016/j.canlet.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 24.Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–369. [PubMed] [Google Scholar]
- 25.Valderrabano M, Singh BN. Electrophysiologic and antiarrhythmic effects of propafenone: focus on atrial fibrillation. J Cardiovasc Pharmacol Ther. 1999;4:183–198. doi: 10.1177/107424849900400308. [DOI] [PubMed] [Google Scholar]
- 26.Capucci A, Villani GQ, Aschieri D, Piepoli M. Safety of oral propafenone in the conversion of recent onset atrial fibrillation to sinus rhythm: a prospective parallel placebo-controlled multicentre study. Int J Cardiol. 1999;68:187–196. doi: 10.1016/s0167-5273(98)00363-5. [DOI] [PubMed] [Google Scholar]
- 27.Hancox JC, Mitcheson JS. Inhibition of Ltype calcium current by propafenone in single myocytes isolated from the rabbit atrioventricular node. Br J Pharmacol. 1997;121:7–14. doi: 10.1038/sj.bjp.0701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JT, Kroemer HK, Silberstein DJ, Funck-Brentano C, Lineberry MD, Wood AJ, Roden DM, Woosley RL. The role of genetically determined polymorphic drug metabolism in the beta-blockade produced by propafenone. N Engl J Med. 1990;322:1764–1768. doi: 10.1056/NEJM199006213222502. [DOI] [PubMed] [Google Scholar]
- 29.McLeod AA, Stiles GL, Shand DG. Demonstration of beta adrenoceptor blockade by propafenone hydrochloride: clinical pharmacologic, radioligand binding and adenylate cyclase activation studies. J Pharmacol Exp Ther. 1984;228:461–466. [PubMed] [Google Scholar]
- 30.Stoschitzky K, Stoschitzky G, Lercher P, Brussee H, Lamprecht G, Lindner W. Propafenone shows class Ic and class II antiarrhythmic effects. Europace. 2016;18:568–571. doi: 10.1093/europace/euv195. [DOI] [PubMed] [Google Scholar]
- 31.Camm AJ, Lip GY, De Caterina R, Savelieva I, Atar D, Hohnloser SH, Hindricks G, Kirchhof P ESC Committee for Practice Guidelines-CPG; Document Reviewers. 2012 focused update of the ESC guidelines for the management of atrial fibrillation: an update of the 2010 ESC guidelines for the management of atrial fibrillation--developed with the special contribution of the European heart rhythm association. Europace. 2012;14:1385–1413. doi: 10.1093/europace/eus305. [DOI] [PubMed] [Google Scholar]
- 32.Bellandi F, Simonetti I, Leoncini M, Frascarelli F, Giovannini T, Maioli M, Dabizzi RP. Longterm efficacy and safety of propafenone and sotalol for the maintenance of sinus rhythm after conversion of recurrent symptomatic atrial fibrillation. Am J Cardiol. 2001;88:640–645. doi: 10.1016/s0002-9149(01)01806-9. [DOI] [PubMed] [Google Scholar]
- 33.Freemantle N, Lafuente-Lafuente C, Mitchell S, Eckert L, Reynolds M. Mixed treatment comparison of dronedarone, amiodarone, sotalol, flecainide, and propafenone, for the management of atrial fibrillation. Europace. 2011;13:329–345. doi: 10.1093/europace/euq450. [DOI] [PubMed] [Google Scholar]
- 34.Teo G, Kim S, Tsou CC, Collins B, Gingras AC, Nesvizhskii AI, Choi H. mapDIA: preprocessing and statistical analysis of quantitative proteomics data from data independent acquisition mass spectrometry. J Proteomics. 2015;129:108–120. doi: 10.1016/j.jprot.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canterbury JD, Merrihew GE, MacCoss MJ, Goodlett DR, Shaffer SA. Comparison of data acquisition strategies on quadrupole ion trap instrumentation for shotgun proteomics. J Am Soc Mass Spectrom. 2014;25:2048–2059. doi: 10.1007/s13361-014-0981-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 37.Shan SW, Do CW, Lam TC, Kong RPW, Li KK, Chun KM, Stamer WD, To CH. New insight of common regulatory pathways in human trabecular meshwork cells in response to dexamethasone and prednisolone using an integrated quantitative proteomics: SWATH and MRM-HR mass spectrometry. J Proteome Res. 2017;16:3753–3765. doi: 10.1021/acs.jproteome.7b00449. [DOI] [PubMed] [Google Scholar]
- 38.Lopez J, Tait SW. Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer. 2015;112:957–962. doi: 10.1038/bjc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166:555–566. doi: 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang LC, Yu YL, Hsieh MT, Wang SH, Chou RH, Huang WC, Lin HY, Hung HY, Huang LJ, Kuo SC. A novel microtubule inhibitor, MT3-037, causes cancer cell apoptosis by inducing mitotic arrest and interfering with microtubule dynamics. Am J Cancer Res. 2016;6:747–763. [PMC free article] [PubMed] [Google Scholar]
- 41.Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC, Han S, Ganesan YT, Ishizawa K, Jebiwott S, Dong Y, Pietanza MC, Hellmann MD, Kris MG, Hsieh JJ, Cheng EH. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat Commun. 2017;8:16078. doi: 10.1038/ncomms16078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng H, Zheng R, Zhang S, Zuo T, Xia C, Zou X, Chen W. Esophageal cancer statistics in China, 2011: estimates based on 177 cancer registries. Thorac Cancer. 2016;7:232–237. doi: 10.1111/1759-7714.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157–170. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.