

Abstract
We provide evidence of a pericellular network of proteases that are elevated and co-expressed in prostate cancer. The network involves the membrane bound serine proteases hepsin and TMPRSS2, the secreted kallikrein-related peptidases KLK4 and KLK14, and the secreted matrix metalloproteinases MMP-3 and MMP-9. Western blot analysis of cell lysates, conditioned cell culture media, immunoprecipitates and cell surface proteins, demonstrates a network of interactions centred largely at the plasma membrane, with the Arg/Lys specific proteases hepsin and TMPRSS2 key regulators of the network. Our data demonstrate that like TMPRSS2, hepsin is able to autoactivate. Active hepsin degrades KLK4, generating a cell associated degradation product with corresponding reduction in levels of cell-free KLK4. In contrast hepsin activates KLK14. TMPRSS2 appears to cleave amino terminal to the KLK4 activation site such that it is available for further processing to generate the active KLK4 protease. In contrast with hepsin, TMPRSS2 degrades KLK14. In addition to these direct mechanisms of regulation, hepsin and TMPRSS2 indirectly modulate KLK4 activity by cleaving the KLK4-activating protease MMP-3. Hepsin and TMPRSS2 also activate MMP-9, which similar to MMP-3, associates with the cell surface. Interestingly our data also show that proteolysis occurs between the membrane spanning and catalytic domains of hepsin and TMPRSS2. Hepsin cleavage occurs via an autoproteolytic mechanism, whereas TMPRSS2 cleavage is mediated by KLK14. Hepsin and TMPRSS2 are not shed from the cell surface but proteolysis likely disrupts domains that regulate the proteolytic activity of these proteases. Immunocytochemical analyses demonstrate that hepsin and TMPRSS2 colocalize on the cell surface with the secreted serine proteases KLK4 and KLK14, only in membrane protrusions, suggesting that reciprocal proteolytic interactions occur in defined cellular structures that are important during cancer dissemination for cell migration, invasion and survival. Also of note, immunohistochemical analysis of serial sections of prostate tumor demonstrated significant overlapping expression of the six proteases in vivo. Collectively these data suggest the possibility that the novel proteolytic network identified by us, will be most important during active dissemination of prostate cancers, and that its disruption could inhibit metastasis.
Keywords: Prostate cancer, serine protease, kallikrein, matrix metalloproteinase, pericellular
Introduction
Aberrant proteolysis contributes to progression of prostate and other cancers by deregulating normal physiological processes, such as angiogenesis, matrix remodelling, and cell invasion and migration [1,2]. Various members of large protease families, including the type II transmembrane serine protease (TTSP), kallikrein-related peptidase (KLK), and matrix metalloproteinase (MMP) families, have demonstrated roles in these processes and have been proposed, and in some cases tested, as therapeutic targets for treatment of cancer [3-7]. Currently there is limited evidence that serine proteases and MMPs are involved in reciprocal interactions. Examples include the ability of the autoactivating TTSP matriptase [8] to activate MMP-3 [9], and the fibrinolytic serine protease plasmin to activate MMP-3 which in turn activates MMP-9 [10].
The TTSPs and KLKs are trypsin-fold serine proteases that comprise 17 and 15 members in humans, respectively. Members of both families are synthesized as single chain zymogens and activated by cleavage at highly conserved activation motifs [3,11]. TTSPs are anchored to the cell surface via an amino terminal membrane-spanning domain. On activation these proteins form two-chain disulfide bond-linked heterodimers with proteolytic activity either remaining tethered to, or actively shed from the cell surface [3]. In contrast, the KLKs are secreted proteins and require removal of a short pro-domain which releases the activated catalytic domain [3,11].
MMPs also comprise a large family of proteases, including 24 mammalian members, that are characterized by a multi-domain structure including a conserved pro-peptide that must be removed for proteolytic activity [12]. These enzymes are metal ion dependent with the catalytic domain incorporating a Zn2+ ion-containing active site [12]. The vast majority of the MMPs are secreted enzymes and activation requires disruption of the interaction between the thiol of a cysteine present within the conserved pro-domain and the zinc ion of the catalytic site [12]. Interestingly, MMP-3 is the only prostate cancer expressed activator of KLK4 [13], a protease that, unique amongst the KLKs, requires activation by cleavage after Gln [14,15].
A limited number of members of these protease families have individually been linked with prostate cancer. This includes the TTSP hepsin which is highly upregulated [16] and also, based on mouse model data, potentially functionally important in progression [17] of this malignancy. Another prostate cancer associated TTSP, TMPRSS2, is differentially expressed and mislocalized in the malignant component of these tumors [18]. This disrupted expression is thought to promote prostate cancer via the ability of TMPRSS2 to initiate tumorigenic signalling through activation of the protease activated receptor PAR-2 [19], and via the pro-HGF/c-Met receptor system [20]. Similarly, KLK4 is significantly elevated in prostate tumors [21], regulates proliferation of prostate cancer cells [22,23], and is a potential mediator of interactions between cancer cells and osteoblasts in prostate cancer bone lesions [24]. Likewise, elevated KLK14 in prostate cancer is associated with elevated risk of prostate-specific antigen relapse [25], and this protease is also an effective regulator of known prostate cancer promoting signalling proteins including components of the HGF/Met axis [26]. Like these TTSPS and KLKs, MMP-3 and MMP-9 are dysregulated in prostate tumors. MMP-3 expression is significantly higher in prostate cancer compared with prostatic intraepithelial neoplasia and normal tissue [27], while MMP-9 is elevated in malignant and stromal cells in prostate tumors and this correlates with disease recurrence [28]. A functional link with advanced prostate cancer is suggested by the observations that MMP-9 expression is increased by the CXCL12/CXCR4 axis, which is known to drive dissemination to bone marrow [29], and this protease is activated by interactions between prostate cancer and bone cells [30].
Focusing on the cell surface as a key site for transduction of cancer promoting signals, here we have used cell based systems to study reciprocal pericelluar interactions that occur between key prostate cancer associated proteases. These include key initiators of proteolysis at the plasma membrane of prostate cancer cells, hepsin and TPRSS2, and secreted proteases known to be important in cell signalling and tissue remodelling in prostate cancer, KLK4, KLK14, MMP-3 and MMP-9. We also performed immunoprecipitation, cell fractionation, immunocytochemical and immunohistochemical analyses to assess the extent to which these various proteases are co-located and interact in cellular contexts. In addition, we examined the co-expression of these proteases in a tumor containing regions broadly representative of prostate pathology. Based on our findings we propose the potential for novel regulatory pericellular networks of prostate cancer expressed proteases that could potentially be selectively targeted to disrupt progression of this malignancy.
Materials and methods
Antibodies and reagents
Antibodies: mouse anti-V5 (GKPIPNPLLGLDST) monoclonal antibody (R96025) was from Invitrogen (Life Technologies, Mulgrave, Vic, Australia); rabbit anti-HA (YPYDVPDYA; H6908), mouse anti-Flag (DYKDDDDK; F1804) monoclonal and rabbit anti-Flag (F7425), and rabbit anti-GAPDH (G9545) antibodies were from Sigma-Aldrich (Castle Hill, NSW, Australia); mouse anti-GAPDH monoclonal antibody was from Millipore (Kilsyth, Vic, Australia); mouse anti-HA (12CA5) monoclonal, mouse anti-Myc (EQKLISEEDL; 9B11) monoclonal and rabbit anti-Myc (71D10) monoclonal antibodies were from Cell Signaling Technology (Danvers, MA, USA); rabbit anti-hepsin antibody from Cayman Chemicals (Ann Arbor, MI, USA); mouse anti-matriptase M69 monoclonal antibody was from Dr C-Y Lin (Georgetown University, Washington, DC) [31]; rabbit anti-TMPRSS2 (NBP1-87210) antibody was from Novus Biologicals (Littleton, CO, USA); and rabbit anti-KLK4 (ab40950), rabbit anti-KLK14 (ab40960), goat anti-MMP-3 (ab18898) and rabbit anti-MMP-9 (ab38898) antibodies were from Abcam (Cambridge, MA, USA). Isotype immunoglobulin (IgG) controls were mouse IgG2a (Sigma-Aldrich) and rabbit IgG (Invitrogen). Secondary antibodies were Alexa Fluor 488 goat anti-mouse, Alexa Fluor 488 goat anti-rabbit, Alexa Fluor 647 goat anti-mouse, Alexa Fluor 647 goat anti-rabbit and Alexa Fluor 680 goat anti-rabbit, antibodies (Invitrogen); IRDye700 donkey anti-goat, IRDye800 donkey anti-mouse, IRDye800CW donkey anti-mouse and IRDye800CW donkey anti-rabbit antibodies (Rockland Immunochemicals, Gilbertsville, PA, USA); DyLight 680 goat anti-rabbit (#5366) and DyLight 800 anti-mouse (#5257) antibodies (Cell Signaling Technology); and biotin conjugated antibodies donkey anti-goat (sc-2042), goat anti-mouse (sc-2039) and goat anti-rabbit (sc-2040) from Santa Cruz Biotechnology (Dallas, TX, USA). Protein A- and protein G-agarose was from Roche Diagnostics (Castle Hill, NSW, Australia); streptavidin agarose resin and cell impermeant EZ-link NHS-SS-biotin were from Thermo Fisher Scientific (Scorsby, Vic); and 4,6-diamidino-2-phenylindole (DAPI) and Alexa Fluor 568 phalloidin were from Invitrogen. Bovine trypsin was from Worthington Biochemical (Lakewood, NJ, USA) and recombinant hepsin and matriptase were from Dr Daniel Kirchhofer (Genentech, South San Franscisco, CA, USA).
Cell culture
Cell culture reagents were from Invitrogen. Monkey kidney COS-7 cells were from ATCC (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, and incubated at 37°C in 5% CO2. At passage cells were detached using EDTA (0.53 mM) in phosphate-buffered saline (PBS).
Expression constructs
A mammalian expression vector encoding KLK4 tagged at the carboxyl terminal with a V5 epitope was previously described [24]. The KLK14-HA expression construct was generated by amplifying the coding sequence from a previously described construct [32], using the proof-reading polymerase Platinum Pfx DNA polymerase (Invitrogen), so as to engineer BamHI and XhoI restriction sites at either end of the coding sequence of KLK14 and also incorporate sequence encoding a carboxyl terminal HA epitope. The final construct was then ligated into a pcDNA3.1 vector (Invitrogen). Hepsin-Flag and -Myc expression constructs were generated by amplifying the coding sequence from a commercially available construct so as to incorporate sequence encoding a carboxyl terminal Flag or Myc epitope then cloning this into a pcDNA3.1 vector. The TMPRSS2-Myc expression construct was generated by amplifying the coding sequence from a previously described construct [19] so as to incorporate sequence encoding a carboxyl terminal Myc epitope then cloning this into a pcDNA3.1 vector. Mutation of the active site serine (S) of KLK4, KLK14, hepsin and TMPRSS2 to an alanine (A) residue was performed by site-directed mutagenesis using Platinum Pfx DNA polymerase. The integrity of all constructs was confirmed by DNA sequencing. Mammalian expression constructs encoding MMP-3 and MMP-9 were from GeneCopoeia (Rockville, MD, USA).
Transfections and collection of lysates
COS-7 cells were transfected using Lipofectamine 2000 or Lipofectamine LTX (Invitrogen) according to the instructions of the manufacturer and cells were cultured for 24 h post-transfection. For co-expression experiments equal quantities of each DNA vector were co-transfected into cells. Cells were washed with PBS then incubated for 15 min in ice cold lysis buffer containing 10 mM Tris pH 8.0, 150 mM NaCl, 1% (v/v) Triton X-100, 5 mM EDTA and Complete EDTA-free protease inhibitor cocktail (Roche Diagnostics) and then detached using a cell scraper. Lysed cells were passed through a syringe with 26 gauge needle 5-6 times, vortexed briefly, placed on ice for 10 min and then vortexed again. After centrifugation at 16,000 g for 10 min at 4°C to pellet insoluble material, the supernatant was collected and the protein concentration determined using a Pierce BCA assay kit (Thermo Fisher).
Incubation of hepsin and matriptase with MMP-3 or MMP-9
Conditioned media from COS-7 cells transiently transfected for 24 h with constructs encoding MMP-3 or MMP-9 were incubated at 37°C for 1 or 14 h with recombinant hepsin (50 nM), recombinant matriptase (50 nM) or bovine trypsin (10 nM). The reactions, stopped with protease inhibitor cocktail and Laemmli sample buffer with or without the reducing agent β-mercaptoethanol, were subjected to SDS-PAGE and examined by Western blot analysis.
Immunoprecipitation
Transiently transfected COS-7 cells were washed with PBS then lysed. Supernatants from lysates pre-cleared against protein A/G-agarose for 1 h at 4°C on a rolling platform, were mixed with the required antibody (anti-V5 (1:1000), -HA (1:1000), -Flag (1 µL/100 µg lysate), -Myc (1:1000), -MMP-3 (1.25 µg/100 µg lysate), -MMP-9 (2.5 µg/100 µg lysate), or control IgG) then incubated overnight at 4°C. Fresh aliquots of protein A/G-agarose beads were then added and the mixture incubated for 4 h at 4°C with gentle agitation. Beads were then washed three times in cell lysis buffer containing protease inhibitor cocktail. Associated proteins were eluted into Laemmli sample buffer containing β-mercaptoethanol and examined by Western blot analysis.
Cell surface biotinylation
Transiently transfected COS-7 cells were washed with PBS, then biotinylated for 10 min on ice with gentle rocking using cell impermeant EZlink NHS-SS-biotin (1.22 mg/ml) as described previously [33]. Cells were then washed three times with PBS prior to preparation of cell lysates, which were incubated with streptavidin agarose resin for 1 h on ice with gentle rocking. The unbound (cytoplasmic) protein fraction was collected by centrifugation (1000 g for 2 min at 4°C) in Pierce Spin Columns (Thermo Fisher). Streptavidin-immobilized biotinylated cell surface (plasma membrane) proteins were washed three times in lysis buffer containing protease inhibitor cocktail then eluted into Laemmli sample buffer containing β-mercaptoethanol. Plasma membrane and cytoplasmic fractions were examined by Western blot analysis under reducing conditions.
Western blot analysis
Lysates (20 µg) and equal volumes of immunoprecipitated and biotinylated proteins were separated by SDS-PAGE as described previously [34] in the presence or absence of the reducing agent β-mercaptoethanol. Separated proteins were transferred to nitrocellulose membranes that were blocked with Odyssey blocking buffer (LI-COR, Lincoln, NE, USA), then incubated from 1 h to overnight at 4°C with antibodies against V5 (1:10,000), HA (1:2500), Flag (1:5000), Myc (1:5000, 9B11; or 1:2000, 71D10), matriptase (1:1000) or GAPDH (1:10,000). Following washes membranes were incubated with species-appropriate fluorescently conjugated secondary antibodies (1:10,000-1:20,000) for 1 h at room temperature. Signals were acquired using an Odyssey Infrared Imaging System (LI-COR).
Confocal microscopy
COS-7 cells plated on sterile glass cover slips were transfected with KLK14-HA or KLK4-V5 and co-transfected with hepsin-Flag or TMPRSS2-Myc. After 24 h, cells were fixed with 4% (v/v) paraformaldehyde for 20 min at room temperature, permeabilized with 0.2% (v/v) Triton X-100 in PBS for 10 min, then internal fluorescence quenched with 50 mM ammonium chloride for 15 min. Non-specific binding sites were blocked with 3% (w/v) bovine serum albumin (BSA) in PBS for 45 min, then cells were stained with anti-HA (2 µg/mL; 12CA5), anti-V5 (1:200; R96025), anti-Flag (1:2500; F7425) or anti-Myc (1:200; 71D10) followed by species-appropriate Alexa Fluor 488 or 647 fluorescent dye-conjugated secondary antibodies (all 1:500). Cell nuclei were stained with DAPI (1:1500) and F-actin with Alexa Fluor 568 phalloidin (1:500). Coverslips were mounted on slides and cells imaged with a Zeiss LSM 510 Meta Confocal imaging system (Zeiss, North Ryde, NSW, Australia). Fluorescence was collected using a 60x magnification objective lens (NA 1.4 Oil), and images recorded using Zen 2009 Light Edition software (Zeiss) then processed using ImageJ and displayed using Microsoft PowerPoint.
Prostate cancer samples and immunohistochemistry
The study was approved by the Mater Health Services Research Ethics Committee under protocol number HREC/13/MHS/35/AM01. Consecutive sections (4 µm) from formalin fixed paraffin embedded tissue from a radical prostatectomy case (58 years old; Gleason 9; extraprostatic extensions) retrieved from the Mater Pathology archive, was sequentially deparaffinized and rehydrated, before antigen retrieval in urea (5% w/v) in 0.1 M Tris buffer (pH 9.5), followed by incubation in H2O2 (3%, v/v) and methanol (20%, v/v) in PBS- 0.05% (v/v) Tween 20 (PBS-T) to quench endogenous peroxidase. Sections were then blocked in BSA (2% w/v) in PBS-T and incubated overnight at 4°C with antibodies against KLK4 (1 µg/ml), KLK14 (0.2 µg/ml), hepsin (1:50), TMPRSS2 (1:100), MMP-3 (1:200) or MMP-9 (1:400). As negative controls, sections were incubated with only biotin conjugated anti-mouse, -rabbit or -goat secondary antibodies. Signal was developed using streptavidin conjugated horseradish peroxidase and 3,3’-diaminobenzidine substrate chromogen (Dako, North Sydney, NSW) and sections were counterstained with Mayer’s hematoxylin (Sigma-Aldrich). Stained slides were examined by a pathologist (BS) to identify regions of tumor showing specific reactivity, and signal visualized by microscopy (Nikon Eclipse 50i microscope, Nikon, Japan). Images were acquired using an Olympus Slide scanner VS120 and associated OlyVIA software, and displayed using Microsoft PowerPoint.
Results
Hepsin autoactivates
An essential step for initiation and amplification of proteolytic signaling is the conversion of an inactive zymogen to an active protease by cleavage at a defined site [35]. Activation drives conformational changes that orient catalytically important residues and permit the formation of substrate binding pockets [36]. Generally, cleavage is mediated by another protease although auto-catalytic mechanisms are not uncommon [2,3,37] with examples including the TTSPs TMPRSS2 [38] and matriptase [8]. As the mechanism of human hepsin activation is currently unknown, we first examined whether this TTSP also has the ability to undergo autoactivation. This was performed by analyzing lysates from cells co-transfected with expression constructs encoding two forms of hepsin that are represented in Figure 1; one tagged at the carboxyl terminal with a Myc epitope, and the other with a Flag tag. As controls we also examined lysates from cells expressing catalytically inactive mutant hepsin (hepsinm) in which the catalytic serine was mutated to alanine. In all assays, to block further proteolysis of proteins after cell disruption, lysates were prepared using a buffer containing a broad spectrum cocktail of protease inhibitors. As shown in Figure 1, Western blot analysis with anti-Myc and -Flag tag antibodies, under reducing conditions, showed a band at 48 kDa for pro-hepsin, and 26 kDa for the hepsin serine protease domain (SPD). Another band was apparent at 30 kDa. As the Myc and Flag tags are located at the carboxyl terminal, this cell associated hepsin fragment was generated by autoproteolysis between the serine protease and transmembrane domains. Two other hepsin fragments apparent at 34 and 36 kDa were generated via the actions of an endogenous COS-7 cell protease as these bands were also produced in cells only expressing catalytically inactive hepsin (Figure 1). These data suggest that hepsin is able to autoactivate and undergo further auto-processing between its catalytic and transmembrane domains.
Figure 1.
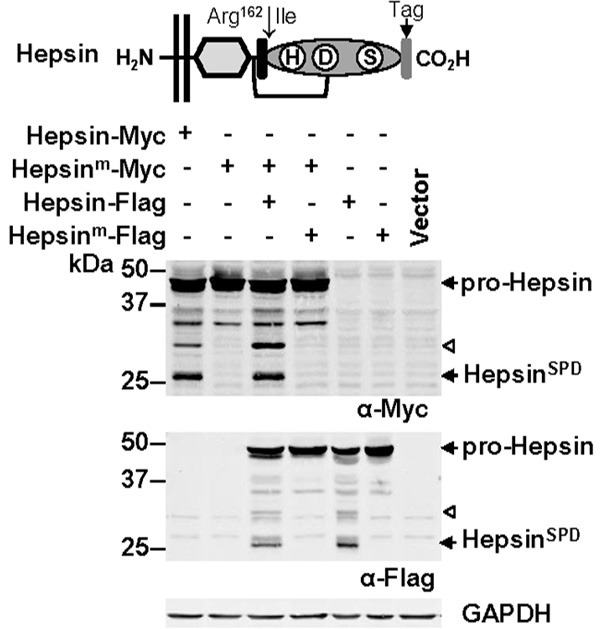
Hespin autoactivation. Top, Hepsin structural features including protease domain (showing residues essential for catalytic activity; histidine (H), aspartate (D) and serine (S)), activation motif (black box), activation site after Arg162, SRCR domain (diamond), transmembrane region (parallel lines), disulfide bond linking the protease domain with the membrane anchored portion of the protein (line), and a carboxyl terminal epitope tag added to facilitate immune-detection. Bottom, Anti-Myc (top), -Flag (middle) and -GAPDH (bottom) Western blot analysis of lysates from COS-7 cells transfected with the indicated vectors. Arrows highlight zymogen hepsin (pro-Hepsin) and the hepsin SPD (HepsinSPD). Arrowhead indicates a ~30 kDa hepsin fragment generated by auto-cleavage within the region between the membrane spanning region and the protease domain.
Reciprocal proteolytic interactions at the cell surface between membrane anchored hepsin and TMPRSS2 and the secreted serine proteases KLK4 and KLK14
As we are interested in mechanisms regulating proteolytic networks in malignant settings, we next examined for reciprocal proteolysis involving prostate cancer expressed membrane anchored and secreted serine proteases. As depicted in Figure 2A, for these assays we studied the prostate cancer associated autoactivating TTSPs hepsin (Figure 1) and TMPRSS2 [38], and secreted kallikreins KLK4 and KLK14. In these assays cells were co-transfected with constructs encoding carboxyl terminal tagged proteases, and included, as controls, constructs encoding catalytically inactive forms of each protease.
Figure 2.
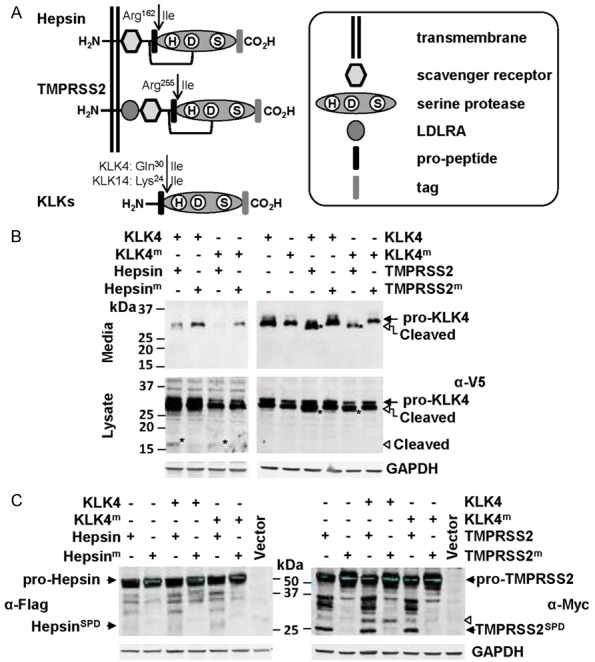
The secreted serine protease KLK4 is cleaved, and potentially transiently activated, at the cell surface by the plasma membrane anchored serine proteases hepsin and TMPRSS2. A. Schematic representation of the structure of hepsin, TMPRSS2 and KLKs. B. Anti-V5 Western blot analysis for KLK4-V5 of conditioned media and lysates from cells co-transfected with wild type or active-site mutated KLK4-V5 (KLK4; KLK4m) and wild type or active-site mutated hepsin-Flag (hepsin; hepsinm) or wild type or active-site mutated TMPRSS2-Myc (TMPRSS2; TMPRSS2m). Arrowhead indicates a 17 kDa KLK4 degradation product. C. Anti-Flag and anti-Myc Western blot analysis for hepsin-Flag and TMPRSS2-Myc of lysates from cells co-expressing KLK4 or KLK4m with hepsin or hepsinm, or KLK4 or KLK4m with TMPRSS2 or TMPRSS2m. Arrowhead indicates a KLK4 generated TMPRSS2 fragment of ~30 kDa.
In the first assay, cells were co-transfected with expression constructs encoding KLK4-V5 and Hepsin-Flag or TMPRSS2-Myc. Anti-V5 antibody Western blot analysis of conditioned media suggests that hepsin causes KLK4 degradation, as levels of both KLK4 and catalytically inactive KLK4m were much lower in media of cells co-expressing hepsin compared with cells co-expressing active site mutant hepsinm (Figure 2B, left). In cell lysates, predominant KLK4 and KLK4m bands were apparent at the predicted molecular weight of the zymogen of ~34 kDa. However, lysates from cells co-transfected with hepsin showed an additional band at 17 kDa for both KLK4 and KLK4m, which was not observed from cells co-transfected with catalytically inactive hepsinm. These data suggest that hepsin is proteolytically active in these transfected cells, and that it cleaves KLK4 resulting in a cell-associated 17 kDa fragment, and reduced levels in conditioned media.
In contrast with hepsin, which appears to degrade KLK4, anti-V5 antibody Western blot analysis indicates that TMPRSS2 mediates partial proteolysis of KLK4 converting it from ~34 to 31 kDa (Figure 2B, right). In these assays, uncleaved KLK4 is apparent as a previously reported doublet of glycosylated forms [26], and it is only when co-expressed with active TMPRSS2 that a reduction in molecular mass to 31 kDa is apparent. While this corresponds to the predicted molecular mass of active KLK4, it is unlikely that TMPRSS2, an Arg/Lys-specific protease [39], would cleave after Gln30, the canonical activation site of pro-KLK4 (Figure 2A) [14,15]. Interestingly, as there are no Arg or Lys residues in the first 104 amino acids of KLK4, it is possible that TMPRSS2 is generating 31 kDa KLK4 by proteolysis at a site other than at its predicted cleavage sites of Arg and Lys.
Consistent with Figure 1, anti-Flag antibody Western blot analysis of lysates from co-transfected cells, identified the 26 kDa SPD and 30 kDa fragment of hepsin that we demonstrated are generated by autoactivation (Figure 2C left). Western blot analysis of conditioned media failed to detect any forms of hepsin that were shed from the cell surface (data not shown), suggesting that this protease remains tethered to the plasma membrane.
Consistent with a previous report [38], anti-Myc antibody Western blot analysis of cell lysates from co-transfected cells, indicated that TMPRSS2 auto-activates generating a band at ~25 kDa corresponding to the predicted molecular mass of its SPD (Figure 2C, right). An additional TMPRSS2 band at ~34 kDa was also due to auto-proteolysis as it was not present in lysates from cells expressing TMPRSS2m. However, another band at ~36 kDa was likely generated by an endogenous protease as it was detected in TMPRSS2 and TMPRSS2m expressing cells (Figure 2C, right). Surprisingly, the Western blot data also suggest that KLK4 is active in this system and can mediate cleavage of TMPRSS2 generating a band at ~30 kDa. As the Myc tag is located at the carboxyl terminal, this cell associated TMPRSS2 fragment was generated by proteolysis between the serine protease and transmembrane domains. The presence of this KLK4-induced band in both TMPRSS2 and TMPRSS2m expressing cells (Figure 2C, right), suggests that active KLK4 was not generated by the direct proteolytic activity of TMPRSS2, but that membrane anchored TMPRSS2 may have a role in advantageously localising secreted KLK4 in proximity to its activating protease. As for hepsin, Western blot analysis of conditioned media failed to detect any forms of TMPRSS2 in conditioned media (data not shown), suggesting that this protease also remains tethered to the plasma membrane in transfected cells.
We also similarly examined interactions between another secreted protease, KLK14, and hepsin and TMPRSS2. Interestingly, anti-HA antibody Western blot analysis of conditioned media and lysates suggests that hepsin cleaves cell-associated but not secreted KLK14. As shown in Figure 3A (left), whereas there was no shift in the molecular mass of KLK14 in conditioned media, analysis of cell lysates demonstrated conversion of both KLK14 and KLK14m from ~28 to 26 kDa, the predicted molecular weight of the SPD of this secreted protease. In contrast, TMPRSS2 may degrade KLK14 as anti-HA antibody Western blot analysis of conditioned media revealed levels of both KLK4 and catalytically inactive KLK4m that were consistently lower in media of cells co-expressing TMPRSS2 compared with cells co-expressing active site mutant TMPRSS2m (Figure 3A, right). The lack of distinct KLK14 cleavage bands unique to cell lysates from TMPRSS2 expressing cells, indicates that TMPRSS2 cleaved KLK14 does not associate with cells (Figure 3A, right). Also, there was no evidence that KLK14 cleaves hepsin or TMPRSS2 (Figure 3B). Collectively, these data suggest a complex range of regulatory interactions occurring at the cell surface between membrane anchored and secreted serine proteases.
Figure 3.
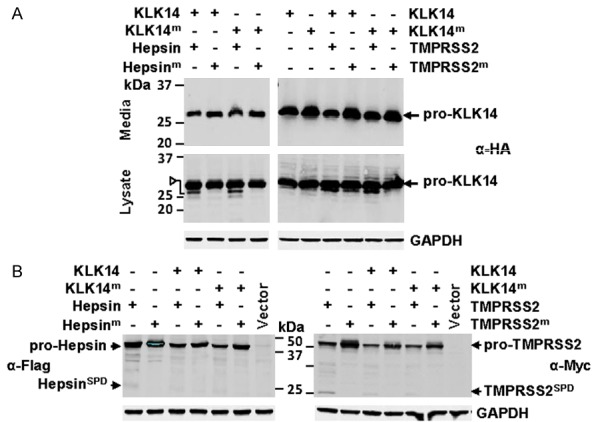
Distinct proteolytic processing of the secreted serine protease KLK14 by plasma membrane anchored serine proteases hepsin and TMPRSS2. A. Anti-HA Western blot analysis for KLK14-HA of conditioned media and lysates from cells co-transfected with wild type or active-site mutated KLK14-HA (KLK14; KLK14m) and wild type or active-site mutated hepsin-Flag (hepsin; hepsinm) or wild type or active-site mutated TMPRSS2-Myc (TMPRSS2; TMPRSS2m). Arrowhead indicates hepsin generated KLK14 fragment of the predicted molecular mass of the KLK14 SPD. B. Anti-Flag and anti-Myc Western blot analysis for hepsin-Flag and TMPRSS2-Myc of lysates from cells co-expressing KLK14 or KLK14m with hepsin or hepsinm, or KLK14 or KLK14m with TMPRSS2 or TMPRSS2m.
Hepsin and TMPRSS2 activate the secreted proteases MMP-3 and MMP-9
As MMP-3 is the only known prostate cancer-expressed activator of KLK4 by proteolysis at Gln30 [13,37,40], we next explored whether this MMP, and the structurally similar prostate cancer expressed protease MMP-9 [28], can be cleaved by hepsin or TMPRSS2, and thereby potentially function in a protease cascade to convert pro-KLK4 to active KLK4. For these assays cells were co-transfected with expression constructs encoding untagged MMP-3 or MMP-9, and hepsin-Myc, TMPRSS2-Myc or, to provide comparison with a known MMP-3 activator, matriptase-Myc [9] (Figure 4A).
Figure 4.
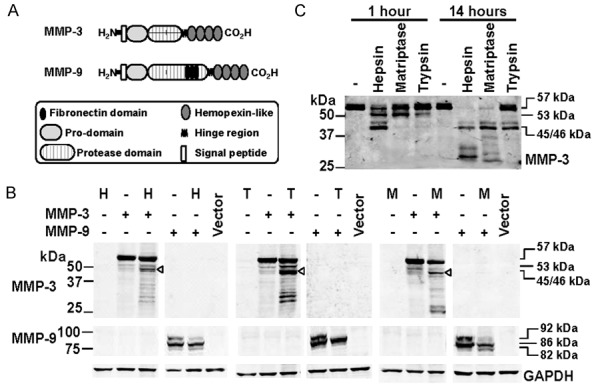
Examination of proteolytic processing of MMP-3 and MMP-9 by hepsin and TMPRSS2. A. Schematic representation of the structure of MMP-3 and MMP-9. B. Anti-MMP-3 (top), -MMP-9 (middle) and -GAPDH (bottom) Western blot analyses of lysates from cells co-transfected with hepsin (H), TMPRSS2 (T) or matriptase (M) and MMP-3 or MM-P-9. ProMMP-3, 57 kDa; intermediate MMP-3, 53 kDa; activated MMP-3, 45/46 kDa. Pro-MMP-9, 92 kDa; intermediate MMP-9, 86 kDa; activated MMP-9, 82 kDa. C. Anti-MMP-3 Western blot analysis of conditioned media from cells transiently expressing MMP-3 or MMP-9 that had been incubated for 1 or 14 h with recombinant hepsin (50 nM), recombinant matriptase (50 nM) or bovine trypsin (10 nM). ProMMP-3, at 57 kDa, intermediate MMP-3 at 53 kDa, and activated MMP-3 at 45/46 kDa are indicated.
Western blot analysis of lysates under reducing conditions indicated that pro-MMP-3 at ~57 kDa is extensively cleaved in the presence of hepsin, TMPRSS2 and matriptase, and to a greater extent than is apparent from cells only expressing MMP-3 (Figure 4B). MMP-3 is activated in a stepwise manner with Arg/Lys specific proteases, including plasma kallikrein and matriptase, first cleaving within a stretch of amino acids (Phe51-Val-Arg-Arg-Lys-Asp) near the middle of the MMP-3 pro-region to yield an intermediate 53 kDa form [41]. This is followed by MMP-3 self-activation by cleaving at His99-Phe to remove the remaining pro-region yielding the 45/46 kDa mature peptide [41]. The presence of this band as the predominant product (Figure 4B, arrowhead) indicates that each of hepsin, TMPRSS2 and matriptase are able to convert MMP-3 to its 53 kDa intermediate, before it autoactivates and is converted to the 45/46 kDa form.
MMP-9 zymogen (92 kDa) is activated in two steps with a first cleavage at Glu59Met generating an 86 kDa intermediate [42] followed by a second auto-proteolytic cleavage at Arg106 Phe to generate an 82 kDa active form [43]. Importantly, it can also be activated directly with a single cleavage at Arg106 Phe by trypsin, KLK1 and other Arg/Lys specific proteases to generate 82 kDa MMP-9 [44,45]. As shown in Figure 4B, Western blot analysis indicated that similar to matriptase, hepsin processed 92 kDa MMP-2. Interestingly, the complete conversion of 92 kDa MMP-9 in the presence of TMPRSS2 suggests that this TTSP is even more robust than hesin and matriptase as a pro-MMP-9 convertase (Figure 4B). Also of note, in contrast with matriptase, in response to hepsin and TMPRSS2 there was no increased accumulation of 82 kDa MMP-9, suggesting that this active form of the protease undergoes further proteolytic processing in the presence of these TTSPs (Figure 4B).
As cleavage of MMP-3 that is mediated by hepsin and TMPRSS2 generates many bands in addition to active 45/46 kDa MMP-3 (Figure 4B), we next examined the kinetics of activation versus degradation of MMP-3. For these assays we incubated conditioned media from COS-7 cells transiently expressing MMP-3, for 1 and 14 hours, with recombinant hepsin (50 nM), or the known activators of MMP-3, matriptase (50 nM) and trypsin (10 nM) [9,46]; unfortunately recombinant TMPRSS2 was not available. Interestingly, Western blot analysis indicated that hepsin was the most efficient activator of MMP-3 converting it to the 53 kDa intermediate followed by autocatalytic conversion to its 45/46 active form within the 1 hour incubation (Figure 4C). In contrast, complete conversion to active MMP-3 was only observed after 14 hours with an equal 50 nM concentration of matriptase and 10 nM trypsin (Figure 4C). Importantly, at this later time point MMP-3 had largely been degraded by hepsin, and partially degraded by matriptase, These data are consistent with the results in Figure 4B, and confirm that hepsin is an efficient activator of MMP-3 that over longer time periods degrades MMP-3.
Hepsin and TMPRSS2 co-immunoprecipitate with KLK4, KLK14, MMP-3 and MMP-9
Our data suggest that interactions occur at the cell surface between membrane anchored serine proteases and secreted KLKs and MMPs. To further examine these interactions, Western blot analyses of proteins immunoprecipitated from cell lysates were performed. Interactions between hepsin-Flag and KLK4-V5 were first assessed, with Western blot analysis indicating that 35 kDa zymogen KLK4 immunoprecipitates with wildtype and mutant catalytically inactive hepsin (Figure 5A). In addition, consistent with Western blot data in Figure 2, wildtype hepsin immunoprecipitated with the hepsin-generated 17 kDa KLK4 cleavage fragment. Western blot analysis also showed that zymogen and active hepsin co-immunopurified with KLK4 (Figure 5A).
Figure 5.
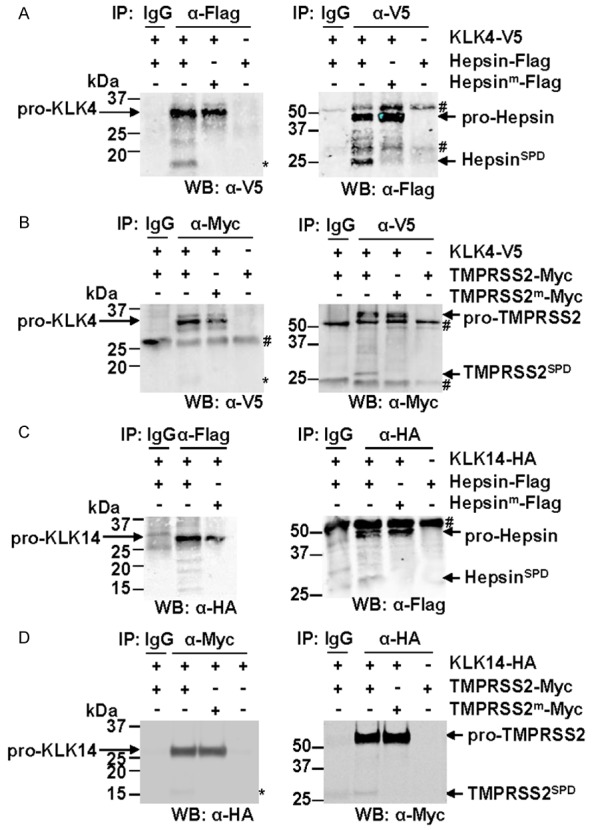
KLK4 and KLK14 immunoprecipitate with hepsin and TMPRSS2. Western blot analyses were performed on immunoprecipitates from lysates of COS-7 cells transiently expressing proteases of interest. A. Cells transiently expressed KLK4-V5 and wild type or active-site mutant hepsin-Flag. Anti-Flag, anti-V5 and control immunoglobulin (IgG) precipitates were examined by anti-V5 (left) and anti-Flag (right) Western blot analysis. Asterisk indicates 17 kDa hepsin-generated KLK4 fragment. B. Cells transiently expressed KLK4-V5 and wild type or active-site mutant TMPRSS2-Myc. Anti-Myc, anti-V5 and control IgG precipitates were examined by anti-V5 (left) and anti-Myc (right) Western blot analysis. Asterisk indicates 15-17 kDa TMPRSS2-generated KLK4 fragment. C. Cells transiently expressed KLK14-HA and wild type or active-site mutant hepsin-Flag. Anti- Flag, anti-HA and control IgG precipitates were examined by anti-HA (left) and anti-Flag (right) Western blot analysis. D. Cells transiently expressed KLK14-HA and wild type or active-site mutant TMPRSS2-Myc. Anti-Myc, anti-HA and control IgG precipitates were examined by anti-HA (left) and anti-Myc (right) Western blot analysis. Asterisk indicates 17 kDa TMPRSS2-generated KLK14 fragment. #, antibody heavy/light chain used for immunoprecipitation.
Similarly, zymogen KLK4 immunoprecipitated both wildtype and mutant catalytically inactive TMPRSS2 (Figure 5B). Consistent with the cleavage of KLK4 by TMPRSS2 apparent in Figure 2B, wildtype TMPRSS2 that immunopurified with KLK4 was in both zymogen and active forms. Interestingly, a faint band of 15-17 kDa representing a degradation product of KLK4 also immunoprecipitated with TMPRSS2 (Figure 5B), suggesting that this TTSP cleaves KLK4 within its catalytic domain as well as amino terminal to its canonical activation site at Gln30. Similar to KLK4, Western blot analysis showed that KLK14 also co-immunoprecipitated with both zymogen and active hepsin and TMPRSS2 (Figure 5C and 5D), with evidence of lower molecular weight KLK14 bands from co-immunoprecipitation with hepsin and TMPRSS2 (asterisk) but not with the mutant TTSPs.
Western blot analysis of immunoprecipitates from transiently transfected cells was also used to examine interactions involving hepsin and TMPRSS2 with MMP-3 and MMP-9. These experiments employed an anti-Myc antibody to precipitate hepsin-Myc and TMPRSS2-Myc, and antibodies against MMP-3 and MMP-9 to precipitate these proteases. These analyses indicated that zymogen, intermediate and active MMP-3 at 57, 53 and 45/46 kDa, respectively, immunoprecipitated with both hepsin and TMPRSS2 (Figure 6A). In addition, while we consistently observed that zymogen, intermediate and active MMP-9 at 92, 86 and 82 kDa, respectively, immunoprecipitated with hepsin, only the zymogen and intermediate forms of MMP-9 consistently co-purified with TMPRSS2 (Figure 6B). Figure 6C and 6D indicated that zymogen and activated hepsin and TMPRSS2 immunopurifiy using the anti-MMP-3 and -MMP-9 antibodies.
Figure 6.
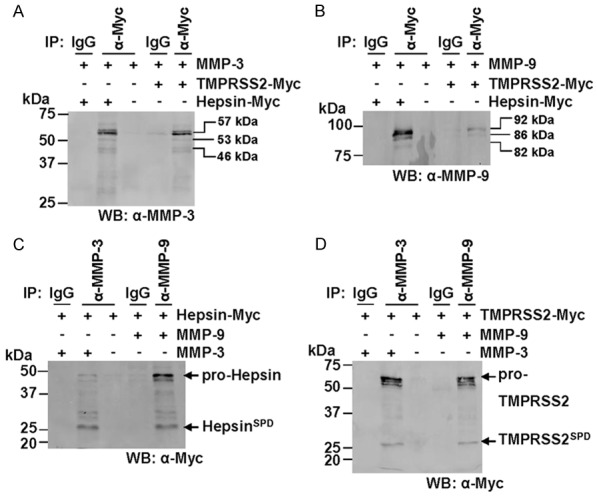
MMP-3 and MMP-9 immunoprecipitate with hepsin and TMPRSS2. Western blot analyses were performed on cell surface biotinylated fractions from COS-7 cells transiently expressing proteases of interest. A. Lysates from COS-7 cells transiently transfected with constructs encoding MMP-3 and hepsin-MYC or TMPRSS2-Myc were subjected to immunoprecipitations using anti-Myc or control immunoglobulins (IgG) then examined by anti-MMP-3 Western blot analysis. ProMMP-3, 57 kDa; intermediate MMP-3, 53 kDa; activated MMP-3, 46 kDa. B. Lysates from COS-7 cells transiently transfected with constructs encoding MMP-9 and hepsin-MYC or TMPRSS2-Myc were subjected to immunoprecipitations using anti-Myc or control immunoglobulins (IgG) then examined by anti-MMP-9 Western blot analysis. Pro-MMP-9, 92 kDa; intermediate MMP-9, 86 kDa; activated MMP-9, 82 kDa. C. Lysates from COS-7 cells transiently transfected with constructs encoding hepsin-MYC and MMP-3 or MMP-9 were subjected to immunoprecipitations using anti-MMP-3, anti-MMP-9 or control immunoglobulins (IgG) then examined by anti-Myc Western blot analysis. D. Lysates from COS-7 cells transiently transfected with constructs encoding TMPRSS2-MYC and MMP-3 or MMP-9 were subjected to immunoprecipitations using anti-MMP-3, anti-MMP-9 or control immunoglobulins (IgG) then examined by anti-Myc Western blot analysis.
Hepsin and TMPRSS2 co-localize with KLK4 and KLK14 on the cell surface
The above data indicate that the secreted proteases KLK4, KLK14, MMP-3 and MMP-9 can associate with and are cleaved by the membrane-anchored proteases hepsin and TMPRSS2 the cell surface. To directly assess the plasma membrane location of these proteins we performed Western blot analysis of cell surface biotinylated proteins, and confocal microscopy analysis.
As MMP-3 and MMP-9 have previously been shown to bind to the cell surface [47], we focused on the plasma membrane location of KLK4 and KLK14, in the presence and absence of hepsin and TMPRSS2, respectively. This was performed by cell surface biotinylation of live COS-7 cells transiently transfected with KLK4-V5 and hepsin-Flag, or KLK14-HA and TMPRSS2-Myc. As shown in Figure 7A, Western blot analysis of biotinylated proteins purified using streptavidin beads, revealed that zymogen and active hepsin, and zymogen KLK4 are located on the cell surface. In addition, a 17 kDa KLK4 fragment, generated through the action of hepsin, was also isolated from the cell surface fraction (Figure 7A). Similarly, zymogen and active TMPRSS2 and zymogen KLK14 were also present in biotinylated fractions although we consistently only obtained small quantities of the TMPRSS2 SPD (Figure 7B). Note that purified fractions were free of GAPDH indicating that cells were intact during biotinylation and that cell surface proteins were not contaminated with cytoplasmic fractions (Figure 7). These data support the proposal that secreted proteases can associate with the cell surface.
Figure 7.
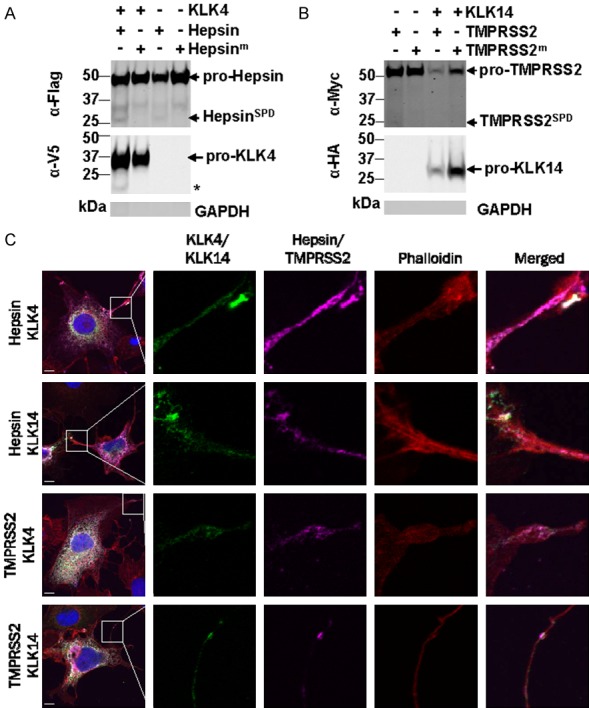
Hepsin, TMPRSS2, KLK4 and KLK14 localize to the cell surface. A. Western blot analysis of cell surface biotinylated fractions from COS-7 cells transiently transfected with KLK4-V5 and hepsin-Flag. Asterisk, 17 kDa KLK4 cleavage products. B. Western blot analysis of cell surface biotinylated fractions from COS-7 cells transiently transfected with KLK14-HA and TMPRSS2-Myc. Purified fractions were free of GAPDH indicating that cells were intact during biotinylation. C. Confocal microscopy analysis of COS-7 cells co-expressing hepsin-Flag or TMPRSS2-Myc (purple) with KLK4-V5 or KLK14-HA (green). Cells were co-stained with DAPI to delineate cell nuclei (blue), and Alexa Fluor 568 conjugated phalloidin to delineate F-actin positive cytoplasm (red). White, regions of co-localisation of hepsin/TMPRSS2 (purple) with KLK4/KLK14 (green). Scale bar = 10 µm.
To further explore the cell surface localization of membrane-anchored hepsin and TMPRSS2 and secreted KLK4 and KLK14, we performed confocal microscopy analysis of transiently expressing COS-7 cells. Fixed and permeabilized cells were stained with antibodies against the epitope tag present on each protease, and with dyes that delineate cell cytoplasm and nuclei. As shown in Figure 8C, the predominant signal for each protease pair was a white signal present in the cytoplasm of cells indicating co-localisation of the proteases during cellular trafficking. Of particular note, as highlighted in the insets in Figure 8C, we also commonly observed distinct co-localization as a white signal of protease pairs on the surface of cellular protrusions. These data support that TTSPs and KLKs co-localize on the cell surface within defined structures, to facilitate reciprocal cleavage events that are necessary for the tight regulation of protease activation and inactivation at sites of interactions with the extracellular milieu.
Figure 8.
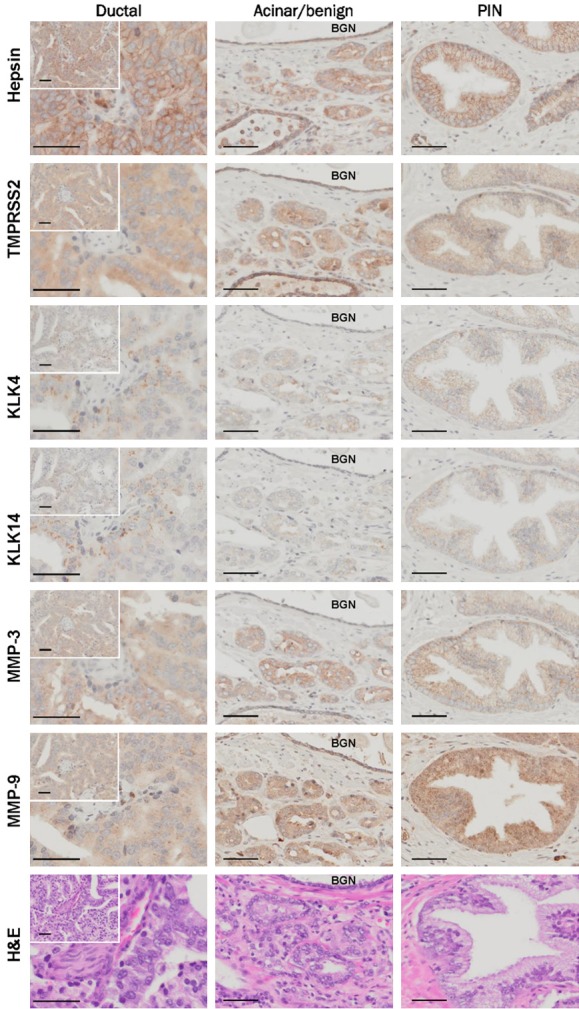
Over-lapping expression of hepsin, TMPRSS2, KLK4, KLK14, MMP-3 and MMP-9 in benign and malignant prostate. Consecutive sections of a prostate tumor were stained with antibodies against hepsin, TMPRSS2, KLK4, KLK14, MMP-3 and MMP-9. Representative regions showing staining in prostatic ductal adenocarcinoma, prostatic acinar adenocarcinoma with adjacent benign prostate, and PIN were photographed. BGN, benign. All images were acquired using a 40× objective unless otherwise indicated.
Over-lapping expression of hepsin, TMPRSS2, KLK4, KLK14, MMP-3 and MMP-9 in benign and malignant prostate
The proteases hepsin [16,48,49], TMPRSS2 [18], KLK4 [21,50], KLK14 [25], MMP-3 [27] and MMP-9 [28] are expressed to various extents in normal and diseased prostate. To explore the extent of the co-expression of these proteases we next performed immunohistochemical analysis. For this purpose we selected a prostate cancer patient sample that contained regions of pathology commonly encountered in the clinical management of prostate disease, including pre-malignant regions of benign prostate and high grade prostatic intraepithelial neoplasia (HG-PIN), as well as prostatic acinar adenocarcinoma, and the less common but aggressive, prostatic ductal adenocarcinoma [51]. Most interestingly, from analysis of consecutive sections, we noted that highest expression for each protease was observed in prostatic ductal adenocarcinoma (Figure 8; left panels). These lesions, while rarer than prostatic acinar adenocarcinoma, follow a more aggressive course, are associated with poor prognosis and have a greater propensity to spread to the testis and penis [52]. In these regions of the tumor, staining for hepsin was predominantly membranous, consistent with its membrane spanning structure, and although TMPRSS2 also has a membrane spanning domain, its expression was predominantly cytoplasmic with some evidence of membrane accentuation in places. Both KLK4 and KLK14 were most obvious as cytoplasmically located punctate structures, while MMP-3 and MMP-9 were located diffusely throughout the cytoplasm with MMP-9 showing cytoplasmic regions of fine granular expression (Figure 8; left panels).
Both hepsin and TMPRSS2 showed predominantly membranous expression in prostatic acinar adenocarcinoma, and it was striking that in these lesions the punctate expression of KLK4 and KLK14 seen in regions of prostatic ductal adenocarcinoma was not apparent (Figure 8; middle panels). Interestingly, in addition to cytoplasmic staining, both MMP-3 and MMP-9 showed evidence of membrane expression in prostatic acinar adenocarcinoma (Figure 8; middle panels). In benign hypertrophic prostate, hepsin was strongly expressed, while in these glands TMPRSS2 was most predominant in basal cells. KLK4 and MMP-3 expression was weak and KLK14 absent in benign prostate. In contrast, MMP-9 levels in benign glands were similar to those seen in prostatic acinar adenocarcinoma (Figure 8; BNG, middle panels). Each protease was expressed in regions of HG-PIN with staining generally more intense than in benign regions but weaker than in areas of prostatic ductal adenocarcinoma (Figure 8C; right panels).
In summary, there is considerable overlap in the expression patterns of hepsin, TMPRSS2, KLK4, KLK14, MMP-3 and MMP-9, in the pathologies commonly seen in prostate tumors, supporting the possibility that these proteases could interact in vivo during progression of prostate cancer, as we have seen from cells in vitro.
Discussion
The data presented here provide evidence of a novel pericellular proteolytic regulatory network of the prostate cancer expressed proteases hepsin, TMPRSS2, KLK4, KLK14, MMP-3 and MMP-9. In summary, as shown in Figure 9, consistent with the ability to function as cell surface initiators of proteolytic networks, we demonstrated that plasma membrane localized hepsin is able to autoactivate, and autoactivation has previously been reported for the structurally related TMPRSS2 [38]. While remaining tethered to the cell surface hepsin and TMPRSS2 proteolyze, to various extents, secreted KLK4, KLK14, MMP-3 and MMP-9. Our data indicate that active hepsin mediates degradation of KLK4 resulting in cell association of a 17 kDa KLK4 degradation product of unknown function. In contrast, active TMPRSS2 mediates limited proteolysis of KLK4, likely generating a precursor that requires further proteolysis at its consensus activation site Gln30 to achieve catalytic activity (Figure 9). Important for the full activation of KLK4 is our observation that both hepsin and TMPRSS2 mediate activation of MMP-3, the only known activator of KLK4 via proteolysis at Gln30 [13,37,40]. Interestingly, active KLK4 itself likely regulates TMPRSS2 activity because it mediates cleavage of this TTSP between its serine protease and transmembrane domains. While this does not release TMPRSS2 proteolysis from the cell surface it will likely disrupt its LDLRA or scavenger receptor regulatory domains. Hepsin undergoes analogous proteolysis within the region between its serine protease and transmembrane domains via an autoproteolytic mechanism (Figure 9). As shown in Figure 9B, in contrast with its degradation of KLK4, hepsin generates the KLK14 SPD which remains plasma membrane associated. Similar to hepsin mediated degradation of KLK4, TMPRSS2 appears to degrade KLK14, resulting in reduced pro-KLK14 in conditioned media.
Figure 9.
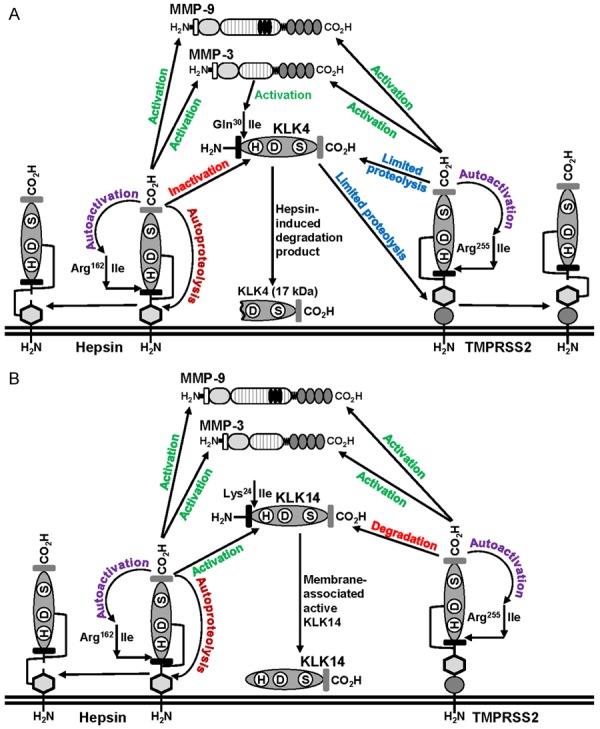
Summary of interactions between membrane anchored hepsin and TMPRSS2, with secreted KLK4, KLK14, MMP-3 and MMP-9. Details are described in the text. A. Proteolytic interactions centred on KLK4. B. Proteolytic interactions centred on KLK14.
While further work is required to validate these results in cells that endogenously express these proteases, two key findings from our study suggest that the identified novel proteolytic interactions at the cell surface, may be important in prostate cancer patients. In particular, we propose that the network will have most importance as the disease progresses during which cells de-differentiate and normal glandular structure is gradually lost. First, our immunohistochemical analysis indicates that in vivo each protease is expressed during progression from benign to malignant prostate. To ensure that we could assess the co-expression of the six proteases in as broad a range as possible of prostate pathologies, we selected a patient sample for this analysis that contained pre-malignant regions of benign prostate and high grade prostatic intraepithelial neoplasia (HG-PIN), as well as prostatic acinar adenocarcinoma and prostatic ductal adenocarcinoma. Highest co-expression was observed in prostatic ductal adenocarcinoma, a less common histological variant than prostatic acinar adenocarcinoma, that follows a more aggressive course, and is associated with poor prognosis with greater propensity to spread to sites other than bone including the testis and penis [52]. Significantly, hepsin displayed predominant cell surface expression throughout all of the regions of the analyzed prostate tumor, while TMPRSS2 was also plasma membrane localized particularly in prostatic acinar adenocarcinoma, the most common histotype for this cancer. Also consistent with our in vitro data, the secreted proteases MMP-3 and MMP-9 also showed evidence of membrane expression in this histotype.
The second key finding points to the importance of the timing of activation of components of the proteolytic network. Our confocal microscopy analysis demonstrates that hepsin and TMPRSS2 colocalize on the cell surface with the secreted serine proteases KLK4 and KLK14, only in membrane protrusions. This suggests that the reciprocal proteolytic interactions identified by us from Western blot analysis of cell lysates, immunoprecipitates and cell surface fractions, occur in defined cellular protrusions. These structures are particularly important during cancer dissemination for cell migration, invasion and survival, that require processing of growth factors and cytokines, activation of receptors, and suppression of immune responses [53]. Coupled with a previous report demonstrating that MMP-3 and MMP-9 also associate with the cell surface [47], these data suggest the possibility that the novel proteolytic network identified by us, will be most important during active dissemination of prostate and potentially other cancers.
This is the first report of cell surface association of the secreted proteases KLK4 and KLK14. While mechanisms regulating these associations have not been defined, cell surface binding of other protease have been defined. These include MMPs where binding occurs via several mechanisms that generally involve the protease hemopexin domain including MMP3 binding via cell surface localized collagen I [47], and MMP9 binding via integrins [54-56] and CD44 [57,58]. Cell surface localization of other secreted serine proteases involves binding to plasma membrane receptors and other membrane linked proteins with examples including factor VIIa and Xa docking to tissue factor to regulate blood coagulation [59,60], uPA binding to its receptor uPAR to regulate tissue remodelling [61], and plasma kallikrein binding via high-molecular weight kininogen which itself binds via negative charges on the cell surface, to regulate blood pressure and inflammation [62]. However, it is important to note that for each of the proteases in these systems, interactions with cell surface proteins is via a non-proteolytic domain. As KLK4 and KLK14 lack non-proteolytic domains it is not clear how plasma membrane association of these proteases occurs, although recent work by Rolland and colleagues [63] revealed the cell surface protein TYRO3 protein tyrosine kinase as a potential binding partner for KLK4. To date it is the only KLK family member to have been associated with a transmembrane/cell-surface binding partner. However, the mechanism regulating interaction between KLK4 and TYRO3 has yet to be determined.
In conclusion, it is important to note that in addition to regulating each other, the proteases examined by us also regulate a range of key signalling systems at the cell surface that promote prostate cancer and other malignancies including serine protease regulation of protease activated receptors [19,24], and the HGF/Met and MSP/RON receptor systems. Thus, a better understanding of the mechanisms that regulate the pericellular proteolytic network of hepsin, TMPRSS2, KLK4, KLK14, MMP-3 and MMP-9, may identify novel approaches to disrupt processes important in cancer progression.
Acknowledgements
This work was supported by funding from the National Health and Medical Research Council of Australia (grant 614206), Cancer Council Queensland (grant APP1021827), Prostate Cancer Foundation of Australia (grant PG 3810) and Australian Research Council (Future Fellowship FT120100917) to J.D.H., a mobility grant (Personalized Medicine) from the German Academic Exchange Service (DAAD) to J.A.C., V.M., and J.D.H., a National Health and Medical Research Council of Australia Principal Research Fellowship to J.A.C., and an Australian Postgraduate Award, and Smart State PhD stipend and Queensland University of Technology top-up and write-up scholarships to J.C.R.
Disclosure of conflict of interest
None.
References
- 1.Bonfil RD, Chinni S, Fridman R, Kim HR, Cher ML. Proteases, growth factors, chemokines, and the microenvironment in prostate cancer bone metastasis. Urol Oncol. 2007;25:407–411. doi: 10.1016/j.urolonc.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011;21:228–237. doi: 10.1016/j.tcb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bugge TH, Antalis TM, Wu Q. Type II transmembrane serine proteases. J Biol Chem. 2009;284:23177–23181. doi: 10.1074/jbc.R109.021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 5.Dong Y, Harrington BS, Adams MN, Wortmann A, Stephenson SA, Lisle J, Herington A, Hooper JD, Clements JA. Activation of membranebound proteins and receptor systems: a link between tissue kallikrein and the KLK-related peptidases. Biol Chem. 2014;395:977–990. doi: 10.1515/hsz-2014-0147. [DOI] [PubMed] [Google Scholar]
- 6.Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 7.Webb SL, Sanders AJ, Mason MD, Jiang WG. Type II transmembrane serine protease (TTSP) deregulation in cancer. Front Biosci (Landmark Ed) 2011;16:539–552. doi: 10.2741/3704. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, Craik CS. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem. 2000;275:26333–26342. doi: 10.1074/jbc.M002941200. [DOI] [PubMed] [Google Scholar]
- 9.Jin X, Yagi M, Akiyama N, Hirosaki T, Higashi S, Lin CY, Dickson RB, Kitamura H, Miyazaki K. Matriptase activates stromelysin (MMP-3) and promotes tumor growth and angiogenesis. Cancer Sci. 2006;97:1327–1334. doi: 10.1111/j.1349-7006.2006.00328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, Nagase H, French DL, Quigley JP. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999;274:13066–13076. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- 11.Lawrence MG, Lai J, Clements JA. Kallikreins on steroids: structure, function, and hormonal regulation of prostate-specific antigen and the extended kallikrein locus. Endocr Rev. 2010;31:407–446. doi: 10.1210/er.2009-0034. [DOI] [PubMed] [Google Scholar]
- 12.Ra HJ, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26:587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beaufort N, Plaza K, Utzschneider D, Schwarz A, Burkhart JM, Creutzburg S, Debela M, Schmitt M, Ries C, Magdolen V. Interdependence of kallikrein-related peptidases in proteolytic networks. Biol Chem. 2010;391:581–587. doi: 10.1515/BC.2010.055. [DOI] [PubMed] [Google Scholar]
- 14.Nelson PS, Gan L, Ferguson C, Moss P, Gelinas R, Hood L, Wang K. Molecular cloning and characterization of prostase, an androgen-regulated serine protease with prostate-restricted expression. Proc Natl Acad Sci U S A. 1999;96:3114–3119. doi: 10.1073/pnas.96.6.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephenson SA, Verity K, Ashworth LK, Clements JA. Localization of a new prostate-specific antigen-related serine protease gene, KLK4, is evidence for an expanded human kallikrein gene family cluster on chromosome 19q13.3-13.4. J Biol Chem. 1999;274:23210–23214. doi: 10.1074/jbc.274.33.23210. [DOI] [PubMed] [Google Scholar]
- 16.Xuan JA, Schneider D, Toy P, Lin R, Newton A, Zhu Y, Finster S, Vogel D, Mintzer B, Dinter H, Light D, Parry R, Polokoff M, Whitlow M, Wu Q, Parry G. Antibodies neutralizing hepsin protease activity do not impact cell growth but inhibit invasion of prostate and ovarian tumor cells in culture. Cancer Res. 2006;66:3611–3619. doi: 10.1158/0008-5472.CAN-05-2983. [DOI] [PubMed] [Google Scholar]
- 17.Tang X, Mahajan SS, Nguyen LT, Beliveau F, Leduc R, Simon JA, Vasioukhin V. Targeted inhibition of cell-surface serine protease Hepsin blocks prostate cancer bone metastasis. Oncotarget. 2014;5:1352–1362. doi: 10.18632/oncotarget.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lucas JM, True L, Hawley S, Matsumura M, Morrissey C, Vessella R, Nelson PS. The androgen-regulated type II serine protease TMPRSS2 is differentially expressed and mislocalized in prostate adenocarcinoma. J Pathol. 2008;215:118–125. doi: 10.1002/path.2330. [DOI] [PubMed] [Google Scholar]
- 19.Wilson S, Greer B, Hooper J, Zijlstra A, Walker B, Quigley J, Hawthorne S. The membraneanchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem J. 2005;388:967–972. doi: 10.1042/BJ20041066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucas JM, Heinlein C, Kim T, Hernandez SA, Malik MS, True LD, Morrissey C, Corey E, Montgomery B, Mostaghel E, Clegg N, Coleman I, Brown CM, Schneider EL, Craik C, Simon J, Bedalov T, Nelson PS. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014;4:1310–25. doi: 10.1158/2159-8290.CD-13-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong Y, Bui LT, Odorico DM, Tan OL, Myers SA, Samaratunga H, Gardiner RA, Clements JA. Compartmentalized expression of kallikrein 4 (KLK4/hK4) isoforms in prostate cancer: nuclear, cytoplasmic and secreted forms. Endocr Relat Cancer. 2005;12:875–889. doi: 10.1677/erc.1.01062. [DOI] [PubMed] [Google Scholar]
- 22.Mize GJ, Wang W, Takayama TK. Prostatespecific kallikreins-2 and -4 enhance the proliferation of DU-145 prostate cancer cells through protease-activated receptors-1 and -2. Mol Cancer Res. 2008;6:1043–1051. doi: 10.1158/1541-7786.MCR-08-0096. [DOI] [PubMed] [Google Scholar]
- 23.Ramsay AJ, Reid JC, Adams MN, Samaratunga H, Dong Y, Clements JA, Hooper JD. Prostatic trypsin-like kallikrein-related peptidases (KLKs) and other prostate-expressed tryptic proteinases as regulators of signalling via proteinase-activated receptors (PARs) Biol Chem. 2008;389:653–668. doi: 10.1515/BC.2008.078. [DOI] [PubMed] [Google Scholar]
- 24.Ramsay AJ, Dong Y, Hunt ML, Linn M, Samaratunga H, Clements JA, Hooper JD. Kallikrein-related peptidase 4 (KLK4) initiates intracellular signaling via protease-activated receptors (PARs). KLK4 and PAR-2 are co-expressed during prostate cancer progression. J Biol Chem. 2008;283:12293–12304. doi: 10.1074/jbc.M709493200. [DOI] [PubMed] [Google Scholar]
- 25.Rabien A, Fritzsche F, Jung M, Diamandis EP, Loening SA, Dietel M, Jung K, Stephan C, Kristiansen G. High expression of KLK14 in prostatic adenocarcinoma is associated with elevated risk of prostate-specific antigen relapse. Tumour Biol. 2008;29:1–8. doi: 10.1159/000132565. [DOI] [PubMed] [Google Scholar]
- 26.Reid JC, Bennett NC, Stephens CR, Carroll ML, Magdolen V, Clements JA, Hooper JD. In vitro evidence that KLK14 regulates the components of the HGF/Met axis, pro-HGF and HGFactivator inhibitor 1A and 1B. Biol Chem. 2016;397:1299–1305. doi: 10.1515/hsz-2016-0163. [DOI] [PubMed] [Google Scholar]
- 27.Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, Pandolfi PP, Saad F, Sonenberg N. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci U S A. 2010;107:14134–14139. doi: 10.1073/pnas.1005320107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oguic R, Mozetic V, Cini Tesar E, Fuckar Cupic D, Mustac E, Dordevic G. Matrix metalloproteinases 2 and 9 immunoexpression in prostate carcinoma at the positive margin of radical prostatectomy specimens. Patholog Res Int. 2014;2014:262195. doi: 10.1155/2014/262195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chinni SR, Sivalogan S, Dong Z, Filho JC, Deng X, Bonfil RD, Cher ML. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12. Prostate. 2006;66:32–48. doi: 10.1002/pros.20318. [DOI] [PubMed] [Google Scholar]
- 30.Wiesner C, Bonfil RD, Dong Z, Yamamoto H, Nabha SM, Meng H, Saliganan A, Sabbota A, Cher ML. Heterogeneous activation of MMP-9 due to prostate cancer-bone interaction. Urology. 2007;69:795–799. doi: 10.1016/j.urology.2007.01.093. [DOI] [PubMed] [Google Scholar]
- 31.Benaud C, Dickson RB, Lin CY. Regulation of the activity of matriptase on epithelial cell surfaces by a blood-derived factor. Eur J Biochem. 2001;268:1439–1447. doi: 10.1046/j.1432-1327.2001.02016.x. [DOI] [PubMed] [Google Scholar]
- 32.Hooper JD, Bui LT, Rae FK, Harvey TJ, Myers SA, Ashworth LK, Clements JA. Identification and characterization of KLK14, a novel kallikrein serine protease gene located on human chromosome 19q13.4 and expressed in prostate and skeletal muscle. Genomics. 2001;73:117–122. doi: 10.1006/geno.2000.6490. [DOI] [PubMed] [Google Scholar]
- 33.Adams MN, Harrington BS, He Y, Davies CM, Wallace SJ, Chetty NP, Crandon AJ, Oliveira NB, Shannon CM, Coward JI, Lumley JW, Perrin LC, Armes JE, Hooper JD. EGF inhibits constitutive internalization and palmitoylation-dependent degradation of membrane-spanning procancer CDCP1 promoting its availability on the cell surface. Oncogene. 2015;34:1375–1383. doi: 10.1038/onc.2014.88. [DOI] [PubMed] [Google Scholar]
- 34.Scarman AL, Hooper JD, Boucaut KJ, Sit ML, Webb GC, Normyle JF, Antalis TM. Organization and chromosomal localization of the murine Testisin gene encoding a serine protease temporally expressed during spermatogenesis. Eur J Biochem. 2001;268:1250–1258. doi: 10.1046/j.1432-1327.2001.01986.x. [DOI] [PubMed] [Google Scholar]
- 35.Rawlings ND, Barrett AJ. Evolutionary families of peptidases. Biochem J. 1993;290:205–218. doi: 10.1042/bj2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neurath H, Walsh KA. Role of proteolytic enzymes in biological regulation (a review) Proc Natl Acad Sci U S A. 1976;73:3825–3832. doi: 10.1073/pnas.73.11.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoon H, Blaber SI, Li W, Scarisbrick IA, Blaber M. Activation profiles of human kallikreinrelated peptidases by matrix metalloproteinases. Biol Chem. 2013;394:137–147. doi: 10.1515/hsz-2012-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afar DE, Vivanco I, Hubert RS, Kuo J, Chen E, Saffran DC, Raitano AB, Jakobovits A. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001;61:1686–1692. [PubMed] [Google Scholar]
- 39.Netzel-Arnett S, Hooper JD, Szabo R, Madison EL, Quigley JP, Bugge TH, Antalis TM. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003;22:237–258. doi: 10.1023/a:1023003616848. [DOI] [PubMed] [Google Scholar]
- 40.Yamakoshi Y, Simmer JP, Bartlett JD, Karakida T, Oida S. MMP20 and KLK4 activation and inactivation interactions in vitro. Arch Oral Biol. 2013;58:1569–1577. doi: 10.1016/j.archoralbio.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagase H, Enghild JJ, Suzuki K, Salvesen G. Stepwise activation mechanisms of the precursor of matrix metalloproteinase 3 (stromelysin) by proteinases and (4-aminophenyl) mercuric acetate. Biochemistry. 1990;29:5783–5789. doi: 10.1021/bi00476a020. [DOI] [PubMed] [Google Scholar]
- 42.Ogata Y, Enghild JJ, Nagase H. Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J Biol Chem. 1992;267:3581–3584. [PubMed] [Google Scholar]
- 43.Shapiro SD, Fliszar CJ, Broekelmann TJ, Mecham RP, Senior RM, Welgus HG. Activation of the 92-kDa gelatinase by stromelysin and 4-aminophenylmercuric acetate. Differential processing and stabilization of the carboxyl-terminal domain by tissue inhibitor of metalloproteinases (TIMP) J Biol Chem. 1995;270:6351–6356. doi: 10.1074/jbc.270.11.6351. [DOI] [PubMed] [Google Scholar]
- 44.Desrivieres S, Lu H, Peyri N, Soria C, Legrand Y, Menashi S. Activation of the 92 kDa type IV collagenase by tissue kallikrein. J Cell Physiol. 1993;157:587–593. doi: 10.1002/jcp.1041570319. [DOI] [PubMed] [Google Scholar]
- 45.Sang QX, Birkedal-Hansen H, Van Wart HE. Proteolytic and non-proteolytic activation of human neutrophil progelatinase B. Biochim Biophys Acta. 1995;1251:99–108. doi: 10.1016/0167-4838(95)00086-a. [DOI] [PubMed] [Google Scholar]
- 46.Peeters-Joris C, Hammani K, Singer CF. Differential regulation of MMP-13 (collagenase-3) and MMP-3 (stromelysin-1) in mouse calvariae. Biochim Biophys Acta. 1998;1405:14–28. doi: 10.1016/s0167-4889(98)00094-9. [DOI] [PubMed] [Google Scholar]
- 47.Murphy G, Nagase H. Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J. 2011;278:2–15. doi: 10.1111/j.1742-4658.2010.07918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–826. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 49.Morrissey C, True LD, Roudier MP, Coleman IM, Hawley S, Nelson PS, Coleman R, Wang YC, Corey E, Lange PH, Higano CS, Vessella RL. Differential expression of angiogenesis associated genes in prostate cancer bone, liver and lymph node metastases. Clin Exp Metastasis. 2008;25:377–388. doi: 10.1007/s10585-007-9116-4. [DOI] [PubMed] [Google Scholar]
- 50.Klokk TI, Kilander A, Xi Z, Waehre H, Risberg B, Danielsen HE, Saatcioglu F. Kallikrein 4 is a proliferative factor that is overexpressed in prostate cancer. Cancer Res. 2007;67:5221–5230. doi: 10.1158/0008-5472.CAN-06-4728. [DOI] [PubMed] [Google Scholar]
- 51.Berman DM, Epstein JI. When is prostate cancer really cancer? Urol Clin North Am. 2014;41:339–346. doi: 10.1016/j.ucl.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 52.Epstein JI. Prostatic ductal adenocarcinoma: a mini review. Med Princ Pract. 2010;19:82–85. doi: 10.1159/000252842. [DOI] [PubMed] [Google Scholar]
- 53.Bravo-Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Curr Opin Cell Biol. 2012;24:277–283. doi: 10.1016/j.ceb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Redondo-Munoz J, Ugarte-Berzal E, Terol MJ, Van den Steen PE, Hernandez del Cerro M, Roderfeld M, Roeb E, Opdenakker G, Garcia-Marco JA, Garcia-Pardo A. Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia b cell survival through its hemopexin domain. Cancer Cell. 2010;17:160–172. doi: 10.1016/j.ccr.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 55.Stefanidakis M, Bjorklund M, Ihanus E, Gahmberg CG, Koivunen E. Identification of a negatively charged peptide motif within the catalytic domain of progelatinases that mediates binding to leukocyte beta 2 integrins. J Biol Chem. 2003;278:34674–34684. doi: 10.1074/jbc.M302288200. [DOI] [PubMed] [Google Scholar]
- 56.Bjorklund M, Heikkila P, Koivunen E. Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J Biol Chem. 2004;279:29589–29597. doi: 10.1074/jbc.M401601200. [DOI] [PubMed] [Google Scholar]
- 57.Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Desai B, Ma T, Zhu J, Chellaiah MA. Characterization of the expression of variant and standard CD44 in prostate cancer cells: identification of the possible molecular mechanism of CD44/MMP9 complex formation on the cell surface. J Cell Biochem. 2009;108:272–284. doi: 10.1002/jcb.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spronk HM, Govers-Riemslag JW, ten Cate H. The blood coagulation system as a molecular machine. Bioessays. 2003;25:1220–1228. doi: 10.1002/bies.10360. [DOI] [PubMed] [Google Scholar]
- 60.Vadivel K, Bajaj SP. Structural biology of factor VIIa/tissue factor initiated coagulation. Front Biosci (Landmark Ed) 2012;17:2476–2494. doi: 10.2741/4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ragno P. The urokinase receptor: a ligand or a receptor? Story of a sociable molecule. Cell Mol Life Sci. 2006;63:1028–1037. doi: 10.1007/s00018-005-5428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Motta G, Tersariol ILS. Modulation of the plasma kallikrein-kinin system proteins performed by heparan sulfate proteoglycans. Front Physiol. 2017;8:481. doi: 10.3389/fphys.2017.00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rolland T, Ta An M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, Kamburov A, Ghiassian SD, Yang X, Ghamsari L, Balcha D, Begg BE, Braun P, Brehme M, Broly MP, Carvunis AR, Convery-Zupan D, Corominas R, Coulombe-Huntington J, Dann E, Dreze M, Dricot A, Fan C, Franzosa E, Gebreab F, Gutierrez BJ, Hardy MF, Jin M, Kang S, Kiros R, Lin GN, Luck K, MacWilliams A, Menche J, Murray RR, Palagi A, Poulin MM, Rambout X, Rasla J, Reichert P, Romero V, Ruyssinck E, Sahalie JM, Scholz A, Shah AA, Sharma A, Shen Y, Spirohn K, Tam S, Tejeda AO, Trigg SA, Twizere JC, Vega K, Walsh J, Cusick ME, Xia Y, Barabasi AL, Iakoucheva LM, Aloy P, De Las Rivas J, Tavernier J, Calderwood MA, Hill DE, Hao T, Roth FP, Vidal M. A proteome-scale map of the human interactome network. Cell. 2014;159:1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]