

Abstract
The present study aimed to explore the role of CXCR4 and protein C system (PCS) in the experimental ulcerative colitis (UC). The expression of CXCR3, CCR10, and CXCR4 in dextran sulfate sodium (DSS)-induced colitis mouse model was measured by immunohistochemistry and western blot analysis. In vitro studies with microvascular endothelial cells (MVECs) were performed. The expression of endothelial protein C receptor (EPCR) and thrombomodulin (TM) were detected by RT-PCR and western blot analysis. Activities of protein C (PC), protein S (PS), activated PC (APC) were evaluated in cells pre-treated with JNK inhibitor SP600125 and c-Jun silencing. DSS mice showed up-regulated expression of CXCR4, higher macroscopic score and histological score (P<0.05), as well as elevated levels of SDF-1α (P<0.05) compared with wild type, CXCR4-/-, or CXCR4-/- +DSS mice. In DSS mice, EPCR expression was down-regulated (P<0.05), accompanied by decreased activity of PC and PS (P<0.05 or P<0.01) with an up-regulated expression of pJNK MAPK and pc-Jun (P<0.05). Moreover, the macroscopic score and histological score index, SDF-1α levels, EPCR expression, PC activity, pJNK, and pc-Jun were reversed in CXCR4-/- +DSS mice (P<0.05). In vitro, SDF-1α-induced inhibition of the PCS was blunted by SP600125 (P<0.05). Meanwhile, down-regulation of c-Jun rescued the inhibition of PCS (P<0.05). MVECs with retrovirus-mediated transfection of c-Jun demonstrated a strong trans-inactivation effect on the EPCR promoter (P<0.05). These findings suggest that CXCR4 is involved in UC pathogenesis and could be a promising therapeutic target for UC treatment.
Keywords: CXCR4, protein C, ulcerative colitis
Introduction
Ulcerative colitis (UC) is the main type of inflammatory bowel disease (IBD) and an unexplained chronic, relapsing inflammation of the digestive tract that alternate with periods of exacerbations and remissions [1]. It is characterized by persistent diarrhea, pain, abdominal cramps, rectal bleeding, loss of weight, but these symptoms vary from one patient to another [1]. The pathogenesis of UC involves a combination of genetic, microbial, immunological, and environmental factors [2], though the underlying mechanisms are not yet fully elucidated.
Recent studies have highlighted that IBD pathogenesis is not mediated by immune cells alone, but also involves microvascular endothelial cells (MVECs) in the process [3-5]. As the pathophysiology of UC involves inflammation and coagulation, a hypercoagulative state is often manifested for micro thrombus formation and microcirculation disturbance in UC patients [6-8]. Endothelial cell damage plays an important role in activating coagulation through protein C system (PCS) [9]. PCS is a complex and effective physiological mechanism of anticoagulation. It is composed of protein C (PC), protein S (PS), endothelial protein C receptor (EPCR), protein C inhibitor (PCI), and thrombomodulin (TM) [10]. PCS plays an important role in regulating intestinal homeostasis, both in human intestinal MVECs and colitis animal models [11-13]. Therefore, PCS might be one of the factors that mediate the pathogenesis of UC, indicating the importance of it to delineate the exact mechanisms underlying in UC pathology and to further identify novel drug targets.
A recent study revealed the inhibitory activity of PCS in dextran sulfate sodium (DSS)-induced UC mice, suggesting a possible mechanism of UC [14]. Furthermore, in DSS-induced mouse colitis, colonic CD14+CD64+ macrophages produced inflammatory cytokines and chemokines in response to immune complex stimulation, resulting in the inhibition of PC pathway via MVECs [14]. This indicated that PCS inhibition in UC involved the interaction between macrophages and MVECs. But how chemokines exert their effects through MVEC is not yet fully elucidated.
Chemokine receptors are a class of G protein-coupled receptors, which play an important role in the inflammatory cells of injured or infected organs. The expression of chemokine receptors was up-regulated in the active stage of UC [15-19]. Uo et al. reported that CXCR4+ IgG plasma cells mediated the activation of CD14+ macrophages via Fcγ receptor, which might lead to UC [20]. CXCR4 expression was significantly up-regulated in patients with UC compared to controls [21]. Animal experiments also showed that CXCR4 antagonist, AMD3100, reduced colon injury in mice [22], and blocking of CXCL12/CXCR4 was beneficial in improving experimental colitis in rodent models [23]. Moreover, a recent study have pointed out that CXCR4 overexpression leads to mobilization and implantation of bone marrow mesenchymal stem cells in the inflammatory colon in rats with TNBS, and acts as a component of anti-inflammatory and immune regulation [24]. Nevertheless, the exact role of CXCR4 in UC is still not yet defined.
Previous studies have revealed that over-activation of mitogen-activated protein kinases (MAPKs) and cascade pathways are involved in numerous physiological and pathological signaling processes. Moreover, MAPKs respond to cellular proliferation and apoptosis regulation and play an important role in the inflammatory process of IBD and progression of UC [25]. The extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 kinases showed extensive cross-talks with other inflammatory and signaling pathways [26]. Hence, these inflammatory mediators are presented as new targets for treating anti-inflammation in inflammatory clinical conditions.
Therefore, the objective of the present study was to explore the role of CXCR4 in PCS changes in UC.
Material and method
Animals
Six-to-eight-week old C57BL/6J mice (specific pathogen-free, 22-24 g, n=78) were obtained from the Animal Center Laboratory of Henan Province. Mice were housed for 1 week under standard conditions (temperature, 23±2°C; humidity, 60%) with free access to water and food. Mice were fasted for 12 hours with free access to water before use. All experimental procedures were performed in accordance with international guidelines for the care and use of laboratory animals and approved by the Animal Ethics Committee of the Henan University School of Medicine, Kaifeng, China. The CXCR4-/- mice was constructed by inserting two loxP sites along with a neomycin cassette into the CXCR4 locus to flank exon 2 (CXCR4fl/wt), and crossed with Tie2-promoted Cre recombinase expression in transgenic mice (Tie2-Cre). Heterozygous mice carrying CXCR4 allele were then bred to CXCR4fl/wt-Cre+ mice.
Induction of experimental mouse colitis model and sampling
Experimental colitis was induced by administrating 4% DSS (Sigma, St Louis, MO, USA) in drinking water for 7 consecutive days (DSS group) according to Schicho et al. [27]. Physical condition, weight, and the presence of gross and occult blood in excreta and at the anus of the mice were examined daily.
At the end of the 7-day period, all the animals were sacrificed by decapitation under anesthesia with inhaled isoflurane. Blood specimens were collected and plasma was prepared by centrifugation at 12,000 g for 10 min at 4°C. Supernatants were collected and stored at -80°C. The colon (starting from 0.5 cm above the anus to the top of the cecum) segment was opened longitudinally, gently cleared of stool, rinsed with normal saline, and put on a Whatman paper for length measurement and macroscopic scoring. The colon was then divided into several portions. One of them was fixed in 10% neutrally-buffered formalin immediately, while the others were flash-frozen in liquid nitrogen and stored at -80°C before use.
Macroscopic scoring
The macroscopic score was documented based on the evaluation system described by Hartmann et al. [28]. In brief, the score consists of: 1) body weight loss from baseline (0% weight loss was scored as 0 point, 1-5% as 1 point, 5-10% as 2 points, 10-20% as 3 points, and >20% as 4 points); 2) stool consistency (0 point was for well formed pellets, 2 points for pasty stools, 3 points for semi-formed stools that did not stick to the anus, and 4 points for liquid stools that remained adhesive to the anus); and 3) bleeding (0 point for negative stool occult blood test, 2 points for positive occult blood test, and 4 points for gross bleeding from the rectum). These scores were summed up and divided by 3, resulting in a total clinical score ranging from 0 (healthy) to 4 (maximal score for DSS-induced colitis).
Histology evaluation
The colon portion fixed in 10% neutral buffered formalin was paraffin-embedded and tissue sections (5 μm) were stained with hematoxylin and eosin (H&E). Five sections at 50 μm apart per colon sample were evaluated in a blinded manner and scored according to Siegmund et al. [29]: 1) cell infiltration of inflammatory cells (0 point for no or rare inflammatory cells in the lamina-propria, 1 point for increased numbers of inflammatory cells, including neutrophils in the lamina propria, 2 points for confluence of inflammatory cells extending into the submucosa, and 3 points for transmural extension of the inflammatory cell infiltrate); and 2) epithelial damage (0 point for no mucosal damage, 1 point for discrete focal epithelial lesions, 2 points for mucosal erosion/ulceration, and 3 points for extensive mucosal damage and/or extension through deeper structures of the bowel wall). The two sub-scores were summed up and the combined histological score ranged from 0 (no changes) to 6 (highest score with extensive cell infiltration and tissue damage).
Immunohistochemistry
De-waxed and hydrated paraffin sections were blocked for endogenous peroxidase ablation by treating with 3% H2O2. After antigen retrieval by microwave, the expression and localization of EPCR and TM in the colon of mice were determined by adding primary antibodies against CXCR4, CXCR3, or CCR10 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:25, 1:50, and 1:50 dilution, respectively). DAB was used for staining. Image analysis of semi-quantitative data from five sections for each specimen was accomplished using the digital Motic Med 6.0 system (Motic, Wetzlar, Germany).
Western blot
Colon tissues were homogenized in lysis buffer containing protease inhibitors (250 μl/20 mg; Beyotime Institute of Biotechnology, Haimen, China) as previously described [30]. The protein concentration was assessed using BCA kit (Pierce Chemical, Dallas, TX, USA), and then were subjected to SDS-PAGE (10-12%) and then transferred onto nitrocellulose membranes. The primary rabbit anti-CXCR3 (1:1000; Abcam, Cambridge, MA, USA), anti-CCR10 (1:1000; Abcam, Cambridge, MA, USA), anti-CXCR4 (1:1000; Abcam, Cambridge, MA, USA), anti-EPCR (1:25; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-TM (1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-pJNK (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-pP38 (1:400; Santa Cruz Bio-technology, Santa Cruz, CA, USA), anti-pERK (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-pc-Jun antibody (1:1000; Abcam, Cambridge, MA, USA) were used along with the loading control of β-actin antibody (1:1000; Abcam, Cambridge, MA, USA). The appropriate secondary antibody HRP-conjugated affinity purified goat anti-rabbit, or rabbit anti-goat, or rabbit anti-mouse lgG was applied at 1:5000 dilution and incubated for 1.5 hours at room temperature. Images were taken with Bio-Rad imaging system (Bio-Rad, Hercules, CA, USA) and analyzed by image analysis system for the optical density of the protein bands.
Enzyme-linked immunosorbent assay
Plasma levels of SDF-1α were determined by commercially available mouse-specific enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocols. Each sample was measured in duplicate using a microplate reader, and data were presented as pg/ml plasma.
Immunoturbidimetry
STAGO automatic blood coagulation analyzer microtubes were used to test the activity of PC and PS by immunoturbidimetry according to the manufacturer’s instructions.
Isolation and culture of colon mucosa MVECs
Colon mucosa MVECs were isolated according to Scaldaferri et al. [31] study. In brief, mice colon tissues were digested by using 0.25% of trypsin, and then cultured in the MCDB131 medium supplemented with 20% FBS, 1×105 IU/L of penicillin, 0.1 g/L of streptomycin, heparin, endothelial cell growth factor, and 0.584 g/L of glutamine for 96 h. After trypsin digestion, the cell suspension was collected and cultured in fresh medium for 72 h. Pure cells were obtained, digested using a cell dissociation buffer, and cultured in fresh medium through the determination of coagulation factor VIII. The third generation cells were used for the subsequent experiments.
siRNA interference
Three pairs of c-Jun gene hairpin oligonucleotide sequences were designed using the web-based siRNA Target Designer-Version 1.51 software (http://www.invitrogen.com), and synthesized by Invitrogen Inc. (Carlsbad, CA, USA). c-Jun knockdown was performed by transfecting MVECs with nonspecific or c-Jun-specific siRNA according to the transfection kit LipofectamineTM 2000 instructions (Invitrogen Inc., Carlsbad, CA, USA). The control and blank groups were performed at the same time. The interference efficiency of the three stealth siRNA to c-Jun was detected and compared. Subsequent experiments were performed using siRNA with the highest interference efficiency. Each index was averaged with parallel wells, and the experiments were repeated at least 5 times in order to explore the survival and functional change of colon MVECs under the interference of the c-Jun gene.
Cells were grouped as normal MVECs as control, SDF-1α-treated cells, cells pretreated with JNK inhibitor, SP600125, and cells treated with c-Jun-siRNA for silencing c-Jun expression.
Transfection and luciferase assay
An upstream fragment (approximately 1100 bp) of EPCR proximal promoters was generated (Table 1) by polymerase chain reaction (PCR) and was subsequently cloned into pGL3.0 firefly luciferase reporter (Promega, Madison, WI, USA) using human genomic DNA as a template in accordance with the manufacturer’s instructions. Cells were infected with c-Jun-expressing retroviruses (Addgene, Cambridge, MA, USA; #40348) or control retroviral vector, pMIEG3, and were sorted for GFP expression as previously described [32]. Transfections and luciferase experiments were performed as described by Zhang et al. [33]. The experiments were performed at least three times in triplicate.
Table 1.
Primers for luciferase assay
| Sequences | |
|---|---|
| EPCR | |
| Forward | 5’-agcgcaaggagaacgtgt-3’ |
| Reverse | 5’-gggttcagagccctcctc-3’ |
| GAPDH | |
| Forward | 5’-tgtgtccgtcgtggatctga-3’ |
| Reverse | 5’-cctgcttcaccaccttcttga-3’ |
Flow cytometry
About 100 μl of colon mucosal MVECs (1×106/ml) from both control and DSS mice were subjected to flow cytometry analysis. After digestion by trypsin, the cell debris were used for testing the expression of CXCR4, CXCR3, and CCR10 (eBioscience, San Diego, CA, USA) by flow cytometry (Bricyte E6; Mindray Medical, Shenzhen, China).
Real time-PCR (RT-PCR)
Total RNA was extracted from mouse colon mucosal MVECs with TRIZOL reagent (Invitrogen Inc., Carlsbad, CA, USA). RNA was converted to cDNA, and cDNA reaction mixture was amplified in an ABI7500 system (Applied Biosystems, Foster City, CA, USA) according to the instructions of the SYBR Primix Ex TaqTM kit (Takara Bio, Otsu, Japan). The parameters were as follows: 1) 95°C for 30 s; and 2) 40 cycles of 95°C for 5 s and 60°C for 31 s. The primers are shown in Table 2.
Table 2.
Primers for RT-PCR
| Sequences | |
|---|---|
| EPCR | |
| Forward | 5’-agcgcaaggagaacgtgt-3’ |
| Reverse | 5’-gggttcagagccctcctc-3’ |
| TM | |
| Forward | 5’-atgcgtggagcatgagtg-3’ |
| Reverse | 5’-ctggcatcgaggaaggtc-3’ |
| GAPDH | |
| Forward | 5’-accacagtccatgccatcac-3’ |
| Reverse | 5’-tccaccaccctgttgctgta-3’ |
Chromogenic substrate assay
According to Zhang et al. [33], colon mucosal MVECs were incubated with SDF-1α for 24 h and washed with TBS (50 mmol/L Tris-HCl, 120 mmol/L NaCl, 2.7 mmol/L KCl, 3 mg/mL BSA) thrice. Then, 200 nmol/L PC (Enzyme Research Laboratories, South Bend, IN, USA) and 0.6 U/mL of TM were added and incubated for 1 h at 37°C. The chromogenic substrate (S-2366; Chromogenix, Instrumentation Laboratory, Bedford, MA, USA) was applied by microplate reader to test the activity of the activated PC (APC) protein.
Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM). All data were analyzed by one-way ANOVA followed by SNK post hoc test. SPSS 13.0 software (SPSS, Chicago, IL, USA) was used for statistical analysis. Two-sided P-values <0.05 were considered statistically significant.
Results
CXCR4 was significantly up-regulated in UC and CXCR4 deletion could partly delay the progression of DSS-induced colitis
The expression levels of CXCR3 and CCR10 were similar in both control as well as DSS mice, while a strong positive immunostaining was observed for CXCR4 in DSS mice, and was mainly located at the vascellum (Figure 1A). In addition, the expression of CXCR4 was significantly up-regulated in DSS mice (P<0.01, Figure 1B and 1C), while no changes were observed for CXCR3 and CCR10 compared to control mice (P>0.05, Figure 1B and 1C). These data suggested that CXCR4 was significantly up-regulated after the establishment of UC. To determine whether CXCR4 was involved in the mice with UC, CXCR4 knockout mice were generated. After intake of 4% DSS as drinking water for several days, mice developed severe colitis, and suffered weight loss (P<0.01, Figure 2A), shortened colon length (P<0.05, Figure 2B), and increased spleen weight (P<0.05, Figure 2C). There was an obvious increase in the macroscopic scores (P<0.05, Figure 2D) with bloody stools, which typically started on day 3-4 and continued till the end of the experimental period (data not shown). This severe colitis was alleviated in CXCR4-/- mice treated with DSS, mainly manifesting as higher weight, longer colon, lower spleen weight (P<0.05), and lower macroscopic score compared with DSS mice (P<0.05). These data suggested that CXCR4 was significantly up-regulated in UC and that CXCR4 deletion could partly delay the progression of DSS-induced colitis.
Figure 1.
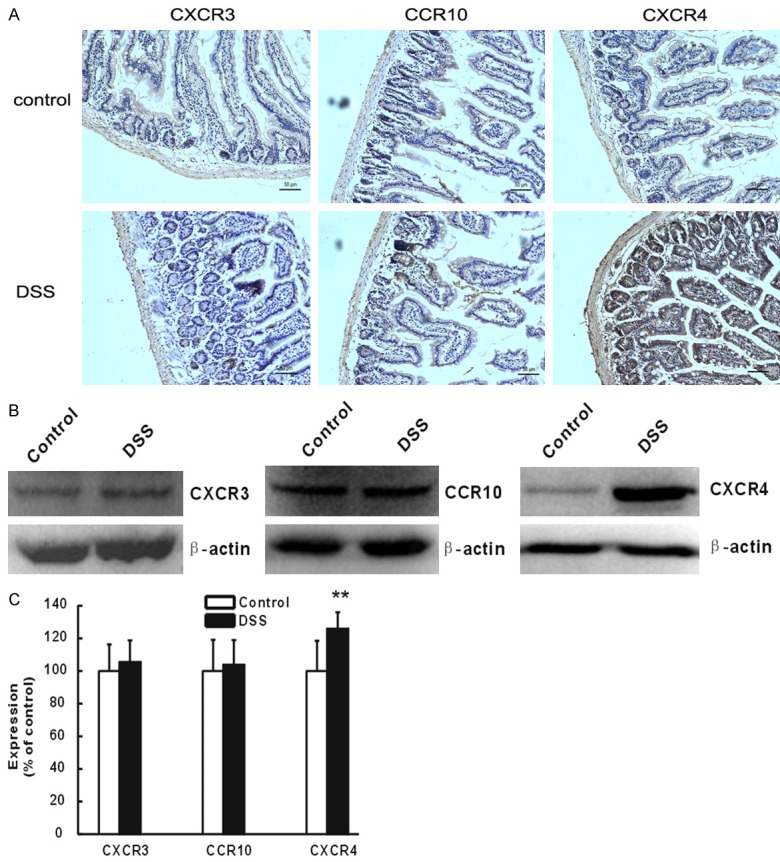
CXCR4, not CXCR3 and CCR10, was highly expressed in DSS-induced ulcerative colitis (UC) mice. A. The expression of CXCR3, CXCR4, and CCR10 were measured by immunohistochemistry 7 days after DSS-induced UC and control mice. B. Protein expression levels of CXCR3, CXCR4, and CCR10 were detected by western blot in DSS-induced UC and control mice. C. Densitometric quantification of protein levels of CXCR3, CXCR4, and CCR10 was performed and normalized to β-actin. **P<0.01 vs. control.
Figure 2.
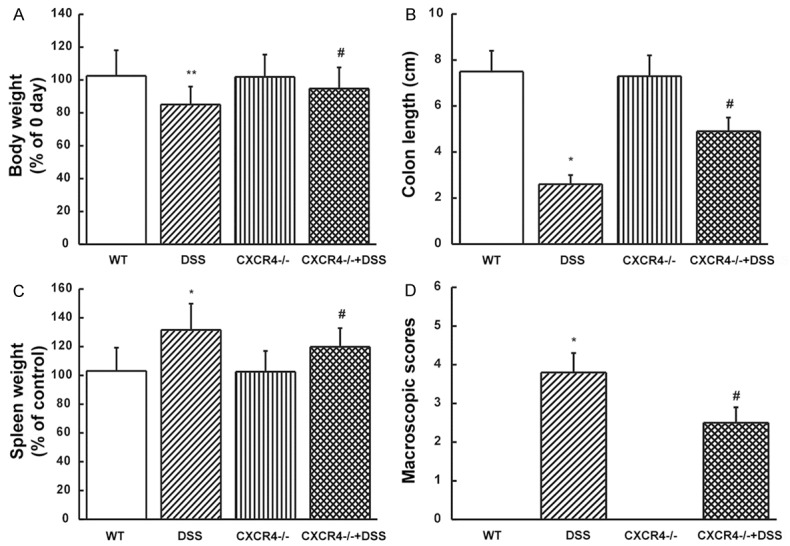
CXCR4 knock-down delayed the progression of DSS-induced colitis. (A) Body weight, (B) colon length, (C) spleen weight, and (D) macroscopic scores in wild type (WT), DSS-induced (DSS), CXCR4 knockout (CXCR4-/-), and DSS-induced CXCR4 knockout (CXCR4-/- +DSS) mice were measured. *P<0.05, **P<0.01 vs. WT. #P<0.05 vs. DSS.
Histological examinations revealed that both wild type and CXCR4-/- mice showed no signs of colitis (Figure 3A and 3C), while DSS mice showed obvious colitis with characteristic ulcers, open sores, multiple erosive lesions, and dropouts of entire crypts in the colon, as well as a marked infiltration of inflammatory cells into the colonic sub-mucosa (Figure 3B). Hence, a significant higher histological score was observed compared with the wild type group (P<0.05, Figure 3E). In CXCR4-/- mice, treatment with DSS induced an attenuated degree of inflammation compared to wild-type mice induced with DSS (P<0.05, Figure 3D and 3E). To further evaluate the degree of systemic inflammation, the plasma levels of SDF-1α, a marker of inflammatory infiltration, were assessed. SDF-1α levels increased significantly when the mice were challenged with DSS (P<0.05), while the SDF-1α levels of CXCR4-/- +DSS mice were lower compared to DSS mice (P<0.05, Figure 3F). These data suggested that CXCR4 silencing could attenuate DSS-induced inflammation.
Figure 3.

CXCR4 deletion attenuated colitis and inflammatory signs in DSS induced ulcerative colitis (UC) mice. A. Wild type (WT) mice showed no signs of colitis and inflammation. B. DSS-induced UC mice (DSS) showed signs of colitis with marked infiltration of inflammatory cells into the submucosa of the colon. C. CXCR4 knockout mice (CXCR4-/-) showed no signs of colitis and inflammation. D. CXCR4-/- mice treated with DSS (CXCR4-/- +DSS) showed attenuated degree of infiltration of inflammatory cells and signs of colitis. E. Histological scores. F. Plasma levels of SDF-1α. *P<0.05 vs. WT. #P<0.05 vs. DSS.
CXCR4 deletion reversed the activity of PCS inhibited by UC
EPCR is the main receptor of PC, and TM combines with thrombin (to form the thrombin-TM-PC complex with the help of Ca2+) to inactivate the coagulation factor [34,35]. Western blot results indicated that EPCR was significantly decreased in the colon of DSS mice compared to the wild type controls (P<0.05, Figure 4A and 4B). EPCR expression was up-regulated in CXCR4-/- +DSS mice when compared to DSS mice (P<0.05, Figure 4A and 4B). TM was not changed either in DSS or CXCR4-/- +DSS mice (P>0.05, Figure 4C and 4D). DSS mice showed significantly decreased PC (P<0.01) and PS (P<0.05) activities, which are another two indexes of PCS, while in CXCR4-/- +DSS mice, this inhibition effect was reversed for PC activity (P<0.05, Figure 4E and 4F). These data suggested that PCS activity was inhibited by UC, and deletion of CXCR4 reversed the inhibition of PCS activity.
Figure 4.
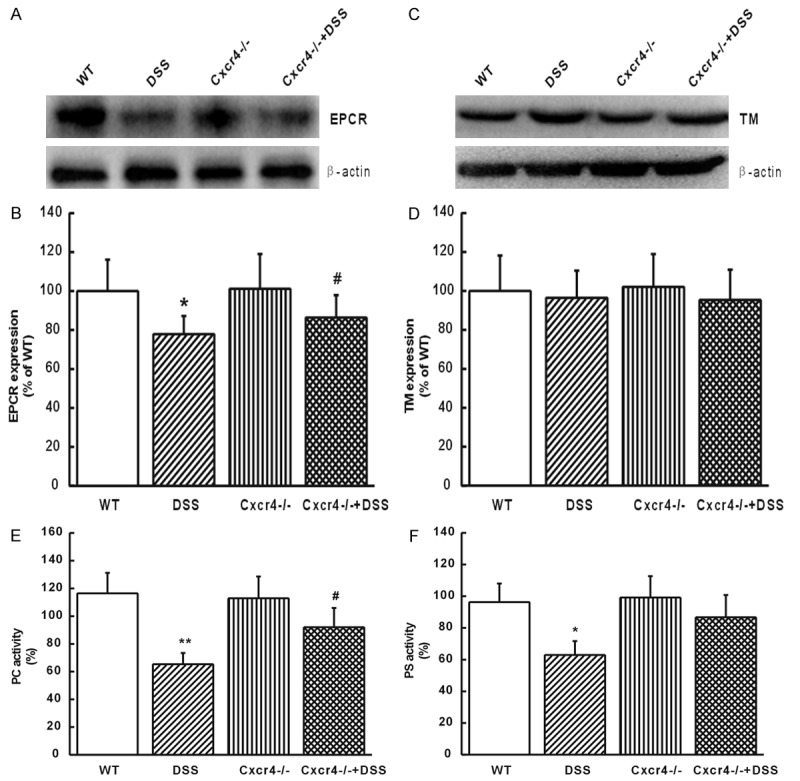
CXCR4 deletion reversed the activity of PCS inhibited by UC. (A) EPCR expression in the colon mucosal cells of WT, DSS, CXCR4-/-, and CXCR4-/- +DSS mice. (B) Protein levels of EPCR were densitometrically quantified and normalized to β-actin. (C) The expression of TM in the colon mucosal cells of WT, DSS, CXCR4-/-, and CXCR4-/- +DSS mice. (D) Protein levels of TM were quantified densitometrically and normalized to β-actin. (E) PC activity and (F) PS activity in WT, DSS, CXCR4-/-, and CXCR4-/- +DSS mice. *P<0.05, **P<0.01 vs. WT. #P<0.05 vs. DSS.
CXCR4 deletion could reverse the expression of pJNK and pc-Jun
Inflammation was determined by the activation of intracellular signaling pathway. Studies revealed that over-activation of MAPK was related to UC progression, suggesting new targets for treating inflammation [36,37]. Results showed that pP38 and pERK MAPKs were not influenced by DSS or CXCR4-/- +DSS, while in UC mice, pJNK and pc-Jun were up-regulated (P<0.05, Figure 5A-D). These enhancements were decreased in the CXCR4-/- +DSS group (P<0.05). These data suggested that CXCR4 deletion could reverse the expression of pJNK/MAPK pathway in UC.
Figure 5.
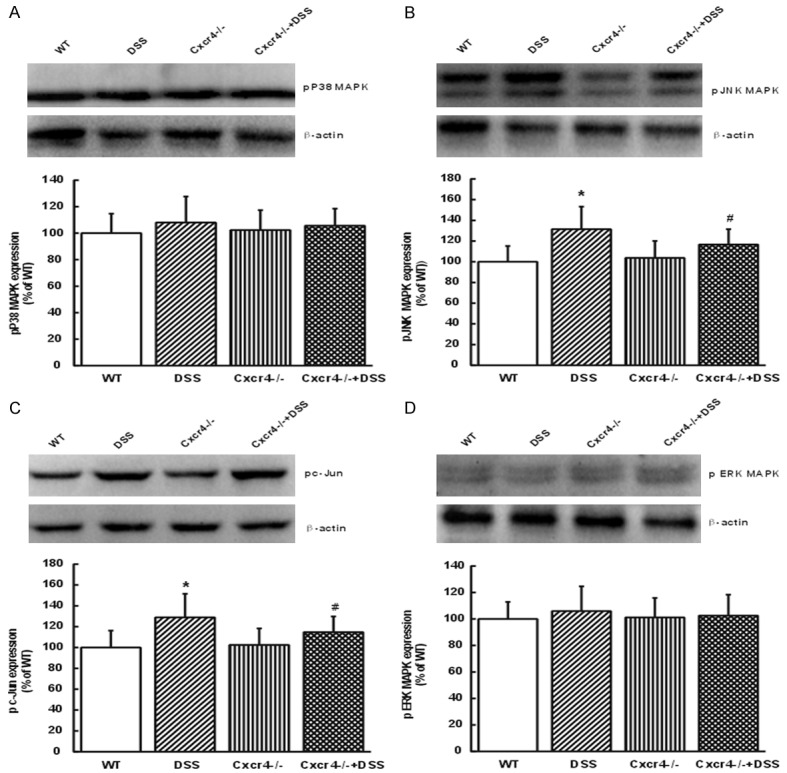
CXCR4 deletion reversed the expression of pJNK and pc-Jun. The protein expression levels of pP38 (A), pJNK (B), pcJun (C), and pERK (D) in WT, DSS, CXCR4-/-, and CXCR4-/- +DSS mice. Protein levels were densitometrically quantified and normalized to β-actin. *P<0.05 vs. WT. #P<0.05 vs. DSS.
CXCR4 was significantly up-regulated in SDF-1α-treated colonic mucosal MVECs
To further determine the role of CXCR4 in UC, SDF-1α was used in vitro to mimic the effects of chemokines on MVECs. Normal colon mucosal MVECs showed cobblestone-like changes, and the positive expression of factor VIII implied the success of cell culture (Figure 6A). Flow cytometry results showed that after stimulation with SDF-1α, CXCR3 and CCR10 expression levels in MVECs were not changed, but CXCR4 was significantly up-regulated (P<0.01, Figure 6B). These data suggested that in vitro CXCR4 was also up-regulated when MVECs were treated with SDF-1α.
Figure 6.

CXCR4 was significantly up-regulated in SDF-1α-treated colonic mucosal MVECs. A. Normal colon mucosal MVECs showed cobblestone-like changes, and positive expression of factor VIII implied the success of cell culture. B. Flow cytometry analysis was performed for the expression levels of CXCR3, CCR10 and CXCR4 after the stimulation of MVECs with SDF-1α. **P<0.01 vs. controls.
Inhibition of JNK MAPK pathway reversed the inhibited activity of PCS by SDF-1α in colon mucosal MVECs
When the cells were pretreated with SDF-1α, the expression of EPCR in normal colon mucosal MVECs was significantly down-regulated (P<0.05 or P<0.01, Figure 7A and 7C), and there was no significant change in TM expression at the mRNA and protein levels (Figure 7B and 7D). To verify whether c-Jun was involved in SDF-1α-induced inhibition of EPCR expression, colon mucosal MVECs were pretreated with 50 nM SP600125 (JNK inhibitor) or SDF-1α-treated c-Jun siRNA cells. Interestingly, SDF-1α-induced inhibition of EPCR expression was blunted by JNK inhibitor and c-Jun siRNA both at the mRNA (P<0.05, Figure 7A) and protein (P<0.05, Figure 7C) levels. These findings suggested that SDF-1α-induced inhibition of EPCR expression was mediated by activation of JNK/c-Jun signaling pathway. Meanwhile, the activity of PC, PS, and APC was also inhibited by SDF-1α (P<0.05, Figure 7F-H), which was reversed by SP600125 or c-Jun siRNA (P<0.05, Figure 7F-H). These data suggested that inhibition of JNK MAPK pathway reversed the inhibited activity of PCS by SDF-1α in colon mucosal MVECs.
Figure 7.
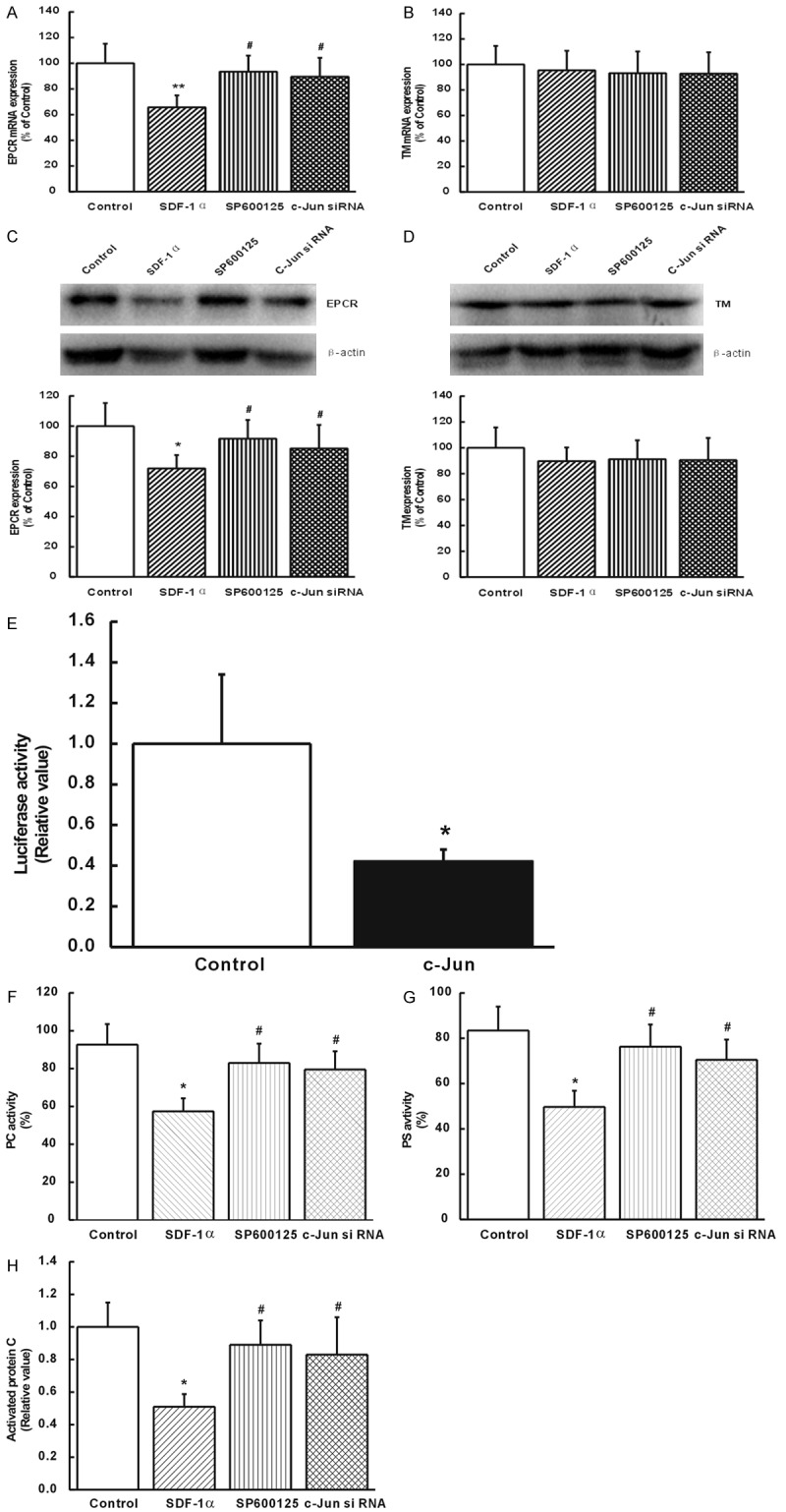
Inhibition of JNK MAPK pathway reversed the inhibited activity of PCS by SDF-1α in colon mucosal MVECs. (A) The mRNA expression levels of Endothelial protein C receptor (EPCR) and (B) thrombomodulin (TM). (C) Protein expression of EPCR and (D) TM. (E) Luciferase assays revealed that c-Jun regulated EPCR by binding to its promoter. The activity of (F) protein C (PC), (G) protein S (PS) and (H) activated PC (APC). *P<0.05 vs. controls. #P<0.05 vs. SDF-1α. Results were obtained from at least three independent experiments performed in triplicate.
c-Jun regulated EPCR by binding to its promoter
In order to understand how c-Jun regulates EPCR, we detected its effects on the promoter activity of EPCR. Results revealed a strong trans-inactivation effect on the EPCR promoter that took place after cells were infected with retroviruses containing c-Jun (P<0.05, Figure 7E). These data suggested that c-Jun regulated EPCR by binding to its promoter.
Discussion
In the present study, we found that CXCR4 was significantly up-regulated and was involved in the pathogenesis of UC. Moreover, deletion of CXCR4 could partly delay the progression of DSS-induced colitis. These results implied that CXCR4 might serve as a novel target for UC treatment.
Previous studies have shown that abnormal coagulation function was closely related to UC activity [34-36]. Generally, PCS serves as the most complex and the most effective physiological mechanism of anticoagulation. Since PCS is an important mediator of vascular endothelial function, and MVECs are the most important non-immune cells in UC, it can be speculated that the PCS plays an important role in the pathological process of UC. A previous study by our team has confirmed that in DSS-induced UC mice, plasma cells and macrophages of colon tissue act upon the mucosal MVECs by secreting inflammatory cytokines and chemokines, and further inhibit the PCS [14]. But the mechanism of how chemokines act on mucosal microvascular endothelial functions and affect the PCS is still unknown.
Uo et al. recently demonstrated that CXCR4 could play a role in UC [20]. Therefore, in order to explore the involvement of CXCR4 in the regulation of PCS in UC, we used both gene knockout mice and ShRNA cells. Results showed that deletion of CXCR4 could partly delay the progression of DSS-induced colitis. These results are supported by a previous study which showed that a CXCR4 antagonist reduced colon injury in mice [22], while another study showed that blocking CXCL12/CXCR4 pathway was beneficial in UC rodents [23]. In addition, our research revealed that deletion of CXCR4 reversed the inhibition of PCS in UC. CXCR4 overexpression leads to increased mobilization of mesenchymal stem cells into the colon, where these cells play a role in inflammatory processes [24]. Taken together, these results suggested that CXCR4 was involved in the pathogenesis of UC, and it may serve as a novel target for UC treatment.
Recently, it has been shown that the JNK MAPK-c-Jun pathway was involved in IBD and UC [25]. Furthermore, ERK, JNK, and p38 are key players in inflammatory processes [26,37-39]. To investigate the mechanism of signal transduction of CXCR4 on MVECs, we examined the activities of pP38, pJNK, pERK, and pc-Jun in mice. Results suggested that pP38 and pERK are probably not involved in UC (at least in the DSS model), but pJNK expression was up-regulated, and this enhancement was inhibited to some extent in the CXCR4 knockout DSS model. As expected, pc-Jun also showed similar results. Furthermore, in the present study, SDF-1α-mediated inhibition of PCS activity was mainly manifested by the down-regulation of EPCR expression, and reduction of PC, PS, and APC activities. All these inhibitions were reversed by JNK MAPK inhibitor, SP600125, and blunted by c-Jun knockdown. Taken together, these results strongly suggested that induction of JNK/c-Jun pathway was involved in the inhibitory effects of SDF-1α on the activity of PCS, and the effects of CXCR4 could be mediated by JNK MAPK-c-Jun pathway.
As the promoter region of EPCR contains a c-Jun-binding TGAGTCA motif, it was speculated that activated c-Jun induced by SDF-1α could act as a transcriptional factor and bind to the promoter region of EPCR to trans-inactivate the expression of EPCR [33]. This hypothesis was confirmed by the luciferase activity, and verified by the indication that JNK-specific inhibitor, SP600125, and c-Jun silencing rescued the reduction of EPCR. These results are supported by a recent study, which showed that free fatty acids could inhibit EPCR expression through JNK pathway in human umbilical vein endothelial cells (HUVECs) [40]. Similarly, another study by Zhang et al. [33] also showed that c-Jun silencing or the use of a JNK inhibitor could attenuate the effects of Aβ1-42 on the activation of PC in mouse brain primary endothelial cells.
Despite of these interesting results, the present study still has some limitations. Only a few mechanisms and inflammatory cytokines were examined, and further comprehensive studies are necessary to get a better grasp on the mechanisms involved in UC. In addition, these results were obtained in mouse models, and further research should be carried out before entering into the clinical trials. Further investigation on specific characterization of these chemokines with their regulatory roles on the mediators of immune response provides a more complete picture regarding the underlying mechanisms involved in UC and other IBDs.
In conclusion, the present study suggested that CXCR4 may influence the function of MVECs in the course of UC, and further inhibit PCS. The mechanism involves JNK MAPK-c-Jun pathway, and acts as a potential therapeutic target for UC treatment.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81500430 and U1304802). The authors thank Dr. Xuequn Ren and Guanchang Cheng for their help in providing reagents/materials/analysis tools.
Disclosure of conflict of interest
None.
References
- 1.Gonzalez-Ramirez AE, Gonzalez-Trujano ME, Orozco-Suarez SA, Alvarado-Vasquez N, Lopez-Munoz FJ. Nerol alleviates pathologic markers in the oxazolone-induced colitis model. Eur J Pharmacol. 2016;776:81–89. doi: 10.1016/j.ejphar.2016.02.036. [DOI] [PubMed] [Google Scholar]
- 2.Zatorski H, Salaga M, Zielinska M, Piechota-Polanczyk A, Owczarek K, Kordek R, Lewandowska U, Chen C, Fichna J. Experimental colitis in mice is attenuated by topical administration of chlorogenic acid. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:643–651. doi: 10.1007/s00210-015-1110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lust M, Vulcano M, Danese S. The protein C pathway in inflammatory bowel disease: the missing link between inflammation and coagulation. Trends Mol Med. 2008;14:237–244. doi: 10.1016/j.molmed.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Kasper JY, Hermanns MI, Cavelius C, Kraegeloh A, Jung T, Danzebrink R, Unger RE, Kirkpatrick CJ. The role of the intestinal microvasculature in inflammatory bowel disease: studies with a modified Caco-2 model including endothelial cells resembling the intestinal barrier in vitro. Int J Nanomedicine. 2016;11:6353–6364. doi: 10.2147/IJN.S92608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danese S, Fiocchi C. Endothelial cell-immune cell interaction in IBD. Dig Dis. 2016;34:43–50. doi: 10.1159/000442925. [DOI] [PubMed] [Google Scholar]
- 6.Zoller B, Li X, Sundquist J, Sundquist K. Autoimmune diseases and venous thromboembolism: a review of the literature. Am J Cardiovasc Dis. 2012;2:171–183. [PMC free article] [PubMed] [Google Scholar]
- 7.Tolstanova G, Deng X, French SW, Lungo W, Paunovic B, Khomenko T, Ahluwalia A, Kaplan T, Dacosta-Iyer M, Tarnawski A, Szabo S, Sandor Z. Early endothelial damage and increased colonic vascular permeability in the development of experimental ulcerative colitis in rats and mice. Lab Invest. 2012;92:9–21. doi: 10.1038/labinvest.2011.122. [DOI] [PubMed] [Google Scholar]
- 8.Senchenkova E, Seifert H, Granger DN. Hypercoagulability and platelet abnormalities in inflammatory bowel disease. Semin Thromb Hemost. 2015;41:582–589. doi: 10.1055/s-0035-1556590. [DOI] [PubMed] [Google Scholar]
- 9.Lin XH, Guo JL, Wen YQ, Li YX, Wei DD, Yang RL, Mu XY, Wang HC. [Role of IgG plasma cells in the change of protein C system in ulcerative colitis] . Sheng Li Xue Bao. 2017;69:172–182. [PubMed] [Google Scholar]
- 10.D’Alessio S, Genua M, Vetrano S. The protein C pathway in intestinal barrier function: challenging the hemostasis paradigm. Ann N Y Acad Sci. 2012;1258:78–85. doi: 10.1111/j.1749-6632.2012.06557.x. [DOI] [PubMed] [Google Scholar]
- 11.Griffin JH, Zlokovic BV, Mosnier LO. Protein C anticoagulant and cytoprotective pathways. Int J Hematol. 2012;95:333–345. doi: 10.1007/s12185-012-1059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 13.Lin X, Wei D, Wang H, Yu Z, Li Y, Li Y, Wen Y, Ren X, Cheng G. Ulcerate colitis and protein C system: is there a link of causality. Int J Colorectal Dis. 2016;31:1255–1256. doi: 10.1007/s00384-015-2453-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin XH, Wang HC, Wei DD, Wang B, Ge QX, Bai CY, Wang YQ, Ren XQ. [Study of the change and role of protein C system in ulcerate colitis] . Sheng Li Xue Bao. 2015;67:214–224. [PubMed] [Google Scholar]
- 15.Papadakis KA, Prehn J, Zhu D, Landers C, Gaiennie J, Fleshner PR, Targan SR. Expression and regulation of the chemokine receptor CXCR3 on lymphocytes from normal and inflammatory bowel disease mucosa. Inflamm Bowel Dis. 2004;10:778–788. doi: 10.1097/00054725-200411000-00013. [DOI] [PubMed] [Google Scholar]
- 16.Oki M, Ohtani H, Kinouchi Y, Sato E, Nakamura S, Matsumoto T, Nagura H, Yoshie O, Shimosegawa T. Accumulation of CCR5+ T cells around RANTES+ granulomas in Crohn’s disease: a pivotal site of Th1-shifted immune response? Lab Invest. 2005;85:137–145. doi: 10.1038/labinvest.3700189. [DOI] [PubMed] [Google Scholar]
- 17.Rivera-Nieves J, Ho J, Bamias G, Ivashkina N, Ley K, Oppermann M, Cominelli F. Antibody blockade of CCL25/CCR9 ameliorates early but not late chronic murine ileitis. Gastroenterology. 2006;131:1518–1529. doi: 10.1053/j.gastro.2006.08.031. [DOI] [PubMed] [Google Scholar]
- 18.Ranganathan P, Jayakumar C, Manicassamy S, Ramesh G. CXCR2 knockout mice are protected against DSS-colitis-induced acute kidney injury and inflammation. Am J Physiol Renal Physiol. 2013;305:F1422–1427. doi: 10.1152/ajprenal.00319.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gren ST, Grip O. Role of monocytes and intestinal macrophages in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2016;22:1992–1998. doi: 10.1097/MIB.0000000000000824. [DOI] [PubMed] [Google Scholar]
- 20.Uo M, Hisamatsu T, Miyoshi J, Kaito D, Yoneno K, Kitazume MT, Mori M, Sugita A, Koganei K, Matsuoka K, Kanai T, Hibi T. Mucosal CXCR4+ IgG plasma cells contribute to the pathogenesis of human ulcerative colitis through FcgammaR-mediated CD14 macrophage activation. Gut. 2013;62:1734–1744. doi: 10.1136/gutjnl-2012-303063. [DOI] [PubMed] [Google Scholar]
- 21.Hosomi S, Oshitani N, Kamata N, Sogawa M, Okazaki H, Tanigawa T, Yamagami H, Watanabe K, Tominaga K, Watanabe T, Fujiwara Y, Maeda K, Hirakawa K, Arakawa T. Increased numbers of immature plasma cells in peripheral blood specifically overexpress chemokine receptor CXCR3 and CXCR4 in patients with ulcerative colitis. Clin Exp Immunol. 2011;163:215–224. doi: 10.1111/j.1365-2249.2010.04290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia XM, Wang FY, Xu WA, Wang ZK, Liu J, Lu YK, Jin XX, Lu H, Shen YZ. CXCR4 antagonist AMD3100 attenuates colonic damage in mice with experimental colitis. World J Gastroenterol. 2010;16:2873–2880. doi: 10.3748/wjg.v16.i23.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mikami S, Nakase H, Yamamoto S, Takeda Y, Yoshino T, Kasahara K, Ueno S, Uza N, Oishi S, Fujii N, Nagasawa T, Chiba T. Blockade of CXCL12/CXCR4 axis ameliorates murine experimental colitis. J Pharmacol Exp Ther. 2008;327:383–392. doi: 10.1124/jpet.108.141085. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Zuo D, Fan H, Tang Q, Shou Z, Cao D, Zou Z. Over-expression of CXCR4 on mesenchymal stem cells protect against experimental colitis via immunomodulatory functions in impaired tissue. J Mol Histol. 2014;45:181–193. doi: 10.1007/s10735-013-9541-4. [DOI] [PubMed] [Google Scholar]
- 25.Sun J, Zhang H, Guan L, Zhou H, Sun M. Alpha-lipoic acid attenuates trinitrobenzene sulfonic acid-induced ulcerative colitis in mice. Int J Clin Exp Med. 2015;8:358–367. [PMC free article] [PubMed] [Google Scholar]
- 26.Waetzig GH, Seegert D, Rosenstiel P, Nikolaus S, Schreiber S. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol. 2002;168:5342–5351. doi: 10.4049/jimmunol.168.10.5342. [DOI] [PubMed] [Google Scholar]
- 27.Schicho R, Bashashati M, Bawa M, McHugh D, Saur D, Hu HM, Zimmer A, Lutz B, Mackie K, Bradshaw HB, McCafferty DM, Sharkey KA, Storr M. The atypical cannabinoid O-1602 protects against experimental colitis and inhibits neutrophil recruitment. Inflamm Bowel Dis. 2011;17:1651–1664. doi: 10.1002/ibd.21538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartmann G, Bidlingmaier C, Siegmund B, Albrich S, Schulze J, Tschoep K, Eigler A, Lehr HA, Endres S. Specific type IV phosphodiesterase inhibitor rolipram mitigates experimental colitis in mice. J Pharmacol Exp Ther. 2000;292:22–30. [PubMed] [Google Scholar]
- 29.Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta-converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li K, Feng JY, Li YY, Yuece B, Lin XH, Yu LY, Li YN, Feng YJ, Storr M. Anti-inflammatory role of cannabidiol and O-1602 in cerulein-induced acute pancreatitis in mice. Pancreas. 2013;42:123–129. doi: 10.1097/MPA.0b013e318259f6f0. [DOI] [PubMed] [Google Scholar]
- 31.Scaldaferri F, Sans M, Vetrano S, Graziani C, De Cristofaro R, Gerlitz B, Repici A, Arena V, Malesci A, Panes J, Grinnell BW, Danese S. Crucial role of the protein C pathway in governing microvascular inflammation in inflammatory bowel disease. J Clin Invest. 2007;117:1951–1960. doi: 10.1172/JCI31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang ZY, Sato H, Kusam S, Sehra S, Toney LM, Dent AL. Regulation of IL-10 gene expression in Th2 cells by Jun proteins. J Immunol. 2005;174:2098–2105. doi: 10.4049/jimmunol.174.4.2098. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Huang L, Lu G, Ge L, Hong Y, Hu Z. Amyloid beta suppresses protein C activation through inhibition of the endothelial protein C receptor (EPCR) J Mol Neurosci. 2014;52:117–123. doi: 10.1007/s12031-013-0123-4. [DOI] [PubMed] [Google Scholar]
- 34.Gan GH, Yang LM, Wang J. Thrombotic mechanism and anticoagulant therapy in inflammatory bowel disease patients. World Chinese Journal of Digestology. 2016;02 [Google Scholar]
- 35.Faioni EM, Ferrero S, Fontana G, Gianelli U, Ciulla MM, Vecchi M, Saibeni S, Biguzzi E, Cordani N, Franchi F, Bosari S, Cattaneo M. Expression of endothelial protein C receptor and thrombomodulin in the intestinal tissue of patients with inflammatory bowel disease. Crit Care Med. 2004;32:S266–270. doi: 10.1097/01.ccm.0000128032.85396.83. [DOI] [PubMed] [Google Scholar]
- 36.Elboudwarej O, Wei J, Siegel R. Diagnosis of an aortic valvular lesion. Non-bacterial thrombotic endocarditis (NBTE) Heart. 2015;101:719, 726. doi: 10.1136/heartjnl-2014-306723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coskun M, Olsen J, Seidelin JB, Nielsen OH. MAP kinases in inflammatory bowel disease. Clin Chim Acta. 2011;412:513–520. doi: 10.1016/j.cca.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 38.Broom OJ, Widjaya B, Troelsen J, Olsen J, Nielsen OH. Mitogen activated protein kinases: a role in inflammatory bowel disease? Clin Exp Immunol. 2009;158:272–280. doi: 10.1111/j.1365-2249.2009.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abarikwu SO. Kolaviron, a natural flavonoid from the seeds of Garcinia kola, reduces LPSinduced inflammation in macrophages by combined inhibition of IL-6 secretion, and inflammatory transcription factors, ERK1/2, NF-kappaB, p38, Akt, p-c-JUN and JNK. Biochim Biophys Acta. 2014;1840:2373–2381. doi: 10.1016/j.bbagen.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Xie W, Zhai Z, Yang Y, Kuang T, Wang C. Free fatty acids inhibit TM-EPCR expression through JNK pathway: an implication for the development of the prothrombotic state in metabolic syndrome. J Thromb Thrombolysis. 2012;34:468–474. doi: 10.1007/s11239-012-0793-8. [DOI] [PubMed] [Google Scholar]