

Abstract
To explore the expression, clinical significance, biological function, and potential mechanism of MEF2D in pancreatic cancer, the expression of MEF2D in human pancreatic cancer tissues and corresponding adjacent normal tissues was analyzed through immunohistochemical staining. The association between MEF2D expression, clinicopathological parameters, overall survival, and disease-free survival was evaluated. Human pancreatic cancer cell lines BxPC-1 and SW1990 were selected to investigate the effect of MEF2D knockdown on cell proliferation, migration, and invasion. Western blot analysis was used to assess the effect of MEF2D expression on the Akt/GSK pathway, as well as the protein expression of cyclin B1, cyclin D1, matrix metalloprotein (MMP)-2, and MMP-9. Our results revealed that the expression of MEF2D was increased in pancreatic cancer tissues compared to adjacent normal tissues and the increased expression of MEF2D was associated with tumor size, histological differentiation, and TNM stage of pancreatic cancer patients. Moreover, the expression of MEF2D was an independent prognostic indicator for pancreatic cancer patients. In addition, knockdown of MEF2D in pancreatic cancer cells inhibited cell proliferation, migration, and invasion by down-regulating the protein expression of cyclin B1, cyclin D1, MMP-2, and MMP-9. Knockdown of MEF2D reduced the levels of phosphorylated Akt and GSK-3β. Our data indicated that MEF2D expression was increased in pancreatic cancer and was an independent molecular prognostic factor for pancreatic cancer patients. Furthermore, we showed that MEF2D controlled cell proliferation, migration, and invasion abilities in pancreatic cancer via the Akt/GSK-3β signaling pathway.
Keywords: Pancreatic cancer, Myocyte enhancer factor 2D (MEF2D), prognosis
Introduction
Pancreatic cancer is one of the most aggressive digestive system cancers and is associated with a poor prognosis. Worldwide, pancreatic cancer is the ninth most common malignant cancer and the fourth cause of cancer-related death [1]. Despite substantial progress in surgical techniques and other anti-cancer treatments, the treatment efficacy for pancreatic cancer has not significantly improved [2]. If untreated, the 5-year overall survival (OS) of pancreatic cancer is still approximately 5% [3].One of the main reasons for this dismal prognosis is the fact that the majority of the pancreatic cancer patients are diagnosed at an advanced stage, when the chance for effective surgical resection has been lost [4]. Furthermore, even those who undergo curative surgical resection will relapse within 2 years [5]. Pancreatic cancer is insensitive to almost all conventional chemotherapy and radiation regimens [6]. Therefore, it is urgent to understand potential mechanisms that contribute to the development of pancreatic cancer and identify reliable prognostic biomarkers and novel therapy targets.
Myocyte enhancer factor 2D (MEF2D), a transcription factor, belongs to the MEF2 family, which includes four different members in mammals: MEF2A, 2B, 2C, and 2D [7]. MEF2 family members were initially identified as vital transcription activators important in the development of muscle and heart [8], and subsequently found to be involved in the initiation and development of human cancers. The first evidence of the important role of MEF2D in human malignancy is from leukemia [9]. The MEF2D/DAZAP1 and DAZAP1/MEF2D fusion proteins resulting from chromosomal translocation in acute lymphoblastic leukemia (ALL) can promote the proliferation of ALL cells. Recently, studies showed that the overexpression of MEF2D can increase the proliferation of hepatocellular carcinoma (HCC) cells via promotion of VEGF signaling, and facilitation of HCC cell invasion and metastasis by promotion of the TGF-β auto-regulation circuitry [7,10]. However, the precise role of MEF2D in the initiation and progression of pancreatic cancer is still unclear.
In this study, multidisciplinary approaches were employed to explore the expression, biological function, and potential mechanism of MEF2D in pancreatic cancer.
Materials and methods
Patients and tissue specimens
In total, 98 paraffin-embedded human pancreatic cancer tissues and their corresponding adjacent normal tissues were collected from patients treated at Shanghai East Hospital, Tongji University, Shanghai China, from January 2006 to December 2010. All patient tissues were obtained before chemical, radiation, and other anti-cancer therapies. The diagnosis of pancreatic cancer was made based on the postoperative pathology evaluation. OS was calculated from the date of surgery to time of death, or the date of last known follow-up. Disease-free survival (DFS) was defined as the period between the date of surgery and the first recurrence or metastasis of pancreatic cancer. Detailed clinicopathological data were obtained from hospital medical records. All subjects signed written informed consent. The current study was approved by Clinical Research Ethics Committee of Shanghai East Hospital, Tongji University, Shanghai, China.
Immunohistochemistry (IHC)
Paraffin-embedded tissues were cut into 4-μm thick sections, deparaffinized in xylene, rehydrated through graded alcohols, and heat treated for 30 min in citrate buffer (pH 6.0; Dako, Carpinteria, CA, USA) for antigen retrieval. Endogenous peroxidase activity was blocked by 3% H2O2 for 10 min. Samples were blocked with 5% normal serum for 2 hours. Afterward, samples were incubated overnight with primary anti-MEF2D antibody (ab32845, Abcam, 1:500 dilution). The next day, the sections were incubated for 1 hour with the appropriate secondary antibody (ab6721, abcam, 1:1000 dilution). Finally, sections were observed under a light microscope. The immunostaining results were evaluated by two independent pathologists blinded to each patient’s status to avoid possible bias.
The staining intensity was scored as 0 points, negative; 1 point, weak intensity; 2 points, moderate intensity; or 3 points, strong intensity. Staining density was scored separately based on the percentage of cells stained as follows: 0 point, 0-5%; 1 point, 6-25%; 2 points, 26-50%; 3 points, more than 50%. The final score was calculated as the sum of the intensity score and the quantity score. Final scores ≥3 points were considered positive, while final scores <3 points were considered negative.
Cell culture
Human pancreatic cancer cell lines BxPC-3 and SW1990 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco). The cells were incubated in a humidified atmosphere at 37°C and 5% CO2.
Small interfering RNA (siRNA) and transient transfection
MEF2D special siRNA (Ruibo, Guangzhou China) was used to silence the MEF2D gene. A scrambled sequence siRNA (siNC) was used as a negative control (Ruibo). The siRNA transfection was optimized using Lipofectamine™-RNAimax (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. Briefly, Lipofectamine and siRNA were diluted separately in serum-free Opti-MEM (Gibco) and incubated at room temperature for 5 min. Afterward, the two solutions were gently mixed and incubated for 20 min. Finally, the mixture was added to plated cells, and after 2 days, the cells were analyzed using the following assays.
RNA isolation and Qrt-PCR analysis
Total RNA was isolated from cells with TRIzol RNA isolation reagent (Invitrogen) according to the manufacturer’s protocol. The qRT-PCR assay was performed as follows. Total cDNA was synthesized from 1 μg purifed RNA with the Reverse Transcription System (Promega, Madison, WI, USA) followed by amplification of MEF2D using the SYBR Premix ExTaq™ kit (Applied Biosystems, Foster City, CA, USA) and ABI PRISM 7500 Sequence Detection System (Applied Biosystems). The relative mRNA level of MEF2D was normalized to GAPDH and was calculated by the comparative Ct method [11]. The sequences of the primers were as follows: MEF2D forward, 5’-AGGGAAATAACCAAAAAACTACCAAA-3’; MEF2D reverse, 5’-GCTACATGAACACAAAAACAGAGACC-3’. GAPDH forward, 5’-AATCCCATCACCATCTTCCA-3’, GAPDH reverse 5’-TGGACTCCACGACGTACTCA-3’. PCR reaction conditions were: denaturation at 95°C for 10 min, followed by 40 PCR cycles at 95°C for 15 s and one cycle at 60°C for 1 min.
Cell proliferation assay
Cells were collected after trypsin treatment when they were in the exponential phase of growth. A 96-well plate was seeded with 2 × 103 cells/well and cultured at 37°C and 5% CO2. Ten μl Cell Counting Kit-8 (CCK-8, Dojindo, Japan) solution was added to individual wells after 24, 48, 72, and 96 hours respectively, and then incubated at 37°C for 1.5 h. The ELX-800 spectrometer reader (BioTek Instruments, Winooski, VT, USA) was used to measure the absorbance at 450 nm.
Plate clone formation assay
Cells were seeded into a 6-well culture plate (500 cells per well) and cultured in 8 ml complete medium for 1 week. Afterwards, the cells were washed twice with PBS, fixed with 4% paraformaldehyde, and stained with crystal violet. Finally, the number of colonies were counted.
Cell cycle analysis
Cells transfected with siNC or MEF2D siRNA were treated with 1 μg/ml aphidicolin. The medium containing aphidicolin (USA, Sigma) was removed after 12 hours and the cells were washed with PBS and incubated in fresh medium containing 50 ng/ml nocodazole (USA, Sigma) for 0, 6, and 12 h. Afterwards, the cells were fixed with 70% ethanol at 4°C overnight and stained with 40 μg/ml propidium iodide in hypotonic fluorochrome buffer for 0.5 h. Finally, the samples were analyzed using a FACSCanto flow cytometer (BD Biosciences, San Francisco, CA, USA). The cell distribution in different phases of the cell cycle was further analyzed with ModFit LT3.0 software.
Wound-healing assay
Six-well plates were seeded with 5 × 105 cells transfected with either MEF2D or control siRNA (Sigma, St. Louis, MO, USA) and incubated until 100% confluence was reached. The layer of cells was scratched with a 10 μl pipette tip (Sigma) and washed with PBS three times. The incubation was continued in fresh serum-free DMEM medium for 24 hours. The distance of cell migration was determined by microscopy.
Transwell migration and invasion assays
Migration assays were performed using a transwell chamber (Corning Inc., Corning, NY, USA). The upper compartment of the chamber was seeded with 1 × 105 BxPC-3 or SW1990 cells in 100 μl serum-free DMEM medium. The bottom compartment of the chamber contained 800 μl DMEM with 10% FBS (v/v), which was the source of chemoattractant. After a 24-h incubation at 37°C in 5% CO2, the non-migrating cells in the upper chamber were removed carefully using a cotton swab. The remaining cells that had traversed the membrane were fixed with 100% methanol and stained with crystal violet. An inverted microscope was adapted to count migrating cells in five randomly selected visual fields. Each assay was repeated at least three times.
Cell invasion was evaluated migration through transwell filters (Corning). The upper chambers of the transwell filters were coated with 100 μL of Matrigel (Corning) followed by 1 × 105 BxPC-3 and SW1990 cells. The medium in the lower chamber was the same as that in the transwell migration assay. After 24 h, filters non-migrating cells were carefully removed from the upper chamber with a cotton swab, and the filters were then fixed and stained as above. Cells that had invaded were counted in five randomly selected visual fields. Each assay was repeated at least three times.
Western blot analysis
Cells were washed twice with cold PBS and lysed with RIPA lysis buffer (radioimmunoprecipitation assay; Beyotime Institute of Biotechnology, China) containing PMSF (protease inhibitors phenylmethanesulfonyl fluoride) (Beyotime Institute of Biotechnology, Jiangsu, China). Protein concentration was determined by bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of Biotechnology). Equal amounts of protein (~30 μg) were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to 0.45 μm polyvinylidine difluoride filter (PVDF) membranes. The membranes were blocked with Tris-buffered saline-Tween 20 (TBST) containing 5% non-fat milk followed by overnight incubation at 4°C with primary antibodies. Next, the membranes were washed three times with TBST for at least 15 minutes, and probed with HRP-linked secondary antibodies for 2 h at room temperature. Protein bands were visualized using the enhanced chemiluminescence detection kit (Thermo Fisher Scientific, Waltham, MA, USA).
The primary antibodies and concentrations used were: MEF2D (ab93257; 1 μg/ml), Akt (ab18785; 2 μg/ml), p-Akt (ab38449; 1/500), GSK (ab32391; 1/5000), p-GSK (ab75745; 1/500), cyclin D1 (ab137875; 1/1000), cyclin B1 (ab32053; 1/3000l), MMP-2 (ab37150; 2 μg/ml), MMP-9 (ab38898; 1/1000) and β-actin (ab8227; 1/5000). The secondary antibody was anti-rabbit-IgG-HRP (926-32210 and 926-32211 licor, 1:5000 dilution). All primary antibodies were purchased from Abcam (Cambridge, UK).
Statistical analysis
Data were expressed as means ± SD. Two-factor analysis of variance procedures followed by Dunnett’s test for multiple comparisons were used to assess the differences within treatment groups. Chi square test was employed to evaluate the association between MEF2D expression and clinicopathological parameters, including age, gender, tumor size, and TNM stage. Survival curves were obtained utilizing the Kaplan-Meier method, and statistical evaluation of survival was analyzed by log-rank test. For cell experiments, Student’s t test was used to analyze the data. All analyses were performed using SPSS 19.0 software (IBM, Armonk, NY). Differences were considered significant when P<0.05 and all tests were two-sided.
Results
Increased expression of MEF2D in pancreatic cancer was associated with tumor size, histological differentiation, and TNM stage of pancreatic cancer patients
To assess whether MEF2D expression was altered in human pancreatic cancer, immunohistochemistry was used to evaluate the MEF2D expression in pancreatic cancer tissues and paired adjacent non-tumor tissues (Figure 1). In matched pancreatic cancer and adjacent non-cancerous tissues, the positive percentage of MEF2D expression was 72.4% (71/98) and 7.1% (7/98). There was significantly higher positive expression of MEF2D in pancreatic cancer compared to the adjacent non-tumor tissues (P<0.05) (Figure 2).
Figure 1.
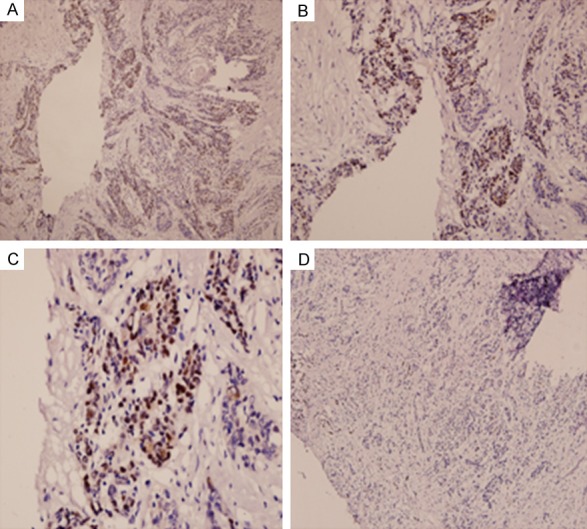
Representative images of MEF2D immunohistochemical staining in pancreatic cancer and adjacent non-cancer tissue. Pancreatic tumor tissue: A. Magnification × 100; B. Magnification × 200; C. Magnification × 400; Normal adjacent tissue: D. Magnification × 100.
Figure 2.
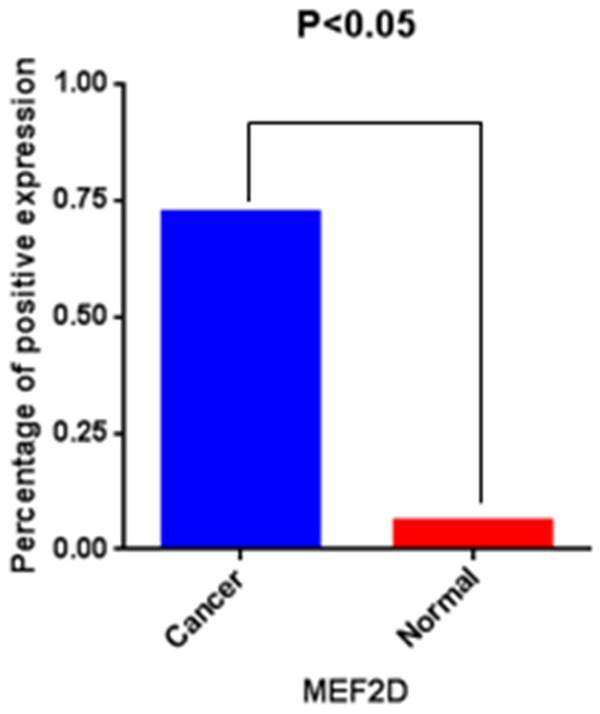
Expression of MEF2D in pancreatic cancer tissues compared to adjacent normal tissues. Results show a significant difference between the groups (Chi-square test; P<0.05 for both).
The association of MEF2D expression with patients’ clinical parameters is shown in Table 1. The results demonstrated that the increased expression of MEF2D in pancreatic cancer was significantly associated with tumor size (χ 2 = 5.567, P = 0.018), histological differentiation (χ 2 = 4.842, P = 0.028), and TNM stage (χ 2 = 5.892, P = 0.015). Nevertheless, there was no significant correlation between MEF2D expression and other clinical parameters, such as patient’s age, gender, tumor location, lymph node metastasis, and nerve invasion (P > 0.05 for all; Table 1).
Table 1.
Expression of MEF2D in relation to pathologic and clinical variables
| Clinicopathological parameters | N2 | MEF2D expression | χ2 | P value | |
|---|---|---|---|---|---|
|
| |||||
| + | - | ||||
| All | 98 | 71 | 27 | ||
| Age (years) | 0.038 | 0.845 | |||
| <60 | 56 | 41 | 15 | ||
| ≥60 | 42 | 30 | 12 | ||
| Gender | 0.155 | 0.694 | |||
| Female | 32 | 24 | 8 | ||
| Male | 66 | 47 | 19 | ||
| Tumor size | 5.567 | 0.018 | |||
| <4 cm | 70 | 46 | 24 | ||
| ≥4 cm | 28 | 25 | 3 | ||
| Histological differentiation | 4.842 | 0.028 | |||
| Well + moderate | 64 | 51 | 13 | ||
| Poor | 34 | 20 | 14 | ||
| Location | 0.092 | 0.762 | |||
| Head-neck | 63 | 45 | 18 | ||
| Body-tail | 35 | 26 | 9 | ||
| Lymph node metastasis | 2.592 | 0.107 | |||
| Absence | 60 | 40 | 20 | ||
| Presence | 38 | 31 | 7 | ||
| TNM stage | 5.892 | 0.015 | |||
| I-II | 57 | 36 | 21 | ||
| III-IV | 41 | 35 | 6 | ||
| Nerve invasion | 0.601 | 0.438 | |||
| Yes | 35 | 27 | 8 | ||
| No | 63 | 44 | 19 | ||
Increased expression of MEF2D was associated with poor prognosis of pancreatic cancer patients
We performed Kaplan-Meier analysis using the log-rank test to further evaluate the prognostic significance of MEF2D expression in pancreatic cancer patients. We found that patients with high MEF2D expression had worse OS and DFS than patients with low MEF2D expression (Figure 3). Simultaneously, univariate and multivariate Cox regression analyses were performed to determine whether MEF2D expression was an independent molecular prognostic indicator for pancreatic cancer patients. Our data revealed that the expression of MEF2D, together with TNM stage, was an independent prognostic indicator for pancreatic cancer patients (Table 2).
Figure 3.
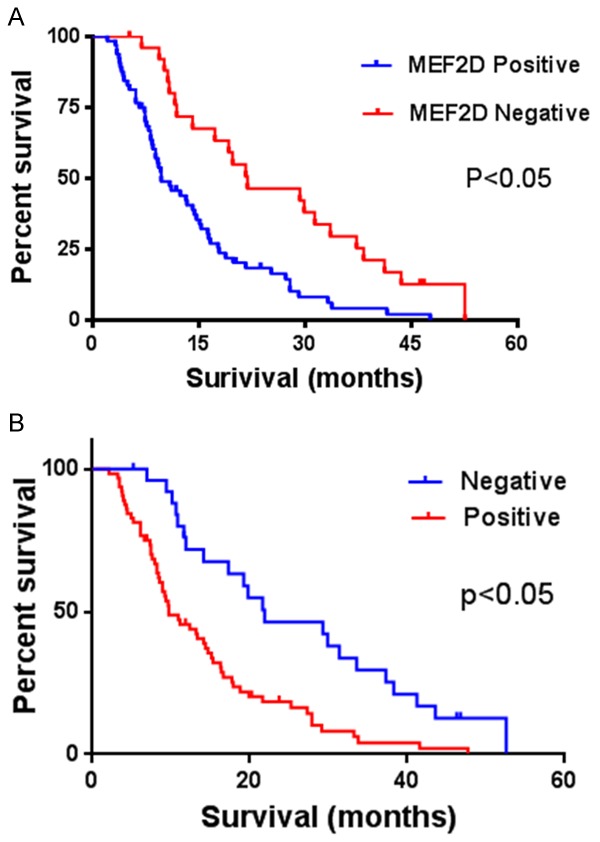
Kaplan-Meier log-rank survival analyses for (A), overall survival and (B), disease-free survival of pancreatic patients according to MEF2D expression.
Table 2.
Analysis of independent correlation factors of pancreatic cancer prognosis with Cox multivariate regression analysis. a, OS; b, DFS. OS: overall survival; DFS: disease-free surbvival
| a | |||||||
|
| |||||||
| Factors | B | S.E. | Ward | Sig. | Exp (B) | 95% CI for Exp (B) | |
|
| |||||||
| Lower | Upper | ||||||
|
| |||||||
| Age | -0.296 | 0.243 | 1.477 | 0.224 | 0.744 | 0.462 | 1.199 |
| Gender | 0.056 | 0.242 | 0.054 | 0.817 | 0.946 | 0.589 | 1.519 |
| Tumor size | -0.285 | 0.280 | 1.035 | 0.309 | 0.752 | 0.43 4 | 1.302 |
| Histological differentiation | 0.226 | 0.306 | 0.546 | 0.460 | 1.254 | 0.688 | 2.286 |
| Location | -0.465 | 0.268 | 3.012 | 0.083 | 0.628 | 0.372 | 1.062 |
| Lymph node metastasis | -0.325 | 0.312 | 1.089 | 0.297 | 0.722 | 0.392 | 1.331 |
| TNM stage | 2.336 | 0.349 | 33.892 | 0.007 | 10.019 | 5.220 | 20.469 |
| Nerve invasion | 0.017 | 0.265 | 0.004 | 0.949 | 1.017 | 0.605 | 1.709 |
| MEF2D | 0.877 | 0.313 | 5.416 | 0.023 | 2.172 | 1.446 | 4.165 |
|
| |||||||
| b | |||||||
|
| |||||||
| Factors | B | S.E. | Ward | Sig. | Exp (B) | 95% CI for Exp (B) | |
|
| |||||||
| Lower | Upper | ||||||
|
| |||||||
| Age | -0.385 | 0.244 | 2.495 | 0.114 | 0.680 | 0.422 | 1.097 |
| Gender | 0.114 | 0.242 | 0.224 | 0.636 | 1.121 | 0.698 | 1.800 |
| Tumor size | -0.164 | 0.285 | 0.334 | 0.563 | 0.848 | 0.486 | 1.482 |
| Histological differentiation | 0.511 | 0.302 | 2.863 | 0.091 | 1.667 | 0.922 | 3.014 |
| Location | -0.282 | 0.270 | 1.095 | 0.295 | 0.754 | 0.445 | 1.279 |
| Lymph node metastasis | -0.615 | 0.317 | 3.779 | 0.052 | 0.540 | 0.291 | 1.005 |
| TNM stage | 2.822 | 0.438 | 21.146 | 0.019 | 8.227 | 4.754 | 17.906 |
| Nerve invasion | 0.011 | 0.262 | 0.002 | 0.966 | 1.011 | 0.605 | 1.690 |
| MEF2D | 1.487 | 0.313 | 6.520 | 0.031 | 4.422 | 2.393 | 8.171 |
MEF2D siRNA knocked down MEF2D mRNA and protein expression
To investigate the role of MEF2D in pancreatic cancer, the expression of MEF2D was knocked down by MEF2D siRNA. Both BxPC-3 and SW1990 were transfected with a scrambled sequence siRNA (siNCtrl) and three different MEF2D siRNA. As shown in Figure 4, both the mRNA (Figure 4A) and the protein (Figure 4B) levels of MEF2D were inhibited significantly by MEF2D_siR1.
Figure 4.
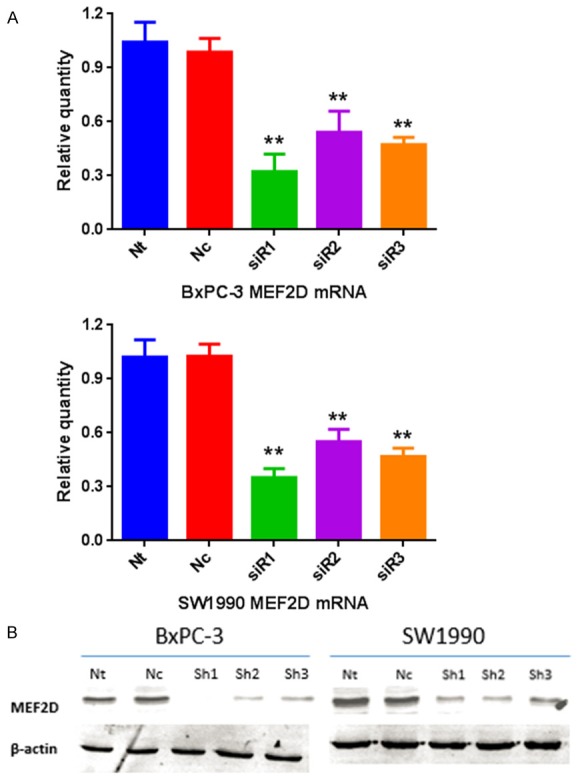
Real-time PCR and western blotting showed that the expression levels of MEF2D mRNA and protein were significantly down-regulated after MEF2D special siRNA infection (**P<0.01). Each experiment was repeated three times.
Down-regulation of MEF2D inhibited cell proliferation in pancreatic cancer
Since the expression level of MEF2D was drastically increased in pancreatic cancer compared to paired adjacent normal tissue, we explored the role of MEF2D in pancreatic cancer cell proliferation. After transient transfection of BxPC-3 and SW1990 cells with control siRNA or MEF2D siRNA1 for 36 hours, cells were subjected to cell proliferation assays and cell cycle analysis. In the CCK-8 cell proliferation assay, we found that the proliferation of BxPC-3 and SW1990 cells was significantly decreased after MEF2D knockdown (Figure 5A, 5B). Similarly, colony formation was decreased after knockdown of MEF2D (Figure 5C, 5D). Flow cytometry analysis was performed to explore whether inhibition of cell proliferation after MEF2D knockdown resulted from cell cycle arrest. Our results revealed that MEF2D knockdown cells have a significantly higher G1 population than control cells in both BxPC-3 and SW1990 cell lines (Figure 5E, 5F).
Figure 5.
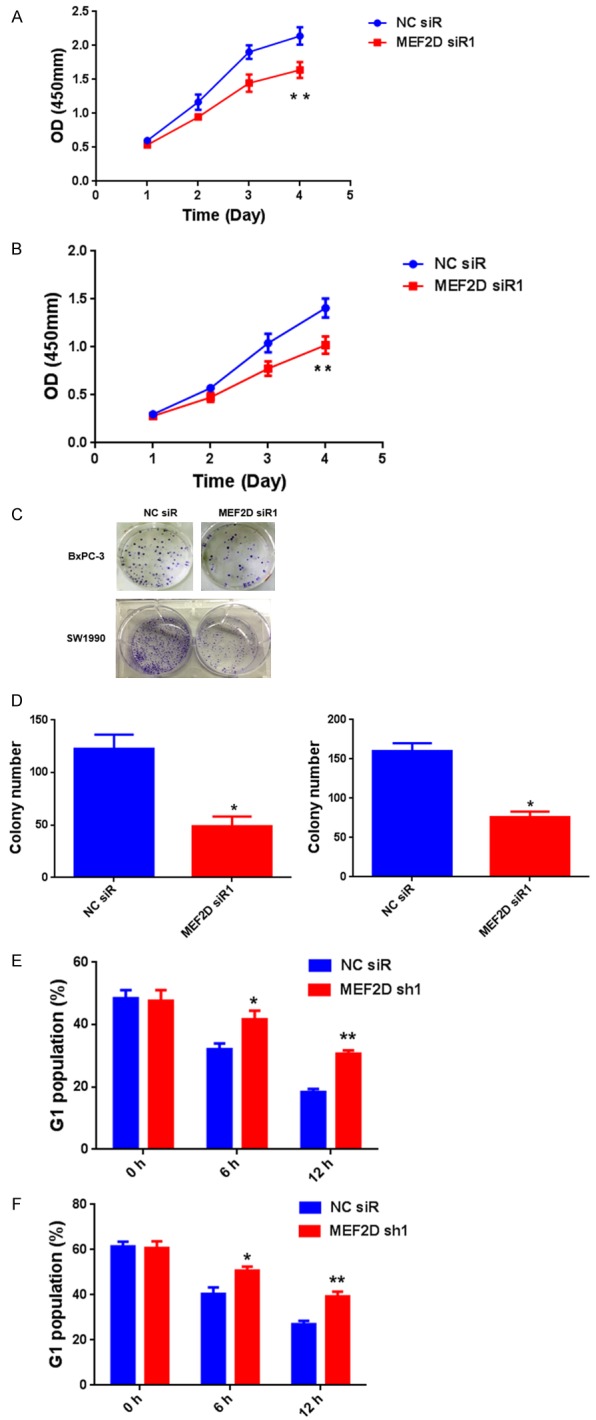
Knockdown of MEF2D in pancreatic cancer cells reduces cell proliferation. A and B. CCK-8 cell proliferation assay after MEF2D knockdown in BxPC-3 and SW1990 cells. C and D. Colony formation assays after MEF2D knockdown in BxPC-3 and SW1990 cells. E and F. The percentage of G1 population cells was measured by flow cytometry after MEF2D knockdown in BxPC-3 and SW1990 cells. *P<0.05, **P<0.01, ***P<0.001.
Down-regulation of MEF2D inhibited pancreatic cancer cell migration and invasion in vitro
Results of the wound-healing and transwell assays indicated that down-regulation of MEF2D expression remarkably inhibited the migration of BxPC-3 and SW1990 cells (Figure 6A-C). In addition, transwell assays showed that down-regulation of MEF2D expression markedly impaired the invasion properties of BxPC-3 cells and SW1990 cells (Figure 6A-F). Collectively, these results revealed that MEF2D may contribute to the progression of pancreatic cancer via enhancing cell migration and invasion.
Figure 6.
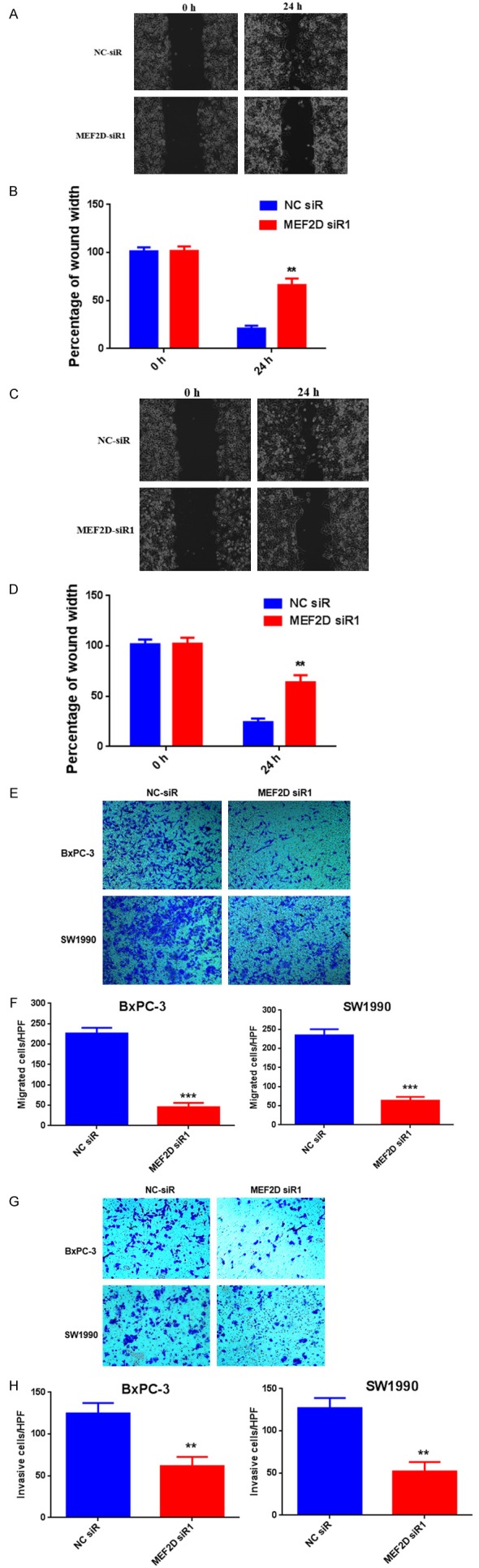
MEF2D knockdown inhibited the migration and invasion of pancreatic cancer cell lines. Wound-healing assay for migration in BxPC3 and SW1990 cells transfected with MEF2D siR1 or NC siR. A. Photomicrograph of the wound-healing assay from control NC siR1 and MEF2D siR1 transfected BxPC3 cells. B. Quantified data for the wound-healing assay that showing less migration by the MEF2D knockdown BxPC-3 cell lines compared to the control presented as the number of migrated cells per high-power field (**P<0.01). C. Photomicrograph of the wound-healing assay from control NC siR1 and MEF2D siR1 transfected SW1990 cells. D. Quantified data for the wound-healing assay that showing less migration by the MEF2D knockdown SW1990 cell lines compared to the control presented as the number of migrated cells per high-power field (**P<0.01). Effects of MEF2D on the cell migration by transwell chamber assay. E. photomicrograph showing crystal violet-stained filters from control NC siR1 and MEF2D siR1 transfected BxPC3 and SW1990 cells. F. Quantified data showing less migration in the MEF2D-knockdown cell lines compared to the control presented as the number of migrated cells per high-power field (***P<0.001). Effects of MEF2D on pancreatic cancer cell invasion by transwell chamber assay. G. Photomicrograph showing crystal violet-stained filters from control NC siR1 and MEF2D siR1 transfected BxPC3 and SW1990 cells. H. Quantified data showing less migration in the MEF2D-knockdown cell lines compared to the control presented as the number of migrated cells per high-power field (**P<0.01).
Down-regulation of MEF2D inhibited the Akt/GSK-3β pathway and suppressed cyclin D1, cyclin B1, and MMP-2/9 expression in pancreatic cancer cells
The Akt/GSK-3β pathway is constitutively active in many human cancers. To investigate whether MEF2D overexpression is accompanied by Akt/GSK-3β activation, the levels of total and phosphorylated Akt and GSK-3β were examined in human pancreatic cancer cells. As shown in Figure 7A, silencing of MEF2D did not obviously influence the total level of Akt although the level of phosphorylated Akt was significantly inhibited. Moreover, the reduction in phosphorylated Akt was accompanied by a significant reduction in the level of phosphorylated GSK-3β, an important downstream target protein of Akt, without obvious change in the total expression of GSK-3β (Figure 7A). These results suggested that overexpression of MEF2D activated the Akt/GSK-3β pathway in pancreatic cancer cells.
Figure 7.
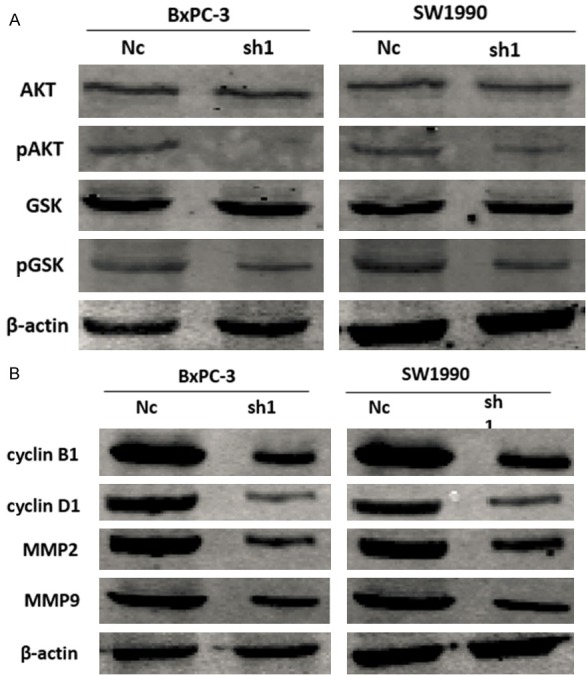
Overexpression of MEF2D activated the Akt/GSK-3β pathway in pancreatic cancer cells measured by western blot. A. The level of phosphorylated Akt and phosphorylated GSK-3β was significantly reduced after knockdown of MEF2D. Knockdown of MEF2D increased expression of markers of cell proliferation and invasion. B. Knockdown of MEF2D dramatically decreased the expression of cyclin D1, cyclin B1, MMP-2 and MMP-9.
It is well known that the cell cycle is strictly controlled by cyclins. We assessed the expression of cyclin D1 and cyclin B1 in pancreatic cancer cells and found that they were decreased upon knockdown of MEF2D (Figure 7B). The invasive capacity of cancer cells can be evaluated by expression of MMPs. Our results showed that knockdown of MEF2D remarkably decreased the protein levels of MMP-2 and MMP-9 (Figure 7B).
Discussion
In the current study, we investigated the expression profile of MEF2D in pancreatic cancer tissues. The results indicated that MEF2D was significantly overexpressed in pancreatic cancer tissues compared to the matched adjacent normal tissues. The increased expression of MEF2D was significantly associated with an aggressive tumor phenotype, larger tumor size, histological de-differentiation, and TNM stage. More importantly, the overexpression of MEF2D was significantly correlated with poor prognosis of pancreatic cancer patients. Univariate and multivariate Cox regression analysis demonstrated that MEF2D expression was an independent molecular prognostic factor for pancreatic cancer patients. These results revealed that the overexpression of MEF2D may be a common factor in the initiation and progression of pancreatic cancer and might serve as an important predictor for poor prognosis of pancreatic cancer patients.
Consistent with this study, overexpression of MEF2D also has been identified in other malignances, including hepatocellular carcinoma, osteosarcoma, gastric cancer, and colorectal cancer [12-15]. Ma et al. showed that overexpression of MEF2D is associated with shorter survival of hepatocellular carcinoma patients. Yu et al. revealed that the overexpression of MEF2D is significantly correlated with shorter survival and can serve as an independent prognostic factor of osteosarcoma cancer patients.
Because overexpression of MEF2D was significantly associated with the aggressive characteristics of pancreatic cancer, we down-regulated the expression level of MEF2D in BxPC-3 and SW1990 cell lines using siRNA to explore the biological function of MEF2D in cell proliferation, migration, and invasion. As expected, our in vitro study showed that knockdown of MEF2D in BxPC-3 and SW1990 markedly suppressed human pancreatic cancer cell proliferation, migration, and invasion. The results from flow cytometry analysis further confirmed that reduced cell proliferation after MEF2D knockdown was due to G1 phase arrest. The G1 phase is regulated by cyclin D1 and the G2/M phase is controlled by cyclin B1 [16]. MEF2D knockdown down-regulated the expression of cyclin D1 and therefore induced cell cycle arrest in G1. In addition, MEF2D knockdown subsequently caused the decrease in number of cells in G2/M and down-regulated cyclin B1. Thus, MEF2D might promote proliferation of pancreatic cancer cell via increased expression of cyclin D1 and cyclin B1. However, whether MEF2D modulated cyclin D1 and cyclin B1 directly is still unclear. Many studies have revealed that cyclin D1 and cyclin B1 are important downstream effectors in a variety of cell signaling pathways [17-19]. Therefore, it is possible that MEF2D regulates the expression of cyclin D1 and cyclin B1 through a specific signaling pathway in pancreatic cancer.
It is widely acknowledged that the degradation of extracellular matrix components plays a critical role in the invasion and metastasis of cancers [20,21]. Growing evidence suggests that MMPs are involved in the degradation of extracellular matrix components. Furthermore, MMP2 and MMP9 have generally been thought of as important biomarkers for the invasion and metastasis of cancer [22,23]. In this study, we found that MEF2D silencing in pancreatic cancer cells led to significant inhibition in cell migration and invasion abilities, and this inhibition was due to the reduced expression of MMP2 and MMP9 after the knockdown of MEF2D.
Many studies have suggested that the Akt/GSK-3β signaling pathway contributes to an aggressive cancer phenotype in some human malignances [24-26]. Thus, we further explored whether MEF2D participates in the initiation and progression of pancreatic cancer via the Akt/GSK-3β signaling pathway. The results showed that knockdown of MEF2D significantly inhibited the amount of phosphorylated Akt and GSK-3β, but did not obviously influence the amount of total Akt and GSK-3β. The biological function of Akt requires the phosphorylation of Akt; therefore, the amount of phosphorylated Akt represents the activity of Akt [27]. In addition, GSK-3β, as an important downstream target of Akt, plays crucial role in the Akt pathway. Therefore, the oncogenic role of MEF2D in pancreatic cancer appeared to involve the Akt/GSK-3β signaling pathway.
In summary, our study suggested that the expression of MEF2D was increased in pancreatic cancer cells and could serve as an independent molecular prognostic factor for pancreatic cancer patients. Knockdown of MEF2D in pancreatic cancer cells reduced cell proliferation, migration, and invasion abilities by down-regulating cyclin B1, cyclin D1, and MMP2/9 expression, which involved the Akt/GSK-3β signaling pathway.
Acknowledgements
This research was supported in part by the National Nature Science Foundation of China (81573008), the Fund of Pudong Health Bureau of Shanghai (PWRd2014-01), and the Project of Key Disciplines Group Construction of Pudong Health Bureau of Shanghai (PWZxq2014-04).
Disclosure of conflict of interest
None.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Ansari D, Andersson R, Bauden MP, Andersson B, Connolly JB, Welinder C, Sasor A, Marko-Varga G. Protein deep sequencing applied to biobank samples from patients with pancreatic cancer. J Cancer Res Clin Oncol. 2015;141:369–380. doi: 10.1007/s00432-014-1817-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartwig W, Werner J, Jager D, Debus J, Buchler MW. Improvement of surgical results for pancreatic cancer. Lancet Oncol. 2013;14:e476–485. doi: 10.1016/S1470-2045(13)70172-4. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Ni Q, Wang X, Zhu H, Wang Z, Huang J. High expression of RAB27A and TP53 in pancreatic cancer predicts poor survival. Med Oncol. 2015;32:372. doi: 10.1007/s12032-014-0372-2. [DOI] [PubMed] [Google Scholar]
- 5.Oettle H, Neuhaus P, Hochhaus A, Hartmann JT, Gellert K, Ridwelski K, Niedergethmann M, Zulke C, Fahlke J, Arning MB, Sinn M, Hinke A, Riess H. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA. 2013;310:1473–1481. doi: 10.1001/jama.2013.279201. [DOI] [PubMed] [Google Scholar]
- 6.Guo S, Contratto M, Miller G, Leichman L, Wu J. Immunotherapy in pancreatic cancer: Unleash its potential through novel combinations. World J Clin Oncol. 2017;8:230–240. doi: 10.5306/wjco.v8.i3.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu W, Huang C, Wang Q, Huang T, Ding Y, Ma C, Ma H, Chen W. MEF2 transcription factors promotes EMT and invasiveness of hepatocellular carcinoma through TGF-beta1 autoregulation circuitry. Tumour Biol. 2014;35:10943–10951. doi: 10.1007/s13277-014-2403-1. [DOI] [PubMed] [Google Scholar]
- 8.McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 9.Prima V, Hunger SP. Cooperative transformation by MEF2D/DAZAP1 and DAZAP1/MEF2D fusion proteins generated by the variant t(1;19) in acute lymphoblastic leukemia. Leukemia. 2007;21:2470–2475. doi: 10.1038/sj.leu.2404962. [DOI] [PubMed] [Google Scholar]
- 10.Bai XL, Zhang Q, Ye LY, Liang F, Sun X, Chen Y, Hu QD, Fu QH, Su W, Chen Z, Zhuang ZP, Liang TB. Myocyte enhancer factor 2C regulation of hepatocellular carcinoma via vascular endothelial growth factor and Wnt/beta-catenin signaling. Oncogene. 2015;34:4089–4097. doi: 10.1038/onc.2014.337. [DOI] [PubMed] [Google Scholar]
- 11.Ma L, Liu J, Liu L, Duan G, Wang Q, Xu Y, Xia F, Shan J, Shen J, Yang Z, Bie P, Cui Y, Bian XW, Prieto J, Avila MA, Qian C. Overexpression of the transcription factor MEF2D in hepatocellular carcinoma sustains malignant character by suppressing G2-M transition genes. Cancer Res. 2014;74:1452–1462. doi: 10.1158/0008-5472.CAN-13-2171. [DOI] [PubMed] [Google Scholar]
- 12.Yu H, Sun H, Bai Y, Han J, Liu G, Liu Y, Zhang N. MEF2D overexpression contributes to the progression of osteosarcoma. Gene. 2015;563:130–135. doi: 10.1016/j.gene.2015.03.046. [DOI] [PubMed] [Google Scholar]
- 13.Xu K, Zhao YC. MEF2D/Wnt/beta-catenin pathway regulates the proliferation of gastric cancer cells and is regulated by microRNA-19. Tumour Biol. 2016;37:9059–9069. doi: 10.1007/s13277-015-4766-3. [DOI] [PubMed] [Google Scholar]
- 14.Su L, Luo Y, Yang Z, Yang J, Yao C, Cheng F, Shan J, Chen J, Li F, Liu L, Liu C, Xu Y, Jiang L, Guo D, Prieto J, Avila MA, Shen J, Qian C. MEF2D transduces microenvironment stimuli to ZEB1 to promote epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2016;76:5054–5067. doi: 10.1158/0008-5472.CAN-16-0246. [DOI] [PubMed] [Google Scholar]
- 15.Qie S, Diehl JA. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl) 2016;94:1313–1326. doi: 10.1007/s00109-016-1475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai J, Mei PJ, Liu H, Li C, Li W, Wu YP, Yu ZQ, Zheng JN. BRG1 expression is increased in human glioma and controls glioma cell proliferation, migration and invasion in vitro. J Cancer Res Clin Oncol. 2012;138:991–998. doi: 10.1007/s00432-012-1172-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Xiao Y, Fan S, Xiao M, Wang X, Zhu X, Chen X, Li C, Zong G, Zhou G, Wan C. Overexpression of DIXDC1 correlates with enhanced cell growth and poor prognosis in human pancreatic ductal adenocarcinoma. Hum Pathol. 2016;57:182–192. doi: 10.1016/j.humpath.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 18.Hu Y, Yang L, Yang Y, Han Y, Wang Y, Liu W, Zuo J. Oncogenic role of mortalin contributes to ovarian tumorigenesis by activating the MAPK-ERK pathway. J Cell Mol Med. 2016;20:2111–2121. doi: 10.1111/jcmm.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao S, Chen SR, Yang XF, Shen DF, Takano Y, Su RJ, Zheng HC. BTG1 might be employed as a biomarker for carcinogenesis and a target for gene therapy in colorectal cancers. Oncotarget. 2017;8:7502–7520. doi: 10.18632/oncotarget.10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 21.Cao J, Yang JC, Ramachandran V, Arumugam T, Deng DF, Li ZS, Xu LM, Logsdon CD. TM4SF1 regulates pancreatic cancer migration and invasion in vitro and in vivo. Cell Physiol Biochem. 2016;39:740–750. doi: 10.1159/000445664. [DOI] [PubMed] [Google Scholar]
- 22.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Liang L, Huang L, Ma X, Li D, Cai S. High expression of protein phosphatase 4 is associated with the aggressive malignant behavior of colorectal carcinoma. Mol Cancer. 2015;14:95. doi: 10.1186/s12943-015-0356-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen R, Yang Q, Lee JD. BMK1 kinase suppresses epithelial-mesenchymal transition through the Akt/GSK3beta signaling pathway. Cancer Res. 2012;72:1579–1587. doi: 10.1158/0008-5472.CAN-11-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang PP, Wang PQ, Qiao CP, Zhang Q, Zhang JP, Chen F, Zhang X, Xie WF, Yuan ZL, Li ZS, Chen YX. Differentiation therapy of hepatocellular carcinoma by inhibiting the activity of AKT/GSK-3beta/beta-catenin axis and TGFbeta induced EMT with sophocarpine. Cancer Lett. 2016;376:95–103. doi: 10.1016/j.canlet.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 26.Mo D, Li X, Li C, Liang J, Zeng T, Su N, Jiang Q, Huang J. Overexpression of AKIP1 predicts poor prognosis of patients with breast carcinoma and promotes cancer metastasis through Akt/GSK-3beta/Snail pathway. Am J Transl Res. 2016;8:4951–4959. [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Wu J, Ling MT, Zhao L, Zhao KN. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol Cancer. 2015;14:87. doi: 10.1186/s12943-015-0361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]