

Abstract
Growing evidence indicates that miR-106a is involved in tumor growth and metastasis of cancers, but the participation of miR-106a in endometrial adenocarcinoma (EC) is not clear. BCL2L11 is a member of the BCL-2 family and is located in the outer membrane of mitochondria, where this protein acts as a key regulator of excitotoxic apoptosis, apoptosis-inducing factor translocation, and mitochondrial depolarization. To identify a novel therapeutic target in EC, we studied the roles of miR-106a in the proliferation, apoptosis, and metastasis of EC. The expression levels of miR-106a were measured in tumor tissues of EC by quantitative real-time PCR, and lentiviral transduction was used to verify the function of miR-106a by silencing. Subcutaneous injection of EC cell lines into athymic mice was used to research EC tumor formation. Bioinformatics tools and a luciferase assay were applied to assess the relation between miR-106a and its target. The protein level of the miR-106a target was measured by western blotting. MiR-106a expression was higher in EC tissues compared with their healthy counterparts. Inhibition of expression of miR-106a reduced EC cell migration and invasion in vitro as well as in vivo tumor growth. BCL2L11 mRNA contains a binding site for miR-106a in the 3’untranslated region. BCL2L11 was found to be one of miR-106a targets. Altogether, our data suggest that miR-106a inhibits proliferation and invasiveness and induces cell cycle arrest and apoptosis in EC cells by targeting BCL2L11, and therefore miR-106a may serve as a prognostic marker of EC.
Keywords: miR-106a, endometrial adenocarcinoma, metastasis, invasion, BCL2L11
Introduction
Endometrial carcinoma (EC) is the most frequent uterine cancer, and the fourth most common cancer among women; it impairs reproductive health and quality of life of women [1]. Recently, EC incidence was increasing significantly due to changes in the environment and abuses of female hormones [2]. The degree of tumor cell differentiation plays a key role in prognosis of EC. Compared with source cells, tumor cells show features of poor differentiation and unlimited proliferation. A poorly differentiated tumor tissue means increased proliferation, high malignancy, and poor prognosis; also, well-differentiated tumors tend to closely resemble healthy endometrial epithelium cells, with limited proliferation and good prognosis . Because differentiation aberrations are closely related to the formation of malignant tumors, the development of methods for inducing tumor cells to differentiate into a cell type similar to that of the source cells is expected to increase the cure rate of cancer. By inducing differentiation, progestin hormone therapy has been successful at slowing the growth of EC in patients with well-differentiated tumor tissue expressing progesterone receptors [5,6]. In contrast, for patients with a poorly differentiated or recurrent cancer or metastasis, at present, the search for an effective cytodifferentiation-inducing agent is still at an early stage.
MicroRNAs (miRNAs) are small (19 to 25 nt) endogenous noncoding RNA molecules that act as gene regulators by binding to partially complementary target sites in an mRNA 3’untranslated region (3’UTR); this action results in degradation of the target mRNAs or translational repression of the encoded proteins [5,6]. Extensive data indicate that miRNAs are closely related to cancer biology; altered miRNA levels can result in the variation of expression of target genes directly, thereby affecting tumorigenesis and cancer progression [9,10]. Although several studies indicate that miRNAs may become a marker for tumor diagnosis or prognosis or contribute to cancer therapy, it is unclear which miRNAs can directly target a relevant gene and promote tumor cell differentiation. Human miR-106a is enriched in the germline and in mesoderm-derived tissues; accordingly, it may play a key role in cell differentiation of the endometrium, which is a mesoderm-derived tissue. BCL2L11 is a member of the BCL-2 family and is located in the outer membrane of mitochondria [11], where this protein is a key regulator of excitotoxic apoptosis, apoptosis-inducing factor translocation, and mitochondrial depolarization [12,13]. Recent studies showed that BCL2L11 is a dual agent that regulates autophagy in drug resistance [14] and is involved in neural apoptosis in methylmalonicacidemia [12,13]. Members of the antiapoptotic BCL2 family contain multiple domains although the proapoptotic members of this family, including BCL2L11, are BH3-domain-only proteins [15,16]. The function of BCL2L11 in these biological processes has been reported, but there is barely any information indicating that BCL2L11 regulates cell growth and apoptosis in cancer, especially in endometrial tumors. The aim of this study was to identify a novel therapeutic target in EC. To this end, we used both in vitro and in vivo experiments to explore the molecular mechanisms of miR-106a’s action in alterations of EC cell phenotype, and bioinformatics analysis was used to predict miR-106a targets in EC. Then, bioinformatics tools and a luciferase reporter assay were applied to determine the relation between miR-106a and its target; this way, the involvement of miR-106a in thepathogenesis of EC can be probedin terms of its target.
Materials and methods
Ethics statement
Human endometrial samples were collected from 24 patients with EC who underwent surgical treatment between January 2010 and December 2013 in Nantong First People’s Hospital (Nantong, China). Tissue samples were snap-frozen in liquid nitrogen immediately after surgical resection and stored at -80°C. They were collected after obtaining written informed consent from all the participating patients. The study protocol was approved by the Institutional Review Board of Nantong First People’s Hospital and conformed to the ethical guidelines of the 1975 Declaration of Helsinki and later amendments. This study did not include patients who had received radiotherapy and/or immunotherapy before or after surgical treatment.
Cell lines and their cultivation
Human endometrial epithelial celllines HEC-1-A, HEC-1B, RL95-2, AN3CA, Ishikawa, and JEC were obtained from the Shanghai Cell Bank, the Chinese Academy of Sciences (Shanghai, China). The cells were cultured inDulbecco’s Modified Eagle’s Medium supplemented with 10% of fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin, and incubated in a humidified atmosphere containing 5% of CO2 at 37°C. Normal human endometrial epithelial cells were generated from human endometrium samples obtained from biopsies of women (22-23 years of age) who underwent surgical treatment of minor gynecological problems and had no underlying endometrial pathology. None of them had received hormonal therapy in the 3 months preceding the sample collection. The endometrial samples were minced into fragments < 1 mm and subjected to mild collagenase digestion. Endometrial epithelial cells were isolated as described previously and cultured to confluence in a steroid-depleted medium composed of 75% Dulbecco’s Modified Eagle Medium and 25% MCDB-105 (Sigma, St. Louis, MO) supplemented with antibiotics, 10% of human albumin, and 5 mg/ml insulin (Sigma).
Lentivirus transduction of an miR-106a inhibitor into EC cells
Lentiviral transduction was applied to downregulate the endogenous expression of miR-106a in the EC cell lines (in vitro). A lentivirus expressing the artificial oligonucleotide of a human mature miR-106a inhibitor, L/miR106aI, and a control lentivirus expressing the nonspecific oligonucleotide, L/I (control), were both purchased from Shanghai GenePharma (Shanghai, China). HEC-1B and RL95-2 cells weretransduced with either L/miR106aI or L/I, along with 8 µg/ml polybrene, for 48 h at multiplicity of infection (MOI) of 20-25. Selection of transduced cell clones was conducted by growing post-transduction HEC-1B and RL95-2 cells in the presence of 1 mg/ml blasticidin for 1 week. After that, well-growing cell colonies were collected and subcultured into a 24-well plate. HEC-1B and RL95-2 cells were then passaged at least 3-5 times, followed by quantitative real-time PCR (qRT-PCR) to evaluate the transduction efficiency.
qRT-PCR
Total RNA was extracted from EC cells using the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. For miRNA analysis, qRT-PCR was carried out by means of the TaqMan MicroRNA Reverse Transcription Kit, TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA), and the corresponding primers. Small nuclear (sn) RNA U6 served as an internal control to normalize miR-106a levels. For mRNA analysis, qRT-PCR was conducted using the TaqMan High-Capacity cDNA Reverse Transcription Kit, TaqMan Fast PCR Master Mix (Applied Biosystems), and the corresponding primers. GAPDH served as an internal control to normalize BCL2L11 expression. qRT-PCR was run in triplicate on a RealPlex4 real-time PCR detection system from Eppendorf Co., Ltd. (Germany). The following primers were used for qRT-PCR: miR-106a forward, 5’-GAGAACAGCAGGTCCAGCAT-3’, and reverse: 5’-CTTCCT CAGCACAGACCGAG-3’; U6 forward, 5’-CTCGCTTCGGCAGCACA-3’, and reverse, 5’-AACGCTTCACGAATTTGCGT-3’; BCL2L11 forward, 5’-CACCAGCACCATAGAAGAA-3’, and reverse, 5’-ATAAGGAGCAGGCACAGA-3’; GAPDH forward, 5’-CCCATGTTCGTCATGGGTGT-3’, and reverse, 5’-CCGTTCAGCTCAGGGATGAC-3’. PCR parameters were as follows: 10 min at 95°C, and then 40 amplification cycles of 15 s at 95°C and 1 min at 60°C. The threshold cycle (Ct) was defined as the fractional cycle number at which the fluorescence passes the fixed threshold, and the relative miRNA or mRNA expression was calculated by the 2-ΔΔCt method.
The cell proliferation assay
RL95-2 cells transfected with a miR-106a-inhibitory lentivirus or a lentivirus expressing negative control miRNA were seeded at a density of 5000 cells per 200 μl of the medium in each well of a 96-well plate. At 0, 1, 2, 3, 4, and 5 days after the transfection, the medium was removed by suction and replaced by serum-free medium containing 1 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and incubated at 37°C for 4 h. After removal of the MTT solution, the formazan precipitate was dissolved in 100 μl of dimethyl sulfoxide, and absorbance was read at 570 and 600 nm on a microplate reader (BioTek, Bad Friedrichshall, Germany). Untransfected cells served as controls.
Cell cycle analysis
Cells were harvested at 48 h after L/I or L/miR106aI transfection. The cells were washed with PBS and fixed in ethanol at -20°C. The cells were then washed with PBS, rehydrated, and resuspended in a propidium iodide (PI)-RNase A solution (Sigma) with incubation at 37°C for 30 min. The stained cells (105) were next analyzed for DNA content on a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Apoptosis analysis
Cells were harvested, washed with ice-cold PBS, and stained with annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kits (KeyGEN Biotech, Nanjing, China). Apoptosis was analyzed on the flow cytometer (BD Biosciences).
The cell migration and invasion assays
Migration of cells was assayed in a chamber with filters (8-μm pores; Corning, New York, NY, USA). Cells were grown to ~50% confluence and transduced with L/I or L/miR106aI vectors. After 24 h, the cells were incubated with a serum-free medium for 24 h. Next, the cells were trypsinized, and 5 × 104 cells in the serum-free medium were added into the upper chamber. After that, a medium with 10% of fetal bovine serum was added into the lower chamber. Cells were incubated for 24 h at 37°C, and nonmigrating cells were completely removed. Cells that migrated to the bottom of the membrane were then fixed in 4% paraformaldehyde and stained with 0.5% crystal violet. Next, the stained cells were visualized under a microscope, counted in five random visual fields, and the average number was calculated. For the invasion assay, the upper chamber was precoated with 1 mg/ml Matrigel (BD Biosciences). The rest of the assay was conducted as the migration assay above.
Bioinformatics methods and a luciferase assay
TargetScan and PicTar software packages were used to predict miR-106 target genes; the target genes predicted by both software packages that turned out to be the same were selected. Target gene BCL2L11 was thus identified preliminarily. The wild-type sequence of BCL2L11 3’UTR was amplified from a human cDNA library. Mutations of the miR-106a-binding site were introduced by site-directed mutagenesis with a Fast Mutation Kit (NEB, US). The PCR product was cloned into the psiCHECK-2 vector downstream of the firefly luciferase-coding region between restriction sites XhoI and NotI (Takara, Japan). The psiCHECK-2-control vector served as an internal control.
Western blot analysis
Total protein in lysates was resolved by SDS-PAGE in a 10% gel and transferred to polyvinyl difluoride membranes (Millipore, Bedford, MA). After blocking in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) with 5% nonfat dry milk for 30 min, the membranes were washed four times in TBS-T and incubated with primary antibodies overnight at 4°C. The primary antibodies were all purchased from Abcam (Cambridge, MA) and were used at the following dilutions: anti-BCL2L11, 1/300; anti-GAPDH, 1/1000. After extensive washes, the membranes were incubated with a horseradish peroxidase-conjugated goat polyclonal anti-rabbit IgG antibody at a dilution of 1:2000 (secondary antibody) for 1 h at room temperature. Immunoreactivity was detected by enhanced chemiluminescence (ECL kit, Pierce Biotechnology) and by exposure of a radiography film to the membranes. GAPDH served as the loading control.
The animal experiment
Male athymic mice (5 weeks old), purchased from the Animal Center of Southern Medi-cal University (Guangzhou, China), were acclimated for 1 week under sterile conditions. Cells transduced with control RNA or the miR-106a inhibitor were collected from the 6-well plates and washed with PBS to obtain a suspension of cells at 2 × 107/ml. We subcutaneously injected 0.1 ml of this suspension into mice, 10 mice per group. Eight weeks later, the mice were euthanized, and the tumors were excised, photographed, and weighed and then were immediately snap-frozen in liquid nitrogen and stored at -80°C until use. To minimize the suffering of the mice, all surgical procedures were conducted under sodium pentobarbital anesthesia.
Statistical analysis
Data were expressed as mean ± SD from at least three independent experiments. Significance of differences was analyzed by Student’s t test. Differences with P < 0.05 or P < 0.01 were considered statistically significant.
Results
The expression of miRNA-106a in patients’ EC tissue and EC cells
To determine miRNA-106a expression in EC, we reanalyzed microarray data from The Cancer Genome Atlas (TCGA) for the EC dataset. We found that miRNA-106a expression was significantly higher in EC tissues when compared with healthy endometrial tissues (P < 0.01, Figure 1A). Furthermore, quantitative RT-PCR analysis was performed on 24 pairs of primary EC RNA samples. Clearly, miRNA-106a was upregulated in 70.8% (17/24) of the tested EC specimens (P < 0.01, Figure 1B). We then measured the expression of miRNA-106a in different EC cell lines (HEC-1-A, HEC-1B, RL95-2, AN3CA, Ishikawa, and JEC). MiR-106a was found to be significantly upregulated in HEC-1B and RL95-2 cells (Figure 1C, P < 0.05).
Figure 1.
Expression of miR-106a in patients’ EC tissue and EC cell lines. A. Expression of miR-106a in patients’ EC tissue according to TCGA database. B. RT-PCR analysis shows the expression of miR-106a in patients’ EC tissue (n = 24). C. Expression of miR-106a in HEC-1-A, HEC-1B, RL95-2, AN3CA, Ishikawa, and JEC cells. D. HEC-1B and RL95-2-7 cell lines were lentivirally transduced with L/I or L/miR106aI, and qRT-PCR was carried out to quantitate the expression of miR-106a. **P < 0.01 as compared to L/I cells.
To elucidate the mechanism of action of miR-106a in EC, we transduced EC cell lines, HEC-1B and RL95-2, with a lentivirus expressing a miR-106a inhibitor (L/miR106aI) to downregulate their endogenous miR-106a. Control HEC-1B and RL95-2 cells were transduced with a lentivirus expressing a nonspecific miRNA (L/I). After transduction was stabilized, transduction efficiency was checked by qRT-PCR, which showed that lentiviral transduction successfully downregulated endogenous miR-106a in both HEC-1B and RL95-2-7 cells (P < 0.05, Figure 1D).
Silencing of miRNA-106a inhibits cell proliferation and growth of xenografts
Lentivirally transduced HEC-1B and RL95-2-7 cellswere then transferred to a 96-well plate to be tested in a 5-day proliferation assay. Every day, the cells were stainedwith the MTT reagent, and their relative proliferation rateswere determined by measurement of optical density at 570 nm. The results revealed that EC cell proliferation in vitro was significantly suppressed by miR-106a downregulation in both HEC-1B and RL95-2 cells (P < 0.05, Figure 2A).
Figure 2.
The knockdown of miR-106a inhibits cell proliferation in vitro and in vivo. A. Viability of HEC-1B and RL95-2-7 cells transduced with L/I or L/miR106aI was assessed at 1, 2, 3, 4, and 5 days by an MTT assay. B. Downregulation of miR-106a significantly inhibited tumor growth in a nude mouse model of a tumor xenograft. Tumor volume was measured for 5 weeks. On day 35, the mice were euthanized, and tumors were weighed (n = 3). **P < 0.01 as compared to the L/I group.
We then evaluated the in vivo effect of miR-106a downregulation on EC. L/miR106aI- or L/I-transduced HEC-1B and RL95-2 cells were subcutaneously injected into left flanks of 4-week-old BALB/C nude mice. In situ HEC-1B and RL95-2 xenografts were allowed to develop for 5 weeks. Weekly live measurements of xenograft volumes indicated that the in vivo development of HEC-1B and RL95-2 xenografts was significantly inhibited by the knockdown of miR-106a (P < 0.05, Figure 2B). At the end of the xenograft experiment, HEC-1B and RL95-2 xenografts were excised and photographed. We confirmed the inhibitory effect of miR-106a downregulation on tumor growth by showing that L/miR106aI-transduced xenografts were significantly smaller than L/I-transduced xenografts (P < 0.05, Figure 2B).
The knockdown of miR-106a induced G1 phase arrest and apoptosis in EC cells
Because cell proliferation decreased when miR-106a was knocked down, we tested whether this inhibition affects cell cycle progression and apoptosis. The cell population at the G0-G1 transition markedly increased, while the proportion of S phase cells notably decreased among HEC-1B and RL95-2 cells transduced with L/miR106aI as compared with the L/I-transduced cells (P < 0.01, Figure 3A). Next, we found that lentiviral transduction with L/miR106aI markedly increased the apoptotic ratio of cells compared to cells transduced with L/I (P < 0.01, Figure 3B).
Figure 3.
The knockdown of miR-106a induces cell cycle arrest in the G1 phase and apoptosis. A. L/miR106aI suppressed the G1-S transition. Cell cycle profiles were analyzed by PI staining and flow cytometry. B. Apoptotic cells were quantified by annexin V-PI staining when HEC-1B and RL95-2-7 cells were transduced with L/miR106aI. **P < 0.01 as compared to the L/I group.
Silencing of miR-106a inhibited migration and invasiveness of EC cells
To determine whether miR-106a influences migration and invasiveness of EC cells, a Transwell assay was performed, and the numbers of migratory and invading cells were assessed after a 24-h culture period. Cells transduced with L/miR106aI showed a significant reduction in migration and invasion as compared to control (L/I-transduced) cells (Figure 4A and 4B).
Figure 4.
Silencing of miRNA-106a suppressed the migration and invasiveness of EC cells. (A) Cell migration and (B) invasion were assessed in a Transwell assay. **P < 0.01 as compared to the L/I group.
BCL2L11 is a target gene of miR-106a in EC
After the function of miR-106a was revealed by this study (by downregulation), we started looking for a target gene of miR-106a in EC. Using online miRNA-binding analysis algorithms such as miRDB (www.miRDB.org) and TargetScan (www.targetscan.org), we noticed that tumor suppressor gene BCL2L11 might be a suitable candidate because miR-106a can bind the 3’-UTR of human BCL2L11 cDNA sequence (Figure 5A). Consequently, we then transfected lentivirally transduced HEC-1B and RL95-2-7 cells with firefly luciferase vectors containing wild-type BCL2L11 3’-UTR (Wt) or mutated BCL2L11 3’-UTR (Mu, without the miR-106a-binding site). After 48 h, analysis by a dual luciferase activity assay showed that in L/I-transduced EC cells, transfection of BCL2L11 3’-UTR (Wt) or BCL2L11 3’-UTR (Mu) did not result in different luciferase activities (Figure 5B). In contrast, in L/miR106aI-transduced EC cells, the difference in luciferase activities was significant between BCL2L11 3’-UTR (Wt) and BCL2L11 3’-UTR (Mu) transfectants (Figure 5B, P < 0.05), thus confirming that BCL2L11 was the target of miR-106a in EC cells. To verify the result of our dual luciferase activity assay, we employed western blot analysis and demonstrated that BCL2L11 protein levels were higher in L/miR106aI-transduced cancer cells than in L/I-transduced cancer cells (Figure 5C and Supplementary Figure 1).
Figure 5.
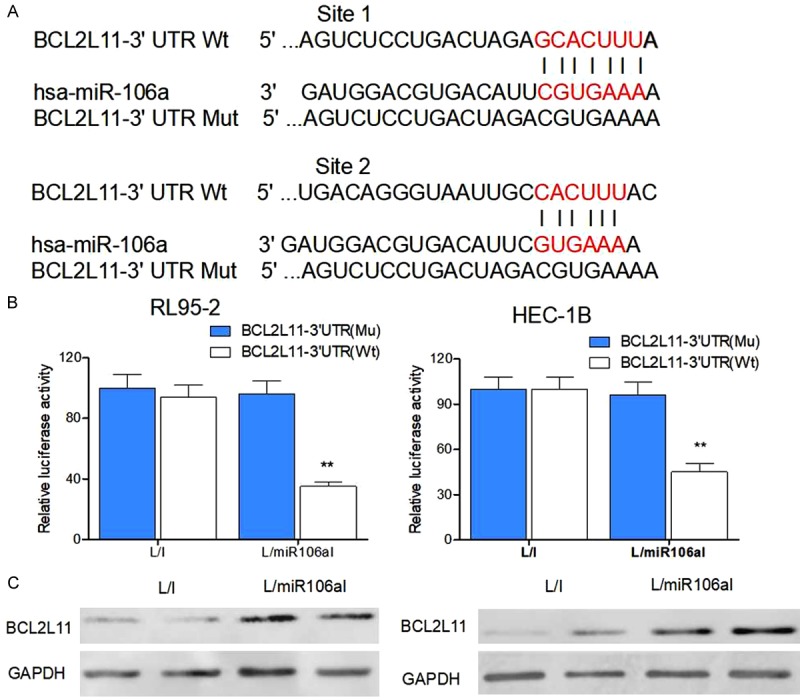
Association of the BCL2L11 gene and miR-106a in EC. A. The wild-type (Wt) human BCL2L11 3’-UTR was shown to bind to human miR-106a. In a mutant (Mu) BCL2L11 3’-UTR, its miR-106a-binding site was point-mutated. B. Lentivirally transduced HEC-1B and RL95-2 cells were and then transfected with either BCL2L11-3’-UTR (Wt) or BCL2L11-3’-UTR (Mu) firefly luciferase vectors or a pSV40-Renilla control luciferase vector. After 48 h, relative luciferase activities were evaluated as the ratio firefly/Renilla signals in the transfected HEC-1B and RL95-2 cells (**P < 0.01). C. Western blotting was performed to compare BCL2L11 protein expression levels between L/I- and L/miR106aI-transduced breast cancer cells (**P < 0.01).
Discussion
In this study, we showed that miR-106a is upregulated and its target gene BCL2L11 is downregulated in EC cell lines, and that a knockdown of miR-106a increases BCL2L11 expression and suppresses cell proliferation, migration, and invasionand induces cell cycle arrest and apoptosis, suggesting that miR-106a acts as an oncogene in EC by inhibiting tumor suppressor BCL2L11 expression. Several miRNAs have been identified that are up- or downregulated in EC. Among miRNA supregulated in EC, miRNAs21, 31, 93, 182, 183, and 205 are strongly associated with tumor progression, growth, and lymph node metastases [17-19]. MiR-106a upregulation was also demonstrated in gastric cancer [20], ovarian cancer [21], human glioblastoma [21], and colorectal carcinoma [22], thereby pointing to a common underlying mechanism of carcinogenesis in these gynecological cancers.
MiR-106a functions as an oncogene in various human cancers. miR-106a is upregulated in gastric cancer tissues and correlates with metastasis, and TIMP2 is a direct target of miR-106a. MiR-106a may be involved in the development of drug resistance and in regulation of PDCD4 expression by modulating CDDP-induced apoptosis in ovarian cancer cells [24]. Besides, miR-106a may be a diagnostic biomarker of colorectal carcinoma [25,26]. Elevated expression of miRNA-106a is deeply involved in the development and progression of glioma by promoting the proliferation and by suppressing apoptosis of glioma cells through the JNK-MAPK signaling pathway [25]. In the present study, miR-106a was found to beoverexpressed in three EC cell lines and to suppress the expression of itstarget BCL2L11, a tumor suppressor gene.
We revealed that miR-106a binds directly to the 3’-UTR of BCL2L11 mRNA, thus inhibiting its translation and the BCL2L11-mediated regulation of essential downstream molecules involvedin cell proliferation and cell cycle progression. To further explore the molecular signaling pathways associated with miR-106a in EC, we performed a dual luciferase activity assay in lentivirally transduced HEC-1B and RL95-2 cells. By transfecting these EC cell lines with firefly luciferase vectors containing either wild-type or mutant (without the miR-106a-binding site) BCL2L11 3’-UTR, we demonstrated that BCL2L11 is indeed the downstream gene of miR-106a in EC. This finding was confirmed by our qRT-PCR and western blot assays of lentivirally transduced HEC-1B and RL95-2 cells, showing that both gene and protein expression levels of BCL2L11 were significantly higher in L/miR106aI-transduced EC cells, likely because of miR-106a downregulation.
In conclusion, this study shows that miR-106a is upregulated and its target gene BCL2L11 is downregulated in EC cell lines. Inhibition of miR106a upregulates BCL2L11 expression, inhibiting cell proliferation, cell migration, and invasion and promoting cell cycle arrest and apoptosis. Our results indicate that miR-106a functions as an oncogenic miRNA in EC by targeting tumor suppressor gene BCL2L11 and that miR-106a as a promising biomarker or therapeutic target in EC.
Acknowledgements
This study was supported by grants from the Youth Research Foundation of Nantong Health Bureau (WQ2015013).
Supporting Information
References
- 1.Li Q, Zhang C, Chen R, Xiong H, Qiu F, Liu S, Zhang M, Wang F, Wang Y, Zhou X, Xiao G, Wang X, Jiang Q. Disrupting MALAT1/miR-200c sponge decreases invasion and migration in endometrioid endometrial carcinoma. Cancer Lett. 2016;383:28–40. doi: 10.1016/j.canlet.2016.09.019. [DOI] [PubMed] [Google Scholar]
- 2.Zeng N, Salker MS, Zhang S, Singh Y, Shi B, Stournaras C, Lang F. 1alpha,25(OH)2D3 induces actin depolymerization in endometrial carcinoma cells by targeting RAC1 and PAK1. Cell Physiol Biochem. 2016;40:1455–1464. doi: 10.1159/000453197. [DOI] [PubMed] [Google Scholar]
- 3.Miyamoto Y, Takechi K. [A case of recurrent endometrial carcinoma with pleural effusion maintained long SD by pegylated liposomal doxorubicin(PLD)chemotherapy] . Gan To Kagaku Ryoho. 2013;40:955–957. [PubMed] [Google Scholar]
- 4.Zhang Y, Qu X, Qu PP. Value of circulating tumor cells positive for thyroid transcription factor-1 (TTF-1) to predict recurrence and survival rates for endometrial carcinoma. J Buon. 2016;21:1491–1495. [PubMed] [Google Scholar]
- 5.Vale CL, Tierney J, Bull SJ, Symonds PR. Chemotherapy for advanced, recurrent or metastatic endometrial carcinoma. Cochrane Database Syst Rev. 2012:D3915. doi: 10.1002/14651858.CD003915.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jutzi L, Hoskins P, Lim P, Aquino-Parsons C, Tinker A, Kwon JS. The importance of adjuvant chemotherapy and pelvic radiotherapy in high-risk early stage endometrial carcinoma. Gynecol Oncol. 2013;131:581–585. doi: 10.1016/j.ygyno.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Zhang L. Members of the microRNA-200 family are promising therapeutic targets in cancer. Exp Ther Med. 2017;14:10–17. doi: 10.3892/etm.2017.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geretto M, Pulliero A, Rosano C, Zhabayeva D, Bersimbaev R, Izzotti A. Resistance to cancer chemotherapeutic drugs is determined by pivotal microRNA regulators. Am J Cancer Res. 2017;7:1350–1371. [PMC free article] [PubMed] [Google Scholar]
- 9.Gallach S, Jantus-Lewintre E, Calabuig-Farinas S, Montaner D, Alonso S, Sirera R, Blasco A, Uso M, Guijarro R, Martorell M, Camps C. MicroRNA profiling associated with non-small cell lung cancer: next generation sequencing detection, experimental validation, and prognostic value. Oncotarget. 2017;8:56143–56157. doi: 10.18632/oncotarget.18603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen GM, Zheng AJ, Cai J, Han P, Ji HB, Wang LL. microRNA-145-3p Inhibits Non-Small Cell Lung Cancer Cell Migration and Invasion by Targeting PDK1 via the mTOR Signaling Pathway. J Cell Biochem. 2017 doi: 10.1002/jcb.26252. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Duan J, Qu Y, Deng T, Liu R, Zhang L, Bai M, Li J, Ning T, Ge S, Wang X, Wang Z, Fan Q, Li H, Ying G, Huang D, Ba Y. Onco-miR-24 regulates cell growth and apoptosis by targeting BCL2L11 in gastric cancer. Protein Cell. 2016;7:141–151. doi: 10.1007/s13238-015-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai Y, Grant S. BCL2L11/Bim as a dualagent regulating autophagy and apoptosis in drug resistance. Autophagy. 2015;11:416–418. doi: 10.1080/15548627.2014.998892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Peng T, Li L, Wang X, Duan R, Gao H, Guan W, Lu J, Teng J, Jia Y. MicroRNA-9 regulates neural apoptosis in methylmalonic acidemia via targeting BCL2L11. Int J Dev Neurosci. 2014;36:19–24. doi: 10.1016/j.ijdevneu.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Nieters A, Conde L, Slager SL, Brooks-Wilson A, Morton L, Skibola DR, Novak AJ, Riby J, Ansell SM, Halperin E, Shanafelt TD, Agana L, Wang AH, De Roos AJ, Severson RK, Cozen W, Spinelli J, Butterbach K, Becker N, de Sanjose S, Benavente Y, Cocco P, Staines A, Maynadie M, Foretova L, Boffetta P, Brennan P, Lan Q, Zhang Y, Zheng T, Purdue M, Armstrong B, Kricker A, Vajdic CM, Grulich A, Smith MT, Bracci PM, Chanock SJ, Hartge P, Cerhan JR, Wang SS, Rothman N, Skibola CF. PRRC2A and BCL2L11 gene variants influence risk of non-Hodgkin lymphoma: results from the InterLymph consortium. Blood. 2012;120:4645–4648. doi: 10.1182/blood-2012-05-427989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mansha M, Hussain A, Kofler A, Grubbauer C, Goetsch K, Ploner C, Kofler R. “Bam”, a novel glucocorticoid-induced BH3-only transcript from the BCL2L11/Bim locus, does not appear to be translated. Leuk Lymphoma. 2013;54:353–358. doi: 10.3109/10428194.2012.708928. [DOI] [PubMed] [Google Scholar]
- 16.Luo S, Rubinsztein DC. BCL2L11/BIM: a novel molecular link between autophagy and apoptosis. Autophagy. 2013;9:104–105. doi: 10.4161/auto.22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitamura T, Watari H, Wang L, Kanno H, Kitagawa M, Hassan MK, Kimura T, Tanino M, Nishihara H, Tanaka S, Sakuragi N. microRNA 31 functions as an endometrial cancer oncogene by suppressing Hippo tumor suppressor pathway. Mol Cancer. 2014;13:97. doi: 10.1186/1476-4598-13-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan H, Liang X, Zhao W, Ma L, Zhao Y. The effects of microRNA-183 promots cell proliferation and invasion by targeting MMP-9 in endometrial cancer. Biomed Pharmacother. 2017;89:812–818. doi: 10.1016/j.biopha.2017.02.091. [DOI] [PubMed] [Google Scholar]
- 19.Qin X, Yan L, Zhao X, Li C, Fu Y. microRNA-21 overexpression contributes to cell proliferation by targeting PTEN in endometrioid endometrial cancer. Oncol Lett. 2012;4:1290–1296. doi: 10.3892/ol.2012.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu M, Zhang N, He S, Lui Y, Lu G, Zhao L. MicroRNA-106a targets TIMP2 to regulate invasion and metastasis of gastric cancer. FEBS Lett. 2014;588:600–607. doi: 10.1016/j.febslet.2013.12.028. [DOI] [PubMed] [Google Scholar]
- 21.Chen L, Zhang F, Sheng XG, Zhang SQ, Chen YT, Liu BW. MicroRNA-106a regulates phosphatase and tensin homologue expression and promotes the proliferation and invasion of ovarian cancer cells. Oncol Rep. 2016;36:2135–2141. doi: 10.3892/or.2016.5010. [DOI] [PubMed] [Google Scholar]
- 22.Feng B, Dong TT, Wang LL, Zhou HM, Zhao HC, Dong F, Zheng MH. Colorectal cancer migration and invasion initiated by microRNA-106a. PLoS One. 2012;7:e43452. doi: 10.1371/journal.pone.0043452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen XH, Ling XM, Shi S. microRNA-106a induces the proliferation and apoptosis of glioma cells through regulating JNK/MAPK pathway. Eur Rev Med Pharmacol Sci. 2015;19:3412–3417. [PubMed] [Google Scholar]
- 24.Li H, Xu H, Shen H, Li H. microRNA-106a modulates cisplatin sensitivity by targeting PDCD4 in human ovarian cancer cells. Oncol Lett. 2014;7:183–188. doi: 10.3892/ol.2013.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He Y, Wang G, Zhang L, Zhai C, Zhang J, Zhao X, Jiang X, Zhao Z. Biological effects and clinical characteristics of microRNA-106a in human colorectal cancer. Oncol Lett. 2017;14:830–836. doi: 10.3892/ol.2017.6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Fan L, Wei J, Weng Y, Zhou L, Shi Y, Zhou W, Ma D, Wang C. Akt mediates metastasis-associated gene 1 (MTA1) regulating the expression of E-cadherin and promoting the invasiveness of prostate cancer cells. PLoS One. 2012;7:e46888. doi: 10.1371/journal.pone.0046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.