

Abstract
Myocardial infarction (MI) is a common cardiovascular disease with high mortality. The aim of the present study was to determine the biological role of miR-145 in MI rats and hypoxia-injured cardiomyocytes and to elucidate the potential mechanism. MI rats were induced by left anterior descending artery (LAD) ligation. qRT-PCR and western blot analysis were performed to determine the mRNA and protein levels, respectively. Compared with sham group, miR-145 levels in MI group were significantly decreased. We observed that lentivirus-mediated overexpression of miR-145 significantly improves cardiac function, reduces infarcted tissue size and prevents post-infarction induced apoptosis in rats after MI. Furthermore, PDCD4 was identified as a novel target of miR-145 in cardiomyocytes, and overexpression of PDCD4 could remarkably restore the miR-145-inhibited cardiomyocytes apoptosis and mitochondrial dysfunction after hypoxia injury. Therefore, our study indicated that miR-145/PDCD4 axis might be potential therapeutic targets for the treatment of MI, and its cardioprotective effect may be attributed to a reduction of mitochondria-mediated apoptosis.
Keywords: Myocardial infarction, left anterior descending, microRNA-145, PDCD4, apoptosis, mitochondria
Introduction
Myocardial infarction (MI) is the most frequent manifestation of coronary heart disease (CHD) and one of the leading causes of morbidity and mortality around the world [1]. Approximately 3-4 million people suffer from MI annually [2].MI is commonly induced by acute occlusion of the coronary artery [3], and the main determinants of outcome in MI patients are myocardial infarct size and left ventricular remodeling [4]. Experimental and clinical studies have shown that cardiomyocyte apoptosis is one of the major pathogenic mechanisms in MI [5]. Over the past decades, great progress has been made in the diagnosis and treatment of MI. However, up to now, the mechanisms underlying ischemia-induced apoptosis in cardiomyocytesremains poorly understood.
MicroRNAs (miRNAs) are a group of small non-protein-coding RNAs that play crucial roles in regulation of gene expression via sequence-specific interaction with the 3’UTR of target mRNA at the post-transcription steps [6]. Since the discovery of the first miRNA, lin-4, in C. elegans, in 1993, it has been estimated that as many as 1000 miRNAs exist in the human genome [7]. In recent years, extensive investigation has demonstrated that dysregulation of miRNAs is frequently involved in MI, includingmiR-223-3p [8], miR-7a/b [9] and miR-16 [10]. It has been shown that miRNAs contribute to MI through regulating the expression of various key elements in cell survival and apoptosis [11].
MiR-145 deregulation has been found to be associated with several types of cardiovascular diseases, indicated by both experimental and clinical studies [12,13]. Recently, link between miRNAs and MI has become increasingly apparent. For example, decreased plasma levels of miR-145 are associated with MI and heart failure [14], and circulating miR-145 was a significant independent predictor of long-term outcome after MI [15]. However, the biological function and molecular mechanism of miR-145 in MI still remain elusive.
Therefore, the present study firstly clarified the protective effect of miR-145 against MI by modulating cardiomyocyte apoptosis. Furthermore, we identified PDCD4 as real targets for miR-145. The results might provide a new insight into early diagnosis or better therapeutic opportunities for patients with MI.
Materials and methods
Recombinant lentivirus construction
The sequences of miR-145 and negative control weresubcloned into Pglv3/h1/gfp+puro plasmid vector (Genepharma, Shanghai) (LV-miR-145, LV-miR-NC). To produce pseudoviral particles, HEK-293T cells (ATCC, Rockefeller, MD, USA) were cotransfected with plasmid vectors and lentivector packaging system (Genepharma, Shanghai).
Animal experiments
A total of forty male Sprague-Dawley (SD) rats weighing 250-300 g, obtained from Shanghai Laboratory Animal Center Co. Ltd, were housed in a temperature-controlled environment with 12 hours light/dark cycles with rodent chow and water available ad libitum. All animal experimental procedures were in accordance with and approved by the Animal Care and Use Committee of Shanghai General Hospital (Shanghai, China).
These rats were subsequently randomized into four experimental groups (n=10/group): sham group, MI group, LV-NC group, and LV-miR-145 group. Three days prior to MI operation, rats were intramyocardially injected with lentivirus containing either miR-145 sequence or NC sequence. Rats were subjected to MI by left anterior descending artery (LAD) ligation, as previously reported [16]. Briefly, the rats were anesthetized with ketamine (50 mg/kg) and xylazine (10 mg/kg), and a left thoracic incision was used to open the chest. The LAD was ligated via using a 6-0 prolene suture. Sham rats underwent the same operation procedures without ligation of LAD.
Echocardiography
24 hours after MI, rats were sedated with 2% isoflurane inhalation and studied on a Vevo-770 high-frequency ultrasound system (Visual Sonics, Toronto, Canada). Left ventricular anterior wall thickness at diastole (LVAWd), left ventricular posterior wall thickness at diastole (LVPWd), left ventricular ejection fraction (LVEF) and left ventricular fraction shortening (LVFS) were thus calculated.
Measurement of infarction area
Two week after MI operation, rats were sacrificed by intraperitoneal injection with pentobarbital (30 mg/kg). Body weight (BW), heart weight (HW) and left ventricle weight (LVW) were recorded. The infarct area was identified by visual inspection under a dissecting microscope, and expressed by a percentage of the area of total LV. The excised heart was then cut for histological analysis and embedded in paraffin. Sections were stained with hematoxylin and eosin (HE) and observed under optical microscopy.
Primary culture of cardiomyocytes and cell transfection
Neonatal rat cardiomyocytes were isolated from 1-3-day-old SD rats. Briefly, rats were killed and the hearts were removed. After washing with cold PBS, the ventricles were cut into small tissue blocks (1-2 mm3) and then digested with pancreatin at 37°C for 8 min with gentle shaking. Discarding the initial supernatant, the precipitates were digested with pancreatin until all tissues were completely digested. Finally, cells were filtered and suspended in DMEM medium containing 10% FBS and maintained in a 5% CO2 environment at 37°C. MiR-145 mimics and its scrambled control microRNA (miR-NC) were obtained from Shanghai Genepharma. Cell transfection was performed by using Lipofectamine 2000 (Invitrogen).
Cell ischemic injury in vitro
Cell ischemic injury was induced by hypoxia in a serum- and glucose-free medium, and reoxygenation. Hypoxia was achieved by placing the cells in a hypoxia chamber at 37°C with 5% CO2 and 0.1% O2 for 6 h. And then, the cells were reoxygenated for 12 h in DMEM containing 10% FBS and normal glucose. The cells that were not subjected to hypoxia/reoxygenation were served as control (normoxia).
Measurement of cell apoptosis
Cell apoptosis was measured by FITC-conjugated Annexin V and propidium iodide (PI) assay. Briefly, cells were washed twice with PBS and then suspended in binding buffer. After incubation with FITC-Annexin V and PI for 10 min in the dark at room temperature, cell apoptosis was detected by flow cytometry (FACS CaliburTM, BD Biosciences, San Jose, CA, USA) and analyzed by Cell Quest Pro software.
Real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA samples were isolated fromheart tissues or cultured cells using Trizol Reagent (Invitrogen). For miRNA quantification, cDNA was synthesized using One Step Prime script miRNAcDNA Synthesis Kit (Qiagen, Valencia, CA). Quantitative RT-PCR was performed using the miRNA-specific TaqMan® MiRNA Assay Kit (Applied Biosystems, Foster City, CA, USA) on the StepOne™ realtime PCR System (Applied Biosystems). For mRNA detection, the Prime Script RT reagent Kit (TaKaRa, Dalian, China)were used to generate cDNA, which was subjected to qRT-PCR using SYBR® GreenMaster Mix (TaKaRa), following the manufacturer’s instructions. All samples were run in triplicate and averaged. The relative expression levels of each gene were calculated using relative quantification (2-ΔΔCt) method [17] with reference to the expression ofU6 or GAPDH. The sequences of primers used for PCR are listed as follows: miR-145RT primer: 5’-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGAACAG-3’; U6 RT primer: 5’-GTGCAGGGTCCGAGGT-3’; miR-145 forward primer: 5’-GTCTGGATTCCTGGAAATA-3’; miR-145 reverse primer: 5’-GTGCAGGGTCCGAGGT-3’; U6 forward primer: 5’-AGGGGCCATGCTAATCTTCT-3’: U6 reverse primer: 5’-TGCTTCGGCAGCACATATAC-3’.
Western blot analysis
Total protein samples were isolated from heart tissues or cultured cells using RIPA lysis buffer (Beyotime, Shanghai, China). Mitochondria and cytosol were isolated using the Cell Mitochondria Isolation Kit (Abcam, Cambridge, UK). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (GE Healthcare, Milan, Italy). The membrane was then blocked in 5% powdered milk at room temperature for 1 h followed by incubation with primary antibodies overnight at 4°C. After washing and incubation with a goat-anti-rabbit secondary antibody conjugated to horseradish peroxidase (HRP), protein bands were detected by a chemiluminescent HRP substrate (Millipore) and imaged on Bio-Rad ChemiDocXRS (Bio-Rad Laboratories, Hercules, CA, USA). GAPDH was used as an internal reference.
Dual luciferase reporter assay
The pGL3-PDCD4 WT 3’-UTR was generated by inserting a fragment of rat PDCD4 3’-UTR into the psiCHECK-2 vector (Promega, Madison, WI, USA). The psiCHECK-2-PDCD4 MUT 3’-UTR construct was generated by mutation of the complementary seed sequence to the miR-145 binding region [18]. HEK293T cells were co-transfected with psiCHECK-2-PDCD4 WT 3’-UTR or psiCHECK-2-PDCD4 MUT 3’-UTR luciferase reporter, miR-145 mimics or mimics control, and Renilla luciferase reporter using Lipofectamine 2000. Cells were incubated for 48 h and luciferase activity was measured. Firefly luciferase units were normalized against Renilla luciferase units to control for transfection efficiency.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was evaluated by cationic dye JC-1 (Beyotime). Brifely, after two PBS washings, cells were incubated with JC-1 10 µg/mL for 15 minutes at 37°C in the dark. Cells were harvested, suspended in PBS, and analyzed by flow cytometry.
Statistical analysis
Data analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA). All data are expressed as mean ± standard error (SD). Statistical analysis was performed using Student’s t-test. A two-tailed P<0.05 was considered significant.
Results
MiR-145 is down-regulated in infarcted myocardial tissues
MI rat model was successfully established by LAD. MI rats showed a reduction in body weight; however, this was not significant (Data not shown). The expression levels of miR-145 in the infarcted myocardial tissues were detected by RT-qPCR analysis. As shown in Figure 1A, the expression levels of miR-145 in the infract areas were significantly reduced, as compared with the sham group. The efficiency of lentivirus-mediatedmiR-145 overexpression was also verified by RT-qPCR analysis (Figure 1B).
Figure 1.
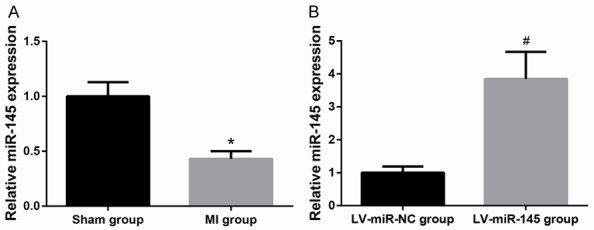
MiR-145 is down-regulated in infarcted myocardial tissues. A. MiR-145 expression was decreased in MI group as compared to sham group. B. MiR-145 expression was increased in LV-miR-145 group as compared to LVmiR-NC group. Data are presented as the mean ± standard deviation. Student’s t-test was used to analyze significant differences. *P<0.05 vs. Sham group, #P<0.05 vs. LV-miR-NC group.
MiR-145 improves cardiac function in rats after MI
24 h of LAD caused severe cardiac dysfunction, as evidenced by decreased LVEF and LVFS with increased LVAWd and LVPWd (Figure 2A). In LV-miR-145-treated rats, the reduction of LVEF and LVFS, and the elevation of LVAWd and LVPWd were significantly attenuated, suggesting that up-regulated miR-145 significantly improved LV systolic function after MI. Additionally, as shown in Figure 2B, significant increases in the HW-to-BWratio and LVW-to-BW ratio were observed in MI group, as compared with Sham group. Transfection of MI rats with LV-miR-145 significantly decreased HW-to-BW ratio and LVW-to-BW ratio.
Figure 2.
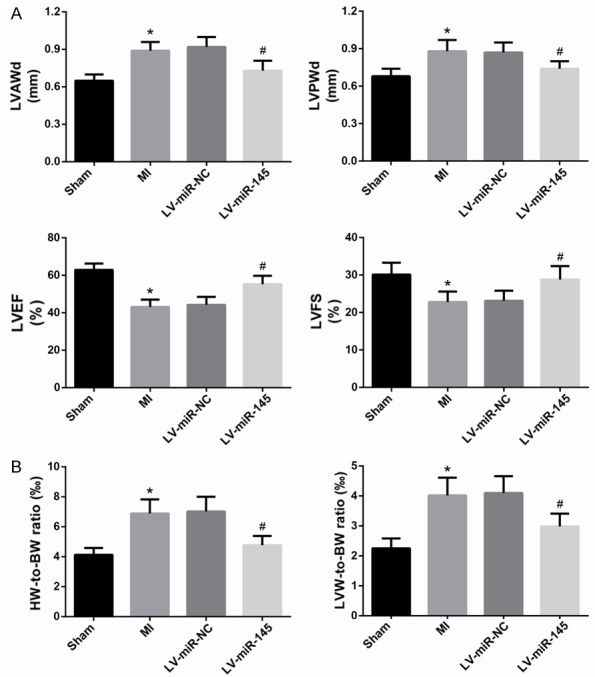
MiR-145 improves cardiac function in rats after MI. A. Echocardiographic analysis of cardiac function was performed. B. The HW-to-BW ratio and LVW-to-BW ratio were assessed. Data are presented as the mean ± standard deviation. Student’s t-test was used to analyze significant differences. *P<0.05 vs. Sham group, #P<0.05 vs. LV-miR-NC group.
MiR-145 prevented post-infarction induced apoptosis
After HE staining, the infarcted cardiac tissues showed structural disorder of myofibril, muscle fiber swelling, and extensive necrotic lesions. However, these pathological changes were attenuated significantly in MI rats treated with LV-miR-145 (Figure 3A). The size of infarcted myocardial tissues was also significantly reduced in MI rats treated with LV-miR-145, as compared with MI rats treated with LV-NC (Figure 3B). In addition, miR-145 overexpression significantly reduced cardiomyocyte apoptosis induced by MI, as determined by Western blot (Figure 3C). LV-miR-145 treatment significantly decreased levels of pro-apoptotic proteins Bax and cleaved caspase-3 and increased the level of anti-apoptotic protein Bcl-2, indicating that more viable cardiomyocytes were presented in the infarcted areas in rats treated with LV-miR-145.
Figure 3.
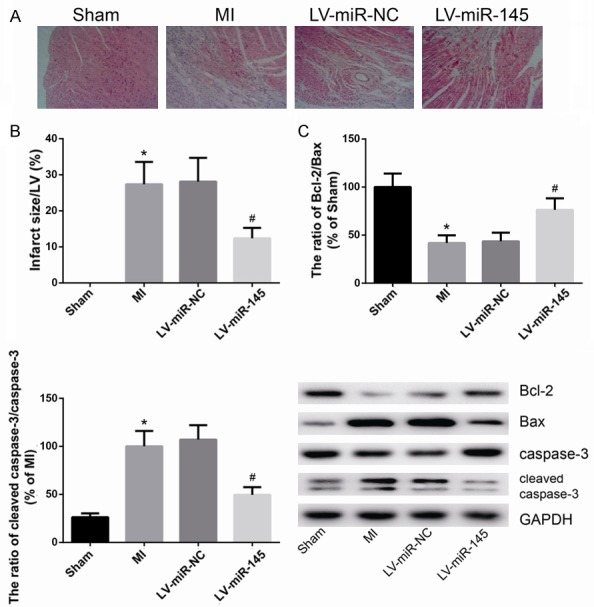
MiR-145 prevented post-infarction induced apoptosis. A. HE staining of myocardium tissues. B. The infarct size was analyzed. C. The protein levels of Bcl-2, Bax and cleaved caspase-3 were determined by western blot analysis. Data are presented as the mean ± standard deviation. Student’s t-test was used to analyze significant differences. *P<0.05 vs. Sham group, #P<0.05 vs. LV-miR-NC group.
PDCD4 is a direct target of miR-145 in cardiomyocytes
In order to further determine the underlying mechanism of miR-145 in MI, we next used several target prediction programs, TargetScan, miRWalk and PicTar, to explore the potential target gene of miR-145. Results of analysis revealed that the 3’-UTR of PDCD4 mRNA has a complementary site for miR-145, which is highly conserved among different species (Figure 4A). Western blot analysis showed that PDCD4 protein expression was reduced in cardiomyocytes with up-regulation of miR-145 (Figure 4B). To further confirm that PDCD4 is a direct target of miR-145, luciferase activity assay was subsequently performed. As showed in Figure 4C, overexpression of miR-145 markedly inhibits the luciferase activity of cells transfected with WT PDCD4-3’UTR but had no significant effect on MUT PDCD4-3’UTR.
Figure 4.
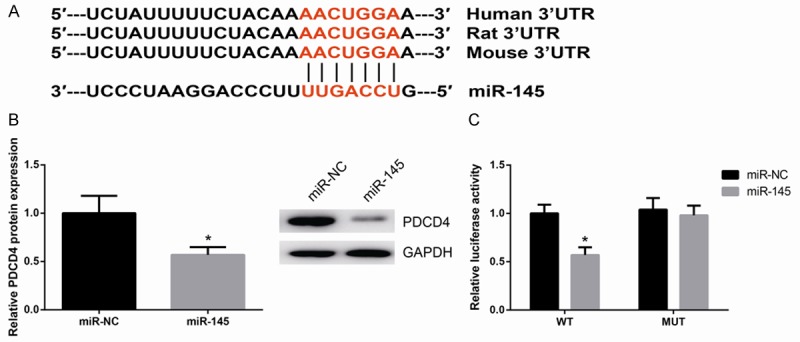
PDCD4 is a direct target of miR-145 in cardiomyocytes. A. Sequence alignment of rat PDCD4-3’UTR and miR-145. B. PDCD4 protein expression was decreased in miR-145 overexpressing cells. C. Luciferase activity of the PDCD4-3’UTR in miR-145 overexpressing cells. Data are presented as the mean ± standard deviation. Student’s t-test was used to analyze significant differences. *P<0.05 vs. NC cells.
MiR-145 attenuates hypoxia-induced cardiomyocyte apoptosis and mitochondrial dysfunction in vitro
Next, the miR-145 expression in hypoxia-treated cardiomyocytes was determined. As indicated in Figure 5A, miR-145 expression was reduced in cardiomyocytes upon hypoxia exposure. Treatment with miR-145 mimics significantly reduced cardiomyocyte apoptosis induced by hypoxia, as determined by flow cytometry analysis (Figure 5B). However, overexpression of PDCD4 could remarkably restore the miR-145-inhibited cardiomyocytes apoptosis after hypoxia injury.
Figure 5.

MiR-145 attenuates hypoxia-induced cardiomyocyte apoptosis and mitochondrial dysfunction in vitro. A. MiR-145 expression was decreased in hypoxia-treated cells. B. Cell apoptosis was measured by FITC-conjugated Annexin V and PI assay. C. The MMP was assessed by cationic dye JC-1. D. The levels of cytochrome c in mitochondria and cytosol were assessed. Data are presented as the mean ± standard deviation. Student’s t-test was used to analyze significant differences. *P<0.05 vs. normoxia cells, #P<0.05 vs. miR-NC cells, ^P<0.05 vs. miR-145 cells.
Mitochondria play a critical role in the process of cell apoptosis. As shown in Figure 5C, hypoxia-treated cardiomyocytes exhibited substantially decreased mitochondrial depolarization compared to control, and treatment with miR-145 mimics obviously stabilized the MMP. Additionally, western blot analysis showed hypoxia exposure promoted mitochondrial cytochrome c release into cytosol, and treatment with miR-145 mimics decreased cytochrome c release (Figure 5D).
Discussion
In recent years, the mechanism of MI has been explored intensively. A large quantity of evidence identified the clinical significance of miRNAs in the diagnosis of MI. Meder et al. [19] assessed the levels of miR-145 in peripheral blood samples of patients with MI, and found that miR-145 levels correlate with infarct sizes estimated by Troponin T release. Investigation of these miRNAs would expand our view to better understand the underlying mechanism in MI. In the present study, the rat MI model was successfully established by LAD, and we confirmed the cardioprotective function of miR-145 in MI rats to understand its underlying molecular mechanisms. We found that lentivirus-mediated overexpression of miR-145 significantly improves cardiac function and attenuates infarct size in rats after MI. Similarly, reduced miR-145 levels were also confirmed in hypoxia-treated cardiomyocytes in vitro.
In the design of this study, we hypothesized that impaired cardiac function might be associated with increased myocardial apoptosis. It is well known that myocardial apoptosis contributes to cardiac dysfunction after myocardial I/R injury [20]. The regulation of apoptosis process is associated with the regulation of many genes, includingthe Bcl-2 family and caspase family. We found that overexpression of miR-145 reduced the expression of cleaved caspase-3 and Bax, whereas it increased Bcl-2 expression in the infarcted myocardial tissues of MI rats. Mitochondria function as central regulators of apoptosis in cell fidelity, which play a critical role in both life and death of cardiomyocytes [21,22]. Myocardial necrosis due to MI leads to increased circulating mitochondrial DNA levels [23]. Li et al. demonstrated that miR-145 plays an important role in regulating mitochondrial apoptotic pathway in heart challenged with oxidative stress [24]. Our in-vitro data showed that miR-145 overexpression causes a reduction of apoptosis in hypoxia-treated cardiomyocytes partly through attenuating mitochondria dysfunction.
As a next step, we used the online websites to identify target genes of miR-145. MiRNAs modulate their biological functions via binding to their target mRNAs and inhibiting their protein expression [25]. In the current study, we identified PDCD4 as a direct target of miR-145 in cardiomyocytes. PDCD4, localized at chromosome band 10q24, was identified as an up-regulated gene during apoptotic process. The involvement of PDCD4 in MI has been well documented. For example, miR-21 protects myocardium against ischemia-induced apoptosis and ischaemia/reperfusion injury through targeting PDCD4 [26,27]. In addition, miR-499-5p protects the cardiomyocytes against apoptosis induced by AMI via PDCD4 [28]. In the present study, we also found that the protective effect of miR-145 against hypoxia-induced mitochondrial dysfunction and cell apoptosis in cardiomyocytes was attenuated by overexpression of PDCD4.
In summary, our data firstly demonstrated the cardioprotective effects of miR-145 in MI through targeting PDCD4. MiR-145 overexpression protects myocardium against ischaemia/reperfusion-induced apoptosis and mitochondrial dysfunction. These findings provided an improved understanding of the mechanism by which miR-145 is involved in the pathogenesis of MI. This information is critical for the development of novel therapies for MI.
Acknowledgements
The work of our research was supported by Shanghai Natural Science Foundation (Grant number: YG2016QN33).
Disclosure of conflict of interest
None.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Baron T, Hambraeus K, Sundstrom J, Erlinge D, Jernberg T, Lindahl B. Type 2 myocardial infarction in clinical practice. Heart. 2015;101:101–6. doi: 10.1136/heartjnl-2014-306093. [DOI] [PubMed] [Google Scholar]
- 3.Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451:914–8. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamas GA, Pfeffer MA. Increased left ventricular volume following myocardial infarction in man. Am Heart J. 1986;111:30–5. doi: 10.1016/0002-8703(86)90549-1. [DOI] [PubMed] [Google Scholar]
- 5.Ji L, Fu F, Zhang L, Liu W, Cai X, Zhang L, Zheng Q, Zhang H, Gao F. Insulin attenuates myocardial ischemia/reperfusion injury via reducing oxidative/nitrative stress. Am J Physiol Endocrinol Metab. 2010;298:E871–80. doi: 10.1152/ajpendo.00623.2009. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Osman A. MicroRNAs in health and disease--basic science and clinical applications. Clin Lab. 2012;58:393–402. [PubMed] [Google Scholar]
- 8.Liu X, Zhang Y, Du W, Liang H, He H, Zhang L, Pan Z, Li X, Xu C, Zhou Y, Wang L, Qian M, Liu T, Yin H, Lu Y, Yang B, Shan H. MiR-223-3p as a novel MicroRNA regulator of expression of voltage-gated K+ channel Kv4.2 in acute myocardial infarction. Cell Physiol Biochem. 2016;39:102–14. doi: 10.1159/000445609. [DOI] [PubMed] [Google Scholar]
- 9.Li R, Geng HH, Xiao J, Qin XT, Wang F, Xing JH, Xia YF, Mao Y, Liang JW, Ji XP. miR-7a/b attenuates post-myocardial infarction remodeling and protects H9c2 cardiomyoblast against hypoxia-induced apoptosis involving Sp1 and PARP-1. Sci Rep. 2016;6:29082. doi: 10.1038/srep29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Sun F, Wang Y, Yang W, Xiao H, Zhang Y, Lu R, Zhu H, Zhuang Y, Pan Z, Wang Z, Du Z, Lu Y. Suppression of microRNA-16 protects against acute myocardial infarction by reversing beta2-adrenergic receptor down-regulation in rats. Oncotarget. 2017;8:20122–32. doi: 10.18632/oncotarget.15391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye Y, Perez-Polo JR, Qian J, Birnbaum Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiol Genomics. 2011;43:534–42. doi: 10.1152/physiolgenomics.00130.2010. [DOI] [PubMed] [Google Scholar]
- 12.Caruso P, Dempsie Y, Stevens HC, McDonald RA, Long L, Lu R, White K, Mair KM, McClure JD, Southwood M, Upton P, Xin M, van Rooij E, Olson EN, Morrell NW, MacLean MR, Baker AH. A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ Res. 2012;111:290–300. doi: 10.1161/CIRCRESAHA.112.267591. [DOI] [PubMed] [Google Scholar]
- 13.Li R, Yan G, Zhang Q, Jiang Y, Sun H, Hu Y, Sun J, Xu B. miR-145 inhibits isoproterenol-induced cardiomyocyte hypertrophy by targeting the expression and localization of GATA6. FEBS Lett. 2013;587:1754–61. doi: 10.1016/j.febslet.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang M, Cheng YJ, Sara JD, Liu LJ, Liu LP, Zhao X, Gao H. Circulating MicroRNA-145 is associated with acute myocardial infarction and heart failure. Chin Med J (Engl) 2017;130:51–6. doi: 10.4103/0366-6999.196573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong YM, Liu XX, Wei GQ, Da YN, Cha L, Ma CS. Prediction of long-term outcome after acute myocardial infarction using circulating miR-145. Scand J Clin Lab Invest. 2015;75:85–91. doi: 10.3109/00365513.2014.981855. [DOI] [PubMed] [Google Scholar]
- 16.Fishbein MC, Maclean D, Maroko PR. Experimental myocardial infarction in the rat: qualitative and quantitative changes during pathologic evolution. Am J Pathol. 1978;90:57–70. [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Nicolas FE. Experimental validation of microRNA targets using a luciferase reporter system. Methods Mol Biol. 2011;732:139–52. doi: 10.1007/978-1-61779-083-6_11. [DOI] [PubMed] [Google Scholar]
- 19.Meder B, Keller A, Vogel B, Haas J, Sedaghat-Hamedani F, Kayvanpour E, Just S, Borries A, Rudloff J, Leidinger P, Meese E, Katus HA, Rottbauer W. MicroRNA signatures in total peripheral blood as novel biomarkers for acute myocardial infarction. Basic Res Cardiol. 2011;106:13–23. doi: 10.1007/s00395-010-0123-2. [DOI] [PubMed] [Google Scholar]
- 20.Yaoita H, Ogawa K, Maehara K, Maruyama Y. Apoptosis in relevant clinical situations: contribution of apoptosis in myocardial infarction. Cardiovasc Res. 2000;45:630–41. doi: 10.1016/s0008-6363(99)00349-1. [DOI] [PubMed] [Google Scholar]
- 21.Gustafsson AB, Gottlieb RA. Heart mitochondria: gates of life and death. Cardiovasc Res. 2008;77:334–43. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 22.Camara AK, Bienengraeber M, Stowe DF. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol. 2011;2:13. doi: 10.3389/fphys.2011.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bliksøen M, Mariero LH, Ohm IK, Haugen F, Yndestad A, Solheim S, Seljeflot I, Ranheim T, Andersen GØ, Aukrust P, Valen G, Vinge LE. Increased circulating mitochondrial DNA after myocardial infarction. Int J Cardiol. 2012;158:132–4. doi: 10.1016/j.ijcard.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 24.Li R, Yan G, Li Q, Sun H, Hu Y, Sun J, Xu B. MicroRNA-145 protects cardiomyocytes against hydrogen peroxide (H2O2)-induced apoptosis through targeting the mitochondria apoptotic pathway. PLoS One. 2012;7:e44907. doi: 10.1371/journal.pone.0044907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brodersen P, Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat Rev Mol Cell Biol. 2009;10:141–8. doi: 10.1038/nrm2619. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. 2009;47:5–14. doi: 10.1016/j.yjmcc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via antiapoptosis through its target PDCD4. Cardiovasc Res. 2010;87:431–9. doi: 10.1093/cvr/cvq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Lu J, Bao X, Wang X, Wu J, Li X, Hong W. MiR-499-5p protects cardiomyocytes against ischaemic injury via anti-apoptosis by targeting PDCD4. Oncotarget. 2016;7:35607–17. doi: 10.18632/oncotarget.9597. [DOI] [PMC free article] [PubMed] [Google Scholar]