

Abstract
Staphylococcus aureus (S. aureus) peptidoglycan (PGN-sa), the major cell wall component of S. aureus, has been demonstrated to be an important virulence factor in the pathogenesis of S. aureus-induced osteomyelitis. However, the exact role of PGN-sa in osteoclastogenesis during S. aureus-induced osteomyelitis and its underlying molecular mechanisms remain unclear. In this study, we found that PGN-sa promoted receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL)-induced osteoclast formation. Quantitative real-time polymerase chain reaction results showed that the mRNA expression of osteoclast-specific marker genes, including tartrate-resistant acid phosphatase, cathepsin K, matrix metalloproteinase-9, and calcitonin receptor was upregulated by PGN-sa treatment. The results of enzyme linked immunosorbent assay showed that PGN-sa promoted the production of proinflammatory cytokines in mouse bone marrow macrophages (mBMMs) treated with RANKL. PGN-sa enhanced RANKL-stimulated protein expression of Toll-like receptor 2 (TLR2), p-IκBα, and nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1). Luciferase reporter assay showed that PGN-sa increased the transcriptional activity of TLR2 and NF-κB in mBMMs treated with RANKL. In addition, we found that downregulation of TLR2 attenuated the effect of PGA-sa on RANKL-induced osteoclastogenesis and activation of the NF-κB/NFATc1 signaling pathway. Taken together, this study revealed that PGN-sa promotes osteoclast formation via TLR2-mediated activation of the NF-κB/NFATc1 signaling pathway, revealing a potential effect of PGN-sa on osteomyelitis. These findings provide new insights into the pathogenic role of PGN-sa in S. aureus-induced osteomyelitis and may help to develop new therapeutic strategies for osteomyelitis.
Keywords: Staphylococcus aureus, peptidoglycan, osteoclastogenesis, TLR2, NF-κB, NFATc1
Introduction
Osteomyelitis is a severe bone infection mainly caused by Staphylococcus aureus (S. aureus). S. aureus not only triggers an immune reaction, but induces osteoblast apoptosis and osteoclastogenesis [1]. Micro-computed tomography has revealed that S. aureus triggers striking alterations in bone turnover, resulting inseverecortical bone destruction [2]. S. aureushas also been shown to induce osteoclastogenesisby producing multiple pathogenic substances, such as peptidoglycan (PGN), lipoteinchoic acid, and coagulase, which are suggested to play a vitalrole in the pathogenesis of bone infections. For example, staphylococcal protein A has been proved to modulate osteoclastogenesis and bone resorption during S. aureus infection [3,4]. Claro et al. suggested that staphylococcal protein A, panton-valentine leukocidin, and coagulase could aggravate bone loss and bone destruction in osteomyelitis [5]. Undoubtedly, understanding the molecular mechanism of osteoclastogenesis induced by S. aureus will contribute to the development of effective treatments for osteomyelitis.
S. aureus PGN (PGN-sa), a virulence factor of S. aureus, is known to be a ligand for Toll-like receptor 2 (TLR2). Several recent researches have shown that PGN-sa contributes to the inflammatory reaction of osteomyelitis and osteoclastogenesis. For instance, Takashi et al. demonstrated that PGN-sa stimulates osteoclast precursors in mouse bone marrow macrophages (mBMMs) in vitro and inducesosteoclastogenesis [6]. Sato et al. suggested that PGN of Actinomyces naeslundii T14V obviously induces osteoclast formation in co-cultures of MCTC3/PA6 cells and BALB/c mouse bone marrow cells [7]. Ren et al. demonstrated that more osteoclast-like cells were induced in RAW 264.7 cells in the presence of PGN-sa than in the absence of PGN-sa [8]. Lactobacillus acidophilus PGN elevated TLR2 level in bovine β-lactoglobulin-sensitized mice, and upregulated nuclear factor-κB (NF-κB) expression [9]. Muramyl dipeptide, the minimal essential structural unit of PGNs, enhances lipopolysaccharide-induced osteoclastformation and bone resorption [10]. These data from previous literature imply that PGN plays a key role in bacteria-induced osteomyelitis. However, the effects of PGN-saon osteoclastogenesis and its exact molecular mechanism during S. aureus-induced osteomyelitis remain unclear.
TLR2 is known to be able to recognize the molecular patterns of bacteria, and plays a potential role in osteoclastogenesis induced by S. aureus. Matsumoto et al. demonstrated that TLR2 heterodimer signaling contributes toosteoclast formation in periodontal disease by inducing prostaglandin E production [11]. Chen et al. reported that S. aureus infection promotescell apoptosis and TLR2 expression, whereas downregulation of TLR2 decreases apoptosis induced by S. aureus in the osteoblastic cell line MC3T3-E1 [12]. Huang et al. suggested that TLR2 stimulation by Pam3CSK4 (a TLR2 agonist) upregulates Trim13 expression, thereby inducing cytokine and chemokine production through activation of NF-κB, and eventually resulting in foam cell formation [13]. Based on these findings, we hypothesize that PGN-sa might enhance osteoclastogenesis via TLR2-mediated activation of NF-κB and other downstream osteoclastogenesis-associated genes, thereby leading to the development of osteomyelitis.
In this study, we aimed to investigate the effects of PGN-saonreceptor activator of NF-κB ligand (RANKL)-induced osteoclastogenesis and explored its underlying molecular mechanisms.
Materials and methods
Animals
Four-week-old C57/BL6 mice were purchased from HuaFuKang Bioscience Co. (Beijing, China) and acclimatized for 1 week. Mice had free access to food and water, and were housed at 25 ± 2°C and 55 ± 5% relative humidity with a 12 h light-dark cycle.
All animal studies were performed in compliance with the Guide for the Care and Use of Laboratory Animals and approved by the Ethical Committee of Tongji Medical College of Huazhong University of Science and Technology.
Cell isolate and culture
mBMMs were isolated from the bones of 6-week-old C57/BL6 mice. For cell proliferation, cells were cultured in culture medium containing 30 ng/mL macrophage-colony stimulating factor (M-CSF; R&D Systems, Minnneapolis, MN, USA) for 3 days. mBMMs were used after washing in order to purify the adherent cells. mBMMs were then cultured in an osteoclastogenic medium supplemented with 100 ng/mL RANKL (R&D Systems) for 3 days, and treated with different concentrations of PGN-sa (Sigma-Aldrich, St. Louis, MO, USA) for 48 h.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was obtained from mBMMs using the Qiagen RNeasy Micro Kit (Qiagen, Hilden, Germany) and cDNA was synthesized with the PrimeScript RT reagent kit (Takara Bio Inc., Tokyo, Japan). RT-PCR was performed using the ABI Prism 7900 sequence detection system (Life Technologies, Carlsbad, CA, USA) with β-actin as an endogenous control gene. The PCR primers used wereas follows: mouse tartrate-resistant acid phosphatase (TRAP) (sense: 5’-CTGCTGGGCCTACAAATCAT-3’ and antisense: 5’-GGT AGT AAG GGC TGG GGA AG-3’); cathepsin K (sense: 5’-AGG CGG CTA TAT GAC CAC TG-3’ and antisense: 5’-CCG AGC CAA GAG AGC ATA TC-3’); matrix metalloproteinase-9 (MMP9) (sense: 5’-CGT CGT GAT CCC CAC TTA CT-3’ and antisense: 5’-AGA GTA CTG CTT GCC CAG GA-3’); calcitonin receptor (sense: 5’-CGC ATC CGC TTG AAT GTG-3’ and antisense: 5’-TCT GTC TTT CCC CAG GAA ATG A-3’); TLR2 (sense: 5’-CTG AGA ATG ATG TGG GCG TG-3’ and antisense: 5’-ATG GGA ATC CTG CTC ACT GTA G-3’); and β-actin (sense: 5’-TTC TAC AAT GAG CTG CGT GT-3’ and antisense: 5’-CTC ATA GCT CTT CTC CAG GG-3’). Relative mRNA expression levels were quantified using the 2-ΔΔCt method.
TRAP assay
mBMMs were washed with phosphate buffered saline, immersed in fixative solution for 5 min, and then incubated in TRAP staining solution for 1 h at 37° C in the dark. TRAP-positive multinucleated cells (MNCs) containing three or more nuclei in each cell were counted as osteoclasts.
Small-interfering RNA (si-RNA) transfection
si-TLR2 and negative control (si-NC) were designed and synthesized by Shenggong Bio. Co., Ltd (Shanghai, China). mBMMs were transfected with si-TLR2 or si-NC using Lipofectamine 2000 reagent (Invitrogen, Inc. Carlsbad, CA, USA) according to the manufacturer’s instructions. At 48 h after transfection, mBMMs were harvested and subjected to western blot analysis.
Luciferase reporter gene assay
To evaluate the impact of PGN-sa on transcriptional activation of TLR2 and NF-κB, luciferase reporter assay was performed according to the manufacturer’s protocol. The promoter sequences of TLR2 or NF-κB were amplified and cloned into Luc-SV40 plasmid (Promega, Madison, WI, USA) to generate Luc-SV40-TLR2 or Luc-SV40-NF-κB reporter plasmids. mBMMs were transiently transfected with luciferase reporter plasmid and then cultured for 48 h, followed by lysis. Transcriptional activation of TLR2 and NF-κB was evaluated using a luminometer (Promega, Madison, WI, USA).
Western blot analysis
Total protein was isolated from mBMMs using ice-cold radioimmunoprecipitation (RIPA) lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China). The extracted proteins were separated on 14% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred ontopolyvinylidene fluoride membranes (Millipore Corporation, Billerica, MA, USA). The membranes were then blocked with Tris-buffered saline containing 5% skim milk and 0.1% Tween-20 (TBST) and cultured at 4°C overnight with the following antibodies: anti-TLR2 (Abcam, Cambridge, MA, USA), anti-p-lκBα (Cell Signaling Technology, Danvers, MA, USA), anti-NFATc1 (Abcam), or anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The membranes were washed with TBST for 30 min and incubated with the corresponding horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) at room temperature for 1 h. The bands were visualized using anEasyBlot ECL kit (Shenggong, Shanghai, China). The intensity of each band was measured by the ImageJ software (National Institutes of Health, NY, USA), and the relative protein expression was compared with β-actin.
Proinflammatory cytokine measurement
In order to assess the production of proinflammatory cytokines by mBMMs, tumor necrosis factorα (TNF-α), interleukin (IL)-1β, IL-6, and IL-12 levels in the cell supernatants were detected usinga commercial enzyme-linked immunosorbent assay (ELISA) kit (Beyotime).
Statistical analysis
Each experiment was repeated at least 3 times. All data are expressed as the mean ± standard deviation, and analyzed using SPSS 20.0 software. Student’s t-test was used to compare the means of two groups. One-Way Analysis of Variance was used to compare the means of three groups. A P-value of 0.05 or less was considered statistically significant.
Results
PGN-sa promotes RANKL-induced osteoclastogenesis in mBMMs
In order to assess the effect of PGN-sa on osteoclast formation, mBMMs were treated with different concentrations of PGN-sa (0, 1, 10, and 100 μM). As shown in Figure 1A and 1B, PGN-sa promoted the formation of TRAP-positive MNCs in a dose-dependent manner. qRT-PCR analysis was performed to examine the expression of osteoclast-specific marker genes, including TRAP, cathepsin K, MMP9, and calcitonin receptor, in the presenceorabsence of PGN-sa. Results showed that PGN-sa promoted the mRNA expression of TRAP, cathepsin K, MMP9, and calcitonin receptor in a concentration-dependent manner (Figure 1C-F). Taken together, these results indicated that treatment with PGN-sa results in dose-dependent promotion of osteoclast formation.
Figure 1.
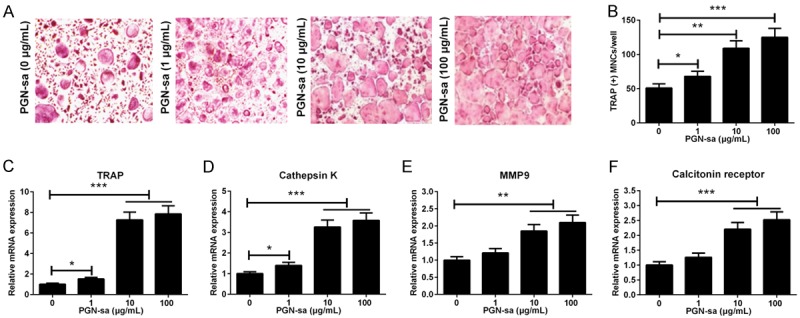
Effect of PGN-sa on RANKL-induced osteoclastogenesis. A. Representative images of TRAP assay were shown. B. The number of TRAP-positive MNCs cells were significantly increased by PGN-sa treatment in a dosedependent manner. C-F. PGN-saincreased the mRNA expression levelsof osteoclastogenesis-associated genes, including TRAP, cathepsin K, MMP9, and calcitonin receptor, in a dose-dependent manner. *P<0.05, **P<0.01, and ***P<0.001.
PGN-sa accelerates the production of proinflammatory cytokines in mBMMs treated with RANKL
To investigate whether PGN-sa accelerates the production of proinflammatory cytokines in mBMMs treated with RANKL, the levels of TNF-α, IL-1β, IL-6, and IL-12 in cell supernatants were determined. The levels of TNF-α (Figure 2A), IL-1β (Figure 2B), IL-6 (Figure 2C), and IL-12 (Figure 2D) were significantly increased in the presence of PGN-sa at concentrations of 10 and 100 μg/mL. These results indicated that PGN-sa evokes inflammation in mBMMs treated with RANKL.
Figure 2.
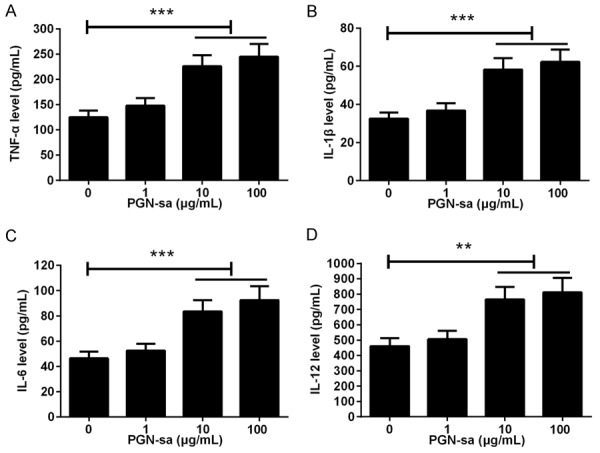
Effect of PGN-sa on the production of proinflammatory cytokines in mBMMs treated with RANKL. mBMM were pretreated with RANKL and then treated with different concentrations of PGN-sa (0, 1, 10, and 100 μg/mL) for 48 h. ELISA tests results showed that PGN-saaccelerated the production of TNF-α (A), IL-1β (B), IL-6 (C), and IL-12 (D) in a dose-dependent manner. **P<0.01 and ***P<0.001.
PGN-sa enhances RANKL-stimulated activation of NF-κB and NFATc1
To clarify the exact molecular mechanismunderlying the effect of PGN-saon osteoclastogenesis, qRT-PCR, western blotting, and luciferase reporter gene assays were performed to evaluate the regulatory effect of PGN-sa on TLR2, NF-κB, and NFATc1. As shown in Figure 3A and 3B, we found that the TLR2 mRNA and protein expression as well as p-lκBα and NFATc1 protein expression levels were upregulated by PGN-sa in a dose-dependent manner. Luciferase reporter gene assay showed that 10 and 100 μg/mL of PGN-sa obviously elevated TLR2 and NF-κB transcriptional activity (Figure 3C and 3D). These data demonstrated that PGN-sa enhances RANKL-stimulated activation of NF-κB and TLR2.
Figure 3.
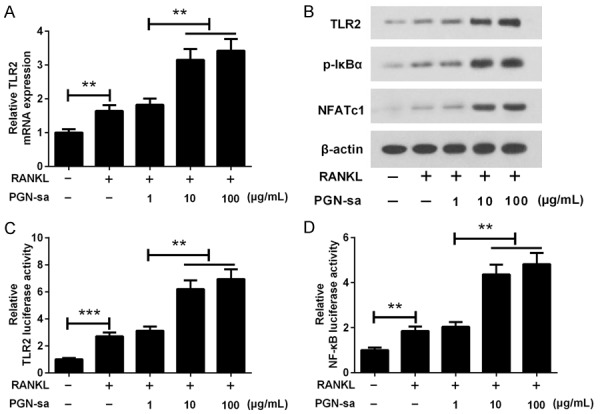
Effect of PGN-sa on RANKL-stimulated activation of NF-κB and NFATc1. A. qRT-PCR assay showed that PGN-saupregulated the expression of TLR2 mRNA in a dose-dependent manner. B. Results of western blot analysis showed that PGN-sa increased the protein expression levels of TLR2, p-IκBα and NFATc1. β-actin was presented as an internal control. C and D. Luciferase reporter gene assay showed that PGN-sapromoted RANKL-stimulated activation of TLR2 and NF-κB. **P<0.01 and ***P<0.001.
Downregulation of TLR2 attenuates the effect of PGA-sa on RANKL-induced osteoclastogenesis
To further explore the role of TLR2 in osteoclastogenesis promoted by PGA-sa, TLR2 knockdown in mBMMs was carried out by transfection with si-TLR2. qRT-PCR and western blot results showed that compared to the si-NC group, TLR2 mRNA and protein expression levels were significantly downregulated in the si-TLR2 group; si-NC had no effect on TLR2 expression (Figure 4A and 4B). TLR2 knockdown attenuated the promotion of RANKL-induced osteoclastogenesis caused by PGA-sa (Figure 4C). To further confirm the role of TLR2, we determined the changes in expression of osteoclast-specific marker genes in response to PGN-sa in RANKL-treated mBMMs. We found that, compared with mBMMs transfected with si-NC, the mRNA expression of TRAP, cathepsin K, MMP9, and calcitonin receptor was obviously decreasedin mBMMs transfected with si-TLR2 (Figure 4D-G), indicating that knockdown of TLR2 attenuates the promotion of RANKL-induced osteoclastogenesis caused by PGA-sa.
Figure 4.
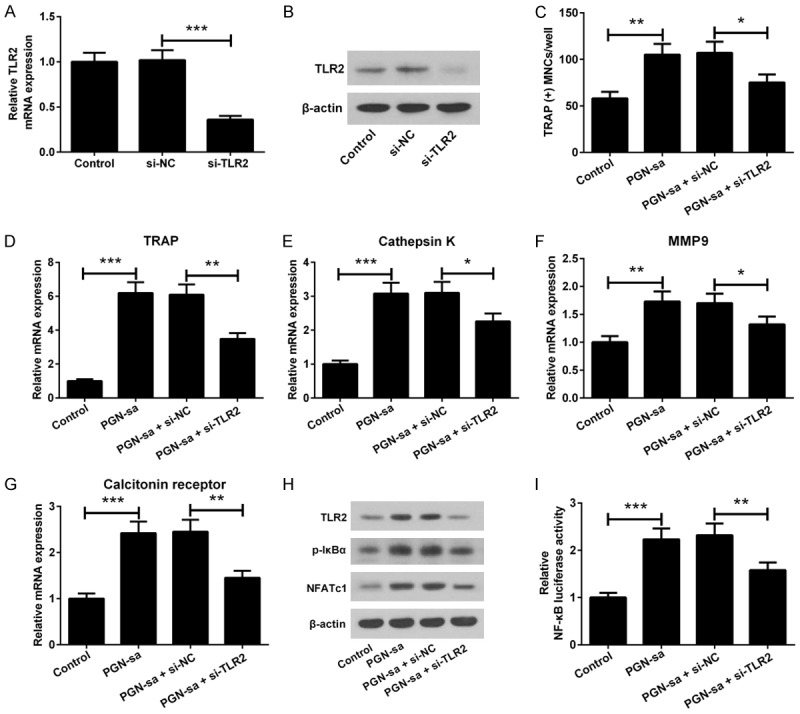
Downregulation of TLR2 attenuates the effect of PGA-sa on RANKL-induced osteoclastogenesis and activation of the NF-κB/NFATc1 signaling pathway. The inhibitory effect of si-TLR2 on TLR2 mRNA and protein expression was validated by qRT-PCR (A) and western blot analysis (B), respectively. (C) The promotoryeffect of PGA-sa on RANKL-induced osteoclastogenesis was attenuated by TLR2 knockdown. (D-G) qRT-PCR assay showed that downregulation of TLR2 by si-TLR2 reversed the upregulatory effect of PGA-saon mRNA expression of TRAP,cathepsin K, MMP9, and calcitonin receptor. (H) The representative image of western blot analysis of TLR2, p-IκBα, and NFATc1 protein expression. (I) Luciferase reporter gene assay showed that si-TLR2 attenuated the promotion of RANKLstimulated activation of NF-κB caused by PGA-sa. *P<0.05, **P<0.01, and ***P<0.001.
Downregulation of TLR2 attenuates the effect of PGN-sa on RANKL-induced activation of theNF-κB/NFATc1 signaling pathway
Compared with the PGN-sa + si-NC group, the protein levels of TLR2, p-lκBα, and NFATc1 were obviously decreased in the PGN-sa + si-TLR2 group (Figure 4H). In addition, NF-κB activity in the PGN-sa + si-TLR2 group was remarkably lower than that in the PGN-sa + si-NC group (Figure 4I). These findings indicated that PGN-sa activates the NF-κB/NFATc1 signaling pathway via a TLR2-dependent pathway, thereby revealing a potential pathogenic role of PGN-sa in S. aureus-induced osteomyelitis.
Discussion
Osteomyelitis is characterized by impaired bone quality and bone mass, and usually accompanied by inflammation and bone destruction. Bone homeostasis is maintained by a dynamic balance betweenthe activities of osteoclasts and osteoblasts [14]. Osteoblasts are involved in bone matrix formation, whereas osteoclasts contribute to bone resorption by expediting acidification and release of lysosomal enzymes [3]. Abnormal osteoclast activation is a major cause of osteomyelitis. Thus, prevention of osteoclastogenesis may have important clinical significance in the treatment of osteomyelitis.
Differentiation of osteoclasts requires two cytokines: M-CSF and RANKL. M-CSF is essential for precursor proliferation and differentiation, while RANKL activates the downstream signaling pathways and regulates osteoclastformation [15]. RANKL binds to its receptor and then recruits TNF receptor-associated factor 6 (TRAF6). RANK/TRAF6 interaction activates the downstream signaling pathways, including NF-κB, mitogen-activated protein kinases (MAPKs), c-Jun N-terminal protein kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 [16,17]. RANK/TRAF6 also induces phosphorylation and degradation of IκBα, and release of p65/p50 heterodimers, along with activation of NF-κB and NFATc1 [18,19]. NFATc1 induces osteoclast differentiation by upregulating osteoclastogenesis-associated genes, including TRAP, cathepsin K, MMP9, and calcitonin receptor, thereby promoting the development of osteoclastogenesis, eventually leading to osteomyelitis [20,21]. Thus, expanding our knowledge on the underlying mechanism of S. aureus-induced osteomyelitis will help develop novel therapeutic strategies. In the present study, we found that PGN-sapromoted RANKL-induced osteoclastogenesis in mBMMs and upregulated the mRNA expression of osteoclastogenesis-associated genes, including TRAP, cathepsin K, MMP9, and calcitonin receptor.
It has been reported that inflammation is closely linked with osteoclastogenesis. The interaction of mesenchymal cells with lymphocytes and cells of the osteoclast lineage can promote osteoclast metabolism and regulate proinflammatory cytokines production and RANKL. S. aureus infection has been shown to play a role in inflammatory response, and increases the production of proinflammatory cytokines, including TNF-α, IL-1β, IL-6, and IL-12 in the early stages of osteomyelitis [22,23]. These proinflammatory cytokines bind to their receptors and activate the inflammatory signaling pathways through activation of the downstream NF-κB signaling pathway, resulting in osteoclastogenesis [24]. Ju et al. reported that IL-1β enhanced osteoclast formation by activating the NF-κB signaling pathway in BMMs treatedwith M-CSF and RANKL [25]. Cao et al. demonstrated that TNF-α exerted stimulatory and inhibitory effects on osteoclastogenesis inmouse monocytes. Their data suggested that TNF-α suppressed osteoclastogenesis by regulating the RANK signaling pathway in mouse monocytes, but this inhibitory effect of TNF-α was prevented by treatment with M-CSF and RANKL [26]. A growing number of evidence has shown that PGN-saactivatesproinflammatory cytokine production and plays a pivotal role in the hostimmune response to S. aureus [27]. Takeda et al. showed that PGN promoted the mRNA and protein expression of IL-6 and IL-8 inhuman dental pulp cells by activating phosphorylated p38 kinase [28]. Irving et al. showed that once PGN was delivered into the host cell cytosol, it could be detected by the intracellular innate immune receptor NOD1, thereby inducing autophagosome formation and inflammatory IL-8 responses in epithelial cells [29]. In this study, we observed that PGN-sacould remarkably enhance the production of proinflammatory cytokines, including TNF-α, IL-1β, IL-6, and IL-12 in a dose-dependent manner.
As aspecific pattern recognition molecule, TLR2 induces inflammation and osteoclastogenesis through the NF-κB/NFATc1 signaling pathway. NF-κB is classified as a Rel protein in terms of the Rel homology domainatits N-terminus. Stimulation of TLR2 leads to degradation of IκB, and the NF-κB complex is then freed to enter the nucleus where it can ‘turn on’ the expression of NFATc1, thuspromoting osteoclast differentiation and fusion [30]. Chen et al. suggested that TLR2 and IL-1 contributed to RANKL-induced activation of osteoclast genes and expression of NFATc1 in a MyD88-dependent manner [31]. In this study, our results showed that PGN-saenhanced RANKL-stimulated activation of TLR2 and NF-κB, as well as the expression of NFATc1. In addition, we found that knockdown of TLR2 attenuated the promotion of RANKL-induced osteoclastogenesis and activation of the NF-κB/NFATc1 signaling pathway, caused by PGN-sa. These results strongly suggested that TLR2 isa critical mediator of RANKL-induced osteoclastogenesis in mBMMs.
In conclusion, our study suggests that PGN-sapromotes osteoclastogenesis by upregulating NF-κB and NFATc1 expression via activation of TLR2. These findings provide new insights into the pathogenic role of PGN-sa in S. aureus-induced osteomyelitis and may help develop new therapeutic strategies to control bone loss caused by osteomyelitis.
Disclosure of conflict of interest
None.
References
- 1.Shi S, Zhang X. Interaction of Staphylococcus aureus with osteoblasts (Review) Exp Ther Med. 2012;3:367. doi: 10.3892/etm.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X, Jiao J, He X, Zhang J, Wang H, Xu Y, Jin T. CHI3L1 regulation of inflammation and the effects on osteogenesis in a staphylococcus aureus-Induced murine model of osteomyelitis. FEBS J. 2017;284:1738–1747. doi: 10.1111/febs.14082. [DOI] [PubMed] [Google Scholar]
- 3.Bertelli AM, Delpino MV, Lattar S, Giai C, Llana MN, Sanjuan N, Cassat JE, Sordelli D, Gomez MI. Staphylococcus aureus protein A enhances osteoclastogenesis via TNFR1 and EGFR signaling. Biochim Biophys Acta. 2016;1862:1975–1983. doi: 10.1016/j.bbadis.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Liu X, Dou C, Cao Z, Liu C, Dong S, Fei J. Staphylococcal protein A promotes osteoclastogenesis through MAPK signaling during bone infection. J Cell Physiol. 2017;232:2396–2406. doi: 10.1002/jcp.25774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claro T, Widaa A, O’Seaghdha M, Miajlovic H, Foster TJ, O’Brien FJ, Kerrigan SW. Staphylococcal protein a, panton-valentine leukocidin and coagulase aggravate the bone loss and bone destruction in osteomyelitis. Cell Physiol Biochem. 2015;32:322–333. doi: 10.1159/000354440. [DOI] [PubMed] [Google Scholar]
- 6.Kishimoto T, Kaneko T, Ukai T, Yokoyama M, Ayon Haro R, Yoshinaga Y, Yoshimura A, Hara Y. Peptidoglycan and lipopolysaccharide synergistically enhance bone resorption and osteoclastogenesis. J Periodontal Res. 2012;47:446–454. doi: 10.1111/j.1600-0765.2011.01452.x. [DOI] [PubMed] [Google Scholar]
- 7.Sato T, Watanabe K, Kumada H, Toyama T, Taniishii N, Hamada N. Peptidoglycan of Actinomyces naeslundii induces inflammatory cytokine production and stimulates osteoclastogenesis in alveolar bone resorption. Arch Oral Biol. 2012;57:1522–1528. doi: 10.1016/j.archoralbio.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Ren L, Yongqing XU, Wang H, Xiaoqing HE, Song M, Chen X. Effect of staphylococcalpeptidoglycanonosteoclast differentiation. Chinese J Rep Reconstr Surg. 2016;8:1244–1248. doi: 10.7507/1002-1892.20160254. [DOI] [PubMed] [Google Scholar]
- 9.Li AL, Sun YQ, Du P, Meng XC, Guo L, Li S, Zhang C. The Effect of Lactobacillus actobacillus Peptidoglycan on Bovine β-Lactoglobulin-Sensitized Mice via TLR2/NF-κB Pathway. Iran J Allergy Asthma Immunol. 2017;16:147–158. [PubMed] [Google Scholar]
- 10.Ishida M, Kitaura H, Kimura K, Sugisawa H, Aonuma T, Takada H, Takanoyamamoto T. Muramyl dipeptide enhances lipopolysaccharide-induced osteoclast formation and bone resorption through increased RANKL expression in stromal cells. J Immunol Res. 2015;2015:e132765. doi: 10.1155/2015/132765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsumoto C, Oda T, Yokoyama S, Tominari T, Hirata M, Miyaura C, Inada M. Toll-like receptor 2 heterodimers, TLR2/6 and TLR2/1 induce prostaglandin E production by osteoblasts, osteoclast formation and inflammatory periodontitis. Biochem Biophys Res Commun. 2012;428:110–115. doi: 10.1016/j.bbrc.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 12.Chen Q, Hou T, Luo F, Wu X, Xie Z, Xu J. Involvement of toll-like receptor 2 and pro-apoptotic signaling pathways in bone remodeling in osteomyelitis. Cell Physiol Biochem. 2014;34:1890–1900. doi: 10.1159/000366387. [DOI] [PubMed] [Google Scholar]
- 13.Huang B, Baek SH. Trim13 potentiates TLR2-mediated NF-κB activation via K29-linked polyubiquitination of TRAF6. Mol Pharmacol. 2017;116:307–316. doi: 10.1124/mol.116.106716. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Xu S, Li K, Tan K, Liang K, Wang J, Shen J, Zou W, Hu L, Cai D, Ding C, Li M, Xiao G, Liu B, Liu A, Bai X. mTORC1 inhibits NF-κB/NFATc1 signaling and prevents osteoclast precursor differentiation, in vitro and in mice. J Bone Miner Res. 2017;32:1829–1840. doi: 10.1002/jbmr.3172. [DOI] [PubMed] [Google Scholar]
- 15.Guo S, Ni Y, Ben J, Xia Y, Zhou T, Wang D, Ni J, Bai H, Wang L, Ma J. Class a scavenger receptor exacerbates osteoclastogenesis by an Interleukin-6-mediated mechanism through ERK and JNK signaling pathways. Int J Biol Sci. 2016;12:1155–1167. doi: 10.7150/ijbs.14654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SY, Lee KS, Yi SH, Kook SH, Lee JC. Acteoside suppresses RANKL-mediated osteoclastogenesis by inhibiting c-Fos induction and NF-κB pathway and attenuating ROS production. PLoS One. 2013;8:e80873. doi: 10.1371/journal.pone.0080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kassem A, Henning P, Lundberg P, Souza PP, Lindholm C, Lerner UH. Porphyromonas gingivalis stimulates bone resorption by enhancing RANKL (receptor activator of NF-κB Ligand) through activation of toll-like receptor 2 in osteoblasts. J Biol Chem. 2015;290:20147–20158. doi: 10.1074/jbc.M115.655787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deepak V, Kasonga A, Kruger MC, Coetzee M. Inhibitory effects of eugenol on RANKL-induced osteoclast formation via attenuation of NF-κB and MAPK pathways. Connect Tissue Res. 2015;56:195–203. doi: 10.3109/03008207.2014.989320. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Z, Hou X, Yin X, Li Y, Duan R, Boyce BF, Yao Z. TNF Induction of NF-κB RelB Enhances RANKL-Induced Osteoclastogenesis by Promoting Inflammatory Macrophage Differentiation but also Limits It through Suppression of NFATc1 Expression. PLoS One. 2015;10:e0135728. doi: 10.1371/journal.pone.0135728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Widaa A, Claro T, Foster TJ, O’Brien FJ, Kerrigan SW. Staphylococcus aureus protein a plays a critical role in mediating bone destruction and bone loss in osteomyelitis. PLoS One. 2012;7:e40586. doi: 10.1371/journal.pone.0040586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gan K, Yang L, Xu L, Feng X, Zhang Q, Wang F, Tan W, Zhang M. Iguratimod (T-614) suppresses RANKL-induced osteoclast differentiation and migration in RAW264.7 cells via NF-κB and MAPK pathways. Int Immunopharmacol. 2016;35:294–300. doi: 10.1016/j.intimp.2016.03.038. [DOI] [PubMed] [Google Scholar]
- 22.Si Y, Chen YB, Chen SJ, Zheng YQ, Liu X, Liu Y, Jiang HL, Xu G, Li ZH, Huang QH. TLR4 drives the pathogenesis of acquired cholesteatoma by promoting local inflammation and bone destruction. Sci Rep. 2015;5:16683. doi: 10.1038/srep16683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Sapkota M, Kim SW, Soh Y. Herbacetin inhibits RANKL-mediated osteoclastogenesis in vitro and prevents inflammatory bone loss in vivo. Eur J Pharmacol. 2016;777:17–25. doi: 10.1016/j.ejphar.2016.02.057. [DOI] [PubMed] [Google Scholar]
- 24.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of roinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7:33–42. doi: 10.1038/nrrheum.2010.196. [DOI] [PubMed] [Google Scholar]
- 25.Moon S, Ahn IE, Jung H, Hyoju YI, Kim J, Kim Y, Kwok S, Park K, Min J, Park S. Temporal differential effects of proinflammatory cytokines on osteoclastogenesis. Int J Mol Med. 2013;31:769–777. doi: 10.3892/ijmm.2013.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao Y, Jansen IDC, Sprangers S, de Vries TJ, Everts V. TNF-α has both stimulatory and inhibitory effects on mouse monocyte-derived osteoclastogenesis. J Cell Physiol. 2017;232:3273–3285. doi: 10.1002/jcp.26024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev. 2005;18:521. doi: 10.1128/CMR.18.3.521-540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda K, Tokunaga N, Aida Y, Kajiya M, Ouhara K, Sasaki S, Mizuno N, Fujita T, Kurihara H. Brain-derived neurotrophic factor inhibits peptidoglycan-induced inflammatory cytokine expression in human dental pulp cells. Inflammation. 2016;40:240–247. doi: 10.1007/s10753-016-0474-4. [DOI] [PubMed] [Google Scholar]
- 29.Irving AT, Mimuro H, Kufer TA, Lo C. The immune receptor NOD1 and kinase RIP2 interact with bacterial peptidoglycan on early endosomes to promote autophagy and inflammatory signaling. Cell Host Microbe. 2014;15:623–635. doi: 10.1016/j.chom.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Aletaha S, Asgary V, Haddad L, Roozbehkia M, Cohan RA, Mahmoudi M, Mirshafiey A. M2000 (β-D-Mannuronic Acid) as a novel antagonist for blocking the TLR2 and TLR4 downstream signaling pathway. Scand J Immunol. 2017;85:122–129. doi: 10.1111/sji.12519. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z, Su L, Xu Q, Katz J, Michalek SM, Fan M, Xu F, Ping Z. IL-1R/TLR2 through MyD88 divergently modulates osteoclastogenesis through regulation of nuclear factor of activated T cells c1 (NFATc1) and b lymphocyte-induced maturation protein-1 (Blimp1) J Biol Chem. 2015;290:30163–30174. doi: 10.1074/jbc.M115.663518. [DOI] [PMC free article] [PubMed] [Google Scholar]