

Abstract
This study was conducted to investigate the role of the cholinergic anti-inflammatory pathway in LPS-induced septic cardiomyopathy in mice. C57BL/6 mice were used to construct septic cardiomyopathy models. The optimal duration of lipopolysaccharide (LPS) treatment was determined by HE staining and TUNEL assay. Blank controls were intraperitoneally injected with saline and models were injected with LPS (10 mg/kg) (LPS), α-bungarotoxin (BT-LPS), BT and dexmedetomidine (BT-DEX-LPS). The pathological examinations were performed on HE- stained myocardium tissues, apoptosis was determined using TUNEL assay, mRNA expression of NF-κB p65, Caspase-3, Caspase-8, Bcl-2, Bax, p53 and α7nACh was quantified using qRT-PCR, protein levels of IL-6, IL-1β, TNF-α and phosphorylated STAT3 (p-STAT3) were analyzed using Western blot analysis. HE staining and TUNEL assays showed that the optimal LPS treatment time for septic cardiomyopathy induction was 16 h. Compared with the blank control, mice in LPS group had significantly higher apoptosis, while DEX and BT reduced apoptosis when they were used separately and increased apoptosis when they were used jointly. In the LPS-treated mice, the levels of NF-κb p65, Caspase-3, Caspase-8, Bax, p53, IL-6, IL-1β, TNF-α and p-STAT3 were significantly increased, while α7nAChR level was decreased significantly (P < 0.01); DEX alone had no impact on the expression of these proteins but significantly up-regulated the expression of these genes except α7nAChR when used jointly with BT (P < 0.01). It is clear that DEX can alleviate heart injury, while α7nAChR-specific blocker BT is antagonistic against the anti-inflammatory effect of DEX on sepsis in mice.
Keywords: Cardiomyopathy, sepsis, apoptosis, gene expression
Introduction
Sepsis is an uncontrollable systemic inflammatory response syndrome (SIRS) caused by pathogenic infections, leading to severe septic shock [1]. In the patients with severe sepsis, approximately 44% have cardiac dysfunction [2,3]. Therefore, it is of great significance to reduce the mortality of patients with sepsis by early diagnosis of cardiac dysfunction and proactive treatments. Recently, it is found that the cholinergic anti-inflammatory pathway (CAP) can stimulate the vague nerve to inhibit the secretion of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) to alleviate excessive systemic inflammatory response [4]. CAP is very important in in vivo immune regulation, because the cholinergic nerve has α7 nicotinic acetylcholine receptor (α7nAChR) to couple with the immune system [5]. The receptor regulates the synthesis and secretion of inflammatory factors such as TNF-α and IL-1 to modulate inflammatory response [6,7]. This physiological mechanism stabilizes the immune system to balance inflammatory and anti-inflammatory responses. Sedative drugs are effective in the treatment of severe septic patients. Dexmedetomidine (DEX) is a highly selective agonist of α2 adrenergic receptor. It acts on α2 adrenergic receptor in the brain and spinal cord to inhibit the activity of sympathetic nerve with analgesic, sedative and antisialogogue functions [8,9]. In addition, it is also shown to protect kidney, brain and heart [10]. α-bungarotoxin (BT) is a neurotoxin, which binds specifically and irreversibly N- type acetylcholine receptor with high affinity. Studies have shown that it has anti-inflammatory and antitumor activities [11]. Therefore, in this study, we investigated the protective role of DEX and BT against myocardial injury using septic cardiomyopathy mouse models induced by LPS. The findings would provide new insights into clinical treatments of septic myocardial injury.
Materials and methods
Animals
Six-week-old specific-pathogen-free C57BL/6 male mice, weighing 23-27 g, were purchased from Slackking Experimental Animal Co, Hunan, China, and all mice were housed under pathogen-free conditions and had access to standard rodent food and water ad libitum. The animal study protocols were approved by Harbin Medical University Animal Care and Use Committee.
Animal model and drug treatment
Septic cardiomyopathy models were constructed by injecting LPS (5 mg/kg, Sigma, USA) via the tail veins. The duration of LPS treatment for successful modelling was determined using HE staining and TUNEL assay according to the severity of sepsis and apoptosis. For drug treatments, the animals were randomly grouped into control, model (LPS) (injected with LPS (5 mg/kg) only), BT-LPS (injected with α-bungarotoxin (BT, 1 µg/kg, Sigma, USA) and LPS), DEX-LPS (injected with dexmedetomidine (DEX, 10 mg/kg, Sigma, USA) and LPS) and BT-DEX-LPS (injected with BT, DEX (10 mg/kg) and LPS). For BT-LPS and DEX-LPS treatments, BT and DEX were injected 1 h before LPS, and for BT-DEX-LPS, BT was injected 1 h before DEX and DEX was injected 1 h before LPS.
HE staining and TUNEL assay
Heart tissue was washed with PBS, fixed in 10% neutral formaldehyde solution, embedded in paraffin and sectioned. The slides were stained with HE stains as described [12] and viewed under light microscope. The sections were also used to detect apoptosis using the TUNEL method as reported [13].
qRT-PCR
Total RNA was isolated from heart tissue by using the TRIzol Reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. RNA quantity was measured by a SmartSpec Plus spectrophotometer (Bio-Rad, Hercules, CA, USA). RNA purity was evaluated by the A260/A280 ratio. Reverse transcription was performed with 200 ng of RNA in a total volume of 10 μl using the High Capacity cDNA Transcriptase Reverse kit (Applied Biosystems by Life Technologies, Carlsbad, California, USA) according to manufacturer’s recommendations. A total of 2.5 μl of the resulting cDNA was subjected to pre-amplification using the SYBR Green qPCR SuperMix (Invitrogen, USA) in a total volume of 12 μl. Non-fluorescent probes were used at 1×. Pre-amplification cycling conditions were 10 min at 95°C followed by 14 cycles, each one consisting of 15 s at 95°C and 4 min at 60°C. RT-qPCR was performed on the 7900HT Fast Real-Time PCR system using TaqMan gene expression assays probes (Applied Biosystems). Human β-actin was used as an internal control. The PCR was carried out in a total volume of 10 μl containing 1.5 μl of diluted and pre-amplified cDNA, 10 μl of TaqMan Gene Expression Master Mix and 1 μl of each fluorescence TaqMan probe using primers listed in Table 1. The cycling conditions were 50°C for 2 min, 95°C for 10 min followed by 40 cycles, each one consisting of 15 s at 95°C and 1 min at 60°C. Samples were run in triplicate and the mean values were calculated for each case.
Table 1.
Primers for qRT-PCR
| Gene | Primer |
|---|---|
| Caspase-8 | F: CCGATTCTCTGGGCAACTGT |
| R: ATCCACATGTGTCCCGTTCC | |
| α7-nAChR | F: ACCTCGTGTGATCCAAAGCC |
| R: GGTTTCCTCTTGCTCAGGGT | |
| Caspase-3 | F: CCACAGCACCTGGTTATT |
| R: ATTCTATCGCCACCTTCC | |
| NF-κbp65 | F: CGACGTATTGCTGTGCCTTC |
| R: TGAGATCTGCCCAGGTGGTAA | |
| Bax | F: CCAGGATGCGTCCACCAA |
| R: AAAGTAGAAGAGGGCAACCAC | |
| BCL-2 | F: GTGGCCTTCTTTGAGTTCG |
| R: ACCCAGCCTCCGTTATCC | |
| P53 | F: TGACGGAGGTCGTGAGAC |
| R: CCAGATACTCGGGATACAAA | |
| β-actin | F: CCTCTATGCCAACACAGTGC |
| R: GTACTCCTGCTTGCTGATCC |
The data were managed using the Applied Biosystems software RQ Manager v1.2.1. Relative expression was calculated by using comparative Ct method and obtaining the fold change value (2-ΔΔCt) according to previously described protocol [14].
Western blot analysis
Tissue was washed twice with cold PBS and lysed with RIPA buffer that contains protease and phosphatase inhibitors cocktail (Roche, UK) for 30 min at 4°C. The supernatants were collected after centrifugation at 12000 rpm for 20 min. The proteins were separated on polyacrylamide gel (SDS-PAGE) by electrophoresis, transferred to PVDF membranes, and then detected by proper primary and secondary antibodies (against IL-6, IL-1β, TNF-α, NF-κB p65, p-STAT3 and α7nAChR, Abcam, USA) before visualization using a chemiluminescence kit. The intensity of blot signals was quantitated using ImageQuant TL analysis software (General Electric, UK).
Statistical analysis
All data were expressed as means ± standard derivation (s.d.) obtained from at least three independent experiments. Statistical comparisons between experimental and control groups were assessed by using the Student’s t-test. P < 0.05 was considered statistically significant.
Results
Optimal time for myocardial injury modelling
We treated the mice with LPS for different times and then examined their myocardial injury. The results showed that the myocardial tissues from the normal mice were evenly stained with normal morphology, regularly arranged fibers and without leakage of red blood cells on the muscle and had no infiltration of inflammatory cells. On other hand, 9 h after LPS treatment, the myocardial gap was widened; 16 h after LPS treatment, the mice had obvious myocardial tissue disorders with severe swelling of myocardial cells, widened muscle gap, hyperemia in the small blood vessels and leakage of a large number of red blood cells in the intermuscular space. Also, fibrosis, necrosis and aggregation were evident with increased number of adherent inflammatory cells. There were even fiber breakages, and these deteriorations were worsen after longer LPS treatment (18 h), which resulted in severely damaged signs of life, rapid temperature decline and increased mortality (Figure 1A). TUNEL assays showed that there was barely apoptosis in control and mice treated with LPS for 9 h. Apoptosis increased from 16 to 18 h after LPS treatment (Figure 1B). However, at 18 h, the animals had a sharply reduced body temperature with very poor mental state. Therefore, mice treated with LPS for 16 h were chosen for subsequent experiments in this study.
Figure 1.
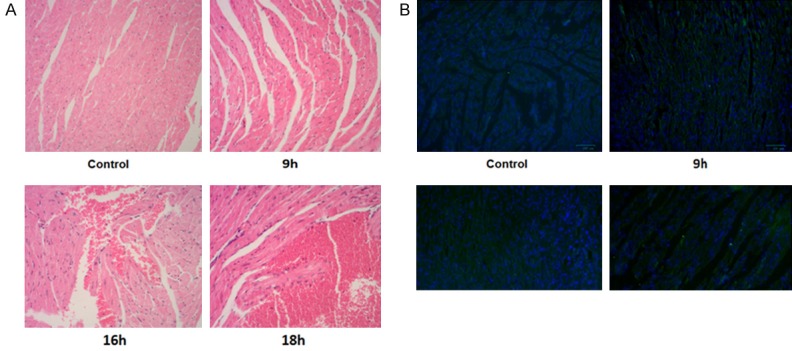
HE staining and TUNEL results of heart tissues following LPS treatments for different times (100×). A. HE staining, showing deformation of muscles and leaking of red blood cells. B. TUNEL assay showing increased number of apoptotic cells following LPS treatment.
Pathological results following drug treatments
We first examined the pathological changes following drug treatments. No red cells and infiltration of inflammatory cells were observed in control mice (Figure 2A). However, the models had obviously disordered heart tissue with swollen myocardial cells, widened muscle gap and marked hyperemia in the small blood vessels. A large number of red blood cells were leaked to the intermuscular space. Fibrosis, necrosis and aggregation were evident with increased number of adherent inflammatory cells. Mice in BT-LPS group showed myocardial disorders, degeneration, dissolution with the accumulation of inflammatory cells between the muscle gaps, focal hemorrhage and infiltration of inflammatory cells. These injuries were more serious than in the LPS group, resulting in severely damaged signs of life, rapid temperature decline and increased mortality. In DEX-LPS group, the mice had widened myocardial muscle gap but not leaked red blood cells, nor the aggregation of inflammatory cells. Their damage was significantly less than that of LPS group. Compared with DEX-LPS group, BT-DEX-LPS group had more damage. TUNEL assays showed that compared with the blank control, the model group had significantly greater apoptosis. DEX and BT reduced the apoptosis when used separately and increased the apoptosis when used jointly (Figure 2B), suggesting that BT can partially block the anti-inflammatory effect of DEX.
Figure 2.
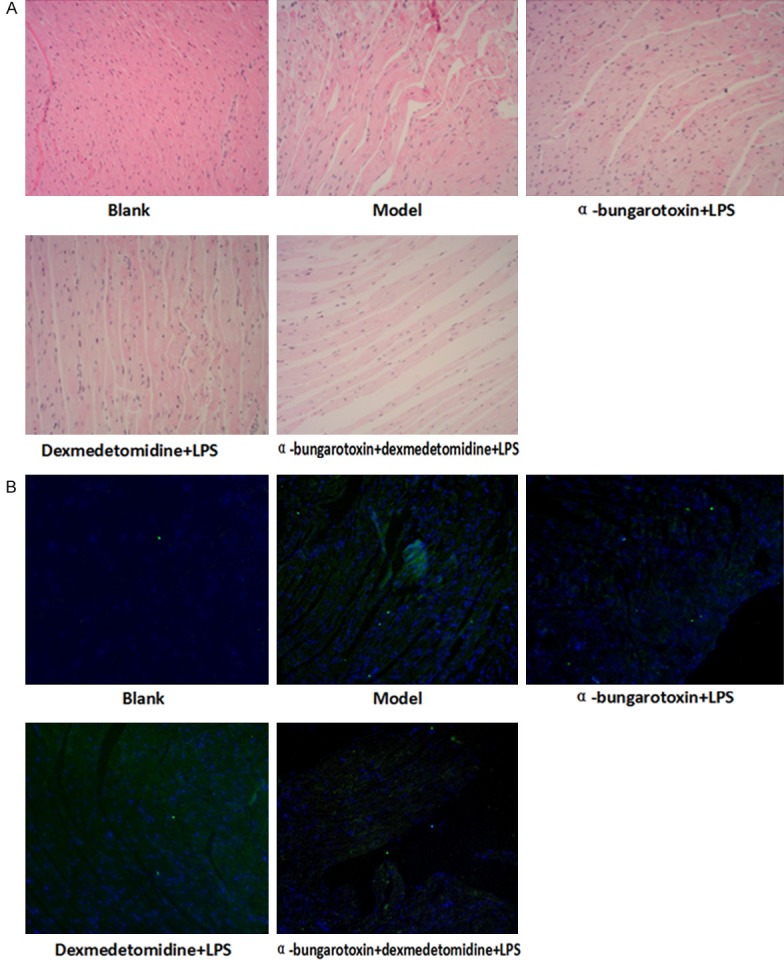
HE staining and TUNEL results of heart tissues following different drug treatments (200×). A. HE staining showing deformation of muscles and leaking of red blood cells. B. TUNEL assay showing apoptotic cells following various treatments.
Expression of NF-κB and α7nAChR mRNA
In the cardiac tissue, the expression of NF-κB p65 and α7nAChR was significantly up- and down-regulated in the model group, respectively (P < 0.01, Figure 3A), as compared with the blank control. DEX alone did not increased the expression further in the model (P > 0.05), but significantly increased NF-κb p65 expression and decreased α7nAChR expression when applied together with BT (P < 0.01, Figure 3A).
Figure 3.
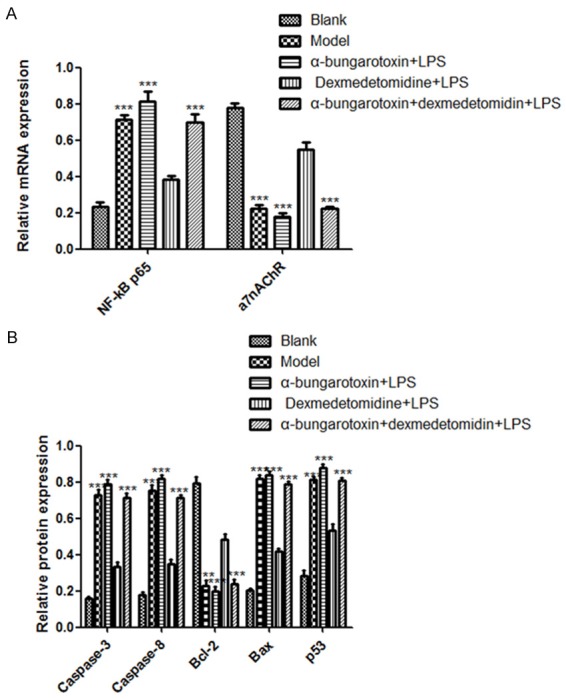
Relative mRNA level of NF-κB, a7nAChR, Caspase-3, Caspase-8, Bcl-2, Bax and p53 mRNA in cardiac tissue following DEX, BT and LPS treatments. A. NF-κB and a7nAChR expression; B. Caspase-3, Caspase-8, Bcl-2, Bax and p53 expression. ***denote P < 0.01 vs control.
Expression of Caspase-3, Caspase-8, Bcl-2, Bax and p53 mRNA
We then assayed the mRNA levels of Caspase-3, Caspase-8, anti-apoptotic protein Bcl-2, pro apoptotic protein Bax and p53 in the cardiac tissue. The results showed that compared with the blank control, LPS significantly increased the mRNA levels of all these genes except Bcl-2, which was decreased significantly (P < 0.01, Figure 3B). Again, DEX alone did not change the expression (P > 0.05), but enhanced the expression of these genes significantly except Bcl-2, which was decreased significantly (P < 0.01, Figure 3B) when it was used with BT.
Expression of IL-6, IL-1β, TNF-α and p-STAT3
Western blot analyses showed that the levels of IL-6, IL-1β, TNF-α and p-STAT3 were significantly up-regulated following LPS treatment. Similarly, DEX alone did not change the expression (P > 0.05), but enhanced the expression of these proteins significantly (P < 0.01, Figure 4, Supplementary Figure 1) when it was used with BT.
Figure 4.
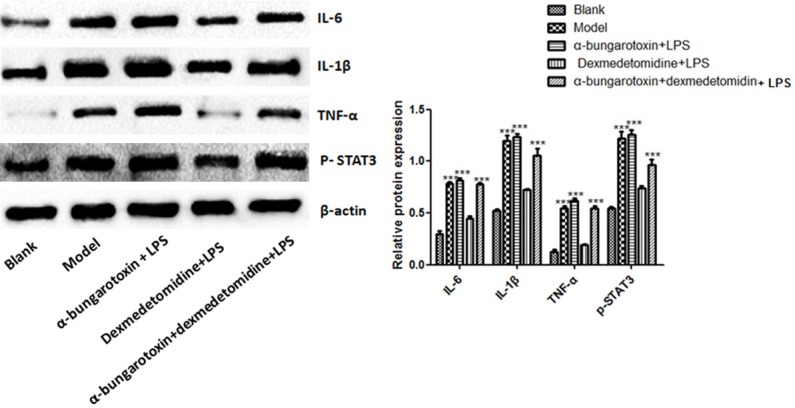
Expression of IL-6, IL-1, TNF- and p-STAT3 proteins in cardiac tissue following DEX, BT and LPS treatments. Left panel: representative Western blots, right panel: relative protein levels. ***denote P < 0.01 vs control.
Discussion
LPS is commonly used to construct model of sepsis. In this study, mice were injected with LPS to produce acute endotoxemia, which eventually leads to systemic sepsis. As a lipopolysaccharide, LPS can elevate the levels of a number of inflammatory factors, causing septic shock and SIRS and resulting in body damage [15]. Since LPS is able to stimulate the release of inflammatory mediators in many kinds of cells to trigger the process of sepsis, we used it to generate the model of sepsis that mimics the clinical features of sepsis.
Although LPS is commonly used for constructing sepsis model, it is critical to identify a suitable duration for LPS treatment to generate model with desirable levels of sepsis for our experiments. Based on our experiments, it was found that 16 h after LPS injection the models had sufficient but not excessive sepsis and were suitable for our experiments.
Using these models, we found that DEX could reduce myocardial tissue damage and apoptosis. However, the improvement disappeared when BT was used following BEX. Early study showed that DEX reduces the apoptosis of myocardial cells and inhibits the apoptosis in sepsis by regulating the expression of Bax/Bcl-2 [16]. DEX injected before LPS also reduced the expression of apoptosis proteins Caspase-3, Caspase-8, Bax and p53 significantly, while increased the expression of anti-apoptotic protein Bcl-2 significantly. Furthermore, BT erased these changes induced by DEX in the expression of apoptosis proteins and anti-apoptotic protein, suggesting that DEX protects myocardium from septic injury via the α7nAChR signal pathway.
During sepsis, inflammatory factors such as IL-6, IL-1β and TNF-α play key roles [17]. Among them, TNF-α is a classical inflammatory factor and is one of the proinflammatory cytokines that appears in the earliest stage of sepsis [18]. DEX has been shown to significantly reduce the expression levels of inflammatory factors such as IL-6, IL-1β and TNF-α to inhibit inflammatory response and exert sedative effect [19]. DEX has anti-inflammatory effects both before and after operation [20]. The occurrence of sepsis is related to the NF-κB signaling pathway. When stimulated, inflammatory cytokines will mediate the cells to release NF-κB. After dissociation into p65 and p50, NF-κB is translocated into the nucleus to regulate the expression of genes coding for inflammatory cytokines, leading to multiple organ injuries [21]. We found that the levels of NF-κB and p65 increased after the occurrence of sepsis, while the expression of α7nAChR decreased; DEX increased the expression of α7nAChR. After injection of LPS, TNF-α expression was significantly increased, suggesting that the release of cytokines plays a very important role in sepsis. Meanwhile, it was found that TNF-α level was significantly lower after DEX injection as compared with the models, suggesting that DEX could reduce the level of TNF-α. IL-6 is also one of the major cytokines involved in inflammatory response. Reduction of IL-6 expression is shown to improve the survival of septic mice [22]. Previous study has also demonstrated that DEX is anti-inflammatory and can improve the survival rate of mice [23]. We showed that the level of IL-6 was significantly increased after injection of LPS and decreased after use of DEX, suggesting that DEX could inhibit the release of cytokines such as IL-6, which is consistent with previous findings that DEX reduces the levels of IL-6, IL-1β and TNF-α [24]. Furthermore, we showed that the use of BT after DEX led to a higher IL-6 level than in DEX-treated mice, suggesting that the anti-inflammatory effect of DEX could be blocked by BT. STAT3 is mainly involved in the signal transduction of IL-6. Studies have shown that after IL-6 treatment, phosphorylation signal of STAT3 is gradually enhanced [25], which is consistent with our results.
Taking together, it is concluded that DEX can reduce heart damage, while α7nAChR-specific blocker BT blocks the anti-inflammatory effect on septic mice by DEX. The anti-inflammatory mechanism of DEX is likely associated with the activation of cholinergic anti-inflammatory pathway.
Acknowledgements
The work was supported by National Natural Science Foundation of China (grant no. 81571871) and Postdoctoral Research Funding of Heilongjiang Province, China (grant no. LBH-Z16147).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kalil AC, Dakroub H, Freifeld AG. Sepsis and solid organ transplantation. Curr Drug Targets. 2007;8:533–541. doi: 10.2174/138945007780362746. [DOI] [PubMed] [Google Scholar]
- 2.Charpentier J, Luyt CE, Fulla Y, Vinsonneau C, Cariou A, Grabar S, Dhainaut JF, Mira JP, Chiche JD. Brain natriuretic peptide: a marker of myocardial dysfunction and prognosis during severe sepsis. Crit Care Med. 2004;32:660–665. doi: 10.1097/01.ccm.0000114827.93410.d8. [DOI] [PubMed] [Google Scholar]
- 3.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 4.Huang JL, Zhang YL, Wang CC, Zhou JR, Ma Q, Wang X, Shen XH, Jiang CL. Enhanced phosphorylation of MAPKs by NE promotes TNF-alpha production by macrophage through alpha adrenergic receptor. Inflammation. 2012;35:527–534. doi: 10.1007/s10753-011-9342-4. [DOI] [PubMed] [Google Scholar]
- 5.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 6.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 8.Taniguchi T, Kidani Y, Kanakura H, Takemoto Y, Yamamoto K. Effects of dexmedetomidine on mortality rate and inflammatory responses to endotoxin-induced shock in rats. Crit Care Med. 2004;32:1322–1326. doi: 10.1097/01.ccm.0000128579.84228.2a. [DOI] [PubMed] [Google Scholar]
- 9.Venn RM, Bradshaw CJ, Spencer R, Brealey D, Caudwell E, Naughton C, Vedio A, Singer M, Feneck R, Treacher D, Willatts SM, Grounds RM. Preliminary UK experience of dexmedetomidine, a novel agent for postoperative sedation in the intensive care unit. Anaesthesia. 1999;54:1136–1142. doi: 10.1046/j.1365-2044.1999.01114.x. [DOI] [PubMed] [Google Scholar]
- 10.Sezer A, Memis D, Usta U, Sut N. The effect of dexmedetomidine on liver histopathology in a rat sepsis model: an experimental pilot study. Ulus Travma Acil Cerrahi Derg. 2010;16:108–112. [PubMed] [Google Scholar]
- 11.daCosta CJ, Free CR, Sine SM. Stoichiometry for alpha-bungarotoxin block of alpha7 acetylcholine receptors. Nat Commun. 2015;6:8057. doi: 10.1038/ncomms9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lillie RD, Pizzolato P, Donaldson PT. Nuclear stains with soluble metachrome metal mordant dye lakes. The effect of chemical endgroup blocking reactions and the artificial introduction of acid groups into tissues. Histochemistry. 1976;49:23–35. doi: 10.1007/BF00490123. [DOI] [PubMed] [Google Scholar]
- 13.Kasagi N, Gomyo Y, Shirai H, Tsujitani S, Ito H. Apoptotic cell death in human gastric carcinoma: analysis by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling. Jpn J Cancer Res. 1994;85:939–945. doi: 10.1111/j.1349-7006.1994.tb02972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Tan Y, Wang Q, She Y, Bi X, Zhao B. Ketamine reduces LPS-induced HMGB1 via activation of the Nrf2/HO-1 pathway and NF-kappaB suppression. J Trauma Acute Care Surg. 2015;78:784–792. doi: 10.1097/TA.0000000000000588. [DOI] [PubMed] [Google Scholar]
- 16.Zhu L, Wang J, Wei T, Gao J, He H, Chang X, Yan T. Effects of naringenin on inflammation in complete freund’s adjuvant-induced arthritis by regulating Bax/Bcl-2 balance. Inflammation. 2015;38:245–251. doi: 10.1007/s10753-014-0027-7. [DOI] [PubMed] [Google Scholar]
- 17.Lee WS, Shin JS, Jang DS, Lee KT. Cnidilide, an alkylphthalide isolated from the roots of cnidium officinale, suppresses LPS-induced NO, PGE2, IL-1beta, IL-6 and TNF-alpha production by AP-1 and NF-kappaB inactivation in RAW 264.7 macrophages. Int Immunopharmacol. 2016;40:146–155. doi: 10.1016/j.intimp.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 18.Wu X, Xu W, Feng X, He Y, Liu X, Gao Y, Yang S, Shao Z, Yang C, Ye Z. TNF-a mediated inflammatory macrophage polarization contributes to the pathogenesis of steroid-induced osteonecrosis in mice. Int J Immunopathol Pharmacol. 2015;28:351–361. doi: 10.1177/0394632015593228. [DOI] [PubMed] [Google Scholar]
- 19.Memis D, Hekimoglu S, Vatan I, Yandim T, Yuksel M, Sut N. Effects of midazolam and dexmedetomidine on inflammatory responses and gastric intramucosal pH to sepsis, in critically ill patients. Br J Anaesth. 2007;98:550–552. doi: 10.1093/bja/aem017. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Miao L, Yao Y, Wu W, Wu X, Gong C, Qiu L, Chen J. Dexmedetomidine ameliorate CLP-induced rat intestinal injury via inhibition of inflammation. Mediators Inflamm. 2015;2015:918361. doi: 10.1155/2015/918361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang WC, Hung MC. Beyond NF-kappa B activation: nuclear functions of Ikappa B kinase alpha. J Biomed Sci. 2013;20:3–5. doi: 10.1186/1423-0127-20-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao T, Pan B, Alam HB, Liu B, Bronson RT, Deng Q, Wu E, Li Y. Protective effect of Clamidine against CLP-induced lethal septic shock in mice. Sci Rep. 2016;6:36696. doi: 10.1038/srep36696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taniguchi T, Kurita A, Kobayashi K, Yamamoto K, Inaba H. Dose- and time-related effects of dexmedetomidine on mortality and inflammatory responses to endotoxin-induced shock in rats. J Anesth. 2008;22:221–228. doi: 10.1007/s00540-008-0611-9. [DOI] [PubMed] [Google Scholar]
- 24.Hofer S, Steppan J, Wagner T, Funke B, Lichtenstern C, Martin E, Graf BM, Bierhaus A, Weigand MA. Central sympatholytics prolong survival in experimental sepsis. Crit Care. 2009;13:R11. doi: 10.1186/cc7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bode JG, Ehlting C, Haussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012;24:1185–1194. doi: 10.1016/j.cellsig.2012.01.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.