

Abstract
Background: Regional or diffuse fibrosis is an early feature of hypertrophic cardiomyopathy (HCM) and is related to poor prognosis. Previous studies have documented low-grade inflammation in HCM. The aim of this study was to examine the relationships between circulating inflammatory markers and myocardial fibrosis, systolic and diastolic dysfunction, and the degree of cardiac hypertrophy in HCM patients. Methods and results: Fifty HCM patients were recruited while 20 healthy subjects served as the control group. Seventeen inflammatory cytokines/chemokines were measured in plasma. Cardiac magnetic resonance imaging and echocardiography were used to assess cardiac phenotypes. Tumour necrosis factor (TNF)-α, interleukin (IL)-6 and serum amyloid P (SAP) were significantly increased in HCM patients compared to controls. IL-6, IL-4, and monocyte chemotactic protein (MCP)-1 were correlated with regional fibrosis while stromal cell-derived factor-1 and MCP-1 were correlated with diffuse fibrosis. Fractalkine and interferon-γ were associated with left ventricular wall thickness. The above associations remained significant in a linear regression model including age, gender, body mass index and family history. TNF-α, IL-6, SAP, MCP-1 and IL-10 were associated with parameters of diastolic dysfunction. White blood cells were also increased in HCM patients and correlated with diffuse fibrosis and diastolic dysfunction. However the associations between parameters of systemic inflammation and diastolic dysfunction were weakened in the linear regression analysis. Conclusions: Systemic inflammation is associated with parameters of the disease severity of HCM patients, particularly regional and diffuse fibrosis. Modifying inflammation may reduce myocardial fibrosis in HCM patients.
Keywords: Hypertrophic cardiomyopathy, systemic inflammation, fibrosis, diastolic dysfunction
Introduction
Hypertrophic cardiomyopathy (HCM) is a genetic myocardial disease, caused by mutations in sarcomeric genes and characterized by asymmetrical septal hypertrophy and myocyte disarray [1,2]. However, the pathology of HCM is not limited to cardiomyocytes and the myocardium. Replacement (regional) or reactive (diffuse) myocardial fibrosis is a common and early feature of HCM [3]. Myocardial fibrosis not only leads to impaired cardiac diastolic function, but is also a major determinant of malignant arrhythmias and end-stage systolic heart failure in HCM and consequently, increases the risk of cardiac death [3-6]. With the introduction of cardiac magnetic resonance imaging (CMR), the diagnosis of myocardial fibrosis noninvasively has greatly advanced [7]. LGE is a well-established method to quantify regional fibrosis [8], while postcontrast myocardial longitudinal relaxation time (T1) mapping is a new technique to evaluate diffuse fibrosis [9]. We and others have reported a lower T1 times in several cardiac disease states associated with diffuse fibrosis [9-11]. However, currently there are still no effective treatments for myocardial fibrosis.
Accumulating evidence has suggested the existence of low-grade systemic and local inflammation in HCM. Mild chronic inflammatory cell infiltration was observed in the myocardium of patients with HCM [12-14]. Several studies also reported increased circulating inflammatory markers in HCM such as tumour necrosis factor (TNF)-α [15,16], and interleukin (IL)-6 [16,17]. Circulating monocyte chemoattractant protein (MCP)-1 levels were also increased in HCM, which were inversely correlated with fractional shortening and correlated with left ventricular end-diastolic pressure [18]. However, the associations between inflammatory markers and myocardial fibrosis have not been well explored. Whilst a previous study showed that circulating levels of C-reactive protein, TNF-α and IL-1RA significantly correlated with maximal LGE [19], the study did not evaluate multiple slices throughout the entire left ventricle which is likely necessary to accurately assess myocardial fibrosis load [19]. Therefore, in this project, we measured 17 inflammatory cytokines/chemokines in plasma samples from 50 HCM patients and 20 controls and examined the relationships between these inflammatory markers and LGE mass (expressed as a percentage of total left ventricular (LV) mass) and T1 times (by CMR), systolic and diastolic function (by CMR and echocardiography) and the degree of left ventricular hypertrophy (by CMR).
Methods
Study population
We recruited 50 patients referred to the Alfred CMR department for the further evaluation of asymmetric septal hypertrophy due to HCM. Asymmetric septal hypertrophy was defined as an interventricular septum thickness of ≥15 mm with a ratio of septal-to-lateral ventricular wall thickness of ≥1.3:1.0 as measured by echocardiography, and the diagnosis of HCM required the absence of any other condition that causes the degree of hypertrophy observed [20]. Exclusion criteria included previous septal reduction therapy, coronary artery disease, atrial fibrillation, valvular heart disease, systemic hypertension, diabetes mellitus, surgery or trauma within previous 6 months, known fibrotic or inflammatory disease or cancer, and contraindications to CMR, including pacemaker and defibrillator implantation, and significant renal dysfunction (estimated glomerular filtration rate (eGFR) <30 ml/min/1.73 m2). Twenty healthy subjects served as a control group.
CMR
CMR was performed using a clinical 1.5-T scanner (Signa HD 1.5-T, GE Healthcare, Waukesha, Wisconsin, USA). Volumetric LV analysis was performed using the summation of disc method with a contiguous short-axis steady-state free precession pulse sequence stack. LGE was used to identify regional fibrosis using a T1-weighted inversion recovery gradient echo technique, while a T1 mapping sequence was used to non-invasively quantify diffuse myocardial fibrosis, as previously described [9]. A region of interest (ROI) was drawn around the entire LV myocardium (excluding papillary muscles) to calculate postcontrast myocardial T1 time. In subjects with regional fibrosis detected by LGE, these areas were excluded from the ROI for the primary analysis of postcontrast myocardial T1 time. T1 times for ROIs including areas of LGE were also calculated. To account for the potential effects of glomerular filtration rate, time delay postcontrast administration, and contrast agent relaxivity on gadolinium pharmacokinetics, corrected values of T1 times were used to normalize postcontrast myocardial T1 times to a matched state (time postcontrast administration =20 minutes, eGFR =90 mL/min per 1.73 m2) [21]. In addition, raw postcontrast T1 times of the LV blood pool (blood T1 times) were calculated. CMR was performed in all HCM patients and 7 healthy controls.
Echocardiography
Transthoracic echocardiography with a standard clinical protocol was performed immediately prior to CMR. Diastolic function was assessed by a combination of mitral inflow pattern (E to A ratio and deceleration time) and mitral annular velocities (e’, measured at the septal and lateral aspects of the mitral annulus in the apical 4-chamber view). Additionally, mitral E/e’ (septal, lateral and mean) was chosen as an index of LV filling pressure.
Blood sample collection
Blood samples were obtained before CMR and collected into EDTA-tubes by venipuncture. Plasma samples (10 min centrifugation at 400 g followed by a further 10 min at 600 g) were stored at -80°C for further assays.
Measurement of 17 inflammatory markers by bioplex
Plasma levels of 17 cytokines/chemokines were measured using multiplex kits from Millipore according to the manufacturer’s instruction, as we previously described [22]. These 17 cytokines/chemokines were TNF-α, IL-1β, IL-6, IL-4, IL-10, IL-12, IL-13, interferon-γ (IFN-γ), transforming growth factor-β (TGF-β), serum amyloid P (SAP), IFN-γ-inducible protein 10 (IP-10, CXCL10), macrophage inhibitory protein (MIP)-1α, MIP-1β, MCP-1, fractalkine, stromal cell-derived factor (SDF)-1 and secondary lymphoid-tissue chemokine (SLC). High-sensitivity cytokine multiplex kits were used for measurement of TNF-α, IL-β, IL-6, IL-10, IL-4, IL-12, IL13, and IFN-γ. The appropriate cytokine/chemokine standards, plasma samples (25 µL), and fluorescent conjugated, antibody-immobilized beads were added to wells of a pre-wet filtered plate and then were incubated overnight at 4°C. The following day, the plate was washed twice with wash buffer and then incubated with secondary detection antibody for 1 h, followed by subsequent incubation with strepavidin-PE for 30 min. After the plate was washed twice again with wash buffer, it was read on the Luminex system (Biorad) with the addition of sheath fluid. Concentrations of different analytes in the plasma samples were determined by using respective standard curves generated in the multiplex assays. Neat plasma samples were used for all assays except for SAP and TGF-β1 (1:2000 and 1:30 dilution, respectively, using assay buffer provided in the kits).
Statistical analysis
Data were expressed as mean ± SD unless otherwise stated. SPSS 17.0 was used for statistical analysis. The normality of data was tested by Kolmogorov-Smirnov test. Chi-square test was used to compare discrete variables among groups. Student t test or Mann-Whitney U test was employed for comparison between the control group and HCM patients when appropriate. Pearson correlation coefficients were computed to assess the correlations between parameters. Multiple linear regression analysis was used to further determine the above associations after adjusting confounders such as age, gender, BMI and family history. A difference of p<0.05 (two-sided) was considered statistically significant.
Results
Patient demographics
Patient demographics are presented in Table 1. There were no significant differences in age, gender, heart rate, systolic and blood pressure, eGFR, haemoglobulin, platelets, haematocrit, and lymphocytes among groups. BMI was significantly greater in HCM patients compared to controls. All patients except 1 had the New York Heart Association (NYHA) class I or II. Counts of white blood cells, neutrophils, and monocytes were significantly higher in HCM patients compared to controls.
Table 1.
Subject characteristics
| Control | HCM | |
|---|---|---|
| n | 20 | 50 |
| Gender (m/f, n) | 14/6 | 38/12 |
| Age (years) | 46±13.0 | 49±13.5 |
| Body mass index (kg/m2) | 22.6±2.2 | 27.4±4.4*** |
| NYHA function class (I/II/III, n) | NA | 25/24/1 |
| Dyspnoea (%) | NA | 52% |
| Chest pain (%) | NA | 28% |
| Presyncope (%) | NA | 36% |
| Syncope (%) | NA | 12% |
| Family history of HCM (%) | NA | 24% |
| Resting heart beat (beats/min) | 62.8±8.7 | 60.5±10.5 |
| Systolic blood pressure (mmHg) | 120.4±10.2 | 128.3±12.5 |
| Diastolic blood pressure (mmHg) | 73.0±9.8 | 72.4±9.0 |
| eGFR (mL/min/1.75 m2) | 86.4±4.5 | 83.6±11.4 |
| Medications | ||
| β-blockers | NA | 54% |
| Calcium channel blockers | NA | 18% |
| Angiotensin convert enzyme inhibitor | NA | 12% |
| Angiotensin receptor blockers | NA | 10% |
| Statin | NA | 18% |
| Full blood count | ||
| Haemoglobin (g/L) | 144.6±9.4 | 146.5±14.3 |
| Platelets (109/L) | 227.3±53.8 | 208.3±56.1 |
| Hematocrit (L/L) | 0.42±0.03 | 0.42±0.04 |
| White blood cells (109/L) | 5.79±1.25 | 7.59±2.30** |
| Neutrophils (109/L) | 3.37±1.16 | 5.01±2.05** |
| Lymphocytes (109/L) | 1.89±0.54 | 1.89±0.53 |
| Monocytes (109/L) | 0.39±0.12 | 0.51±0.15** |
| Monocytes (%) | 6.83±1.84 | 6.90±1.78 |
Data are expressed as mean ± SD. HCM: Hypertrophic cardiomyopathy; NYHA: the New York Heart Association; eGFR: Estimated glomerular filtration rate.
P<0.01, vs. controls;
P<0.001, vs. controls.
CMR and echocardiography data are presented in Table 2. As expected, septal thickness, the ratio of septal to lateral wall thickness, LV mass and LV mass indexed to body surface area (BSA) were significantly increased in the HCM group compared with the control group. Two HCM patients had septal hypertrophy exceeding 30 mm. The HCM group had a significantly higher LV ejection fraction, suggesting hypercontractility in these HCM patients. LV end-diastolic volume, LV end-diastolic volume indexed to BSA and LV stroke volume were not significantly different between both groups, but LV end-systolic volume was significantly smaller in HCM compared with controls. Patients with HCM had significantly shorter global post-contrast myocardial T1 times compared with controls. When regions of LGE were included in the analysis of HCM patients, a further reduction in T1 time was observed. T1 times corrected for GFR and time delay postcontrast administration were also calculated and shown. There was no significant difference in raw blood T1 times between the 2 groups, effectively ruling out contrast kinetics as a confounding factor for the observed differences in myocardial T1 time between the 2 groups. LGE (regional fibrosis) was observed in the majority of HCM patients, generally localized to the ventricular septum or points of RV free wall insertion. Subendocardially-based LGE, consistent with ischemic scar, was not observed in any patient. The mean quantity of LGE, expressed as a percentage of LGE mass to total LV mass, was 5.1±6.7%.
Table 2.
Echocardiography and CMR data
| Control | HCM | |
|---|---|---|
| n | 20 | 50 |
| Echocardiography | ||
| Left atrial volume (ml/BSA) | 27.6±8.9 | 45.3±15.5*** |
| E/A ratio | 1.38±0.36 | 1.35±0.56 |
| Deceleration time (ms) | 182.1±27.0 | 213.2±57.3* |
| Septal e’ (cm/s) | 10.6±2.9 | 6.1±1.6*** |
| Lateral e’ (cm/s) | 13.7±3.5 | 8.3±2.8*** |
| Mean e’ (cm/s) | 12.1±3.0 | 7.2±2.0*** |
| Septal E/e’ | 8.5±2.6 | 13.9±5.5*** |
| Lateral E/e’ | 6.3±1.9 | 10.7±5.0*** |
| Mean E/e’ | 7.8±2.6 | 12.2±4.8*** |
| Resting LVOT gradient (mmHg) | 5.2±1.2 | 38.5±45.1** |
| CMR | ||
| Septal thickness (mm) | 8.3±1.8 | 19.2±4.8*** |
| Lateral wall thickness (mm) | 7.9±1.5 | 8.6±1.8 |
| Septal/lateral wall thickness | 1.1±0.1 | 2.3±0.6*** |
| LV mass (g) | 113.1±20.6 | 171.1±61.6*** |
| LV mass index (g/BSA) | 57.5±8.0 | 86.2±28.0*** |
| LV ejection fraction (%) | 60.9±5.8 | 69.5±7.3** |
| LVEDV (mL) | 180.4±29.1 | 160.3±38.6 |
| LVEDV indexed (mL/BSA) | 91.9±11.5 | 78.0±21.8 |
| LVESV (mL) | 72.4±22.6 | 48.3±20.4*** |
| LVSV (mL) | 108.0±7.8 | 106.5±35.2 |
| Presence of LGE (%) | NA | 86% |
| Quantity of LGE (% of LV) mass | NA | 5.1±6.7 |
| T1 times (ms), excluding LGE | 578.4±45.4 | 497.5±79.2* |
| T1 times (ms), including LGE | 578.4±45.4 | 481.2±83.8** |
| T1 times (ms, corrected values) | 566.3±49.6 | 478.6±80.9** |
| Blood T1 times (ms) | 303.1±13.7 | 334.6±31.5 |
Data are expressed as mean ± SD. CMR: Cardiac magnetic resonance; LVOT: Left ventricular outflow tract; LV: Left ventricular; LVEDV: Left ventricular end-diastolic volume; LVESV: Left ventricular end-systolic volume; LVSV: Left ventricular stroke volume; LGE: Late gadolinium enhancement; BSA: Body surface area; T1 times (corrected values): T1 times were normalized to a matched state (time post-contrast administration =20 minutes, eGFR =90 mL/min/1.73 m2) to account for the potential effects of glomerular filtration rate, time delay post-contrast administration, and contrast agent relaxivity on gadolinium pharmacokinetics.
P<0.05, vs. controls;
P<0.01, vs. controls;
P<0.001, vs. controls.
Echocardiography showed diastolic dysfunction in patients with HCM. Left atrial volume indexed to BSA (LAVI), deceleration time, septal, lateral and mean E/e’ and resting left ventricular outflow tract (LVOT) gradient were significantly higher while septal, lateral and mean e’ were significantly lower in HCM patients compared to controls (Table 2).
Changes of circulating inflammatory markers in HCM
Plasma levels of TNF-α, IL-6, and SAP were significantly increased in HCM patients compared to healthy controls (Figure 1), but plasma levels of TGF-β1 were significantly decreased in HCM patients compared to healthy controls (Figure 1). However, the differences in other inflammatory markers between HCM patients and healthy controls did not reach significance (Figure 1).
Figure 1.
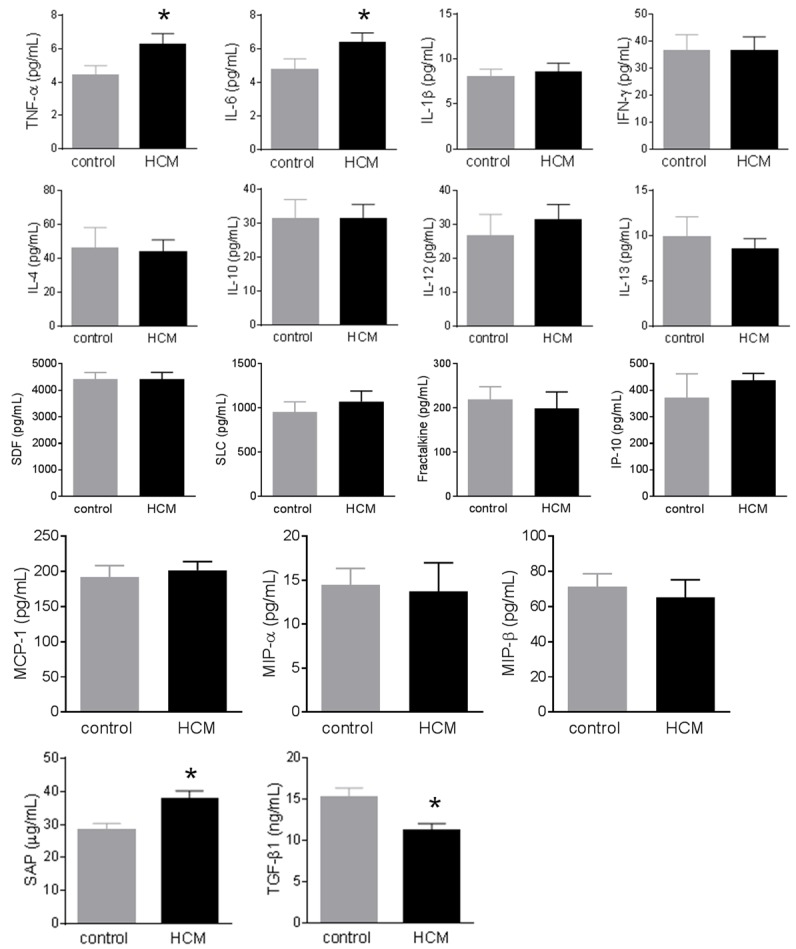
Change of circulating inflammatory markers in hypertrophic cardiomyopathy (HCM). Plasma cytokines/chemokines were measured using multiplex kits in 20 healthy controls and 50 patients with HCM. Plasma levels of TNF-α, IL-6, and SAP were significantly increased in HCM patients compared to healthy controls, but plasma levels of TGF-β1 were significantly decreased in HCM patients compared to healthy controls. However, the differences in other inflammatory markers between HCM patients and healthy controls did not reach significance. Data were expressed as mean ± SEM. *: P<0.05 vs. the control group. IFN-γ: interferon-γ; IL: interleukin; IP-10: IFN-γ-inducible protein 10; MCP-1: monocyte chemotactic protein-1; MIP: macrophage inhibitory protein; SAP: serum amyloid P; SDF: stromal cell-derived factor; SLC: secondary lymphoid-tissue chemokine; TGF-β1: transforming growth factor-β1; TNF-α: tumor necrosis factor-α.
Inflammatory markers and LVOT obstruction
There were no significant differences in plasma inflammatory markers between HCM patients with a LVOT obstruction (≥30 mmHg at rest) compared to those without an obstruction (<30 mmHg at rest) (data not shown).
Compared to non-obstructive HCM patients, obstructive HCM patients had increased lateral E/e’ (12.7±5.2 vs. 9.3±4.5, p=0.045), borderline higher mean E/e’ (13.9±4.2 vs. 11.2±4.9, p=0.073) and borderline reduced E/A (1.16±0.40 vs. 1.46±0.61, p=0.067), as well as higher LVEF (72.5±5.1 vs. 66.9±8.1, p=0.007). These results indicate that obstructive HCM patients display diastolic dysfunction but hypercontractility compared to non-obstructive HCM patients in this study.
Inflammatory markers and LV wall thickness
In the whole population, inflammatory markers were not significantly correlated with LV wall thickness. However, in the HCM group alone, several inflammatory markers were positively correlated with LV wall thickness. Fractalkine (r=0.310, p=0.034) was positively correlated with LV septal wall thickness, while fractalkine (r=0.351, p=0.016) and IFN-γ (r=0.308, p=0.037) were positively correlated with the ratio of septal/lateral wall thickness. The above associations remained significant after adjusting for age, gender, BMI and family history in a multiple linear regression model.
Inflammatory markers and myocardial fibrosis
We then examined the relationship between inflammatory markers and regional (LGE) and diffuse fibrosis (reduced T1 times). In the whole population, circulating levels of SDF and MCP-1 were inversely correlated with T1 times, while circulating levels of IL-6, IL-4 and MCP-1 were positively correlated with the quantity of LGE (Figure 2). Similar findings for the above inflammatory markers were obtained when associations were assessed within HCM group alone (data not shown). In addition, IL-10 was positively correlated with LGE if assessed within HCM group alone (r=0.289, p=0.05). The above associations remain significant after adjusting for age, gender, BMI and family history in a multiple linear regression model.
Figure 2.
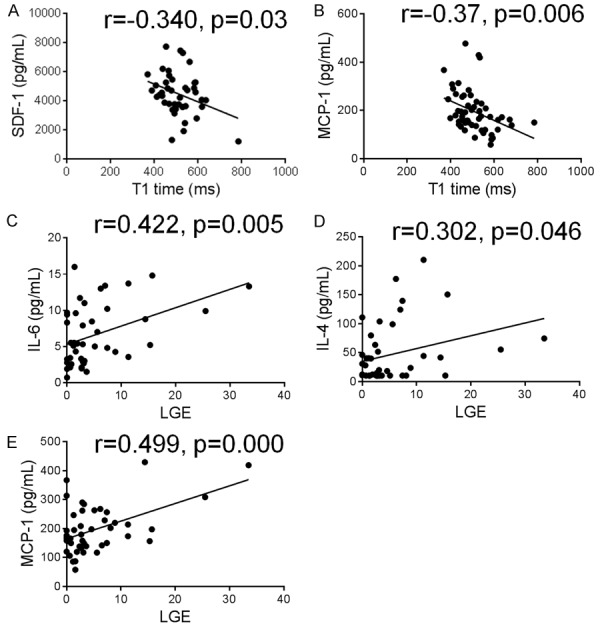
Correlations between circulating inflammatory markers and diffuse and regional myocardial fibrosis. Plasma cytokines/chemokines were measured using multiplex kits in 20 healthy controls and 50 patients with hypertrophic cardiomyopathy. Diffuse fibrosis was evaluated by post-contrast T1 time. T1 time used in this figure indicated values excluding LGE (late gadolinium enhancement). Regional myocardial fibrosis was evaluated by LGE and expressed as a percentage of LGE mass to total LV mass. Plasma levels of stromal cell-derived factor (SDF)-1 and monocyte chemotactic protein (MCP)-1 were inversely correlated with T1 times, while circulating levels of interleukin (IL)-6, IL-4 and MCP-1 were positively correlated with the quantity of LGE.
Inflammatory markers and diastolic function
Several circulating inflammatory markers were also linked to diastolic dysfunction. In the whole population, SAP, IL-6, TNF-α, IL-10, and MCP-1 were positively correlated with diastolic dysfunction, but TGF-β was negatively correlated with diastolic dysfunction (Table 3). However, only a few associations with diastolic dysfunction remain significant after adjusting for age, gender, BMI and family history (Table 3, figures in bold). Correlations between inflammation and diastolic function were also assessed within HCM group. Notably, IP-10 was positively correlated with septal (r=0.321, p=0.049), lateral (r=0.417, p=0.011) and mean E/e’ (r=0.378, p=0.015) while negatively correlated with lateral e’ (r=-0.387, p=0.02) and mean e’ (r=-0.359, p=0.029). IL-10 was positively correlated with LAVI (r=0.356, p=0.03) whereas TGF-β1 was again negatively correlated with LAVI (r=-0.339, p=0.04). However, these associations lost significance after adjusting for age, gender, BMI and family history.
Table 3.
Correlations between circulating inflammatory markers and diastolic dysfunction in the whole population
| LAVI | Septal e’ | Lateral e’ | Mean e’ | Septal E/e’ | Lateral E/e’ | Mean E/e’ | |
|---|---|---|---|---|---|---|---|
| SAP | 0.296 | ||||||
| TGF-β | -0.460 | 0.315 | 0.320 | 0.333 | -0.278 | ||
| IL-6 | 0.338 | -0.338 | -0.296 | 0.284 | 0.360 | 0.318 | |
| TNF-α | 0.260 | -0.266 | 0.271 | ||||
| MCP-1 | -0.267 | 0.278 | |||||
| IL-10 | 0.291 | 0.29 |
SAP: Serum amyloid P; TGF-β: Transforming growth factor-β; IL-6: Interleukin-6; TNF-α: Tumor necrosis factor-α; MCP-1: Monocyte chemotactic protein-1; IL-10: Interleukin-10; LVAI: Left atrial volume indexed to body surface area. Figures in the table represent pearson correlation coefficients. Only significant correlations were shown in the table. Figures in bold indicate that the correlations remain signficant after adjusting for age, gender, BMI and family history in a multiple linear regression model.
Inflammatory markers and systolic function
Inflammatory markers were not significantly correlated with systolic function in the whole population or in HCM group alone (data not shown).
Relationship between peripheral blood cells and cardiac phenotypes
In addition, we examined the connections between peripheral blood cell counts and cardiac phenotypes. In the whole population, total white blood cell count was positively correlated with septal wall thickness (r=0.308, 0.05) and septal/lateral wall thickness (r=0.321, p=0.041), and also correlated with reduced T1 times (r=-0.309, p=0.049), but not with LGE. Moreover, white blood cells, neutrophils, and monocytes were associated with a number of parameters of diastolic dysfunction (Table 4), but not with systolic function (data not shown). However, only a few associations with diastolic dysfunction remain significant after adjusting for age, gender, BMI and family history (Table 4, figures in bold). There were also some correlations between peripheral blood cell counts and inflammatory markers. Percentage of monocytes were correlated with TNF-α (r=0.339, p=0.016) while lymphocyte count was correlated with IP-10 (r=0.393, p=0.005).
Table 4.
Correlations between peripheral blood cells and diastolic dysfunction in the whole population
| LAVI | Septal e’ | Lateral e’ | Mean e’ | LVOT gradient | |
|---|---|---|---|---|---|
| WBC | 0.322 | -0.331 | -0.332 | 0.326 | |
| Neutrophils | 0.318 | -0.345 | -0.337 | 0.328 | |
| Monocytes | 0.333 | -0.395 | -0.409 | -0.437 |
WBC: White blood cells; LAVI: Left atrial volume indexed to body surface area; LVOT: Left ventricular outflow tract. Figures in the table represent pearson correlation coefficients. Only significant correlations were shown in the table. Figures in bold indicate that the correlations remain signficant after adjusting for age, gender, BMI and family history in a multiple linear regression model.
Relationship between BMI and cardiac phenotypes and systemic inflammation
We finally examined the associations between BMI and all these parameters. Higher BMI was associated with diffuse fibrosis (reduced T1 times), cardiac dysfunction (reduced septal, lateral and mean e’, increased lateral and mean E/e’, LVOT, LV stroke volume, and LV end-diastolic volume), and LV hypertrophy (LV septal and lateral wall thickness), but not with regional fibrosis (LGE). BMI was also positively correlated with counts of total white blood cells, neutrophils and monocytes, as well as TNF-α.
Discussion
Previous studies have documented infiltration of leukocytes in the myocardium and elevation of several circulating inflammatory cytokines/chemokines in patients with HCM [15-18]. In the present study, we measured 17 inflammatory cytokines/chemokines and correlated them with the degree of hypertrophy, diffuse and regional fibrosis and diastolic and systolic dysfunction measured by CMR and echocardiography. We found that TNF-α, IL-6, and SAP were significantly elevated while TGF-β1 was significantly decreased in the plasma of HCM patients. The changes of these inflammatory markers were modest, which were accompanied by the increased counts of peripheral white blood cells, neutrophils, and monocytes. Thus, low-grade systemic inflammation exists in patients with HCM. We further found that several circulating inflammatory markers and peripheral inflammatory cells were associated with myocardial fibrosis, the degree of hypertrophy and diastolic dysfunction in patients with HCM, suggesting that markers of systemic inflammation may serve as biomarkers for the disease severity of HCM patients.
The elevation of TNF-α and IL-6 in HCM is in consistent with previous studies [15-17], although IL-6 but not TNF-α was elevated in the study by Hogye et al [17]. TNF-α and IL-6 are two important inflammatory cytokines, which may play a pathogenic role in HCM. It was shown that cardiac overexpression of TNF-α caused LV hypertrophy, dilated cardiomyopathy and premature death [23]. In addition, the uncommon allele of TNF-α-308G/A polymorphism, known to produce more TNF-α, was associated with greater LV mass and clinical diagnosis at a younger age in patients with HCM [24]. IL-6 has been shown to be an important mediator of LV hypertrophy, myocardial fibrosis and LV dysfunction in response to pressure overload [25]. SAP is another inflammatory cytokine and plasma SAP is associated with increased cardiovascular disease [26]. However, the contribution of SAP to the pathology of HCM needs to be further explored. Notably, plasma TGF-β1 was decreased in patients with HCM in our study. TGF-β has been known to be involved in cardiac remodeling including hypertrophy and fibrosis in HCM patients [27]. Previous studies showed that myocardial [28,29] and plasma TGF-β levels [30] were higher in patients with HCM compared with controls. The discrepancy in plasma TGF-β may be due to different disease status, or medications, amongst other factors. However, our result may indicate that TGF-β is not an early mediator of the pathology of HCM since TGF-β also inhibits inflammation.
Accumulating evidence has suggested that fibrosis is linked to a danger-triggered inflammatory response during wound repair [31] and that chronic low-grade inflammatory activity may be involved in myocardial fibrosis [32]. In the present study, we found close correlations between several inflammatory markers and both regional and diffuse fibrosis assessed by CMR in HCM patients. A recent paper documented the infiltration of inflammatory cells in the myocardium in cats with pre-clinical HCM [33]. Therefore, inflammatory response is likely to trigger myocardial fibrosis in HCM. As myocardial fibrosis is a major determinant of malignant arrhythmias and end-stage systolic heart failure in HCM [4,5], modifying the inflammatory cascade might reduce myocardial fibrosis, which could be of critical importance for preventing cardiac death.
LGE reflects collagenous scar formation [34] while reduced T1 times indicate diffuse interstitial fibrosis in HCM. Until now, there has been no data available on whether there are different mechanisms for the development of regional versus diffuse fibrosis. We demonstrated that regional and diffuse myocardial fibrosis were correlated with different circulating inflammatory markers, with LGE being correlated with IL-6, IL-4, and MCP-1 while reduced T1 times being correlated with SDF and MCP-1. SDF raises the retention of infiltrated T cells via inducing the SDF/CXCR4 axis [35] and SDF-1/CXCR4 axis is also an important mediator of fibrocyte migration [36]. So we speculate that T cells and fibrocytes may be more important in diffuse fibrosis while regional fibrosis is probably more related to residential fibroblasts since LGE was in close correlations with IL-6, and IL-4, which are 2 profibrotic cytokines. IL-6 increases fibroblast proliferation and collagen and glycosaminoglycan production in fibroblasts [37,38]. In vitro data demonstrate that IL-4 upregulates procollagen genes and stimulates collagen production in mouse cardiac fibroblasts [39]. MCP-1 is correlated with both regional and diffuse fibrosis. MCP-1 not only recruits monocytes, but also stimulates collagen expression and endogenous up-regulation of TGF-β expression in fibroblasts, leading to autocrine and/or juxtacrine stimulation of collagen gene expression [40]. In addition, the finding that total white blood cell count is correlated with T1 times but not LGE might further support the notion that diffuse fibrosis is more related to inflammatory cells. However, the different pathogenic mechanisms for diffuse and regional fibrosis warrant further investigations with proof of concept studies.
In our study, HCM patients had diastolic dysfunction but well-preserved systolic function. We did not find correlations between systemic inflammation and systolic function. We found that a number of inflammatory markers and peripheral inflammatory cells were correlated with diastolic dysfunction. However, the correlations were weakened after adjusting for age, gender, BMI and family history in a multiple linear regression model. From the multiple linear regression model, BMI contributed most to the parameters of diastolic dysfunction. These data suggest that it is important to advise HCM patients to control their BMI to improve their diastolic function.
We also found that fractalkine was associated with septal wall thickness while fractalkine and IFN-γ were associated with the ratio of septal/lateral wall thickness within HCM patients. It was reported that fractalkine-neutralizing antibody [41] attenuated cardiac hypertrophy in animal models and in vitro, fractalkine induced the expression of markers of cardiac hypertrophy in neonatal cardiomyocytes [42]. IFN-γ deficiency [43] attenuated cardiac hypertrophy in animal models, while SAP-IFN-γ transgenic mice developed chronic myocarditis and cardiomyopathy [44]. However, the effects of IFN-γ on cardiac hypertrophy in animal experiments and in vitro cardiomyocyte culture are conflicting [45]. Taken together, inflammatory markers may play a role in potentiating septal wall thickness in patients with HCM.
This study has several limitations. Firstly, our findings were based on the 17 cytokines/chemokines measured in this study, potentially omitting other important cytokines/chemokines which mediate the pathology of HCM. Secondly, we found that systemic inflammation was associated with myocardial fibrosis and diastolic dysfunction. Given the cross-sectional nature of the study, it is impossible to determine whether these associations are causal. However, this study sheds more light on the associations between systemic inflammation and cardiac phenotypes in HCM. More studies are required to further define the role of inflammation and specific inflammatory markers in HCM prior to the application of effective anti-inflammatory therapies in patients with HCM. This is really important since there is currently lack of effective treatments for HCM.
In conclusion, several inflammatory markers and inflammatory cells in peripheral blood are increased in HCM patients compared with healthy controls. Systemic inflammation is associated with myocardial fibrosis, septal wall thickness and LV diastolic dysfunction. Thus, markers of systemic inflammation may serve as biomarkers for the disease severity of HCM patients. Modulating inflammatory reactions in HCM patients could be a useful therapeutic approach for managing myocardial fibrosis.
Acknowledgements
This study was supported in part by the Victorian Government’s Operational Infrastructure Support Program. Prof. Dart is an NHMRC fellow. Associate Prof. Murphy is an NHMRC Career Development Fellow and a National Heart Foundation Future Leader Fellow.
Disclosure of conflict of interest
None.
References
- 1.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 2.Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–1891. doi: 10.1016/S0140-6736(04)16358-7. [DOI] [PubMed] [Google Scholar]
- 3.Varnava AM, Elliott PM, Sharma S, McKenna WJ, Davies MJ. Hypertrophic cardiomyopathy: the interrelation of disarray, fibrosis, and small vessel disease. Heart. 2000;84:476–482. doi: 10.1136/heart.84.5.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adabag AS, Maron BJ, Appelbaum E, Harrigan CJ, Buros JL, Gibson CM, Lesser JR, Hanna CA, Udelson JE, Manning WJ, Maron MS. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008;51:1369–1374. doi: 10.1016/j.jacc.2007.11.071. [DOI] [PubMed] [Google Scholar]
- 5.Keren A, Syrris P, McKenna WJ. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med. 2008;5:158–168. doi: 10.1038/ncpcardio1110. [DOI] [PubMed] [Google Scholar]
- 6.Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000;35:36–44. doi: 10.1016/s0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
- 7.Mewton N, Liu CY, Croisille P, Bluemke D, Lima JA. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011;57:891–903. doi: 10.1016/j.jacc.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim RJ, Judd RM. Gadolinium-enhanced magnetic resonance imaging in hypertrophic cardiomyopathy: in vivo imaging of the pathologic substrate for premature cardiac death? J Am Coll Cardiol. 2003;41:1568–1572. doi: 10.1016/s0735-1097(03)00190-6. [DOI] [PubMed] [Google Scholar]
- 9.Iles L, Pfluger H, Phrommintikul A, Cherayath J, Aksit P, Gupta SN, Kaye DM, Taylor AJ. Evaluation of diffuse myocardial fibrosis in heart failure with cardiac magnetic resonance contrastenhanced T1 mapping. J Am Coll Cardiol. 2008;52:1574–1580. doi: 10.1016/j.jacc.2008.06.049. [DOI] [PubMed] [Google Scholar]
- 10.Chan W, Duffy SJ, White DA, Gao XM, Du XJ, Ellims AH, Dart AM, Taylor AJ. Acute left ventricular remodeling following myocardial infarction: coupling of regional healing with remote extracellular matrix expansion. JACC Cardiovasc Imaging. 2012;5:884–893. doi: 10.1016/j.jcmg.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Mascherbauer J, Marzluf BA, Tufaro C, Pfaffenberger S, Graf A, Wexberg P, Panzenbock A, Jakowitsch J, Bangert C, Laimer D, Schreiber C, Karakus G, Hulsmann M, Pacher R, Lang IM, Maurer G, Bonderman D. Cardiac magnetic resonance postcontrast T1 time is associated with outcome in patients with heart failure and preserved ejection fraction. Circulation Cardiovascular Imaging. 2013;6:1056–1065. doi: 10.1161/CIRCIMAGING.113.000633. [DOI] [PubMed] [Google Scholar]
- 12.Lamke GT, Allen RD, Edwards WD, Tazelaar HD, Danielson GK. Surgical pathology of subaortic septal myectomy associated with hypertrophic cardiomyopathy. A study of 204 cases (1996-2000) Cardiovasc Pathol. 2003;12:149–158. doi: 10.1016/s1054-8807(03)00036-x. [DOI] [PubMed] [Google Scholar]
- 13.Baandrup U, Olsen EG. Critical analysis of endomyocardial biopsies from patients suspected of having cardiomyopathy. I: morphological and morphometric aspects. Br Heart J. 1981;45:475–486. doi: 10.1136/hrt.45.5.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phadke RS, Vaideeswar P, Mittal B, Deshpande J. Hypertrophic cardiomyopathy: an autopsy analysis of 14 cases. J Postgrad Med. 2001;47:165–170. [PubMed] [Google Scholar]
- 15.Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br Heart J. 1994;72:561–566. doi: 10.1136/hrt.72.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zen K, Irie H, Doue T, Takamiya M, Yamano T, Sawada T, Azuma A, Matsubara H. Analysis of circulating apoptosis mediators and proinflammatory cytokines in patients with idiopathic hypertrophic cardiomyopathy: comparison between nonobstructive and dilated-phase hypertrophic cardiomyopathy. Int Heart J. 2005;46:231–244. doi: 10.1536/ihj.46.231. [DOI] [PubMed] [Google Scholar]
- 17.Hogye M, Mandi Y, Csanady M, Sepp R, Buzas K. Comparison of circulating levels of interleukin-6 and tumor necrosis factor-alpha in hypertrophic cardiomyopathy and in idiopathic dilated cardiomyopathy. Am J Cardiol. 2004;94:249–251. doi: 10.1016/j.amjcard.2004.03.078. [DOI] [PubMed] [Google Scholar]
- 18.Iwasaki J, Nakamura K, Matsubara H, Nakamura Y, Nishii N, Banba K, Murakami M, Ohta-Ogo K, Kimura H, Toh N, Nagase S, Oka T, Morita H, Kusano KF, Ohe T. Relationship between circulating levels of monocyte chemoattractant protein-1 and systolic dysfunction in patients with hypertrophic cardiomyopathy. Cardiovasc Pathol. 2009;18:317–322. doi: 10.1016/j.carpath.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 19.Kuusisto J, Karja V, Sipola P, Kholova I, Peuhkurinen K, Jaaskelainen P, Naukkarinen A, Yla-Herttuala S, Punnonen K, Laakso M. Lowgrade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart. 2012;98:1007–1013. doi: 10.1136/heartjnl-2011-300960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH 3rd, Spirito P, Ten Cate FJ, Wigle ED. American college of cardiology/european society of cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American college of cardiology foundation task force on clinical expert consensus documents and the european society of cardiology committee for practice guidelines. J Am Coll Cardiol. 2003;42:1687–1713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 21.Gai N, Turkbey EB, Nazarian S, van der Geest RJ, Liu CY, Lima JA, Bluemke DA. T1 mapping of the gadolinium-enhanced myocardium: adjustment for factors affecting interpatient comparison. Magn Reson Med. 2011;65:1407–1415. doi: 10.1002/mrm.22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang L, Moore XL, Chan W, White DA, Chin-Dusting J, Dart AM. Decreased fibrocyte number is associated with atherosclerotic plaque instability in man. Cardiovasc Res. 2012;95:124–133. doi: 10.1093/cvr/cvs156. [DOI] [PubMed] [Google Scholar]
- 23.Bryant D, Becker L, Richardson J, Shelton J, Franco F, Peshock R, Thompson M, Giroir B. Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation. 1998;97:1375–1381. doi: 10.1161/01.cir.97.14.1375. [DOI] [PubMed] [Google Scholar]
- 24.Patel R, Lim DS, Reddy D, Nagueh SF, Lutucuta S, Sole MJ, Zoghbi WA, Quinones MA, Roberts R, Marian AJ. Variants of trophic factors and expression of cardiac hypertrophy in patients with hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:2369–2377. doi: 10.1006/jmcc.2000.1267. [DOI] [PubMed] [Google Scholar]
- 25.Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, Girgis M, Elias HK, Zhu Y, Davani A, Yang Y, Chen X, Ye S, Wang OL, Chen L, Hauptman J, Vincent RJ, Dawn B. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res. 2016;118:1918–1929. doi: 10.1161/CIRCRESAHA.116.308688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jenny NS, Arnold AM, Kuller LH, Tracy RP, Psaty BM. Serum amyloid P and cardiovascular disease in older men and women: results from the cardiovascular health study. Arterioscler Thromb Vasc Biol. 2007;27:352–358. doi: 10.1161/01.ATV.0000254150.97741.fe. [DOI] [PubMed] [Google Scholar]
- 27.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li RK, Li G, Mickle DA, Weisel RD, Merante F, Luss H, Rao V, Christakis GT, Williams WG. Overexpression of transforming growth factorbeta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation. 1997;96:874–881. doi: 10.1161/01.cir.96.3.874. [DOI] [PubMed] [Google Scholar]
- 29.Li G, Borger MA, Williams WG, Weisel RD, Mickle DA, Wigle ED, Li RK. Regional overexpression of insulin-like growth factor-I and transforming growth factor-beta1 in the myocardium of patients with hypertrophic obstructive cardiomyopathy. J Thorac Cardiovasc Surg. 2002;123:89–95. doi: 10.1067/mtc.2002.118275. [DOI] [PubMed] [Google Scholar]
- 30.Ayça B, Sahin I, Kucuk SH, Akin F, Kafadar D, Avşar M, Avci II, Gungor B, Okuyan E, Dinckal MH. Increased transforming growth factor-beta levels associated with cardiac adverse events in hypertrophic cardiomyopathy. Clin Cardiol. 2015;38:371–377. doi: 10.1002/clc.22404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. 2007;127:1009–1017. doi: 10.1038/sj.jid.5700811. [DOI] [PubMed] [Google Scholar]
- 32.Kalogeropoulos AP, Georgiopoulou VV, Butler J. From risk factors to structural heart disease: the role of inflammation. Heart Fail Clin. 2012;8:113–123. doi: 10.1016/j.hfc.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Khor KH, Campbell FE, Owen H, Shiels IA, Mills PC. Myocardial collagen deposition and inflammatory cell infiltration in cats with pre-clinical hypertrophic cardiomyopathy. Vet J. 2015;203:161–168. doi: 10.1016/j.tvjl.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 34.Moon JC, Reed E, Sheppard MN, Elkington AG, Ho SY, Burke M, Petrou M, Pennell DJ. The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;43:2260–2264. doi: 10.1016/j.jacc.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 35.Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J Exp Med. 1996;184:1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duncan MR, Berman B. Stimulation of collagen and glycosaminoglycan production in cultured human adult dermal fibroblasts by recombinant human interleukin 6. J Invest Dermatol. 1991;97:686–692. doi: 10.1111/1523-1747.ep12483971. [DOI] [PubMed] [Google Scholar]
- 38.Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D. Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol. 2005;204:428–436. doi: 10.1002/jcp.20307. [DOI] [PubMed] [Google Scholar]
- 39.Peng H, Sarwar Z, Yang XP, Peterson EL, Xu J, Janic B, Rhaleb N, Carretero OA, Rhaleb NE. Profibrotic role for interleukin-4 in cardiac remodeling and dysfunction. Hypertension. 2015;66:582–589. doi: 10.1161/HYPERTENSIONAHA.115.05627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem. 1996;271:17779–17784. doi: 10.1074/jbc.271.30.17779. [DOI] [PubMed] [Google Scholar]
- 41.Xuan W, Liao Y, Chen B, Huang Q, Xu D, Liu Y, Bin J, Kitakaze M. Detrimental effect of fractalkine on myocardial ischaemia and heart failure. Cardiovasc Res. 2011;92:385–393. doi: 10.1093/cvr/cvr221. [DOI] [PubMed] [Google Scholar]
- 42.Husberg C, Nygard S, Finsen AV, Damas JK, Frigessi A, Oie E, Waehre A, Gullestad L, Aukrust P, Yndestad A, Christensen G. Cytokine expression profiling of the myocardium reveals a role for CX3CL1 (fractalkine) in heart failure. J Mol Cell Cardiol. 2008;45:261–269. doi: 10.1016/j.yjmcc.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 43.Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Muller DN. Interferongamma signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60:1430–1436. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 44.Reifenberg K, Lehr HA, Torzewski M, Steige G, Wiese E, Kupper I, Becker C, Ott S, Nusser P, Yamamura K, Rechtsteiner G, Warger T, Pautz A, Kleinert H, Schmidt A, Pieske B, Wenzel P, Munzel T, Lohler J. Interferon-gamma induces chronic active myocarditis and cardiomyopathy in transgenic mice. Am J Pathol. 2007;171:463–472. doi: 10.2353/ajpath.2007.060906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levick SP, Goldspink PH. Could interferon-gamma be a therapeutic target for treating heart failure? Heart Fail Rev. 2014;19:227–236. doi: 10.1007/s10741-013-9393-8. [DOI] [PMC free article] [PubMed] [Google Scholar]