

Abstract
Esophageal squamous cell carcinoma (ESCC) is widely regarded as one of the most lethal types of cancer around the world. The fact that early detection of ESCC could dramatically improve the treatment outcome of the patients has sparked considerable interest in searching for reliable and accurate diagnostic biomarkers. Recently, circular RNAs (circRNA) have emerged as a new type of non-coding RNAs with significant RNase resistance, wide abundance and remarkable internal diversity. There is also increasing evidence suggesting that circRNAs could be implicated in the pathogenesis of cancer and other diseases. In this study, we performed a comparative analysis of the global circRNA expression profiles in normal and malignant esophageal epithelial cell lines by a combination of RNA sequencing and bioinformatics analysis. We identified 813 significantly up-regulated and 445 down-regulated circRNA candidates, of which 32 were subsequently validated by quantitative real-time reverse transcription polymerase chain reaction analysis. The differentially expressed circRNAs were found to be associated with pathways involved in metabolism, cell apoptosis, proliferation and migration, which are commonly altered in cancer cells. Based on the obtained data, we constructed a circRNA-miRNA interaction network, in which circRNA9927-NBEAL1 represented the biggest node. Our study could lay the groundwork for further investigation concerning the pathological roles of circRNAs in ESCC.
Keywords: Transcriptome, circular RNAs, miRNA, SHEE, SHEEC
Introduction
Esophageal squamous cell carcinoma (ESCC) is considered to be one of the deadliest malignancies globally and particularly in China, where it accounts for over 95% of all esophageal cancer cases and is the 4th leading cause of cancer death [1]. The high mortality rate of ESCC is largely attributable to the difficulty in diagnosing the disease at an early stage [2]. Patients with advanced ESCC face a grim five-year survival rate below 20% [3] due to a lack of viable treatment options, as the tumor has often become too advanced for surgical resection. Additionally, clinical statistics shows that tumor metastasis can occur in around half of all late-stage ESCC patients at the point of diagnosis, which presents further obstacles to successful treatment [4]. In contrast, early detection of ESCC can significantly improve the patients’ clinical outcome, in which the overall ten-year survival rate can exceed 95% following the surgery [5]. As a result, it is argued that the key to improving the long-term prognosis of ESCC patients lies in the development of novel diagnostic methods that can detect or even predict tumor presence at an earlier stage with greater accuracy and sensitivity.
Circular RNAs (circRNA), which feature a loop architecture with a 5’-to-3’ linkage, have emerged as a new type of non-coding RNAs with potential roles in a wide range of biological processes [6]. Although initially regarded as insignificant abnormalities from splicing events, advances in sequencing methods and bioinformatics analysis techniques have led to the rediscovery of circRNAs as a unique and internally diverse group of genetic molecules with intriguing structural features and biological functions. It is now established that circRNAs are produced by a process called back-splicing, in which a downstream 5’ splice site and an upstream 3’ splice site are covalently joined together by a spliceosome [6,7]. The prevalence of circRNAs is further compounded by the tremendous variation in their origins, lengths and expression levels [6,8]. Studies have suggested that the circularization of RNAs can confer several advantages including enhanced exonuclease resistance, greater genetic variability and improved suitability as amplification template [6]. These findings suggested that circRNAs might play regulatory roles similar to those of microRNAs (miRNA) and long non-coding RNAs.
Recently, Hansen and colleagues reported that a circRNA, ciRS-7, could inhibit the activity of miRNA-7 and resultantly de-repress its downstream mRNA targets [9]. In addition, the same group also observed an interaction between another circRNA, sex-determining region Y circRNA, and miRNA-138 [9]. Taken together, the results implied that circRNAs could play important roles in regulating miRNAs and other RNA molecules. On the other hand, the expression patterns of circRNAs have been shown to vary among different types of cells and tissues. In particular, there is evidence that circRNAs are often less abundant in tumor tissues compared to their normal counterparts, possibly due to a dilutive effect caused by the fast division and proliferation of cancer cells [10]. Sand et al. performed a microarray analysis on cutaneous squamous cell carcinoma tissue samples and found 322 circRNAs to show aberrant expression patterns [11]. In another study, Li et al. compared the circRNA expression profiles between 101 gastric tumors and the surrounding noncancerous tissues, from which they found hsa_circ_002059 to be significantly down-regulated [12]. These findings highlighted the feasibility of using circRNAs as novel diagnostic biomarkers for early cancer detection.
Herein we report our comparative analysis of the global circRNA expression profiles between the immortalized human esophageal epithelial cell line SHEE and the malignantly transformed esophageal carcinoma cell line SHEEC, using a combination of RNA sequencing, bioinformatics analysis and quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) validation. The downstream miRNA targets and the putative biological functions of the differentially expressed circRNA candidates were then predicted. Based on these results, a circRNA-miRNA interaction network was generated to provide a new perspective on the involvement of various non-coding RNAs in the development and progression of ESCC.
Methods and materials
Cell culture
SHEE and SHEEC cells were grown at 37°C under a humidified atmosphere of 5% CO2 in minimum essential medium (MEM, Gibco, Thermo Fisher Scientific, Waltham, USA) containing 10% (v/v) fetal bovine serum, 100 μg/mL streptomycin and 100 μg/mL penicillin. Once the cells formed a full monolayer, they were harvested and immediately stored at -70°C until use.
Total RNA isolation
Total RNA extraction was conducted using a TRK-1001 Total RNA Purification Kit (LC Sciences, Houston, USA) according to the manufacturer’s instructions. The concentration of the obtained RNA was measured on a NanoDrop ND-2000 UV-Vis Spectrometer (Thermo Fisher Scientific, Waltham, USA). RNA integrity was analyzed using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, USA).
cDNA library construction and high-throughput sequencing
For cDNA library construction, roughly 3 μg of the extracted total RNA was used. Depletion of ribosomal RNA was conducted by using the Human/Mouse/Rat Ribo-Zero rRNA Removal Kit (Epicentre, Madison, USA) according to the manufacturer’s protocol. Next, fragmentation of the poly(A)- or poly(A)+RNA fractions was performed using divalent cations at elevated temperatures. The resultant RNA fragments were reverse-transcribed using the First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, USA) and the cDNA libraries were constructed following the dUTP method as described previously [13]. The average insert size of the paired-end libraries was 300 ± 50 bp. RNA libraries were then sequenced on the Illumina HiSeq 2500 platform with 125-bp paired-end reads.
Detection and annotation of circRNAs
Detection of circRNA was performed based on a previously described protocol with minor modifications [14]. Sequence information of the human reference genome hg19 (Feb 2009, GRCh37) was obtained from the GENCODE project (http://www.gencodegenes.org/) and used for all subsequent analysis. Raw RNA-seq reads were mapped using Bowtie2 (version 2.1.0) [15]. First, the reads that were not mapped to any rRNA were retained for genome mapping. Subsequent refinement and analysis of the non-rRNA reads was performed based on a computational pipeline called find_circ, developed by Memczak et al. for detecting and identifying circular RNAs [14]. Briefly, all remaining reads that mapped to the genome by aligning the whole read without any trimming (end-to-end mode) were neglected. Reads not mapping continuously to the genome were used for circRNA candidate detection. In the next step, the 20-nucleotide sequences derived from both ends of each retained read, also known as anchors, were re-aligned independently to the genome. The anchor alignments were subsequently extended to span each corresponding full read sequence. The order in which both terminal anchors of each read aligned to the genome was used to identify potential circRNA candidates. Specifically, if the two anchors aligned consecutively to the genome but in a reverse orientation, it would signify a head-to-tail splicing event characteristic of a circRNA. Conversely, sequential anchor alignment in the same orientation denoted a linear splicing event. A read was considered to contain a putative circRNA if it met the following screening criteria, including 1) the presence of at least two independent reads as evidence for a head-to-tail splice junction, 2) the presence of splice sites separated by a sequence that contains no more than 100 kilobases and is flanked by the GT/AG signal, 3) the detection of unambiguous breakpoints located no more than two nucleotides inside the anchor alignment, 4) the detection of no more than two mismatches within the extended anchor alignments, 5) a difference greater than 35 between the Bowtie2-calculated alignment score of the best and that of the second best anchor alignment.
Annotation of all identified circRNAs and the prediction of their exon-intron structures were performed based on a previously described method [14]. Known introns in circRNAs were assumed to be spliced out. Each circRNA was assigned to a gene structure category if it overlaps fully or partially with the respective features.
Validation of circRNA by qRT-PCR
A pair of divergent primers were designed for each putative circRNA candidate and used to amplify both genomic DNA and cDNA. First-strand cDNA synthesis was performed using Superscript II Reverse Transcriptase (Invitrogen, Carlsbad, USA) based on the manufacturer’s instructions with minor modifications. Briefly, the reverse transcription mixture, consisting of 1-2 μg of the RNA and 2 μL of the reverse transcriptase, was incubated sequentially at 42°C for 50 min, 46°C for 10 min and then 75°C for 15 min. Subsequently, the RNA templates were digested by RNase H at 37°C for 20 min, followed by the inactivation of the enzyme at 65°C for 10 min. The resultant cDNA was then used as template for real-time PCR amplification of each circRNA candidate. Specifically, each PCR reaction mixture was set up to contain 30 ng cDNA sample, 5 μM of each primer and 1 × SYBR R Green PCR Master Mix (Thermo Fisher Scientific, Waltham, USA) in nuclease-free sterile water. PCR amplification was performed in the Applied Biosystems 7900 HT Fast Real-Time PCR System (Thermo Fisher Scientific, Waltham, USA) according to manufacturer’s recommended protocol with optimizations. Meanwhile, convergent primers were used in similar PCR reactions to amplify the reference glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene and the linear transcripts. Ct values were converted to fold changes in RNA expression. A complete list of primers used in this study was provided in Table S1.
Statistical analysis
The back-spliced junction reads and linear mapped reads were combined and scaled to reads per million mapped reads (RPM) to quantify circRNA expression levels. All qRT-PCR amplifications were performed in triplicate. Differences in circRNA expression levels between SHEEC and SHEE cell lines were analyzed using the student’s t-test. P < 0.05 was considered statistically significant.
Result
We have previously reported a comparative analysis on the transcriptional changes that occurred between SHEEC and SHEE cell lines by a combination of high-throughput RNA sequencing and microarray [16]. The obtained sequencing data, which contained a total of 166 million valid reads derived from ribosomal RNA-depleted total RNA of both cell lines, were analyzed using python scripts [14] to identify putative circRNA candidates. Roughly 79.5% of all valid reads were mapped to the human GRCh38 genome (available at http://www.gencodegenes.org/releases/21.html), of which 68.5% were identified as unique linear mapping. The unmapped reads were then re-aligned, from which we found 685988 and 332722 reads from SHEEC and SHEE cell lines, respectively, to span back-spliced junctions, representing a combined 0.61% of all valid reads. With de novo assembly of the circular RNA transcripts, we identified 9867 putative circRNA candidates from the SHEEC genome with at least one back-spliced junction read, and 748 from that of SHEE. In addition, a total of 367 circRNAs were detected in both genomes. The identified circRNAs mentioned above were edited from a total of 4057 host genes encoded in the SHEEC genome and 647 in the SHEE genome, which was consistent with the notion that a single gene locus can give rise to multiple circRNAs at different back-splice sites [17].
An examination of the genomic origins of the identified circRNAs revealed that around 70.28% of those found in SHEEC cells were derived from protein-coding exons. In comparison, approximately 25.71% of all SHEEC circRNAs showed alignment with introns and the remaining 4.01% were intergenic. On the other hand, 78.02%, 2.88% and 19.01% of all SHEE circRNAs were of exonic, intergenic and intronic origins, respectively (Figure 1).
Figure 1.
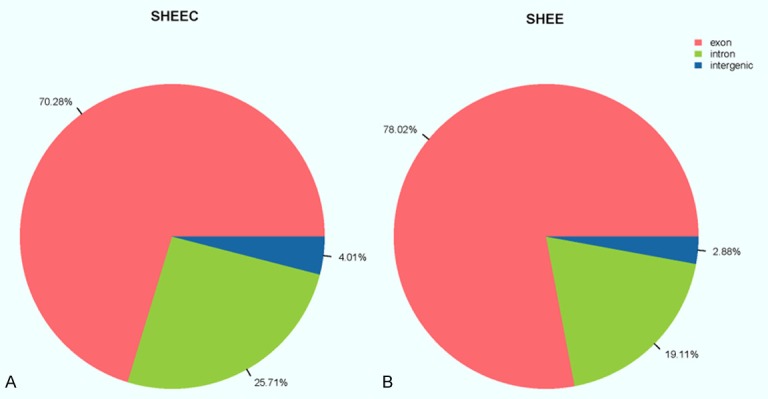
Genomic distribution of the detected SHEEC (A) and SHEE (B) circRNAs. Exonic, intronic and intergenic circRNAs are colored in red, green and blue, respectively.
We then compared the expression levels of all circRNAs candidates between SHEEC and SHEE cells. Using CuffDiff [18], a total of 1258 differentially expressed circRNA candidates were identified based on the criteria of P < 0.05 and a minimum of two-fold change between the two cell lines. Among these circRNAs, 813 and 445 were found to be up-regulated and down-regulated in SHEEC cells, respectively, in comparison to SHEE cells. Notably, 776 circRNAs were detected in SHEEC but not in SHEE cells, leading to infinite fold-change values. Because of this, these circRNAs were further arranged based on their expression levels in SHEEC cells, as indicated by the SHEEC values (Table S2). Similarly, 370 circRNAs were only found in SHEE cells and thus ranked according their expression levels. The results showed the top five most up-regulated circRNAs to be ciRNA11, circRNA904, circRNA3594, ciRNA101 and circRNA1241. Meanwhile, the top five most down-regulated candidates were circRNA9864, circRNA9650, circRNA9865, circRNA9671 and circRNA9930.
We subsequently performed GO and KEGG analyses on all differentially expressed circRNA candidates according to their coding genes using Web Gestalt [19]. The predicted GO functions were classified into three subcategories, namely biological process, cellular component as well as molecular function, and ranked according to the number of related genes (Figure 2, Table S3). Based on the results, the main dysregulated pathways that could be meaningfully interpreted included the ones implicated in the regulation of gene transcription, cell proliferation, metabolism and apoptosis. Meanwhile, KEGG analysis of the identified circRNA candidates revealed a wide variety of pathways related to proteostasis, cell cycle, as well as amino acid biosynthesis and metabolism (Figure 3, Table S4). In addition, several pathways with significant enrichment scores were involved in cell-cell and cell-extracellular matrix interactions (focal adhesion, adherens junction and tight junction), which are known to play crucial roles in modulating cell differentiation, migration and proliferation. Particularly, two of the enriched pathways, the Hippo pathway and the 5’-adenosine monophosphate-activated protein kinase pathway, were frequently implicated in cancer development.
Figure 2.
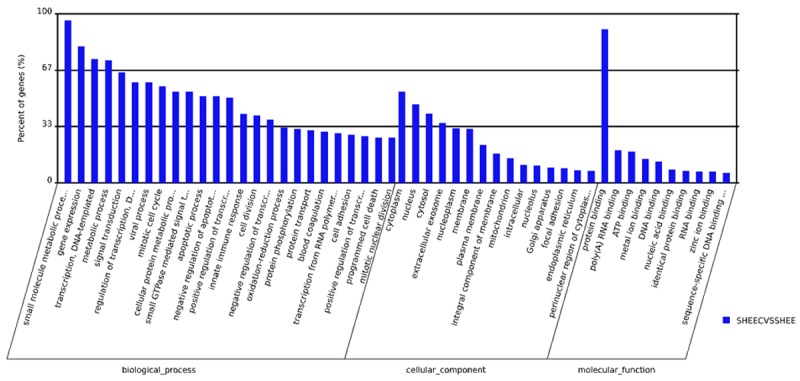
GO enrichment analysis of the differentially expressed circRNA candidates between SHEEC and SHEE cells.
Figure 3.
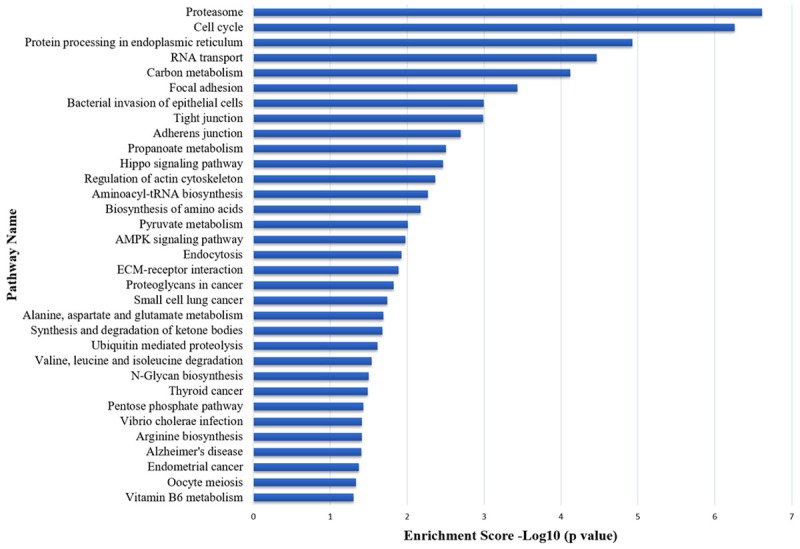
KEGG enrichment analysis of the differentially expressed circRNA candidates. Enrichment scores are graphed on a logarithmic scale.
The constructed circRNA-miRNA interaction network (Figure 4 and Table S5) comprised 979 circRNAs, 85 miRNAs and 3473 associations. In addition, we specifically created an interaction network based on all aberrantly expressed circRNAs involved in the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, based on our previous finding that it was one of the most significantly enriched pathways in the competing endogenous RNA network generated from SHEE and SHEEC cells [16]. Cytoscape analysis indicated that circRNA9953-PKN2, circRNA3706-LAMA3, circRNA5431-PRKAA1, circRNA7681-FN1, circRNA8507-LAMC2, circRNA3703-LAMA3, circRNA3341-HSP90AA1, circRNA4628-EGFR, circRNA7574-ITGA6, circRNA7683-FN1 and circRNA8506-LAMC1 contained more than 4 nodes. In particular, circRNA9953-PKN2 could potentially interact with 18 miRNA targets (Figure 5).
Figure 4.

Construction of the circRNA-miRNA interaction network.
Figure 5.
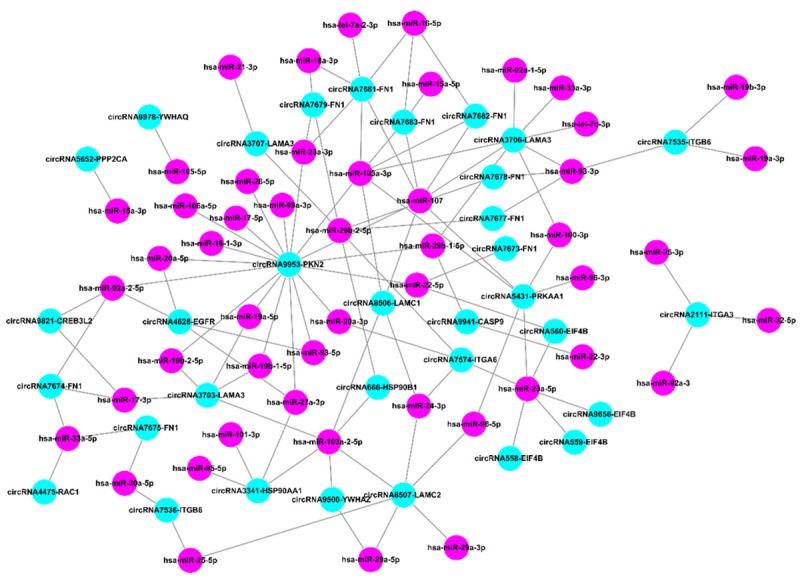
CircRNA-miRNA network showing the interactions between all the circRNAs putatively involved in the PI3K/Akt signaling pathway and their miRNA targets.
Based on the abovementioned analysis results, we experimentally verified the expression levels of the 26 most up-regulated and the six most down-regulated circRNAs in SHEEC and SHEE cells by qRT-PCR. As depicted in Figure 6, 27 of all the tested circRNA candidates exhibited consistent dysregulation patterns as shown by both qRT-PCR and RNA sequencing. In the overwhelming majority of these cases, qRT-PCR showed a higher fold-change compared to sequencing. However, the two methods contradicted each other on circRNA3341, circRNA378, circRNA4628, circRNA666 and circRNA904. Taken together, these data lent further credence to the validity of our sequencing and bioinformatics analysis method.
Figure 6.
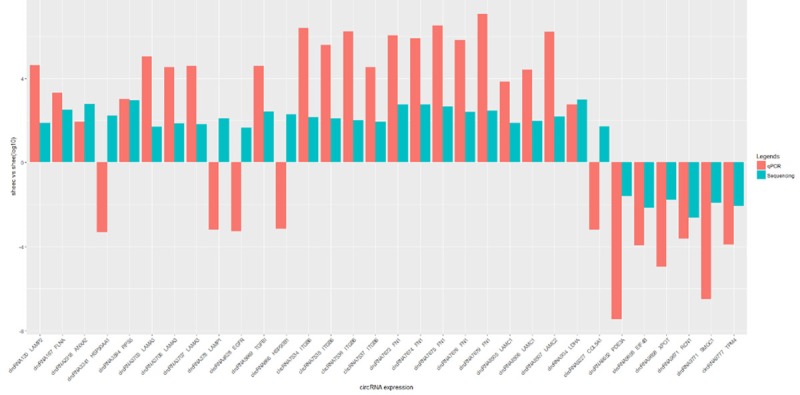
qRT-PCR validation of the 26 most up-regulated and the six most down-regulated circRNAs. In each instance, the fold enrichment score was calculated as a ratio of the expression level of the circRNA candidate in SHEEC cells to that in SHEE cells and graphed on a logarithmic scale. Data obtained by qRT-PCR and RNA sequencing were illustrated in the form of red and blue columns, respectively.
Discussion
To the best of our knowledge, our study provides the first global circRNA expression profiling of any type of esophageal tumor cells. Despite increasing attention and research efforts, the distribution and molecular roles of circRNAs in tumors remain poorly understood, even when compared to other classes of noncoding RNAs such as miRNAs and lncRNAs. Bachmayr-Heyda and colleagues reported the presence of at least 10,758 back-splice junctions supported by two or more reads in their sequencing of enriched circRNA samples derived from 31 normal and colorectal tumor tissues [10]. The results strongly suggested that the actual abundance of circRNAs in nonmalignant and cancerous tissues could still have been greatly underestimated despite the best enrichment and sequencing efforts. The authors also detected a global reduction in the quantity ratio of circRNAs to linear transcripts in tumoral samples compared to their normal counterparts [10]. In another study, a microarray-based comparative analysis of circRNA profiles between laryngeal squamous cell cancer tissues and their adjacent nonmalignant tissues led to the identification of 698 aberrantly expressed candidates, of which 302 were up-regulated and 396 underwent down-regulation [20]. Similar results were obtained in another microarray profiling study on bladder carcinoma samples, in which a total of 469 dysregulated circRNAs were found [21]. In addition, the authors showed that one of the most significantly up-regulated circRNAs, circTCF25, could play a stimulatory role in cell migration and proliferation by acting as a sponge for miR-103a-3p and miR-107 [21]. In the current study, our comparative analysis of the circRNA expression profiles between SHEE and SHEEC cells revealed 813 significantly up-regulated and 445 down-regulated candidates. In total, we identified 10248 potential circRNA candidates from all qualifying sequencing reads, which was largely in line with those previously published studies. Interestingly, our data indicated that there were 13-times more circRNAs in SHEEC cells compared to SHEE cells. Similar correlation between the number of circRNAs and the extent of malignancy has been noted in Ahmed’s study [22]. Needless to say, the vast diversity of circRNAs in SHEEC cells and other cancer tissues could potentially serve as molecular fingerprints to aid in tumor genotyping for the development of more tailored treatment strategies.
It has come to our attention that two research groups have recently reported differential circRNA expression profiles in esophageal cancer cells. Su and colleagues detected a total number of 3752 circRNAs from a radioresistant esophageal squamous cancer cell line KYSE-150R, from which 57 circRNAs were found to be up-regulated and 17 down-regulated [23]. On the other hand, Xia et al. described the identification of hsa_circ_0067934 based on a comparative analysis of circRNA profiles between paired ESCC and adjacent normal tissues [24]. Subsequently, the authors confirmed that in vitro silencing of the circRNA could inhibit the proliferation and migration of ESCC cells [24]. Compared to these studies, which both employed circRNA microarrays, the current study adopted the RNA sequencing method, which could in theory examine the expression of all circRNAs from the genome. Indeed, our method identified a significantly greater number of aberrantly expressed circRNA candidates in SHEEC cells, which was consistent with the emerging awareness that the abundance of these non-coding RNA molecules has been grossly underestimated due to the lack of suitable detection and analytic tools [10]. On the other hand, our GO and KEGG analysis have largely echoed the abovementioned studies by indicating that the differentially expressed circRNAs in SHEEC cells could contribute to malignant development by affecting cell apoptosis, differentiation, proliferation and migration through multiple signaling pathways. Combined with our previous study on the competing endogenous RNAs in SHEEC cells [16], our results significantly furthered the understanding of the regulatory roles of different non-coding RNAs in esophageal cancer. Further studies would be necessary to determine the exact downstream miRNA and gene targets of the circRNAs that we identified.
Like miRNAs and lncRNAs, the realization that numerous circRNAs exhibit distinct expression patterns between cancerous and nonmalignant tissues hints at their clinical potential as diagnostic biomarkers. Indeed, circRNAs possess several desirable biochemical properties that render them well-suited for this purpose. There is substantial evidence indicating that the covalently circular structure of these RNA molecules allows them to be much more resistant to RNase R and RNA exonuclease degradation than their linear counterparts [25]. Indeed, a recent study demonstrated that the average half-life of circRNAs was significantly greater than that of mRNAs [26]. Furthermore, circRNAs are also universally present in high abundance in cells, and are readily accessible thanks to the rapid advancement of RNA sequencing technologies. Due to these properties, circRNAs have been suggested as a new group of diagnostic biomarkers for the early detection of cancer and other diseases. In this regard, Shang et al. investigated the global circRNA expression profile of hepatocellular carcinoma (HCC) tissues and found hsa_circ_0005075 to be dysregulated [27]. Further statistical analysis showed a significant correlation between hsa_circ_0005075 expression and tumor size, hinting at the possibility of its use in HCC detection [27]. Another potential application of circRNAs as cancer biomarkers involves tumor sub-classification. As mentioned earlier, the large number and diversity of circRNAs present in tumoral tissues could be exploited to distinguish between various cancer subtypes on a molecular level. This not only could provide an objective method for assessing the severity of tumor malignancy, but could also offer useful guidance in the choice of targeted cancer therapies.
Conclusion
We have provided the first comprehensive analysis of the circRNA expression patterns in esophageal cancer. Employing a combination of RNA sequencing and bioinformatics analysis, we identified a large repertoire of dysregulated circRNA candidates in SHEEC cells. The use of TargetScan and MiRanda enabled us to determine the potential miRNA targets of the detected circRNAs. GO and KEGG analyses revealed that the aberrantly expressed circRNAs were primarily associated with signaling pathways governing cell cycle, apoptosis, proliferation and gene transcription, all of which were frequently altered as a result of cancer development and progression. qRT-PCR validation of the expression levels of selected circRNAs produced results that were largely consistent with those produced by sequencing, further supporting the robustness of our method. We envision that our study could provide the framework for future research on the molecular implications of circRNAs in esophageal cancer and other types of tumors.
Acknowledgements
We acknowledge anonymous reviewers and academic editor for their constructive suggestions on the previous version of our manuscript. And, we thank Dr. Jianning Liu of KEGENE Biotech for his help of bioinformatics analysis. This work was supported by the National Natural Science Fund (Grant No. U1304809) and the key scientific research projects of Henan Provincial Department of Education.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of eighteen major cancers in 1985. Int J Cancer. 1993;54:594–606. doi: 10.1002/ijc.2910540413. [DOI] [PubMed] [Google Scholar]
- 3.Shin D, Protano MA, Polydorides AD, Dawsey SM, Pierce MC, Kim MK, Schwarz RA, Quang T, Parikh N, Bhutani MS, Zhang F, Wang G, Xue L, Wang X, Xu H, Anandasabapathy S, Richards-Kortum RR. Quantitative analysis of highresolution microendoscopic images for diagnosis of esophageal squamous cell carcinoma. Clin Gastroenterol Hepatol. 2015;13:272–279. e272. doi: 10.1016/j.cgh.2014.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enzinger PC, Ilson DH, Kelsen DP. Chemotherapy in esophageal cancer. Semin Oncol. 1999;26:12–20. [PubMed] [Google Scholar]
- 5.Kranzfelder M, Büchler P, Friess H. Surgery within multimodal therapy concepts for esophageal squamous cell carcinoma (ESCC): the MRI approach and review of the literature. Adv Med Sci. 2009;54:158. doi: 10.2478/v10039-009-0044-1. [DOI] [PubMed] [Google Scholar]
- 6.Lasda E, Parker R. Circular RNAs: diversity of form and function. Rna. 2014;20:1829–1842. doi: 10.1261/rna.047126.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilusz J. Circular RNA and splicing: skip happens. J Mol Biol. 2015;427:2411–2413. doi: 10.1016/j.jmb.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 8.Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta. 2016;1859:163–168. doi: 10.1016/j.bbagrm.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 10.Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D. Correlation of circular RNA abundance with proliferation-exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015;5:8057. doi: 10.1038/srep08057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sand M, Bechara FG, Gambichler T, Sand D, Bromba M, Hahn SA, Stockfleth E, Hessam S. Circular RNA expression in cutaneous squamous cell carcinoma. J Dermatol Sci. 2016;83:210–218. doi: 10.1016/j.jdermsci.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 12.Li P, Chen S, Chen H, Mo X, Li T, Shao Y, Xiao B, Guo J. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta. 2015;444:132–136. doi: 10.1016/j.cca.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 13.Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F, Rajewsky N. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 15.Langmead B, Salzberg SL. Fast gappedread alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun J, Yan J, Yuan X, Yang R, Dan T, Wang X, Kong G, Gao S. A computationally constructed ceRNA interaction network based on a comparison of the SHEE and SHEEC cell lines. Cell Mol Biol Lett. 2016;21:21. doi: 10.1186/s11658-016-0022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–147. doi: 10.1016/j.cell.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with tophat and cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005:33. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xuan L, Qu L, Zhou H, Wang P, Yu H, Wu T, Wang X, Li Q, Tian L, Liu M, Sun Y. Circular RNA: a novel biomarker for progressive laryngeal cancer. Am J Transl Res. 2016;8:932–9. [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong Z, Lv M, Chen J. Screening differential circular RNA expression profiles reveals the regulatory role of circTCF25-miR-103a-3p/miR-107-CDK6 pathway in bladder carcinoma. Sci Rep. 2016;6:30919. doi: 10.1038/srep30919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed I, Karedath T, Andrews SS, Alazwani IK, Mohamoud YA, Querleu D, Rafii A, Malek JA. Altered expression pattern of circular RNAs in primary and metastatic sites of epithelial ovarian carcinoma. Oncotarget. 2016;7:36366–36381. doi: 10.18632/oncotarget.8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su H, Lin F, Deng X, Shen L, Fang Y, Fei Z, Zhao L, Zhang X, Pan H, Xie D, Jin X, Xie C. Profiling and bioinformatics analyses reveal differential circular RNA expression in radioresistant esophageal cancer cells. J Transl Med. 2016;14:225. doi: 10.1186/s12967-016-0977-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia W, Qiu M, Chen R, Wang S, Leng X, Wang J, Xu Y, Hu J, Dong G, Xu PL, Yin R. Circular RNA has_circ_0067934 is upregulated in esophageal squamous cell carcinoma and promoted proliferation. Sci Rep. 2016;6:35576. doi: 10.1038/srep35576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A. Characterization of RNase Rdigested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006;34:e63. doi: 10.1093/nar/gkl151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32:453–461. doi: 10.1038/nbt.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shang X, Li G, Liu H, Li T, Liu J, Zhao Q, Wang C. Comprehensive circular RNA profiling reveals that hsa_circ_0005075, a new circular RNA biomarker, is involved in hepatocellular crcinoma development. Medicine (Baltimore) 2016;95:e3811. doi: 10.1097/MD.0000000000003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.