

Abstract
The potential role of macrophages in pulmonary fibrosis (PF) prompted us to evaluate the roles of CX3CR1, a chemokine receptor abundantly expressed in macrophages during bleomycin (BLM)-induced PF. Intratracheal BLM injection induced infiltration of leukocytes such as macrophages into the lungs, which eventually resulted in fibrosis. CX3CR1 expression was mainly detected in the majority of macrophages and in a small portion of α-smooth muscle actin-positive cells in the lungs, while CX3CL1 was expressed in macrophages. BLM-induced fibrotic changes in the lungs were reduced without any changes in the number of leukocytes in Cx3cr1 −/− mice, as compared with those in the wild-type (WT) mice. However, intrapulmonary CX3CR1+ macrophages displayed pro-fibrotic M2 phenotypes; lack of CX3CR1 skewed their phenotypes toward M1 in BLM-challenged lungs. Moreover, fibrocytes expressed CX3CR1, and were increased in BLM-challenged WT lungs. The number of intrapulmonary fibrocytes was decreased in Cx3cr1 −/− mice. Thus, locally-produced CX3CL1 can promote PF development primarily by attracting CX3CR1-expressing M2 macrophages and fibrocytes into the lungs.
Introduction
When tissue injuries occur due to microbial infection, toxic substances, or mechanical damages, inflammatory responses are initiated. Tissue repair occurs through the interplay between various hematopoietic and non-hematopoietic cells1. Deposition of extracellular matrix (ECM) components, such as collagen, is a crucial but reversible process in most cases of wound healing. However, if the tissue injury is severe or repetitive, or if the inflammatory response becomes dysregulated, progressive and irreversible accumulation of ECM may occur. The subsequent development of fibrosis can lead to organ failure, as seen in pulmonary fibrosis (PF), end-stage liver, kidney diseases, and heart failure1.
CX3CL1, also known as fractalkine, is a member of the CX3C chemokine family. It can bind to its specific receptor, CX3CR1, in a 1:1 ratio2,3. CX3CL1 is expressed by epithelial cells in the lungs, kidneys, and intestines as a membrane-bound molecule; its chemokine domain attaches to the cell surface through a mucin-like stalk in the resting state. CX3CR1 is abundantly expressed by monocytes/macrophages, T cells, natural killer cells, and smooth muscle cells2,3.
Accumulating evidence has demonstrated differential roles of CX3CL1-CX3CR1 interactions in various types of human diseases4. The CX3CL1-CX3CR1 system is suggested to promote the development of both atherosclerosis and coronary artery diseases5,6. It has also been reported that serum CX3CL1 levels correlate positively with disease severity in rheumatoid arthritis patients7. Furthermore, CX3CL1-CX3CR1 interactions could contribute to the pathogenesis of lung diseases, including asthma8,9 and emphysema10,11. On the contrary, CX3CL1-CX3CR1 signaling can either be beneficial or detrimental in carcinogenesis and cancer metastasis via alternative pathways4. Interestingly, studies have shown that the CX3CL1-CX3CR1 axis could protect against epilepsy, Parkinson’s disease, and amyotrophic lateral sclerosis12. Furthermore, the CX3CL1-CX3CR1 axis could maintain β cell function to preserve sufficient insulin secretion13. We have previously demonstrated that CX3CL1 protects against polymicrobial sepsis and toxin A-induced enteritis by binding to CX3CR1-expressing macrophages14,15.
PF is characterized pathologically by diffuse interstitial inflammation and fibrosis16. Moreover, PF can develop from various conditions, such as irradiation injury, oxygen toxicity-related pneumonitis, and scleroderma; however, approximately half of the PF cases are idiopathic. Environmental exposures to various substances17, as well as genetic factors18, have been proposed to be implicated in the development of idiopathic PF (IPF). However, their contribution to disease pathogenesis remains to be investigated. Aberrant tyrosine kinase activation in IPF has incited the clinical application of nintedanib, a multi-tyrosine kinase inhibitor. Moreover, pirfenidone, an agent with anti-inflammatory and anti-oxidant activities, was also examined for its effectiveness against IPF19. While both agents can delay the decline in respiratory function in IPF, they are unable to cure the disease. As a consequence, it is necessary to establish a novel treatment strategy based on the molecular and cellular mechanisms of IPF pathogenesis.
Bleomycin (BLM) can induce lung injuries that mimic the pathological features observed in human IPF. Analysis of BLM-induced lung injuries revealed the contribution of pulmonary macrophages to disease onset20–24. The expression of CX3CR1 on monocytes/macrophages prompted us to investigate the roles of the CX3CL1-CX3CR1 axis in BLM-induced lung injury in CX3CR1-deficient (Cx3cr1 −/−) mice. We revealed that genetic ablation of Cx3xr1 significantly attenuated BLM-induced lung fibrosis.
Results
Intrapulmonary expression of CX3CL1 and CX3CR1 following BLM treatment
We first evaluated gene expression of Cx3cl1 and Cx3cr1 in lungs of WT mice following intratracheal BLM administration. Both Cx3cl1 and Cx3cr1 mRNAs were detected in lungs of untreated WT mice. Expression of Cx3cl1 was significantly upregulated at 1 day and 3 days after the BLM challenge, followed by augmentation in Cx3cr1 expression (Fig. 1a and b). Double-color immunofluorescence analysis demonstrated that F4/80+ cells, but not α-SMA+ fibroblasts and epithelial cells, were major cellular sources of CX3CL1 (Fig. 1c and Supp. Figure 1). Moreover, CX3CR1 proteins were detected in F4/80+ macrophages (Fig. 1d) and in a small proportion of α-SMA+ fibroblasts (Fig. 1e). Thus, intratracheal BLM injection induced F4/80+ cells mainly in the lungs to produce CX3CL1. Upregulation of CX3CL1 can attract F4/80+ macrophages and α-SMA+ fibroblasts through interactions with CX3CR1 expressed on these cells.
Figure 1.

(a) and (b) Expression of Cx3cl1 (a) and Cx3cr1 (b) in the lung of WT mice after BLM challenge. Quantitative RT-PCR analyses of Cx3cl1 and Cx3cr1 gene expression was carried out. Values represent mean ± SEM (n = 6). *P < 0.05; **P < 0.01, vs. unchallenged lungs (time = 0). (c) to (e) Cell types expressing CX3CL1 and CX3CR1 in lungs of WT mice at 3 days after BLM exposure. Double-color immunofluorescence analyses were performed; representative images from six individual animals are shown here. Signals were merged digitally.
Pathogenic roles of the CX3CL1-CX3CR1 axis in BLM-induced PF
In order to determine the pathophysiological roles of the CX3CL1-CX3CR1 axis in BLM-induced PF, we challenged WT and Cx3cr1 −/− mice with intratracheal BLM. No significant histological differences were observed between the lungs of untreated WT and Cx3cr1 −/− mice (Fig. 2a). At 21 days after BLM treatment, WT mice exhibited severe destruction of pulmonary alveolar structures and massive collagen deposition in the lungs, as evidenced by positive Masson staining (Fig. 2a). On the contrary, these fibrotic changes were markedly attenuated in Cx3cr1 −/− mice (Fig. 2a). Moreover, Col1a1 mRNA expression was significantly elevated in the lungs of WT mice at 7 and 14 days after BLM treatment; this enhancement was reduced in Cx3cr1 −/− mice (Fig. 2b). The intrapulmonary content of hydroxyproline (Hyp), a major component of collagen, was also consistently increased in WT, but not Cx3cr1 −/− mice, at 21 days after BLM treatment (Fig. 2c). We further challenged bone marrow (BM) chimeric mice generated from WT and Cx3cr1 −/− mice with BLM, as CX3CR1 is expressed by BM- and non-BM-derived cells25. BLM treatment increased intrapulmonary Hyp content to a similar level as that in WT and Cx3cr1 −/− mice with WT-derived BM (Fig. 2d). On the contrary, the increased Hyp content was reduced to a similar level in WT and Cx3cr1 −/− mice with Cx3cr1 −/–-derived BM. Therefore, BM-derived CX3CR1+ cells may play a major role in the pathogenesis of BLM-induced PF.
Figure 2.

(a) Histopathological analysis of lungs from WT and Cx3cr1 −/− mice after BLM treatment. Representative results from six animals at each time point are shown (HE stain and Masson’s stain). (b) Quantitative RT-PCR analyses of Col1a1 in the lungs of WT and Cx3cr1 −/− mice at the indicated time intervals after BLM treatment. All values represent mean ± SEM (n = 6). **P < 0.01, WT vs. Cx3cr1 −/− mice. (c) Intrapulmonary Hyp content in WT and Cx3cr1 −/− mice at 21 days after BLM challenge. All values represent mean ± SEM (n = 6). **P < 0.01, WT vs. Cx3cr1 −/− mice. (d) BLM-induced PF in BM chimeric mice. Recipient female mice were transplanted with BM cells from Cx3cr1 −/− or WT male donor. BM chimeric mice were injected with BLM at 60 days after BM transplantation. Hyp content in the lungs of BM chimeric mice was determined at 21 days after BLM exposure. All values represent mean ± SEM (n = 6). **P < 0.01, recipient with Cx3cr1 −/−BM cells vs. recipients with WT-BM cells.
Involvement of the CX3CL1-CX3CR1 axis in intrapulmonary M2-macrophage infiltration
As leukocytes are hypothesized to play essential roles in the development of PF26, we wanted to evaluate leukocyte infiltration following BLM challenge. We did not observe any differences in the number of granulocytes, macrophages, and T cells between untreated WT and Cx3cr1 −/− mice (Fig. 3). BLM challenge induced infiltration of granulocytes, macrophages, and T cells to similar extents in WT and Cx3cr1 −/− mice (Fig. 3), which was confirmed at 5 days after BLM injection by flow cytometric analyses of bronchoalveolar lavage fluids (BALFs) (Supp. Figure 2). Therefore, absence of the CX3CL1-CX3CR1 axis may trigger compensatory enhancements in the expression of other chemokines, which lead to subsequent maintenance of inflammatory cell infiltration. However, this assumption was negated by the observation that absence of CX3CR1 had few impacts on the intrapulmonary expression of several chemokine and chemokine receptor genes, which were proposed to be involved in the pathogenesis of BLM-induced PF (Supp. Figure 3)26. Several lines of evidence demonstrated that functionally polarized macrophage subtypes, M1 and M2, are crucial for inflammation and tissue repair, respectively27. M2-macrophages are known to be important for tissue fibrosis28. As CX3CR1 was expressed by macrophages recruited into the lungs (Fig. 1d), we evaluated the phenotypes of BALF macrophages in WT and Cx3cr1 −/− mice following BLM challenge. In the BALF of BLM-treated WT mice, the number of CD206+ macrophages (M2-type) dominated over CD86+ macrophages (M1-type). However, in BLM-treated Cx3cr1 −/− mice, the opposite trend was observed (Fig. 4a). Immunofluorescence analysis demonstrated that CD206 and CD68 were simultaneously expressed by CX3CR1-expressing cells in the lung parenchyma after BLM challenge (Fig. 4b and c). Thus, the CX3CL1-CX3CR1 axis can play a crucial role in the intrapulmonary recruitment of pro-fibrotic M2-macrophages that eventually contribute to BLM-induced PF.
Figure 3.

Immunohistochemical analyses of leukocyte numbers in WT and Cx3cr1 −/− mouse lungs. (a) to (c) Immunohistochemical analyses were conducted using anti-Ly-6G (a) anti-F4/80 (b) or anti-CD3 Abs (c). Representative results from six individual animals are shown. (d) to (f) Number of granulocytes (d) macrophages (e) and T cells (f) in lung tissues was determined. All values represent mean ± SEM (n = 6).
Figure 4.

(a) Flow cytometric analysis of CD206+ M2-macrophages and CD86+ M1-macrophages among CD68+ macrophages in BALF from WT and Cx3cr1 −/− mice at 5 days following BLM challenge. Representative results from three independent experiments with four animals in each group are shown. (b) Detection of CD206 on CX3CR1+ cells in the lungs of WT mice at 3 days after BLM exposure. (c) Detection of CX3CR1 on CD68+CD206+ M2-macropahges in the lungs of WT mice at 3 days after BLM exposure. Representative results from six individual animals are shown. Signals were merged digitally.
Reduced intrapulmonary fibrocyte accumulation in Cx3cr1−/− mice
Given the crucial involvement of BM-derived CX3CR1+ cells in lung fibrosis, we focused on an additional cell type present in the BM, the fibrocytes. These cells were identified as CD45+Col-I+ BM cells and hypothesized to be essential for tissue repair and abnormal fibrosis29–31. We generated BM chimeric mice by transplanting lethally irradiated WT mice with GFP-transgenic (Tg) mouse-derived BM cells. When WT mice bearing GFP+ BM cells were intratracheally injected with BLM, GFP+ cells was detected in the lungs, indicating that BM cells were recruited into the lungs after BLM treatment. In addition, Col-I was also detected in a substantial proportion of GFP+ cells (Fig. 5a and b). Moreover, immunofluorescence analyses revealed that both Col-I and CD45 were present in a large proportion of GFP+ cells recruited from the BM following BLM treatment (Fig. 5c). These observations suggested that BM-derived CD45+Col-I+ fibrocytes migrate to lungs after BLM treatment. Flow cytometry analysis revealed that CX3CR1 was expressed in approximately 60% of CD45+Col-I+ fibrocytes in the BM of WT mice after BLM treatment (Fig. 5d). We next determined the number of fibrocytes in the BM, peripheral blood, and lung tissues; there were no significant differences in fibrocyte numbers between untreated WT and Cx3cr1 −/− mice (Fig. 6 and Supp. Figure 4). BLM challenge increased the number of fibrocytes to similar extents in the BM and peripheral blood of WT and Cx3cr1 −/− mice (Supp. Figure 4). Number of fibrocytes in the lungs were markedly increased in WT mice at 12 days after BLM treatment. However, this enhancement in the number of fibrocytes was suppressed in Cx3cr1 −/− mice (Fig. 6). Collectively, these results suggested that locally-produced CX3CL1 can contribute to BLM-induced PF by recruiting CX3CR1-expressing fibrocytes that produce Col-I from the BM into the lungs.
Figure 5.

(a) Detection of BM-derived cells in the lungs after BLM treatment. BM chimeric mice were generated by transplanting BM cells of GFP-Tg mice to lethally irradiated WT mice. The resultant cells were challenged with BLM, and the lungs were obtained at 14 days after the challenge. Representative results from six individual animals are shown here. Nuclear staining is shown in blue. (b) Characterization of GFP+ BM cells recruited into the lungs after BLM challenge (at 14 days). Cells were immunostained with anti-Col-I and DAPI staining. (c) Characterization of GFP+ cells in the lung of WT mice at 5 days after BLM exposure. GFP+ cells were further treated with anti-Col-I and anti-CD45 antibodies. Representative results from 6 individual animals are shown. (d) Flow cytometric analysis of CX3CR1+ fibrocytes among CD45+Col-I+ fibrocytes in the BM of WT mice prior to BLM challenge. Representative results from 6 individual animals are shown here.
Figure 6.

Quantitative evaluation of intrapulmonary fibrocytes by flow cytometric analyses at 12 days after BLM challenge. (a) Representative results from six independent experiments are shown here. (b) Changes in percentage of fibrocytes in lungs are shown. Values represent mean ± SEM (n = 6). **P < 0.01, WT vs. Cx3cr1 −/−.
Reduced TGF-β1 expression in mice lacking CX3CR1
TGF-β1 is a potent fibrogenic growth factor in various organs. We examined the cell types expressing TGF-β1 in the lungs after BLM treatment. Immunofluorescence analyses showed expression of TGF-β1 in CX3CR1+ cells (Fig. 7a), which suggested that CX3CR1-expressing cells, fibrocytes, and M2-macrophages could be a major source of TGF-β1 in the lungs. Moreover, BLM treatment progressively enhanced TGF-β1 expression at both the gene and protein levels in the lungs of WT mice; this enhancement was markedly suppressed in the lungs of Cx3cr1 −/− mice (Fig. 7b and c). As a result, the CX3CL1-CX3CR1 axis can have an indispensable role in BLM-induced lung fibrosis by inducing the migration of TGF-β1-producing fibrocytes and M2-macrophages into the lungs.
Figure 7.
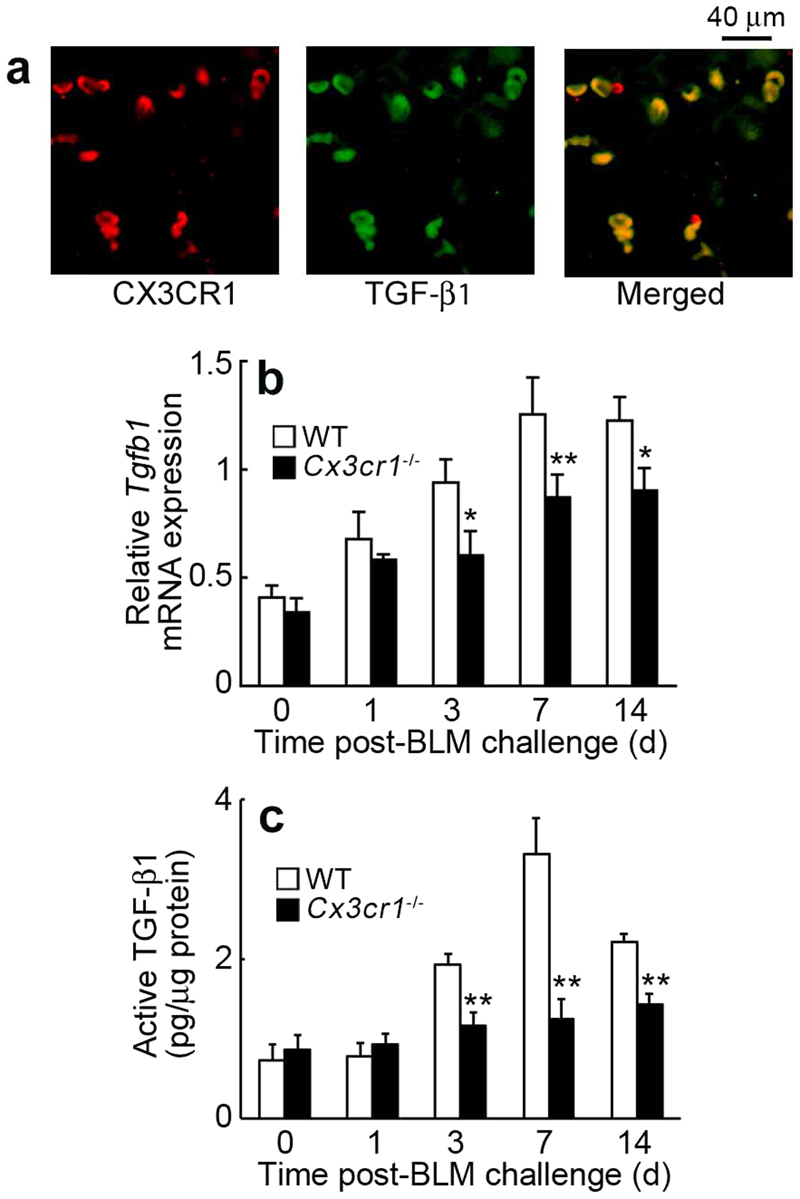
Intrapulmonary TGF-β1 expression in the lungs after BLM treatment. (a) Cell types expressing TGF-β1 in the lungs of BLM-treated WT mice. Double-color immunofluorescence analyses were performed. Representative results from six individual animals are shown here. Signals were merged digitally. (b) Intrapulmonary TGF-β1 expression in WT and Cx3cr1 −/− after BLM challenge. Tgfb1 mRNA expression was analyzed by quantitative RT-PCR. (c) Active TGF-β1 protein content in the lungs was determined with ELISA. Values represent mean ± SEM (n = 6). *P < 0.05; **P < 0.01, WT vs. Cx3cr1 −/− mice.
Discussion
It is highly evident from previous studies that macrophages are indispensable in tissue repair and the subsequent fibrotic changes32,33. Robust CX3CR1 expression in macrophages2,3 prompted several scientific groups to investigate the roles of the CX3CL1-CX3CR1 axis in fibrosis in various organs. Studies have shown that hypertension- and unilateral ureteral obstruction-induced renal fibrosis are attenuated by Cx3cr1 gene deficiency34–36. On the contrary, the interplay between CX3CL1 and CX3CR1 can prevent carbon tetrachloride-induced liver injury37,38. These discrepancies may be explained by organ-specific differences in macrophage populations; CX3CR1-expressing macrophages may exert anti-inflammatory activities, as observed in corneal tissues39. Hence, we examined the effect of CX3CR1 deficiency on BLM-induced PF. Our results revealed that CX3CR1 deficiency inhibits BLM-induced lung pathologies, but does not exert any effect on infiltration of inflammatory cells, including macrophages.
Macrophages in the lungs belong to two subpopulations, interstitial macrophages (IMs) and alveolar macrophages (AMs), residing in the interstitial and alveolar spaces, respectively40. Ly6highCCR2+ inflammatory monocytes in the circulation are recruited into lung tissues, most likely in a CCL2-dependent manner, similar to lung metastasis in breast cancer41. On the contrary, AMs originate from fetal liver monocytes, and self-maintain throughout their life cycle under steady state42. Allergen challenges reduced the number of embryo-derived AMs43 and enhanced CCL2 expression-induced migration of CCR2-expressing monocyte-derived AMs to replenish the AM loss44. Therefore, total number of intrapulmonary macrophages may not differ significantly between BLM-treated WT and Cx3cr1 −/− mice, since BLM may be able to enhance the intrapulmonary expression of the chemoattractant CCL2 for both IMs and AMs to similar extents.
Alternatively, there remains a possibility that CX3CR1 deficiency may have profound impact on macrophage functions. Macrophages can be functionally polarized into two phenotypes, M1 and M2. Skewing of macrophage polarization can contribute to pathogenesis of inflammatory responses, allergic diseases, tissue repair, and tumor progression45–47. Several lines of evidence indicated that macrophages with the M2 phenotype could promote tissue fibrosis48,49. Indeed, CX3CR1-expressing CD68+ cells in the lungs were also positive for CD20650,51, a M2-specific marker. This indicated that CX3CR1+ macrophages are M2-macrophages. Moreover, M2-macrophages were reduced in BLM-challenged lungs of Cx3cr1 −/− mice. This was concomitant with a reciprocal increase in CD68+ M1-macrophages. Therefore, absence of CX3CR1 can have profound effects on macrophage polarization rather than total macrophage numbers in the lungs after BLM challenge.
Myofibroblasts display characteristics of both fibroblasts and smooth muscle cells, as represented by their ECM-synthesizing capacity and contractile cytoskeleton, respectively52. As a result, myofibroblasts have indispensable roles in connective tissue remodeling and the subsequent development of fibrosis in various organs, including the lung. Myofibroblasts can be differentiated from several cell types, including locally residing mesenchymal cells (e.g., fibroblasts and smooth muscle cells), other local sources such as epithelial and endothelial cells, and BM-derived fibrocytes53. Studies on BM chimeric mice have revealed the crucial role of radiosensitive BM-derived CX3CR1-expressing cells in BLM-induced collagen deposition in the lung. Moreover, injection of BLM into chimeric mice with BM derived from GFP Tg mice resulted in cellular expression of GFP+ and Col-I+ in the lung. Therefore, fibrocytes may be a major source of collagen-producing myofibroblasts in BLM-induced PF.
Fibrocytes are defined as BM-derived cells that are intermediate between hematopoietic and mesenchymal cells, as they simultaneously express leukocyte markers such as CD45 and mesenchymal cell markers such as Col-I. Fibrocytes, once generated in the BM, circulate in the bloodstream, and migrate to organs in response to tissue-specific cues29–31,54–62. BLM was found to increase fibrocyte numbers in the BM, peripheral blood, and lungs, which is consistent with our previous report26. Moreover, fibrocytes were hypothesized to express several chemokine receptors, including CXCR4, CCR3, CCR5, and CCR731,63,64, which allow them to migrate to fibrotic organs in response to their cognate ligands. It was found that inhibition of CXCR4 function31, or deficiencies in Ccr3 65 or Ccr5 26 attenuated BLM-induced pathologies, together with reduced intrapulmonary fibrocyte numbers. Our present observations suggested that CX3CR1 is one of the chemokine receptors involved in migration of fibrocytes into fibrotic lungs.
We have previously demonstrated that Ccl3 −/− and Ccr5 −/− mice exhibit reduction in BLM-induced fibrosis and the number of CD45+Col-I+CCR5+ fibrocytes in the lungs, together with suppressed CXCL12 expression26. These observations indicated that the CCL3-CCR5 axis can mediate BLM-induced fibrocyte migration into the lungs, partly via its interactions with the CXCL12-CXCR4 axis. BLM-induced increase in intrapulmonary fibrocyte numbers was not completely nullified in Cx3cr1 −/− mice, suggesting the contribution of other chemokines such as CXCL12 and CCL3 to fibrocyte infiltration. The decreased BLM-induced fibrosis and fibrocyte infiltration into the lungs of Cx3cr1 −/− mice, however, were not accompanied with changes in intrapulmonary expression of CXCL12 and ligands for CCR5, CCL3, CCL4, and CCL5. Thus, the CX3CL1-CX3CR1 axis can regulate BLM-induced fibrocyte migration and subsequent fibrosis development independently of other chemokines.
CX3CL1 has distinct structural and functional features from other chemokines. Most chemokines are produced as a secretory molecule, and bind to proteoglycan and glycosaminoglycan in the ECM through their carboxy-terminal α-helix portion66; this binding is reversible and unstable. On the contrary, CX3CL1 is expressed as a membrane-bound molecule with a mucin-like stalk2,3. As compared with other chemokines, CX3CL1 can be firmly retained on the cell surface, and can consequently affect its target cells in a more stable manner. Moreover, in addition to chemotactic activities, CX3CL1-CX3CR1 interactions can promote the survival and/or proliferation of non-hematopoietic cells, such as neuronal cells67, and hematopoietic cells, such as monocytes68 and helper T cells9. If CX3CL1 is able to support survival and/or proliferation of fibrocytes as well as non-hematopoietic cells, it can attract CX3CR1-expressing fibrocytes into the lung. This can eventually promote the survival and/or proliferation of fibrocytes, thereby inducing fibrotic changes.
Collectively, our observations suggested that the CX3CL1-CX3CR1 axis is essential in the development of BLM-induced PF by regulating fibrocytes and M2-macrophags, which can exert pro-fibrotic activities. Absence of CX3CR1 impairs wound healing in the skin with reduced macrophage recruitment and fibroblast accumulation10. To summarize, the interactions between CX3CL1 and CX3CR1 may provide potential candidate molecule(s) for the treatment of IPF.
Methods
Reagents and antibodies (Abs)
BLM was purchased from Sigma Chemical Co. (St. Louis, MO). The following monoclonal antibodies (mAbs) and polyclonal antibodies (pAbs) were used for immunohistochemical and immunofluorescence analyses in this study: goat anti-mouse CX3CL1 pAbs and goat anti-mouse CX3CR1 pAb (Santa Cruz Biotechnology, Santa Cruz, CA); rabbit anti-human CX3CR1 pAbs, which cross-reacts with mouse CX3CR1 (Abnova, Walnut, CA); rat anti-mouse F4/80 mAb (clone, BM8; BMA Biomedicals, Switzerland); mouse anti-human α-smooth muscle actin (α-SMA) mAb (clone, 1A-4), rabbit anti-mouse pan-Cytokeratin pAbs (Santa Cruz Biotechnology), rabbit anti-CD68 (BIO RAD, Hercules, CA), rat anti-CD206 (Abcam, Cambridge, UK), rabbit anti-human CD3 pAbs, which cross-reacts with mouse CD3 (Dako Cytomation, Kyoto, Japan); rat anti-mouse Ly-6G mAb, (clone, 1A8; BD Biosciences, San Jose, CA); rat anti-mouse F4/80 mAb (clone, A3-1; AbD Serotec, Oxford, UK); rabbit anti-mouse collagen type I (Col-I) pAbs (Merck Millipore, Billerica, MA); rat-anti mouse TGF-β1 mAb (clone, 860206; R&D, Minneapolis, MN), Cy3-conjugated donkey anti-rat, -goat, -mouse, and -rabbit IgG pAbs, and FITC-conjugated donkey anti-goat, -rat, and -rabbit IgG pAbs (Jackson ImmunoResearch Laboratories, West Grove, PA). For flow cytometric analysis, the following antibodies (Abs) were commercially obtained: PerCP-conjugated rat anti-mouse CD45 mAb (clone, 30-F11) and FITC-conjugated rat anti-mouse CD34 mAb (clone, RAM34; BD Biosciences); rabbit anti-mouse Col-I pAbs (Merck Millipore, Billerica, MA); PE-conjugated rabbit anti-mouse CX3CR1 (PromoKine, Heidelberg, Germany); FITC-conjugated rat anti-mouse CD68 (BIO RAD, Hercules, CA); PE-conjugated rat anti-mouse CD86, FITC-conjugated rat anti-mouse CD8, FITC-conjugated Armenian hamster anti-mouse CD3, PE-conjugated mouse anti-mouse NK1.1, FITC-conjugated rat anti-mouse CD11b (BD Biosciences, Franklin Lakes, NJ); APC-conjugated rat anti-mouse CD206 (R&D, Minneapolis, MN); and FITC-conjugated rat anti-mouse Gr-1, PE-conjugated rat anti-mouse CD4 (Thermo Fisher Scientific, Waltham, MA).
Mice
Pathogen-free 8-week-old male C57BL/6 mice were obtained from Sankyo Laboratories (Tokyo, Japan), and were designated as wild-type (WT) mice. Cx3cr1 −/− mice on the C57BL/6 genetic background were a generous gift from Drs. P.M. Murphy and J.L. Gao (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD)25. Tg mice on the C57BL/6 genetic background were obtained from RIKEN BRC (Tsukuba, Japan). All animals were housed individually in cages under specific pathogen-free conditions during the experiments. Age- and sex-matched mice were used for the experiments. All animal experiments complied with the standards set out in the guidelines for the Care and Use of Laboratory Animals at the Wakayama Medical University.
BLM-induced lung Injury
Mice were put under deep anesthesia with an intraperitoneal injection of pentobarbital (50 µg/g weight). A cervical midline incision was made, and the trachea was exposed. Thereafter, BLM (0.075 U) in 50 µl sterile saline was intratracheally administered with a 26-guage needle. At the indicated time intervals following BLM administration, mice were sacrificed by pentobarbital overdose, and both lungs were removed for subsequent analyses.
Generation of bone BM chimeric mice
BM chimeric mice were prepared as previously described26. Briefly, male Cx3cr1 −/− BM → female WT mice, male WT BM → female Cx3cr1 −/− mice, male WT BM → female WT mice, and male Cx3cr1 −/− BM → female Cx3cr1 −/− mice. BM cells were collected from femurs of donor mice by aspiration and flushing. Recipient mice were irradiated to 15 Gy with a RX-650 irradiator (Faxitron X-ray Inc., Wheeling, IL). The animals then received intravenous injections of 5 × 106 BM cells in 200 μl sterile PBS(−) from donor mice under anesthesia. Thereafter, the mice were housed in sterilized microisolator cages, and were fed normal chow and autoclaved hyperchlorinated water for 60 days. Similarly, BM chimeric mice were generated by transplanting GFP-Tg mouse-derived BM cells into lethally irradiated WT mice. To verify successful engraftment and reconstitution of the BM in transplanted mice, genomic DNA was isolated from peripheral blood and tail tissues of each chimeric mouse at 30 days after BMT with a NucleoSpin tissue kit (Macherey-Nagel, Duren, Germany). Polymerase chain reaction (PCR) was used to detect the Sry gene in the Y chromosome (forward primer, 5′-TTGCCTCAACAAAA-3′; reverse primer, 5′-AAACTGCTGCTTCTGCTGGT-3′). The amplified PCR products were fractionated on a 2% agarose gel, and were visualized by ethidium bromide staining. After durable BM engraftment was confirmed, mice were treated with BLM, as described above.
Histopathological and immunohistochemical analyses
Lung tissues were fixed in 10% formalin buffered with PBS (pH 7.2), and were embedded in paraffin. Tissues were sectioned (6 µm-thick) and stained with hematoxylin and eosin or Masson’s trichrome to detect collagen deposition. Immunohistochemical analyses were also performed using anti-Ly6G, anti-F4/80, or anti-CD3 Abs, as previously described26. Cells were quantified in 10 randomly chosen visual fields at 200× magnification, and the average of the 10 selected microscopic fields was calculated. All measurements were performed by an examiner without prior knowledge of the experimental procedures.
Double- and triple-color immunofluorescence analysis
Double-color immunofluorescence analysis was conducted to identify the types of CX3CL1- or CX3CR1-expressing cells in the lung, as previously described69. Briefly, deparaffinized sections were incubated with PBS containing 1% normal donkey serum and 1% BSA to reduce nonspecific reactions. Tissue sections were then incubated with a combination of anti-CX3CR1 and anti-F4/80 Abs, anti-CX3CL1 and anti-F4/80 Abs, or anti-CX3CR1 and anti-α-SMA Abs at 1 µg/ml at 4 °C overnight. Similarly, triple-color immunofluorescence analysis was conducted to identify the molecules expressed on BM-derived fibrocytes (GFP+Col-I+ cells), such as TGF-β1 and CX3CR1. Sections containing GFP+ cells were incubated with a combination of anti-Col-I and anti-CD45 Abs, anti-Col-I and anti-TGF-β1 Abs, or anti-Col-I and anti-CX3CR1 Abs at a concentration of 1 µg/ml at 4 °C overnight. In order to identify the subtypes of macrophages recruited into the lungs, tissue sections were incubated with a combination of anti-CD68, anti-CX3CR1, and anti-CD206 Abs, or anti-CD206 and anti-CX3CR1 Abs at a concentration of 1 mg/ml at 4 °C overnight. Following incubation with fluorochrome-conjugated secondary Abs, tissues were imaged under a fluorescence microscope.
Determination of Hyp content
Lung tissues were removed at 21 days after BLM administration to determine the content of Hyp, a major component of collagen, as previously described26. Data were expressed as the amount (μg) of Hyp per lung.
Flow cytometric analysis of leukocytes in BALF
BALF was collected as previously described with some modifications26. Briefly, mice were sacrificed by abdominal aorta dissection. The trachea was cannulated, and the airway lumen was washed two times with 1 ml ice-cold PBS. Single cell suspensions were incubated with 25 µg/ml Fc block (BD Biosciences, Piscataway NJ) for 15 min on ice to prevent nonspecific binding. Cells were stained with anti-CD68, anti-CD86, CD-206, anti-CD3, anti-CD4, anti-CD8, anti-NK1.1, anti-Gr-1, and anti-CD11b. Analysis was performed on a FACScan flow cytometer (BD Biosciences) using the FlowJo software (Tommy Digital Biology, Tokyo, Japan).
Quantitative RT-PCR analysis
Total RNA was extracted from lung tissue using ISOGEN (Nippon Gene, Toyama, Japan), according to the manufacturer’s instructions. Total RNA (3 µg) was reverse transcribed to cDNA using PrimeScript™ Reverse Transcriptase (Takara Bio, Shiga, Japan) with Oligo(dT)15 primers. Quantitative PCR was carried out with the resultant cDNA as templates and SYBR® Premix Ex Taq™ II (Takara Bio) with specific primer sets, as previously described69 (Supplemental Table 1). Primers were purchased from Takara Bio. Amplification and detection of mRNA were conducted using the Thermal Cycler Dice® Real Time System (Takara Bio, TP800), according to the manufacturer’s instructions. Expression levels of target genes were analyzed using the ΔΔCt comparative threshold method. The β-actin gene was used as internal control.
Determination of fibrocytes
Mononuclear cells were isolated from the lungs, the BM, and peripheral blood, as previously described25. Nonspecific binding was blocked by incubation with 25 µg/ml Fc block (BD Biosciences Pharmingen, Piscataway, NJ) for 15 min at 4 °C. Cells were stained with PerCP-labeled anti-CD45 and FITC-labeled anti-CD34. After extensive washing with PBS(−), cells were permeabilized using cytofix/cytoperm (BD Biosciences Pharmingen), stained with anti-Col-I pAbs, and incubated with PE-conjugated goat anti-rabbit Ig (Life Technologies, Grand Island, NY), as previously described26. After two PBS(−) washes, cells were fixed in 2% paraformaldehyde. Isotype-matched control immunoglobulins were used to detect non-specific binding. Stained cells were analyzed on the FACS Calibur flow cytometer. In parallel, BM cells from BLM-treated WT mice were stained with anti-Col-I, anti-CD45, and anti-CX3CR1 to determine the proportion of CX3CR1+ fibrocytes. Flow cytometry was performed on a FACS Calibur flow cytometer, and data were analyzed using the FlowJo software.
ELISA
Lung samples from WT mice were homogenized with PBS containing complete protease inhibitor cocktail (Roch Diagnostics, Mannheim, Germany). Homogenates were centrifuged at 10,000 × g for 10 min. Supernatants were used to quantify the active form of TGF-β1 with a commercial ELISA kit (R&D Systems), according to manufacturer’s instructions. The detection limits was 7 pg/ml. Total protein in the supernatant was measured with a commercial kit (BCA protein assay kit, Pierce, Rockford, IL). Data were expressed as TGF-β1 (pg) per total protein (µg) for each sample.
Statistical analysis
The means and SEMs were calculated for all investigated parameters in the study. Statistical significance was evaluated by ANOVA or the Mann-Whitney’s U-test. P < 0.05 was accepted as being statistically significant.
Study approval
All animal experiments were approved by the Committee on Animal Care and Use at the Wakayama Medical University. All methods were performed in accordance with relevant guidelines and regulations.
Data availability
Data that support the findings of this study are available from the corresponding author upon reasonable request.
Electronic supplementary material
Acknowledgements
This work was supported in part by Grants-in-Aids for Scientific Research (A) (grant 20249040, to T. Kondo) and for Young Scientists (A) (grant 20689015, to Y. Ishida) from the Ministry of Education, Culture, Science, and Technology of Japan, the Extramural Collaborative Research Grant of Cancer Research Institute, Kanazawa University (to Y. Ishida), and by Research Grant on Priority Areas (to T. Kondo) from Wakayama Medical University.
Author Contributions
Y.I. and T.Ko. formulated the hypothesis and designed the project; Y.I. performed the main experiments; A.K. provided technical assistance and discussion; Y.K. and M.N. helped with some experimental procedures; H.H., I.S., and T.Ka. helped in classification of macrophages; N.M. and T.Ko. supervised the experiments and provided the main funding for the project; Y.I., N.M. and T.Ko. participated in writing the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-17007-8.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Julia V. CX3CL1 in allergic diseases: not just a chemotactic molecule. Allergy. 2012;67:1106–1110. doi: 10.1111/j.1398-9995.2012.02870.x. [DOI] [PubMed] [Google Scholar]
- 3.White GE, Greaves DR. Fractalkine: a survivor’s guide: chemokines as antiapoptotic mediators. Arterioscler. Thromb. Vasc. Biol. 2012;32:589–594. doi: 10.1161/ATVBAHA.111.237412. [DOI] [PubMed] [Google Scholar]
- 4.Liu W, et al. Role of CX3CL1 in Diseases. Arch. Immunol. Ther. Exp. (Warsz). 2016;64:371–83. doi: 10.1007/s00005-016-0395-9. [DOI] [PubMed] [Google Scholar]
- 5.McDermott DH, et al. Genetic polymorphism in CX3CR1 and risk of HIV disease. Science. 2000;290:2031. doi: 10.1126/science.290.5499.2031a. [DOI] [PubMed] [Google Scholar]
- 6.Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1−/− mice reveals a role for fractalkine in atherogenesis. J. Clin, Invest. 2003;111:333–40. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Odai T, et al. Correlation of CX3CL1 and CX3CR1 levels with response to infliximab therapy in patients with rheumatoid arthritis. J. Rheumatol. 2009;36:1158–65. doi: 10.3899/jrheum.081074. [DOI] [PubMed] [Google Scholar]
- 8.El-Shazly A, et al. Fraktalkine produced by airway smooth muscle cells contributes to mast cell recruitment in asthma. J. Immunol. 2006;176:1860–8. doi: 10.4049/jimmunol.176.3.1860. [DOI] [PubMed] [Google Scholar]
- 9.Mionnet C, et al. CX3CR1 is required for airway inflammation by promoting T helper cell survival and maintenance in inflamed lung. Nat. Med. 2010;16:1305–12. doi: 10.1038/nm.2253. [DOI] [PubMed] [Google Scholar]
- 10.Xiong Z, Leme AS, Ray P, Shapiro SD, Lee JS. CX3CR1+ lung mononuclear phagocytes spatially confined to the interstitium produce TNF-α and IL-6 and promote cigarette smoke-induced emphysema. J. Immunol. 2011;186:3206–14. doi: 10.4049/jimmunol.1003221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McComb JG, et al. CX3CL1 up-regulation is associated with recruitment of CX3CR1+ mononuclear phagocytes and T lymphocytes in the lungs during cigarette smoke-induced emphysema. Am. J. Pathol. 2008;173:949–61. doi: 10.2353/ajpath.2008.071034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardona AE, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006;9:917–24. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 13.Lee YS, et al. The fractalkine/CX3CR1 system regulates β cell function and insulin secretion. Cell. 2013;153:413–25. doi: 10.1016/j.cell.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishida Y, et al. Essential involvement of CX3CR1-mediated signals in the bactericidal host defense during septic peritonitis. J. Immunol. 2008;181:4208–18. doi: 10.4049/jimmunol.181.6.4208. [DOI] [PubMed] [Google Scholar]
- 15.Inui M, et al. Protective roles of CX3CR1-mediated signals in toxin A-induced enteritis through the induction of heme oxygenase-1 expression. J. Immunol. 2011;186:423–31. doi: 10.4049/jimmunol.1000043. [DOI] [PubMed] [Google Scholar]
- 16.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 17.Taskar V, Coultas D. Exposures and idiopathic lung disease. Semin. Respir. Crit. Care Med. 2008;29:670–679. doi: 10.1055/s-0028-1101277. [DOI] [PubMed] [Google Scholar]
- 18.Kropski JA, Lawson WE, Young LR, Blackwell TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis. Model Mech. 2013;6:9–17. doi: 10.1242/dmm.010736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-de la Mora DA, et al. Role and New Insights of Pirfenidone in Fibrotic Diseases. Int. J. Med. Sci. 2015;12:840–847. doi: 10.7150/ijms.11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satoh T, et al. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature. 2013;495:524–8. doi: 10.1038/nature11930. [DOI] [PubMed] [Google Scholar]
- 21.Satoh T, et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature. 2017;541:96–101. doi: 10.1038/nature20611. [DOI] [PubMed] [Google Scholar]
- 22.Vannella KM, Wynn TA. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017;79:593–617. doi: 10.1146/annurev-physiol-022516-034356. [DOI] [PubMed] [Google Scholar]
- 23.Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune Mechanisms in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016;55:309–22. doi: 10.1165/rcmb.2016-0121TR. [DOI] [PubMed] [Google Scholar]
- 24.Byrne AJ, Maher TM, Lloyd CM. Pulmonary Macrophages: A new therapeutic pathway in fibrosing lung disease? Trends. Mol. Med. 2016;22:303–16. doi: 10.1016/j.molmed.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Ishida Y, Gao JL, Murphy PM. Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. J. Immunol. 2008;180:569–79. doi: 10.4049/jimmunol.180.1.569. [DOI] [PubMed] [Google Scholar]
- 26.Ishida Y, et al. Essential roles of the CC chemokine ligand 3-CC chemokine receptor 5 axis in bleomycin-induced pulmonary fibrosis through regulation of macrophage and fibrocyte infiltration. Am. J. Pathol. 2007;170:843–54. doi: 10.2353/ajpath.2007.051213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017;18:E1545. doi: 10.3390/ijms18071545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray LA, et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS. One. 2010;5:e9683. doi: 10.1371/journal.pone.0009683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomperts BN, Strieter RM. Fibrocytes in lung disease. J. Leukoc. Biol. 2007;82:449–56. doi: 10.1189/jlb.0906587. [DOI] [PubMed] [Google Scholar]
- 30.Reese C, et al. Fibrocytes in the fibrotic lung: altered phenotype detected by flow cytometry. Front. Pharmacol. 2014;5:141. doi: 10.3389/fphar.2014.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips RJ, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Invest. 2004;114:438–46. doi: 10.1172/JCI200420997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mescher AL. Macrophages and fibroblasts during inflammation and tissue repair in models of organregeneration. Regeneration (Oxf). 2017;4:39–53. doi: 10.1002/reg2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wynn TA, Vannella Kevin M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimizu K, et al. Fractalkine and its receptor, CX3CR1, promote hypertensive interstitial fibrosis in the kidney. Hypertens. Res. 2011;34:747–752. doi: 10.1038/hr.2011.23. [DOI] [PubMed] [Google Scholar]
- 35.Engel DR, et al. CX3CR1 Reduces Kidney Fibrosis by Inhibiting Local Proliferation of Profibrotic Macrophages. J. Immunol. 2015;194:1628–1638. doi: 10.4049/jimmunol.1402149. [DOI] [PubMed] [Google Scholar]
- 36.Peng X, Zhang J, Xiao Z, Dong Y, Du J. CX3CL1-CX3CR1 Interaction Increases the Population of Ly6C-CX3CR1hi Macrophages contributing to unilateral ureteral obstruction-induced fibrosis. J. Immunol. 2015;195:2797–2805. doi: 10.4049/jimmunol.1403209. [DOI] [PubMed] [Google Scholar]
- 37.Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52:1390–1400. doi: 10.1002/hep.23795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karlmark KR, et al. The fractalkine receptor CX3CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52:1769–1782. doi: 10.1002/hep.23894. [DOI] [PubMed] [Google Scholar]
- 39.Lu P, et al. Protective roles of the fractalkine/CX3CL1-CX3CR1 interactions in alkali-induced corneal neovascularization through enhanced antiangiogenic factor expression. J. Immunol. 2008;180:4283–4291. doi: 10.4049/jimmunol.180.6.4283. [DOI] [PubMed] [Google Scholar]
- 40.Lumeng CN. Lung Macrophage Diversity and Asthma. Ann. Am. Thorac. Soc. 2016;13(Suppl 1):S31–34. doi: 10.1513/AnnalsATS.201506-384MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitamura T, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015;212:1043–1059. doi: 10.1084/jem.20141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guilliams M, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gibbings SL, et al. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood. 2015;126:1357–1366. doi: 10.1182/blood-2015-01-624809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee YG, et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am. J. Respir. Cell. Mol. Biol. 2015;52:772–784. doi: 10.1165/rcmb.2014-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sica, A. & Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, (2012). [DOI] [PMC free article] [PubMed]
- 46.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 47.Locati M, Mantovani A, Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol. 2013;120:163–84. doi: 10.1016/B978-0-12-417028-5.00006-5. [DOI] [PubMed] [Google Scholar]
- 48.Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013;93:875–81. doi: 10.1189/jlb.1012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Braga TT, Agudelo JS, Camara NO. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front Immunol. 2015;6:602. doi: 10.3389/fimmu.2015.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Movahedi K, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6Chigh monocytes. Cancer Res. 2010;70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 51.Arnold L, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007;204:1057–69. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hinz B, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hinz B, et al. The myofibroblast: one function, multiple origins. Am. J. Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Falk E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006;47:C7–12. doi: 10.1016/j.jacc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 55.Cummins EP, et al. Prolyl hydroxylase-1 negatively regulates IĸB kinase-β, giving insight into hypoxia-induced NFĸB activity. Proc. Natl. Acad. Sci. USA. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kisseleva T, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol. 2006;45:429–38. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 57.Wang CH, et al. Increased circulating fibrocytes in asthma with chronic airflow obstruction. Am. J. Respir. Crit. Care. Med. 2008;178:583–91. doi: 10.1164/rccm.200710-1557OC. [DOI] [PubMed] [Google Scholar]
- 58.Mehrad B, Burdick MD, Strieter RM. Fibrocyte CXCR4 regulation as a therapeutic target in pulmonary fibrosis. Int. J. Biochem. Cell. Biol. 2009;41:1708–18. doi: 10.1016/j.biocel.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niedermeier M, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc. Natl. Acad. Sci. USA. 2009;106:17892–7. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mathai SK, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab. Invest. 2010;90:812–23. doi: 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murray LA, et al. TGF-β driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int. J. Biochem. Cell. Biol. 2011;43:154–62. doi: 10.1016/j.biocel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 62.Nikam VS, et al. Treprostinil inhibits the adhesion and differentiation of fibrocytes via the cyclic adenosine monophosphate-dependent and Ras-proximate protein-dependent inactivation of extracellular regulated kinase. Am. J. Respir. Cell. Mol. Biol. 2011;45:692–703. doi: 10.1165/rcmb.2010-0240OC. [DOI] [PubMed] [Google Scholar]
- 63.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J. Immunol. 2001;166:7556–62. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 64.Moore BB, et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am. J. Pathol. 2005;166:675–84. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huaux F, et al. Role of Eotaxin-1 (CCL11) and CC chemokine receptor 3 (CCR3) in bleomycin-induced lung injury and fibrosis. Am. J. Pathol. 2005;167:1485–1496. doi: 10.1016/S0002-9440(10)61235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernandez EJ, Lolis E. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 2002;42:469–499. doi: 10.1146/annurev.pharmtox.42.091901.115838. [DOI] [PubMed] [Google Scholar]
- 67.Tong N, et al. Neuronal fractalkine expression in HIV-1 encephalitis: roles for macrophage recruitment and neuroprotection in the central nervous system. J. Immunol. 2000;164:1333–1339. doi: 10.4049/jimmunol.164.3.1333. [DOI] [PubMed] [Google Scholar]
- 68.Landsman L, et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113:963–972. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- 69.Ishida Y, et al. Pivotal role of the CCL5/CCR5 interaction for recruitment of endothelial progenitor cells in mouse wound healing. J. Clin. Invest. 2012;122:711–21. doi: 10.1172/JCI43027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that support the findings of this study are available from the corresponding author upon reasonable request.