

Abstract
Although prognostic markers for early estrogen receptor (ER)‐positive breast cancer have been extensively developed, predictive markers for adjuvant endocrine therapy are still lacking. Focusing on the mechanisms underlying endocrine resistance, we investigated whether the endocrine sensitivity of ER‐positive breast cancer cells was correlated with the expression of aspartate‐β‐hydroxylase (ASPH), which is involved in the development of hepatocellular carcinoma. ASPH expression in ER‐positive and tamoxifen‐resistant breast cancer cells was upregulated by the MAPK and phosphoinositide‐3 kinase (PI3K) pathways, which both play pivotal roles in endocrine resistance. In the clinical setting, ASPH expression was negatively correlated with recurrence‐free survival of luminal B breast cancer patients that received adjuvant endocrine therapy, but not in patients that did not receive adjuvant endocrine therapy. Luminal B breast cancer is one of the intrinsic molecular subtypes identified by the Prediction Analysis of Microarray 50 (PAM50) multiple gene classifier, and because of its poor response to endocrine therapy, chemotherapy in addition to endocrine therapy is generally required after surgical resection. Our results suggest that the endocrine sensitivity of luminal B breast cancer can be assessed by examining ASPH expression, which promotes the consideration of a prospective study on the association between ASPH expression at the mRNA and protein levels in luminal B breast cancer and subsequent response to endocrine therapy.
Keywords: Aspartate‐β‐hydroxylase, breast neoplasm, mitogen‐activated protein kinase, phosphatidylinositol 3‐kinase, tamoxifen
Estrogen receptor‐positive breast cancer accounts for 75% of breast cancer cases, and endocrine treatment, including estrogen antagonists, aromatase inhibitors, and luteinizing hormone‐releasing analogues are effective against the disease. Giving postoperative estrogen antagonist tamoxifen for 5 years decreases the recurrence of ER‐positive breast cancer by 39%.1 Giving adjuvant aromatase inhibitor letrozole for 5 years further decreases ER‐positive breast cancer recurrence by 19% over tamoxifen administration.2
ER‐positive breast cancer generally has a favorable prognosis compared with ER‐negative disease, although a portion of patients with ER‐positive disease exhibit poor outcomes. Recent advances in multiple gene expression assays have enabled the classification of ER‐positive breast cancer into favorable and unfavorable prognoses. Representative examples of gene expression assays are Oncotype DX, MammaPrint, and Curebest 95GC,3, 4, 5 which were developed to detect patient subpopulations that do not require adjuvant chemotherapy. Moreover, Prediction Analysis of Microarray 50 (PAM50) which classifies breast cancer into five intrinsic subtypes (luminal A, luminal B, receptor tyrosine protein kinase ERBB2 [HER2]‐enriched, claudin‐low, and basal‐like) based on their biological and histological characteristics,6 is widely used, as each identifiable subtype has a distinct prognosis and distinct therapeutic strategy, and clinical markers, including ER, PR, HER2, and Ki‐67 can be used as surrogate markers to classify breast cancer types.7 Nearly all ER‐positive breast cancer is classified as luminal A or luminal B. Luminal B breast cancer has a worse prognosis than luminal A, and thus patients with luminal B breast cancer are often treated with chemotherapy in addition to endocrine therapy.8, 9
Although prognostic indicators have been extensively studied and developed for breast cancer, prospectively evaluated predictive markers for endocrine therapy are limited. The IMPACT trial prospectively demonstrated that the RFS of patients with ER‐positive breast cancer was negatively correlated with Ki‐67 labeling in tumor tissues treated with neoadjuvant endocrine therapy for 2 weeks.10 However, this approach is not applicable to breast cancer patients who receive only post‐surgical endocrine therapy. Therefore, a predictive marker that can be applied to tumor tissues untreated with endocrine therapy would be beneficial for ER‐positive breast cancer.
Focusing on the mechanism underlying resistance to endocrine therapy, we investigated whether ASPH was a predictive marker for endocrine therapy against ER‐positive breast cancer. ASPH is a transmembrane protein that hydroxylates aspartyl and asparaginyl residues in the epidermal growth factor‐like domains of some proteins, including Notch and Jagged.11 ASPH is upregulated in various cancer types, including breast cancer and HCC. Patients with HCC that express high levels of ASPH exhibit worse post‐surgical outcomes than those with lower levels.12 In HCC cells, upregulation of ASPH is mediated by MAPK and PI3K/Akt pathways.13 Activation of the MAPK and PI3K/Akt pathways by activated receptor tyrosine kinases, including IGF1R and the ERBB family, is a major cause of endocrine resistance, thus reversing the effects of ER inhibition.14 Moreover, activated MAPK and Akt phosphorylate ER, leading to estrogen‐independent activation and transcription of ER‐responsive genes.15, 16 Based on these findings, we hypothesized that ASPH expression could be used to predict the response of ER‐positive breast cancers to endocrine therapy.
Materials and Methods
Cell culture
Cell lines used in the present study were purchased from the American Type Culture Collection (Manassas, VA, USA) except for TamR MCF7 cells, which were generated by culturing the parental MCF7 cell line with 0.1–10 μM 4‐OHT (Sigma‐Aldrich, St Louis, MO, USA) for 1 year. Cells were maintained in DMEM/F12 with 10% FBS (both Sigma‐Aldrich); TamR MCF7 cell media also included 10 μM 4‐OHT. For serum‐free culture, cells grown in six‐well plates were extensively washed with PBS and cultured in HEPES‐buffered DMEM/F12 (EMD‐Millipore, Billerica, MA, USA) for 24 h. Then, cells were stimulated with 100 ng/mL recombinant human IGF1 (R&D Systems, Minneapolis, MN, USA) or 10% FBS in DMEM/F12 for the indicated time periods. Stimulated cells were used for RNA or protein analysis. As indicated, 2 or 20 μM PD98059 (Cell Signaling Technology, Danvers, MA, USA), or 1, 10 or 20 μM LY294002 (Cell Signaling Technology) was added 2 h prior to IGF1/FBS stimulation.
Western blotting
Cells were lysed with RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with phosphatase and protease inhibitor cocktails (Roche Diagnostics, Basel, Switzerland). SDS‐PAGE (10%) and semi‐dry transfer were carried out using standard procedures. Membranes were incubated at 4°C overnight with primary monoclonal antibodies including mouse anti‐ASPH (FB‐50),17 rabbit anti‐β actin (D6A8; Cell Signaling Technology), rabbit anti‐ERK1/2 (137F5; Cell Signaling Technology), rabbit anti‐P‐ERK1/2 (Thr202/Tyr204; D13.14.4E; Cell Signaling Technology), rabbit anti‐Akt (C67E7; Cell Signaling Technology), and rabbit anti‐P‐Akt (Ser473; D9E; Cell Signaling Technology). All primary antibodies were raised against human proteins. Membranes were incubated with HRP‐conjugated anti‐mouse IgG antibody (Jackson ImmunoResearch, West Grove, PA, USA) or anti‐rabbit IgG antibody (Cell Signaling Technology), followed by chemiluminescence detection. Acquired images were processed using ImageJ (1.48v; National Institutes of Health, Bethesda, MD, USA), and ACTB/β actin (D6A8; Cell Signaling Technology) was used as a loading control.
RNA preparation and qRT‐PCR
cDNA (40 μL) was synthesized from 1 μg total RNA prepared using TRIzol reagent (Thermo Fisher Scientific) and a ReverTra Ace kit (Toyobo Life Science, Osaka, Japan). Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and the LightCycler 480 Real‐Time PCR System (Roche Diagnostics) were used for qRT‐PCR in accordance with the manufacturers’ instructions. The real‐time PCR amplification protocol was as follows: (i) 95°C for 10 min for initial denaturation; (ii) 45 cycles of 95°C for 15 s and 60°C for 1 min for amplification. Standards prepared by serial dilution of the cDNA solution containing the most abundant target cDNA were routinely included in each plate. Target mRNA expression was normalized to 18S rRNA expression. The primer sequences used were as follows: 18S‐forward, GGACACGGACAGGATTGACA; 18S‐reverse, ACCCACGGAATCGAGAAAGA; ASPH‐forward, CGCAAATACCCTCAGAGTCC; ASPH‐reverse, GCTCCACGTAGCACCTCATT.
Tamoxifen sensitivity assay
Cells were plated in 96‐well plates at 3000 cells per well in maintenance medium and cultured overnight. Subsequently, the cells were cultured in maintenance medium supplemented with 0–40 μM 4‐OHT for 48 h. The cells were then fixed with 4% paraformaldehyde, stained with Hoechst 33342 (Thermo Fisher Scientific), and counted using an IN Cell Analyzer 6000 (GE Healthcare, Chicago, IL, USA). Multiple samples (N = 6) were evaluated for each condition. IC50 values were calculated using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA).
Analyses of clinical data
KM Plotter (http://kmplot.com/analysis/index.php?p=background) was used to estimate associations between gene expression data and recurrence‐free survival.18 KM Plotter is a Web‐based tool to compare two Kaplan–Meier curves of patient subgroups stratified by the expression levels of a gene of interest that were evaluated with Affymetrix gene expression microarrays including the HG‐U133A, HG‐U133 Plus 2.0, and HG‐U133A 2.0 chips. Multiple gene expression datasets were collected from the Gene Expression Omnibus database. Patients’ clinicopathological backgrounds can be arbitrarily selected. The website was accessed on 20 July 2017, when data derived from 3951 breast cancer patients were available. There are seven Affymetrix probes for ASPH mRNA. We used probe ID 205808_at for the detection of full‐length ASPH transcripts (isoforms a and f), because the other six probes detect full‐length ASPH as well as Humbug and Junctate, two short splice variants of the ASPH gene that are not relevant to cancer progression. Patients were divided into two ASPH expression groups based on a cut‐off of the upper tertile. For validation, we used a previously described19 Affymetrix HG‐U133 Plus 2.0 microarray data set derived from 105 patients with ER‐positive, node‐negative breast cancer who had received adjuvant endocrine therapy but not chemotherapy in our institution. Association of ASPH status with various clinicopathological characteristics were evaluated using Fisher's exact tests. Univariate and multivariate analyses of various clinicopathological characteristics in association with RFS were conducted by the Cox proportional hazard model using JMP Pro 13.0.0 (SAS Institute, Cary, NC, USA). All 105 participants provided informed consent, and the Ethical Review Board of Osaka University Hospital approved the study protocols.
Results
IGF1 mediates ASPH upregulation by the MAPK and PI3K pathways
To examine whether ASPH is upregulated in ER‐positive breast cancer cells after stimulation with IGF1, serum‐starved MCF7 cells were treated with IGF1 for 24 h. Both ASPH transcript (Fig. 1a) and protein (Fig. 1b) were upregulated by IGF1. To examine whether the MAPK and PI3K pathways mediate ASPH upregulation, serum‐starved MCF7 cells were stimulated with IGF1 for 24 h in the presence or absence of the MEK1/2 inhibitor PD9805920 or the PI3K inhibitor LY294002.21 Both PD98059 and LY294002 inhibited upregulation of ASPH mRNA in a dose‐dependent way upon stimulation with IGF1 (Fig. 1c). Western blot analysis of lysates extracted from cells stimulated with IGF1 for 20 min showed that PD98059 and LY294002 specifically inhibited phosphorylation of the MEK1/2 substrate ERK1/2 and the PI3K substrate Akt, respectively (Fig. 1d). These results indicate that the MAPK and PI3K pathways, both of which contribute to endocrine resistance in ER‐positive breast cancer cells, transcriptionally regulate ASPH.
Figure 1.
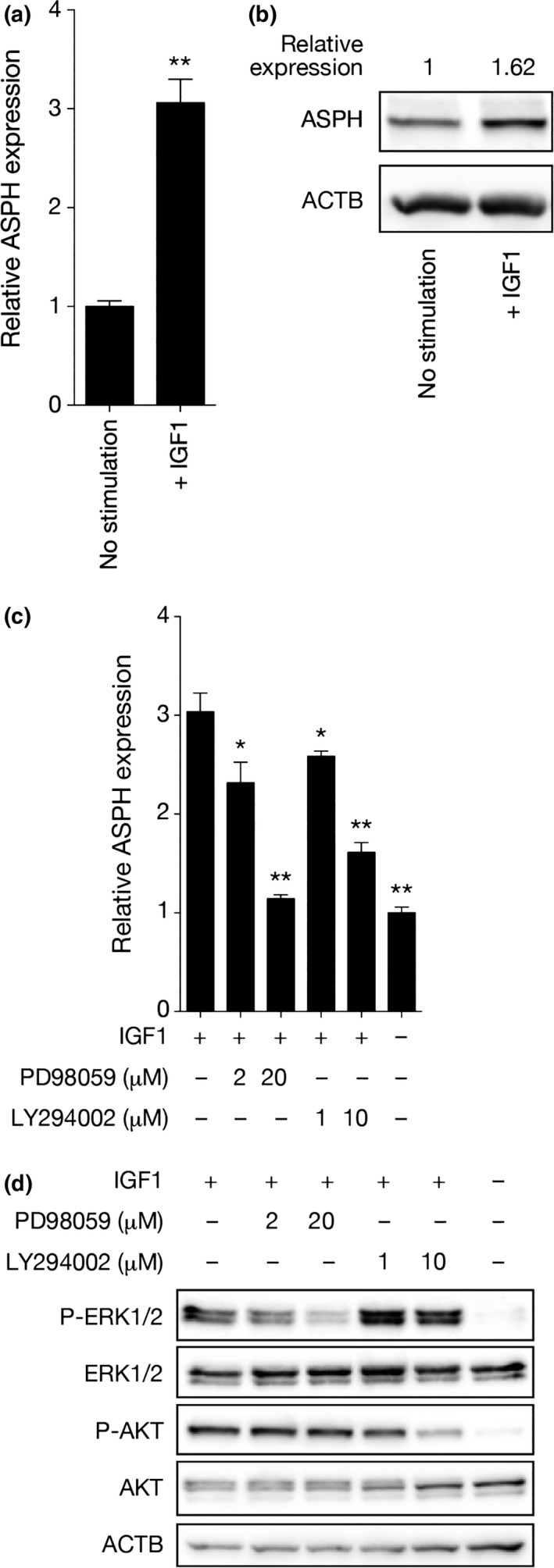
MAPK and PI3K pathways mediate aspartate‐β‐hydroxylase (ASPH) upregulation by insulin‐like growth factor 1 receptor (IGF1). (a) Quantitative RT‐PCR (qRT‐PCR) analysis of ASPH mRNA expression in MCF7 cells stimulated with IGF1 for 24 h. Mean ± standard deviation (SD) of triplicate samples is shown. (b) Western blot analysis of ASPH protein in MCF7 cells stimulated with IGF1 for 24 h. ASPH expression is shown as quantified by densitometry. Values were normalized to β actin (ACTB) expression. (c) qRT‐PCR analysis of ASPH mRNA expression in MCF7 cells stimulated with IGF1 for 24 h in the presence or absence of PD98059 or LY294002. Mean ± SD of triplicate samples is shown. (d) Western blot analysis of phosphorylated ERK1/2 and Akt in MCF7 cells stimulated with IGF1 for 20 min in the presence or absence of PD98059 or LY294002. *P < 0.05, **P < 0.01 (Student's t‐test).
Expression of ASPH is upregulated when tamoxifen sensitivity is reduced
Tamoxifen is the most widely used drug for adjuvant endocrine therapy in ER‐positive breast cancer patients. It is metabolized in the liver to 4‐OHT, which acts as an estrogen antagonist. Using TamR MCF7 cells generated by growing cells with 4‐OHT for 1 year, we compared ASPH expression and ERK1/2 and Akt phosphorylation in TamR and parental MCF7 cells in maintenance medium containing 10% FBS. IC50 values of 4‐OHT in TamR and parental MCF7 cells were >40 and 14.4 μM, respectively, confirming that TamR MCF7 cells were resistant to 4‐OHT (Fig. 2a). Both ASPH mRNA (Fig. 2b) and protein (Fig. 2c) were upregulated in TamR cells compared with the parental line. As expected, increased ERK1/2 and Akt phosphorylation was observed in TamR cells compared to the parental line (Fig. 2c), suggesting greater activation of the MAPK and PI3K pathways in TamR cells compared with parental cells. Next, we examined whether MAPK and PI3K pathways regulated ASPH expression in TamR MCF7 cells. Serum‐deprived cells preincubated with PD98059 and LY294002 for 2 h were stimulated with maintenance medium containing 10% FBS for 6 h, followed by lysate preparation. Similar to parental cells, ASPH transcripts in TamR cells were downregulated by inhibition of MEK1/2 and PI3K (Fig. 2d), suggesting that these pathways mediate ASPH upregulation, although it is unclear whether IGF1 was involved in pathway activation. These results indicate that when the sensitivity to tamoxifen is reduced, ASPH expression is upregulated through the activation of the MAPK and PI3K pathways.
Figure 2.
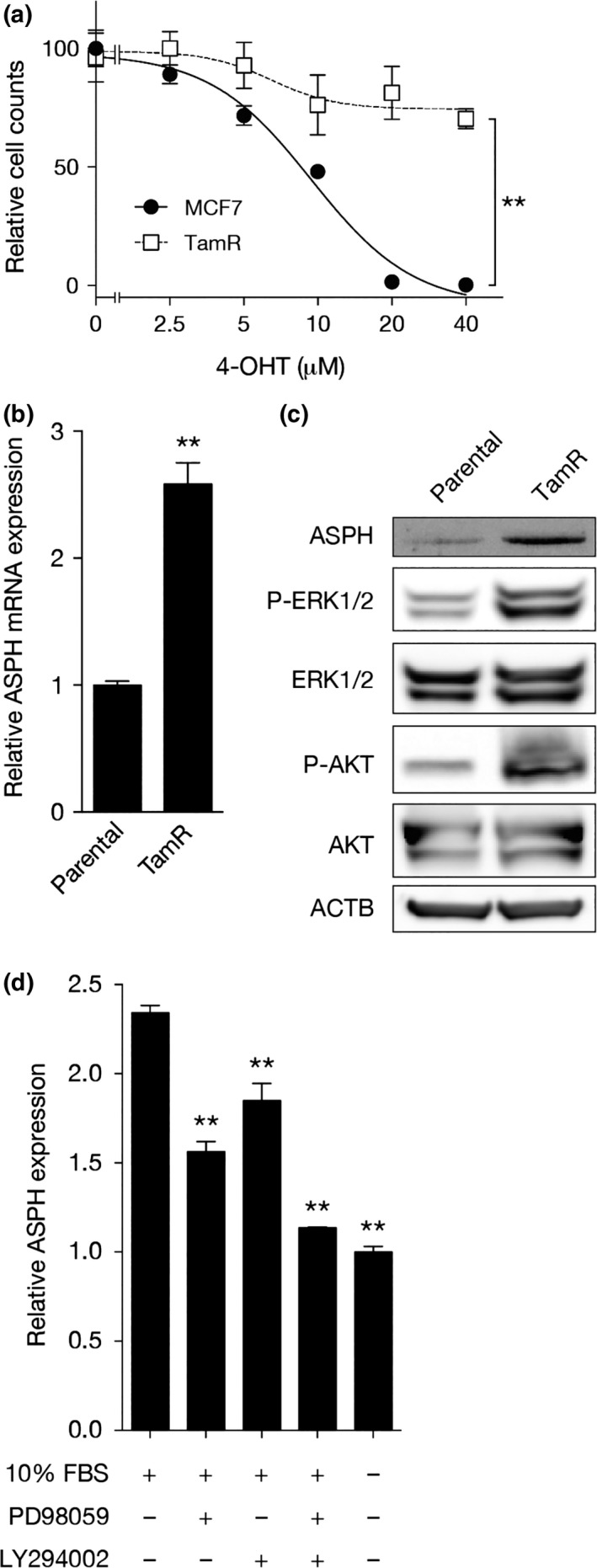
Expression of aspartate‐β‐hydroxylase (ASPH) is upregulated when tamoxifen sensitivity is reduced. (a) Dose–response curves of 4‐hydroxytamoxifen (4‐OHT) in parental (●) and TamR (□) MCF7 cells. The x‐axis is expressed on a logarithmic scale. Mean ± SD of multiple samples (N = 6) is shown. (b) Quantitative RT‐PCR (qRT‐PCR) analysis of ASPH mRNA expression in parental and TamR MCF7 cells grown in maintenance medium. Mean ± SD of triplicate samples is shown. (c) Western blot analysis of ASPH and phosphorylated ERK1/2 and Akt in parental and TamR MCF7 cells grown in maintenance medium. (d) qRT‐PCR analysis of ASPH mRNA expression in MCF7 cells stimulated with maintenance medium for 12 h in the presence or absence of PD98059 or LY294002. Mean ± SD of triplicate samples is shown. **P < 0.01 (Student's t‐test).
Tamoxifen sensitivity of ER‐positive breast cancer cells is negatively correlated with ASPH expression
We examined whether tamoxifen sensitivity of ER‐positive breast cancer cell lines was negatively correlated with ASPH expression. The ASPH transcript was abundant in MDA‐MB‐361 cells, moderately expressed in T47D and ZR75‐1 cells, and minimally expressed in ZR75‐30 and MCF7 cells (Fig. 3a). Tamoxifen sensitivity assays revealed the lowest 4‐OHT IC50 values in MCF7 and ZR75‐30 cells, intermediate values in ZR75‐1 and T47D cells, and the highest IC50 in MDA‐MB‐361 cells (Fig. 3b). Single regression analysis of relative ASPH expression versus 4‐OHT IC50 values showed that these parameters were significantly correlated (R 2 = 0.956, P = 0.004; Fig. 3c). These results indicate that tamoxifen sensitivity of ER‐positive breast cancer cell lines is negatively correlated with ASPH expression.
Figure 3.
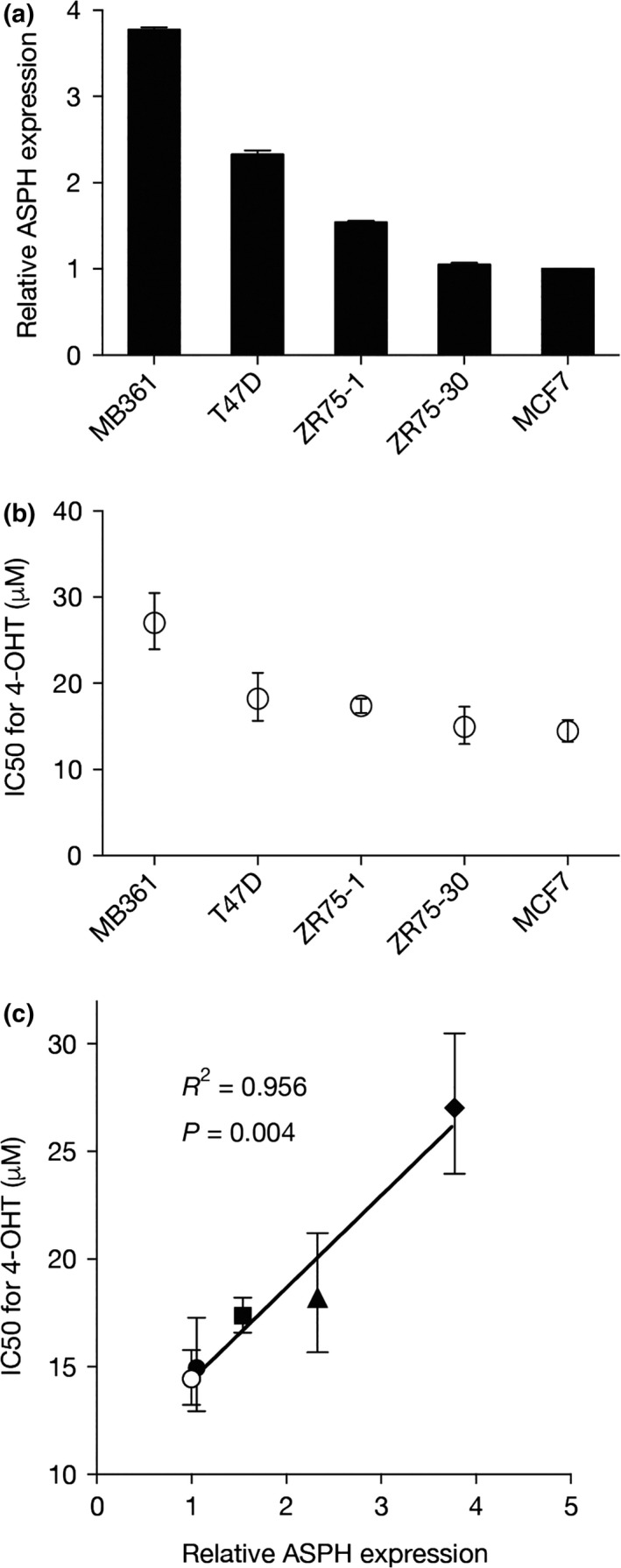
Tamoxifen sensitivity of estrogen receptor (ER)‐positive breast cancer cells is negatively correlated with aspartate‐β‐hydroxylase (ASPH) expression. (a) Quantitative RT‐PCR (qRT‐PCR) analysis of ASPH mRNA expression in five ER‐positive breast cancer cell lines grown in maintenance medium. Mean ± SD of triplicate samples is shown. (b) IC 50 values for 4‐hydroxytamoxifen (4‐OHT) in the five cell lines determined by tamoxifen sensitivity assay. Mean IC 50 values with 95% confidence intervals are shown. (c) Scatter plot of relative ASPH expression versus 4‐OHT IC 50 values. ○, MCF7; ●, ZR75‐30; ■, ZR75‐1; ▲, T47D; ♦, MDA‐MB‐361 (MB361).
Clinical implication of ASPH expression in ER‐positive breast cancer
To extend our experimental findings into the clinical setting, we examined whether ASPH mRNA expression in ER‐positive breast cancer was associated with patient outcome. Multiple data sets curated by Györffy et al.18 were analyzed, which contained Affymetrix global gene expression profiles and survival information derived from 3951 breast cancer cases. To ensure consistency regarding patient background with our in‐house data set curated by Naoi et al.,19 ER‐positive, node‐negative breast cancer patients who had received adjuvant endocrine therapy but not chemotherapy were extracted. No difference in RFS between the ASPH‐high and ASPH‐low groups was observed when all ER‐positive breast cancer patients were analyzed (N = 488, P = 0.14; Fig. 4a). ER‐positive breast cancer can be classified into luminal A and B subtypes using PAM50 gene signatures.6 Clinically, ER‐positive, HER2‐negative, and low Ki‐67 index breast cancer is classified as the luminal A‐like subtype, which shows a good response to endocrine therapy; the remaining ER‐positive breast cancers (ER‐positive, high Ki‐67 index and/or HER2‐positive) fall into the luminal B‐like subtype, which shows a relatively poor response to endocrine therapy.22, 23 As in clinical subtyping, the platform developed by Györffy et al.24 classifies luminal A and luminal B subtypes based on the expression levels of ESR1, MKI67, and ERBB2. There was no difference in RFS between the ASPH‐high and ASPH‐low groups of patients with luminal A‐like breast cancer (N = 347, P = 0.95; Fig. 4b); however, ASPH‐high luminal B‐like breast cancer patients showed significantly poorer outcomes than ASPH‐low luminal B‐like breast cancer patients (N = 132, P = 0.0055; Fig. 4c). Interestingly, there was no difference in RFS between the ASPH‐high and ASPH‐low groups of patients with luminal B‐like breast cancer who had not received adjuvant endocrine therapy and chemotherapy (N = 155, P = 0.34; Fig. 4d), suggesting that ASPH is not a prognostic marker.
Figure 4.
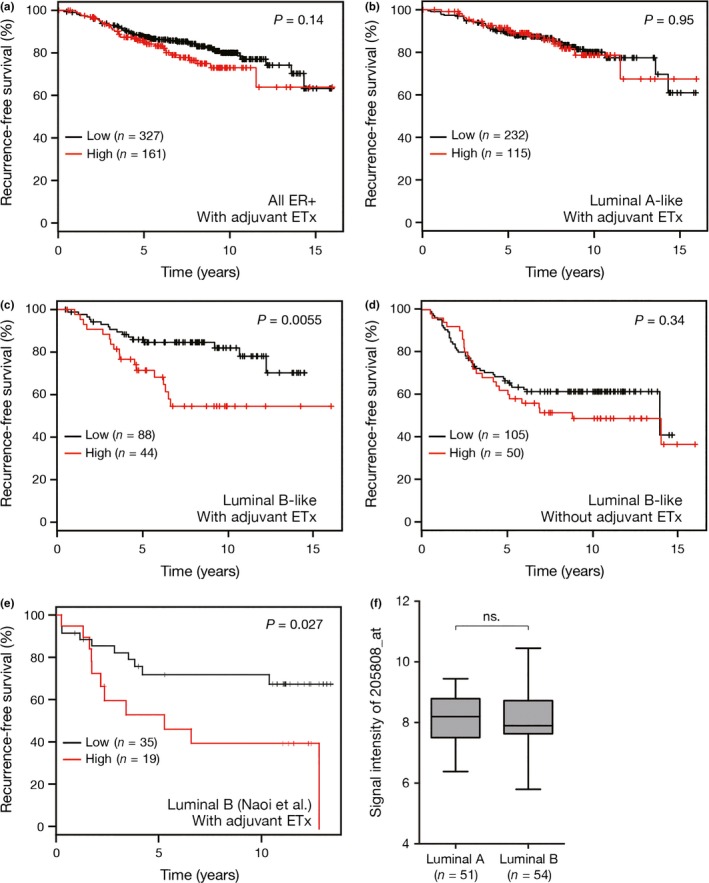
Clinical implications of aspartate‐β‐hydroxylase (ASPH) expression in estrogen receptor (ER)‐positive breast cancer (a–d) recurrence‐free survival (RFS) of patients with ER‐positive, node‐negative breast cancer in multiple data sets curated by Györffy et al.18 All patients who had received chemotherapy were excluded from analysis. (a) RFS of all patients with ER‐positive breast cancer who had received adjuvant endocrine therapy (N = 488). (b) RFS of patients with luminal A‐like (ER‐positive, HER2‐negative, and Ki‐67‐low) breast cancer who had received adjuvant endocrine therapy (N = 347). (c) RFS of patients with luminal B‐like (ER‐positive, Ki‐67‐high and/or HER2‐positive) breast cancer who had received adjuvant endocrine therapy (N = 132). (d) RFS of patients with luminal B‐like breast cancer who had not received adjuvant endocrine therapy (N = 155). (e) RFS of patients with node‐negative luminal B breast cancer who had received adjuvant endocrine therapy but not chemotherapy, using our in‐house data set curated by Naoi et al.19 (N = 54). (f) Boxplots of the signal intensities of the ASPH probe 205808_at in luminal A and luminal B breast cancer. Ends of the whiskers represent minimum and maximum. ns., not significant. P‐values in panels (a–e) were estimated by log–rank tests, and those in panel (f) were estimated by Student's t‐tests.
In general, Internet‐based large expression profiling databases use multiple datasets of different institutions, and the quality of RNA preparation can vary extensively. Thus, our in‐house data set, derived from 105 patients with ER‐positive, node‐negative disease that had received adjuvant endocrine therapy but not chemotherapy, was analyzed for validation. In this cohort, breast cancer subtyping with PAM50 was applied, and 54 patients were classified as luminal B, while the remaining 51 patients were classified as luminal A. As expected, the ASPH‐high group showed a significantly poorer outcome than the ASPH‐low group (P = 0.027; Fig. 4e). ASPH status of the 54 patients with luminal B breast cancer was not associated with any clinicopathological features including menopausal status, tumor size, and histological grade as well as Ki‐67, PR, and HER2 statuses (Table 1). Univariate and multivariate analyses of the clinicopathological characteristics and ASPH status in association with RFS revealed that ASPH status was a factor independently associated with RFS (Table 2). These results suggest that response to adjuvant endocrine therapy is negatively correlated with ASPH expression in luminal B breast cancer. No differences in the signal intensities of the ASPH probe 205808_at were observed between the luminal A and luminal B subtypes (unpaired t‐test P = 0.62; Fig. 4f).
Table 1.
Association of ASPH status with clinicopathological characteristics
| ASPH low (N = 35) | ASPH high (N = 19) | P‐valuea | |
|---|---|---|---|
| Menopausal status | |||
| Premenopausal | 16 | 11 | 0.57 |
| Postmenopausal | 18 | 8 | |
| Unknown | 1 | 0 | |
| T status | |||
| T1 | 17 | 6 | 0.26 |
| T2 or T3 | 18 | 13 | |
| Histological grade | |||
| 1 or 2 | 27 | 15 | 1.0 |
| 3 | 8 | 4 | |
| Ki‐67 | |||
| Low | 25 | 14 | 1.0 |
| High | 10 | 5 | |
| Progesterone receptor | |||
| Negative | 10 | 5 | 1.0 |
| Positive | 25 | 14 | |
| HER2 | |||
| Negative | 29 | 14 | 0.49 |
| Positive | 6 | 5 | |
Fisher's exact test. ASPH, aspartate‐β‐hydroxylase.
Table 2.
Univariate and multivariate analyses of clinicopathological characteristics and ASPH status in association with RFS
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| Menopausal status (post vs pre) | 0.74 | 0.29–1.79 | 0.51 | 0.99 | 0.35–2.71 | 0.98 |
| T status (T1 vs T2 and T3) | 0.53 | 0.20–1.28 | 0.16 | 0.49 | 0.17–1.27 | 0.14 |
| Histological grade (1 and 2 vs 3) | 0.62 | 0.25–1.75 | 0.35 | 0.67 | 0.22–2.31 | 0.51 |
| Ki‐67 (low vs high) | 0.73 | 0.28–2.26 | 0.55 | 1.44 | 0.43–5.70 | 0.57 |
| Progesterone receptor (negative vs positive) | 1.09 | 0.35–2.82 | 0.87 | 1.54 | 0.42–5.27 | 0.50 |
| HER2 (negative vs positive) | 0.31 | 0.12–0.82 | 0.020 | 0.35 | 0.12–1.08 | 0.068 |
| ASPH (low vs high) | 0.40 | 0.16–0.94 | 0.037 | 0.37 | 0.14–0.98 | 0.045 |
ASPH, aspartate‐β‐hydroxylase; CI, confidence interval; HR, hazard ratio; RFS, recurrence‐free survival.
Discussion
In the present study, we demonstrated four findings; first, ASPH expression in ER‐positive breast cancer cells depends on both the MAPK and PI3K pathways. Second, ASPH expression is upregulated in tamoxifen‐resistant breast cancer cells as a result of MAPK and PI3K pathway activation. Third, ASPH expression is negatively correlated with tamoxifen sensitivity in various ER‐positive breast cancer cell lines. Fourth, ASPH expression is negatively correlated with the RFS of luminal B breast cancer patients receiving adjuvant endocrine therapy, but not with the RFS of patients not receiving adjuvant endocrine therapy. These findings suggest that ASPH mRNA levels may be a useful and possibly predictive biomarker of endocrine treatment for luminal B breast cancer. However, a weakness of this study was the lack of quantitative ASPH protein expression in the breast tumor tissue and further studies will be required.
Luminal B breast cancer is one of five intrinsic subtypes classified by multiple gene expression analysis with PAM50.6, 25 Nearly all ER‐positive breast cancer falls into luminal A or luminal B subtypes, and these subtypes are truly distinct populations in terms of clinicopathological and biological features.22, 26 Clinically, luminal A breast cancer responds well to endocrine therapy and has a favorable prognosis. In contrast, luminal B breast cancer has a poorer response to endocrine therapy, requiring adjuvant chemotherapy, and has an unfavorable prognosis.9, 22 Biologically, the luminal B subtype consistently displays upregulation of proliferation‐related genes, including MKI67 (which encodes Ki‐67).7 Signals that drive the proliferation of luminal B breast cancer cells are transduced through several receptors, including IGF1R, ERBB family members, fibroblast growth factor receptor etc. Importantly, most of these signals converge on the MAPK and PI3K pathways in the cytoplasm, resulting in multiple cellular reactions such as proliferation, resistance to cell death, and promotion of metabolism.27, 28 Moreover, both the MAPK and PI3K pathways play pivotal roles in the resistance to endocrine therapy.14, 29, 30, 31 In this study, we confirmed that ASPH is upregulated in tamoxifen‐resistant cells as a result of activation of the MAPK and PI3K pathways. This observation suggests that luminal B breast cancer with upregulation of ASPH may be resistant to endocrine therapy because of MAPK and PI3K pathway activation.
The evidence that luminal B breast cancer is more resistant to endocrine therapy than the luminal A subtype is still inconclusive as a result of insufficient prospective research. The ACOSOG Z1031 trial, in which the association of the preoperative endocrine prognostic index score with luminal subtypes was investigated with neoadjuvant use of endocrine therapy, showed that the luminal B subtype had a significantly higher preoperative endocrine prognostic index score, predicting worse prognosis, than the luminal A subtype. However, differences between the two subtypes in both the responses to endocrine therapy and the rates of breast‐conserving surgery were not significant, although trends towards worse outcomes were observed for luminal B breast cancer.32 Considering that luminal B breast cancer clearly has an unfavorable prognosis compared with the luminal A subtype,8 it is likely that luminal B breast cancer shows a wide spectrum of sensitivity to endocrine therapy. Thus, a predictive marker of response or poor response to adjuvant endocrine therapy for patients with luminal B breast cancer would be valuable. In the present study, we have demonstrated that the outcomes of patients with luminal B breast cancer who received adjuvant endocrine therapy but not chemotherapy was negatively correlated with ASPH expression, whereas no correlation was found for patients who did not receive adjuvant endocrine therapy and chemotherapy. These results clinically support the experimental finding that endocrine sensitivity is negatively associated with ASPH expression in ER‐positive breast cancer cells, and raise the hypothesis that ASPH mRNA expression levels could be used as a predictive marker of adjuvant endocrine therapy for patients with luminal B breast cancer. The reason why ASPH expression is negatively correlated with sensitivity to endocrine therapy in luminal B but not luminal A breast cancer is currently unknown; however, studies have demonstrated that the mechanism regulating ASPH expression may be different between luminal A and luminal B breast cancer, which have been shown to be molecularly distinct subtypes, as described above.22, 26
A high level of ASPH expression in endocrine‐resistant breast cancer cells reflects the activation of the MAPK and PI3K pathways. Thus, one can expect that inactivation of these pathways in endocrine‐resistant breast cancer with high ASPH expression may lead to restoration of endocrine sensitivity. Everolimus is an inhibitor of mTOR, which mediates PI3K/Akt signaling. Everolimus, in addition to exemestane, a steroidal aromatase inhibitor, significantly improves progression‐free survival over exemestane alone in patients with hormone receptor‐positive metastatic breast cancer already resistant to non‐steroidal aromatase inhibitors.33 To date, no biomarkers to predict everolimus efficacy have been established, including PIK3CA mutations. Our findings encourage the evaluation of genes upregulated by the PI3K pathway, such as ASPH, to evaluate whether these genes can predict everolimus efficacy in patients with endocrine‐resistant ER‐positive breast cancer.
In conclusion, our results suggest that ASPH expression correlates with the sensitivity of luminal B breast cancer to adjuvant endocrine therapy, supporting further investigation as to whether ASPH can be used as a predictive marker of response to adjuvant endocrine therapy. If so, it could be used to identify a population of patients with luminal B breast cancer that do not require adjuvant chemotherapy, significantly improving the quality of life of these patients.
Disclosure Statement
Shinzaburo Noguchi has received honoraria from Chugai Pharmaceutical, Novartis Pharmaceuticals, AstraZeneca, and Taiho Pharmaceutical. Shinzaburo Noguchi has received research grants from Chugai Pharmaceutical, Novartis, AstraZeneca, Takeda Pharmaceutical, Pfizer Japan, and Taiho Pharmaceutical. Masafumi Shimoda and Jack R. Wands hold a patent on dendritic cell vaccines for asparaginyl‐β‐hydroxylase‐expressing tumors. All other authors declare no conflicts of interest for this article.
Abbreviations
- 4‐OHT
4‐hydroxytamoxifen
- ASPH
aspartate‐β‐hydroxylase
- ER
estrogen receptor
- HCC
hepatocellular carcinoma
- IGF1R
insulin‐like growth factor 1 receptor
- PAM50
Prediction Analysis of Microarray 50
- PI3K
phosphoinositide‐3 kinase
- PR
progesterone receptor
- qRT‐PCR
quantitative RT‐PCR
- RFS
recurrence‐free survival
- TamR
tamoxifen‐resistant
Acknowledgments
We thank Rolf Carlson for administrative work and Yoko Chihara for help with experiments.
Cancer Sci 108 (2017) 2454–2461
Funding Information
This work was supported in part by a grant‐in‐aid for scientific research (C) from the Japan Society for the Promotion of Science (26461945).
References
- 1. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) , Davies C, Godwin J et al Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient‐level meta‐analysis of randomised trials. Lancet 2011; 378: 771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Regan MM, Neven P, Giobbie‐Hurder A et al Assessment of letrozole and tamoxifen alone and in sequence for postmenopausal women with steroid hormone receptor‐positive breast cancer: the BIG 1‐98 randomised clinical trial at 8.1 years median follow‐up. Lancet Oncol 2011; 12: 1101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paik S, Shak S, Tang G et al A multigene assay to predict recurrence of tamoxifen‐treated, node‐negative breast cancer. N Engl J Med 2004; 351: 2817–26. [DOI] [PubMed] [Google Scholar]
- 4. Cardoso F, van't Veer LJ, Bogaerts J et al MINDACT Investigators: 70‐Gene signature as an aid to treatment decisions in early‐stage breast cancer. N Engl J Med 2016; 375: 717–29. [DOI] [PubMed] [Google Scholar]
- 5. Naoi Y, Kishi K, Tsunashima R et al Comparison of efficacy of 95‐gene and 21‐gene classifier (Oncotype DX) for prediction of recurrence in ER‐positive and node‐negative breast cancer patients. Breast Cancer Res Treat 2013; 140: 299–306. [DOI] [PubMed] [Google Scholar]
- 6. Parker JS, Mullins M, Cheang MC et al Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009; 27: 1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheang MC, Chia SK, Voduc D et al Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009; 101: 736–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gnant M, Filipits M, Greil R et al Predicting distant recurrence in receptor‐positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 postmenopausal patients of the ABCSG‐8 trial treated with adjuvant endocrine therapy alone. Ann Oncol 2014; 25: 339–45. [DOI] [PubMed] [Google Scholar]
- 9. Coates AS, Winer EP, Goldhirsch A et al Tailoring therapies–improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol 2015; 26: 1533–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dowsett M, Smith IE, Ebbs SR et al Short‐term changes in Ki‐67 during neoadjuvant treatment of primary breast cancer with anastrozole or tamoxifen alone or combined correlate with recurrence‐free survival. Clin Cancer Res 2005; 11: 951s–8s. [PubMed] [Google Scholar]
- 11. Cantarini MC, de la Monte SM, Pang M et al Aspartyl‐asparagyl β hydroxylase over‐expression in human hepatoma is linked to activation of insulin‐like growth factor and notch signaling mechanisms. Hepatology 2006; 44: 446–57. [DOI] [PubMed] [Google Scholar]
- 12. Wang K, Liu J, Yan ZL et al Overexpression of aspartyl‐(asparaginyl)‐β‐hydroxylase in hepatocellular carcinoma is associated with worse surgical outcome. Hepatology 2010; 52: 164–73. [DOI] [PubMed] [Google Scholar]
- 13. de la Monte SM, Tamaki S, Cantarini MC et al Aspartyl‐(asparaginyl)‐β‐hydroxylase regulates hepatocellular carcinoma invasiveness. J Hepatol 2006; 44: 971–83. [DOI] [PubMed] [Google Scholar]
- 14. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 2009; 9: 631–43. [DOI] [PubMed] [Google Scholar]
- 15. Kato S, Endoh H, Masuhiro Y et al Activation of the estrogen receptor through phosphorylation by mitogen‐activated protein kinase. Science 1995; 270: 1491–4. [DOI] [PubMed] [Google Scholar]
- 16. Simoncini T, Hafezi‐Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol‐3‐OH kinase. Nature 2000; 407: 538–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lavaissiere L, Jia S, Nishiyama M et al Overexpression of human aspartyl(asparaginyl)β‐hydroxylase in hepatocellular carcinoma and cholangiocarcinoma. J Clin Invest 1996; 98: 1313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Györffy B, Lanczky A, Eklund AC et al An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 2010; 123: 725–31. [DOI] [PubMed] [Google Scholar]
- 19. Naoi Y, Kishi K, Tanei T et al Development of 95‐gene classifier as a powerful predictor of recurrences in node‐negative and ER‐positive breast cancer patients. Breast Cancer Res Treat 2011; 128: 633–41. [DOI] [PubMed] [Google Scholar]
- 20. Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen‐activated protein kinase cascade. Proc Natl Acad Sci USA 1995; 192: 7686–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3‐kinase, 2‐(4‐morpholinyl)‐8‐phenyl‐4H‐1‐benzopyran‐4‐one (LY294002). J Biol Chem 1994; 269: 5241–8. [PubMed] [Google Scholar]
- 22. Ades F, Zardavas D, Bozovic‐Spasojevic I et al Luminal B breast cancer: molecular characterization, clinical management, and future perspectives. J Clin Oncol 2014; 32: 2794–803. [DOI] [PubMed] [Google Scholar]
- 23. Tran B, Bedard PL. Luminal‐B breast cancer and novel therapeutic targets. Breast Cancer Res 2011; 13: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mihály Z, Györffy B. Improving pathological assessment of breast cancer by employing array‐based transcriptome analysis. Microarrays 2013; 2: 228–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perou CM, Sørlie T, Eisen MB et al Molecular portraits of human breast tumours. Nature 2000; 406: 747–52. [DOI] [PubMed] [Google Scholar]
- 26. Prat A, Pineda E, Adamo B et al Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015; 24(Suppl 2): S26–35. [DOI] [PubMed] [Google Scholar]
- 27. Pearson G, Robinson F, Beers Gibson T et al Mitogen‐activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22: 153–83. [DOI] [PubMed] [Google Scholar]
- 28. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129: 1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Osborne CK, Shou J, Massarweh S, Schiff R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 2005; 11: 865s–70s. [PubMed] [Google Scholar]
- 30. Gee JM, Robertson JF, Ellis IO, Nicholson RI. Phosphorylation of ERK1/2 mitogen‐activated protein kinase is associated with poor response to anti‐hormonal therapy and decreased patient survival in clinical breast cancer. Int J Cancer 2001; 95: 247–54. [DOI] [PubMed] [Google Scholar]
- 31. Miller TW, Hennessy BT, González‐Angulo AM et al Hyperactivation of phosphatidylinositol‐3 kinase promotes escape from hormone dependence in estrogen receptor‐positive human breast cancer. J Clin Invest 2010; 120: 2406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ellis MJ, Suman VJ, Hoog J et al Randomized phase II neoadjuvant comparison between letrozole, anastrozole, and exemestane for postmenopausal women with estrogen receptor‐rich stage 2 to 3 breast cancer: clinical and biomarker outcomes and predictive value of the baseline PAM50‐based intrinsic subtype–ACOSOG Z1031. J Clin Oncol 2011; 29: 2342–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baselga J, Campone M, Piccart M et al Everolimus in postmenopausal hormone‐receptor‐positive advanced breast cancer. N Engl J Med 2012; 366: 520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]