

Abstract
We investigated the combinatorial effects of pentoxifylline (PTX) on the efficacy of gemcitabine (GEM) in a human pancreatic tumor xenograft model. PTX significantly improved the efficacy of GEM, as shown by a 50% reduction in tumor growth rate at 4 weeks of treatment compared with that in animals given GEM alone. The fluorescent drug doxorubicin (DOX) was used to test whether drug delivery was improved by PTX, contributing to the improved efficacy of GEM. PTX given for 2 weeks prior to giving DOX improved drug distribution by 1.8‐ to 2.2‐fold with no changes in vessel density, suggesting that improvement in drug delivery was not related to the vascular mechanism. Instead, collagen I content in tumor stroma was significantly reduced, as was the expression of alpha‐smooth muscle actin of cancer‐associated fibroblasts and connective tissue growth factor (CTGF) by PTX pretreatment. Overall, our data demonstrated that increased efficacy of GEM by PTX was associated with improved drug delivery to tumor tissue, which may be attributed to decreased expression of CTGF and subsequent reduction in the stromal collagen matrix in the pancreatic ductal adenocarcinoma tumor. These results support the usefulness of PTX in combination with chemotherapy for targeting drug delivery barriers associated with the stromal matrix, which should be further evaluated for clinical development.
Keywords: Connective tissue growth factor, desmoplasia, gemcitabine, pancreatic ductal adenocarcinoma, pentoxifylline
Pancreatic ductal adenocarcinoma is the fourth leading cause of cancer‐related deaths worldwide, with an overall 5‐year survival rate of approximately 5%.1 GEM, a nucleoside analog of cytidine, has been used as the current standard treatment in patients with PDAC, but only extends median survival by a few weeks.2 Numerous GEM‐based combination trials with cytotoxic drugs and/or biological targeted agents have been carried out; however, positive results have not been achieved, likely owing to intrinsic drug resistance, a unique feature of PDAC.3 Such chemoresistance is a major problem in the treatment of human solid tumors, including PDAC.
Studies have shown that the ECM within the tumor microenvironment plays an important role in altering the drug sensitivity of solid cancers.4, 5 Poor drug penetration is considered a type of EMDR as it is attributed to the dense fibrotic stroma (desmoplasia) generated by dysregulation of the ECM and hypovascularity in solid tumor tissues,6, 7 both of which are hallmark features of PDAC.9 Type I collagen, a main ECM component, is known to contribute to chemoresistance by acting as a drug penetration barrier10 and by inducing adhesion‐mediated activation of various prosurvival pathways.11 Depleting the stromal collagen matrix may therefore be a potentially effective method for controlling drug resistance by improving drug delivery to tumor tissues and by modulating tumor microenvironmental factors that can reduce the drug sensitivity of cancer cells. Several approaches have been reported for targeting stromal collagen matrix. CAF are important stromal components that augment the production of stromal ECM; hence, inhibition of collagen synthesis by CAF has been attempted by blockade of pathways associated with TGF‐β, CTGF, and sonic hedgehog signaling.12, 13, 14 ECM‐degrading enzymes, including hyaluronidase and collagenase, are alternative approaches used to modulate EMDR.15, 16, 17
Pentoxifylline (PTX), a non‐selective phosphodiesterase inhibitor with hemorrheological and immunomodulating properties, has been available for over 30 years for the treatment of intermittent claudication in peripheral vascular disease.18 The antifibrotic effects of PTX have been reported for radiation‐induced fibrosis in human patients and for liver and peritoneal fibrosis in rat models.19, 20, 21 PTX has been reported to have conventional sensitizing effects when combined with conventional anticancer agents and radiation in patients and preclinical models.18 However, potential antitumor effects of PTX have not been evaluated in relation to the antifibrotic effects.
Here, we reported the synergistic antitumor activity of PTX when combined with GEM in a human pancreatic tumor xenograft model for the first time. The synergistic effects of PTX were attributable to improved drug distribution in tumor tissue, as demonstrated using the autofluorescent drug DOX. Reduced stromal collagen network and inhibition of CAF activity were confirmed. Interference with CTGF (also known as CCN2) signaling seemed to be involved in this ECM remodeling effect of PTX. Overall, our study provides a basis for further evaluation of PTX in combination with chemotherapeutics and studies on the underlying mechanisms of the synergy related to EMDR in stroma‐rich fibrotic tumors, such as PDAC.
Materials and Methods
Cell lines and chemical reagents
The human pancreatic cancer cell line, Capan‐1, was obtained from the Korea Cell Line Bank (Seoul, Korea). Cells were maintained in RPMI‐1640 medium (Gibco BRL, Grand Island, NY, USA) supplemented with 100 μg/mL streptomycin, 100 units/mL penicillin (Sigma Aldrich, St Louis, MO, USA) and 10% heat‐inactivated FBS (Welgene, Seoul, Korea). PTX powder was kindly provided by Handok Pharmaceutical. Co. Ltd (Seoul, Korea). PTX was dissolved in sterile, pyrogen‐free normal saline and then filtered through a 0.2‐μm‐pore‐size filter (Corning Costar, New York, NY, USA) before use. DOX and GEM were a kind gift from Ildong Pharmaceutical. Co. Ltd and Boryung Pharmaceutical. Co. Ltd (Seoul, Korea), respectively. Liposomal formulation of DOX (LP‐DOX) was prepared as described in a previous study.22 Primary antibodies for immunohistochemistry were purchased from Abcam (Cambridge, UK) unless otherwise indicated.
Xenograft experiment
Female BALB/c nu/nu mice (5–6 weeks old) were obtained from Orient Bio Inc. (Seongnam‐si, Gyeonggi‐do, Korea). Animal care and experimental protocol was in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of College of Medicine, The Catholic University of Korea (CUMC‐2010‐0141‐01). For tumor induction, 5 × 106 viable Capan‐1 cells were injected s.c. into the right flank of mice. Drug treatment was started when tumor size reached approximately 200 mm3. For the combination study, PTX and GEM were given i.p. at 100 mg/kg per day and 50 mg/kg twice a week, respectively, for 4 weeks. For the combination of PTX and DOX (or LP‐DOX), PTX was given i.p. at 100 mg/kg per day for 2 weeks prior to i.v. administration of DOX and LP‐DOX at 8 mg/kg by tail vein once. For the drug distribution experiment, a higher dose of 20 mg/kg as DOX was given after PTX pretreatment, and tumor tissue was harvested after 5 min (DOX) or 24 h (LP‐DOX) when the maximum drug distribution was expected in each group. Excised tumors were divided into several pieces and were either fixed overnight in 10% formalin solution for immunohistochemistry or embedded in OCT compound (Sakura, Tissue‐Tek®, Torrance, CA, USA) for immunofluorescence staining. For biochemical analysis, some tumor pieces were snap‐frozen and stored at −70°C until analysis.
Immunofluorescence detection and imaging
For examination of the tissue distribution of DOX, tissue samples were snap‐frozen using OCT compound, cut into 10 μm cryosections, and examined under an inverted fluorescence microscope (Axiovert 200M; Carl Zeiss AG, Oberkochen, Germany) at λEx/Em = 540/580 nm. Tissue sections were then stained for CD31 and type I collagen using conventional procedures. Briefly, sections were fixed in cold acetone for 10 min and non‐specific binding was blocked with 5% normal goat serum for 1 h. Primary antibodies of a rabbit anti‐collagen I (1:200, Ab292, anti‐human/mouse) and a rat anti‐CD31 (1:100, anti‐mouse; BD Pharmingen, San Diego, CA, USA) were applied, at 4°C in a humidified chamber overnight. Visualization was done with secondary antibodies of Alexa‐488‐conjugated goat anti‐rabbit IgG (1:100; Invitrogen, Carlsbad, CA, USA) or Cy3‐conjugated goat anti‐rat IgG (1:100; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Image overlay was done to evaluate the relative distribution of drug and blood vessels using Media Cybernetics Image Pro PLUS (version 5.0).
Immunohistochemistry
Detection of proteins, α‐SMA, TGF‐β1, CTGF and MT1‐MMP, were carried out using respective primary antibody and Dako EnVision™ Detection System (K5007; DAKO, Glostrup, Denmark). Briefly, paraffin‐embedded samples were cut into 3‐μm‐thick sections, deparaffinized and rehydrated. After antigen retrieval using Target Retrieval Solution (S2375; DAKO), non‐specific binding was blocked with 10% normal goat serum for 1 h. Sections were then incubated with primary antibodies against α‐SMA (1:200), TGF‐β1 (1:200), CTGF (1:400) and MT1‐MMP (1:40) in dilution buffer overnight at 4°C in a humidified chamber. Slides were counterstained with hematoxylin and examined under a microscope (Olympus AX70; Olympus, Tokyo, Japan). For quantitative analysis, % area of stained region was calculated using image analysis software OPTIMAS version 6.5 (Media Cybernetics, Silver Spring, MD, USA).
ELISA for TGF‐β1 in tumor tissue
Frozen tumors were cut into small pieces and homogenized using Precellys 24 homogenizer (Bertin Technologies, Rockville, MD, USA) in Tissue Extraction Reagent I (Invitrogen) containing protease inhibitor cocktail (Complete Mini; Roche, Basel, Switzerland). Supernatant of the homogenate was obtained by centrifugation and stored at −70°C until analysis. Total and active TGF‐β1 concentrations were determined using TGF‐β1 ELISA kit (eBioscience, Vienna, Austria) according to the manufacturer's protocol and total protein concentration using BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA).
Quantitative real‐time PCR analysis
Total RNA was isolated from tumor homogenates using Trizol. After 1 μg total RNA was reverse transcribed with AccuPower CycleScript RT PreMix (Bioneer, Daejeon, Korea), quantitative real‐time PCR was carried out using the LightCycler 480 Real‐Time PCR System II (Roche) with SYBR Green Master mix (Roche) according to the manufacturer's recommendation. Primers for mouse GAPDH, human GAPDH, mouse α‐SMA, mouse TGF‐β1, mouse CTGF, mouse COL1A1 (type I collagen, alpha 1) and human MT1‐MMP were as follows: mouse GAPDH primers, forward 5′‐TGCTGAGTATGTCGTGGAGTCTA‐3′, reverse 5′‐AGTGGGAGTTGCTGTTGAAG‐TC‐3′; human GAPDH primers, forward 5′‐CCACCCATGGCAAATTCCATGGCA‐3′, reverse 5′‐TCTAGACGGCAGGTCAGGTCCACC‐3′; mouse α‐SMA primers, forward 5′‐ACTGGGACGACATGGAAAAG‐3′, reverse 5′‐CATCTCCAGAGTCCAGCACA‐3′; mouse TGF‐β1 primers, forward 5′‐CAACAATTCCTGGCGTTACCTTGG‐3′, reverse 5′‐GAAAGCCCTGTATTCCGTCTCCTT‐3′; mouse CTGF primers, forward 5′‐TCCCGAGAAGGGTCAAGCT‐3′, reverse 5′‐TCCTTGGGCTCGTCACACA‐3′; mouse COL1A1 primers, forward 5′‐GAGCGGAGAGTACTGGATCG‐3′, reverse 5′‐TACTCGAACGGGAATCCATC‐3′. All expression data were normalized using mouse or human GAPDH expression. Quantitation of relative gene expression was carried out using the ΔΔC t method (2−ΔΔCt).
Statistical analysis
Three to six mice per group were used in all experiments and data were expressed as mean ± standard error (SE). Statistical significance tests were carried out using ANOVA with LSD or t‐test.
Results
Combination of PTX and GEM synergistically inhibited tumor growth
The growth inhibitory effects of GEM in combination with PTX were examined in nude mice bearing Capan‐1 xenografts. Capan‐1 tumor showed rapid growth with a tumor volume doubling time (TD) of 10 days in the control group, and no changes in tumor growth were observed in the group given PTX alone (Fig. 1a). Tumor growth delay was observed in mice given GEM treatment (50 mg/kg i.p. twice a week for 4 weeks), i.e., 4‐ and 7‐day delays in the time to reach tumor volumes of 400 and 600 mm3, respectively. When PTX (100 mg/kg, i.p., daily) was added to GEM treatment, a strong synergistic interaction between the two drugs was observed as tumor growth was inhibited by 50% at 4 weeks post‐treatment (P < 0.05 at day 21, 24 and 28, Fig. 1a). Notably, no toxicity was noted, as observed by the lack of significant changes in bodyweights in all treatment groups (Fig. 1b). These results indicated that low‐dose PTX significantly enhanced the antitumor effects of GEM against the growth of Capan‐1 xenograft tumors without exerting its own antitumor effects.
Figure 1.
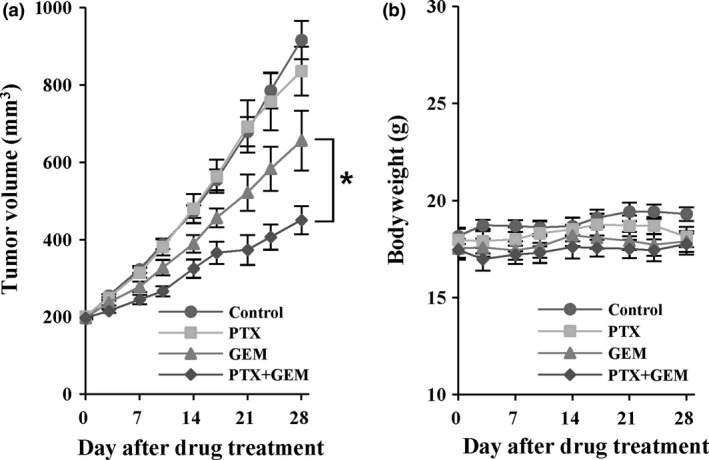
Combination treatment with pentoxifylline (PTX) and gemcitabine (GEM) in Capan‐1 xenografts. Mice harboring Capan‐1 xenografts (n = 7) were randomized to receive PTX (100 mg/kg per day, i.p.), GEM (50 mg/kg, i.p., twice a week), i.p., twice a week), or a combination of PTX plus GEM for 4 weeks. (a) Mean tumor volume in each group was compared. (b) Mean bodyweight of each group. Data are expressed as means ± standard errors. *P < 0.05 vs GEM group.
PTX pretreatment improved the distribution and efficacy of DOX and LP‐DOX
In order to investigate the synergistic effects of PTX, changes in drug distribution in the tumor tissue were evaluated using DOX as a model drug owing to the ease of fluorescent signal detection. Mice harboring Capan‐1 xenografts were pretreated with PTX for 2 weeks (100 mg/kg, i.p., daily) and then given a bolus dose of DOX in solution or in a liposomal formulation (LP‐DOX; Fig. 2a). Mice were either killed right after DOX dosing for evaluation of drug distribution or further monitored for tumor growth delay. No significant differences in tumor growth rates were observed among the treatment groups, except for the combination group, until 3 weeks of treatment. Similar to the results of the first experiment, PTX alone did not have antitumor effects. Significant changes in tumor growth curves were noted in mice pretreated with PTX between 18 and 27 days (P < 0.05), e.g. a 23.7% decrease on day 23 (1.52‐fold tumor growth for DOX only group vs 1.16‐fold for PTX plus DOX group; Fig. 2b). Mice given LP‐DOX also showed increased inhibition of tumor growth at days 25, 27 and 29 (P < 0.05), e.g. a 31.1% decrease in relative tumor volume on day 29 (1.74‐fold tumor growth for LP‐DOX group vs 1.39‐fold for PTX plus LP‐DOX group; Fig. 2c). Although the antitumor effects of a single dose of DOX and LP‐DOX were transient and tumor growth resumed thereafter, improved efficacy was obvious when combined with PTX pretreatment, whereas DOX or PTX alone did not affect tumor growth. No toxicity was noted either during 14 days of PTX pretreatment or for the remainder of the days following a one‐time, single dose of DOX or LP‐DOX (data not shown).
Figure 2.
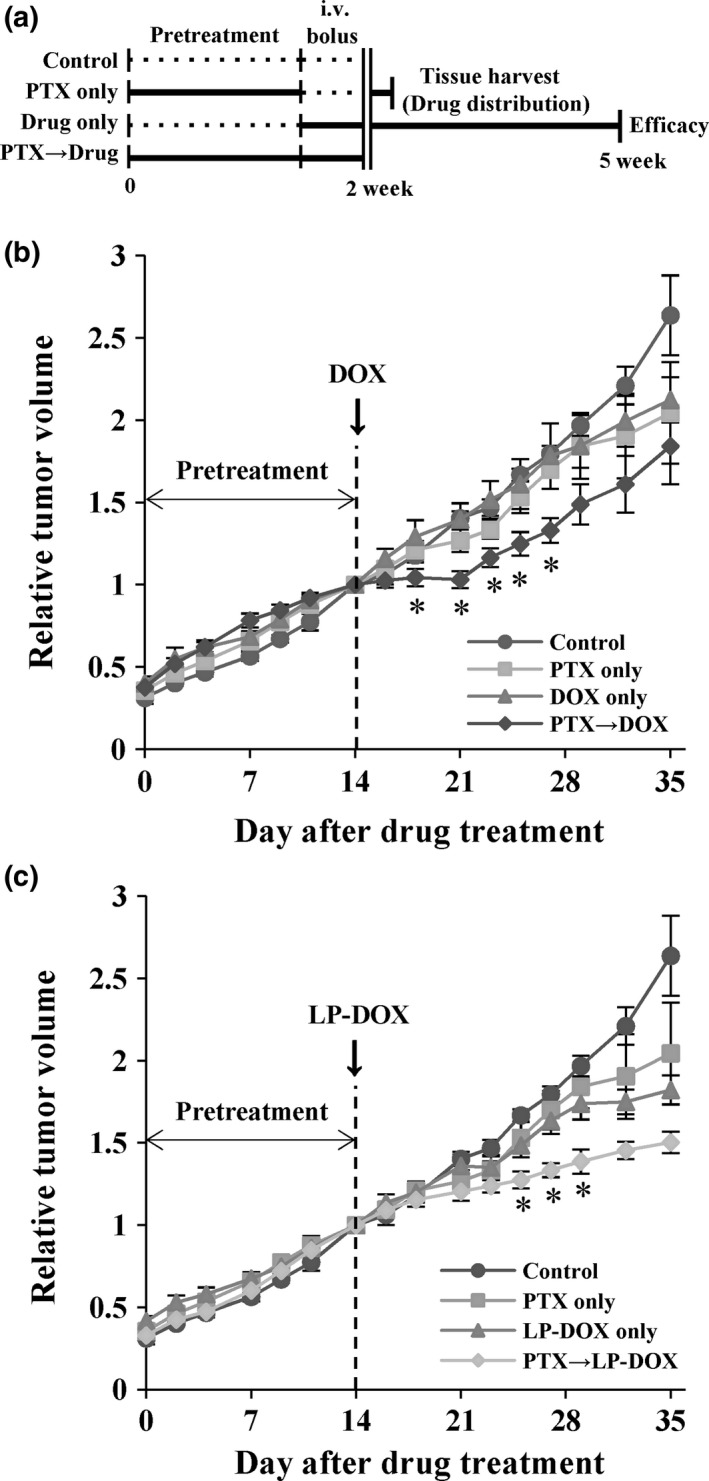
Effects of pentoxifylline (PTX) pretreatment on doxorubicin (DOX) and liposomal formulation of DOX (LP‐DOX) in Capan‐1 xenografts. (a) Treatment schedule for the combination of PTX with either DOX or LP‐DOX. Comparison of tumor growth (n = 3–5) for the combination with (b) DOX or (C) LP‐DOX. PTX was given i.p. at 100 mg/kg per day for 2 weeks followed by a dose of DOX and LP‐DOX i.v. at 8 mg/kg. Relative tumor volume was expressed as a ratio to the tumor volume at day 14. Data are expressed as means ± standard errors. *P < 0.05 vs DOX only or LP‐DOX only.
When given without PTX pretreatment either as DOX solution or LP‐DOX, DOX showed a limited distribution in images showing blood vessels (represented by CD31 staining) and DOX autofluorescence (Fig. 3a). When quantified as the percent area of DOX fluorescence in tumor sections, improved drug distribution was observed in a region farther away from blood vessels by 2.2‐ and 1.8‐fold for the DOX and LP‐DOX groups with PTX pretreatment, respectively (P < 0.001; Fig. 3b). This improved drug distribution was not attributed to new vessel formation as CD31 immunostaining showed no differences between groups (Fig. 3b).
Figure 3.
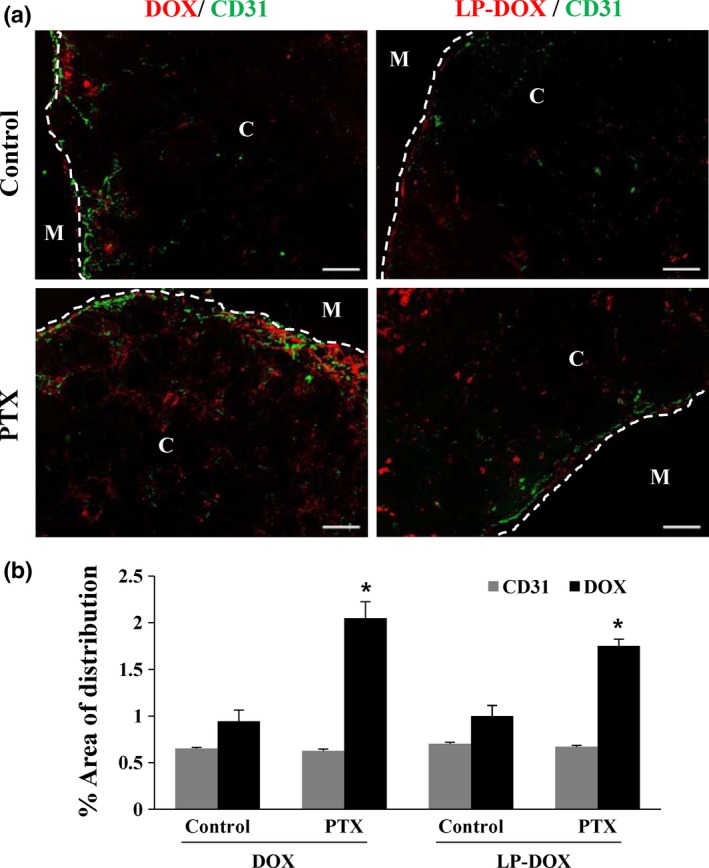
Effects of pentoxifylline (PTX) on the distributions of doxorubicin (DOX) and liposomal formulation of DOX (LP‐DOX) in Capan‐1 xenografts. (a) Distribution of DOX (red) was evaluated in CD31‐positive vessels (green) in frozen tumor sections (n = 3). PTX was given i.p. at 100 mg/kg per day for 2 weeks followed by a dose of DOX or LP‐DOX i.v. at 20 mg/kg. C and M indicate central and marginal regions in tumor sections, respectively (50× magnification, scale bar, 200 μm). (b) Percent distribution area of DOX and CD31 staining. Data are expressed as means ± standard errors. *P < 0.001 vs the control.
PTX reduced type I collagen contents in tumor tissue
The ECM has been shown to play an important role as a barrier to drug distribution in solid tumors; thus, we next examined the effects of PTX pretreatment on type I collagen content. Type I collagen content was reduced by PTX treatment. That is, collagen fibers showed a sparse network in both the central and marginal regions of tumors after PTX treatment for 2 weeks (Fig. 4a). Treatment with a high dose of PTX (100 mg/kg) resulted in a greater reduction in collagen content than that with a lower dose (50 mg/kg; P < 0.05; Fig. 4b), suggesting a concentration‐dependent activity. Change in collagen expression was confirmed at the mRNA level, with a 72% decrease in collagen expression compared with that in the control group (P < 0.05; Fig. 4c). These results clearly indicated that PTX induced collagen matrix reduction, which may be involved in the increased DOX distribution observed in Figures 2 and 3 and the increased efficacy of GEM shown in Figure 1.
Figure 4.

Effects of pentoxifylline (PTX) on the distribution of type I collagen in Capan‐1 xenografts. Mice carrying Capan‐1 xenografts (n = 3) received PTX at 50 or 100 mg/kg per day for 2 weeks. (a) Immunofluorescence staining of type I collagen in marginal and central regions of frozen sections of tumors. Arrows indicate regions showing decreased collagen content. (b) Percent area of collagen distribution. (c) mRNA expression of type I collagen, as assessed by real‐time RT‐PCR. All expressed data were normalized using mouse GAPDH. Data are expressed as means ± standard errors. *P < 0.05, **P < 0.001 vs the control (50 × magnification, scale bar: 200 μm).
PTX inhibited expression of profibrotic growth factors in tumors
We examined changes in the expression of profibrotic growth factors, such as TGF‐β1 and CTGF, and the fibroblast marker α‐SMA. PTX pretreatment significantly reduced the expression levels of α‐SMA and CTGF by 66% (P < 0.001) and 25% (P < 0.05), respectively, as determined by IHC (Fig. 5a). Consistent with these findings, analysis of mRNA expression using quantitative real‐time RT‐PCR showed that α‐SMA and CTGF expression levels decreased by 77% and 50%, respectively (P < 0.05; Fig. 5b). For TGF‐β1, neither IHC nor ELISA showed any differences in expression following PTX pretreatment (Fig. 5a,c), although mRNA expression level was significantly different (Fig. 5b). These results indicated that the antifibrotic effects of PTX resulting in reduced collagen matrix may be attributed mainly to decreased expression of CTGF, which was closely related to the reduced activity of CAF, a main cell type responsible for CTGF production, in Capan‐1 xenografts.
Figure 5.
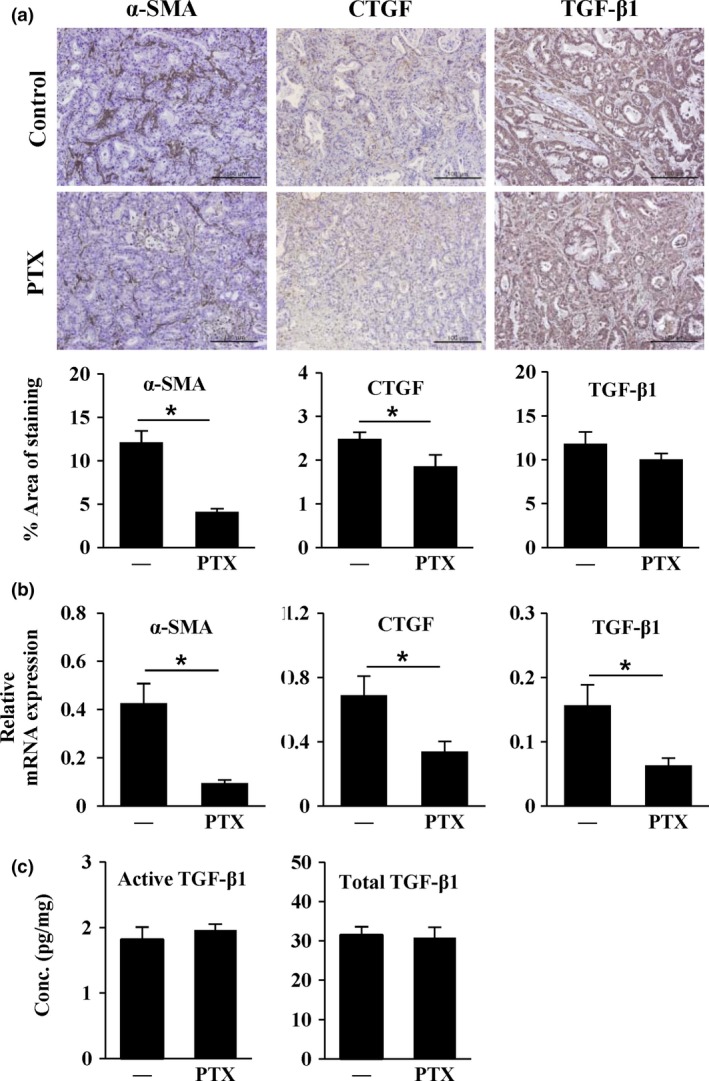
Pentoxifylline (PTX) decreased the expression of fibrosis‐related proteins in Capan‐1 xenografts. Mice harboring Capan‐1 xenografts (n = 3) received PTX 100 mg/kg per day for 2 weeks. (a) Immunohistochemical staining of alpha‐smooth muscle actin (α‐SMA), connective tissue growth factor (CTGF), and transforming growth factor beta (TGF‐β)1 in paraffin sections of tumors. (b) mRNA expression of α‐SMA, CTGF, and TGF‐β1, as assessed by real‐time RT‐PCR. All expressed data were normalized using mouse GAPDH. (c) Active and total TGF‐β1 levels (pg/mg) measured by ELISA in tumor homogenates. —, control group. Data are expressed as means ± standard errors. *P < 0.05 vs the control (200× magnification, scale bar, 100 μm).
PTX did not influence the expression of MT1‐MMP, a GEM resistance factor
MT1‐MMP has been reported to have a role in GEM resistance in a type I collagen‐dependent way in PDAC.10 Hence, in the present study, we examined the possibility that reduced expression of type I collagen following PTX pretreatment may result in increased GEM sensitivity by suppression of MT1‐MMP expression. MT1‐MMP expression, however, did not show any significant changes at either the protein or mRNA level following 2 weeks of PTX treatment (Fig. 6). This result indicated that the mechanism underlying our observations of the synergy between PTX and GEM was associated with collagen network reduction by an MT1‐MMP‐independent pathway.
Figure 6.
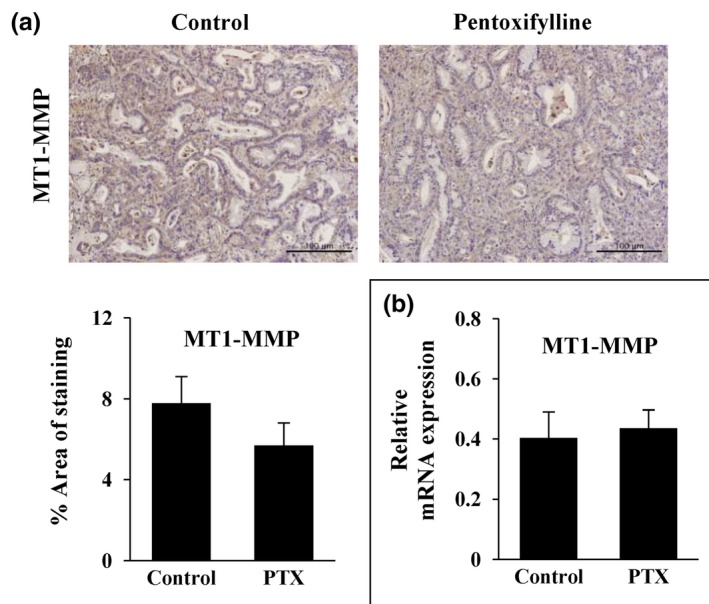
Effects of pentoxifylline (PTX) on the expression of membrane type I matrix metalloproteinase (MT1‐MMP) in Capan‐1 xenografts. Mice harboring Capan‐1 xenografts (n = 3) were given PTX 100 mg/kg per day for 2 weeks. (a) Immunohistochemical staining of MT1‐MMP in paraffin‐embedded tumor sections. (b) Expression of MT1‐MMP mRNA, as assessed by real‐time RT‐PCR. All expressed data were normalized using mouse GAPDH. Data are expressed as means ± standard errors (200× magnification, scale bar, 100 μm).
Discussion
Modulation of intracellular mechanisms involved in desmoplasia has been suggested as a promising strategy for improving drug delivery (pharmacokinetics).23, 24 Strategies to target desmoplasia in PDAC can be implemented either by disintegration of the ECM network using direct enzymatic digestion16 or by modulation of signaling pathways (e.g. TGF‐β or sonic hedgehog pathways) involved in the proliferation or activation of CAF, major players in ECM deposition.13 The potent antifibrotic effects of PTX (e.g. inhibition of proliferation or ECM synthesis in human renal and lung fibroblasts25, 26 and peritoneal mesothelial cells27) have been reported in several in vitro studies. Moreover, the antifibrotic effects of PTX have also been demonstrated using in vivo models of glomerulonephritis,28 biliary fibrosis,29 and liver fibrosis.30 In the present study, we reported, for the first time, the synergistic effects of GEM and PTX and suggest improved drug distribution induced by dysregulation of the ECM network as an underlying mechanism for the observed synergism.
Our study showed that PTX exerted synergistic antitumor effects when combined with GEM or DOX in a Capan‐1 xenograft model (Figs 1 and 2). Similar synergistic interaction with GEM or DOX was reported for several other agents having antifibrotic activities, including halofuginone,31 losartan,13, 32 and hedgehog signaling inhibitors;12, 33 however, the dose levels used were high enough for these compounds to exhibit their own antitumor activities. Another substantial difference was that giving PTX was started during later phases of tumor growth (when the tumor volume had already reached ~200 mm3) rather than during an early stage of tumor growth, which supports the clinical relevancy of our data. Notably, the synergistic interactions observed in the present study were induced at a low dose of 100 mg/kg PTX, which did not induce either toxicity or its own inhibitory effects on tumor growth (Fig. 1). In addition, potential protective effects of PTX towards DOX‐induced side‐effects have been reported, based on the capacity of PTX to sequester aromatic compounds (DOX) in staking complexes.34, 35
In the present study, we tested the hypothesis that the synergistic interaction between PTX and GEM (Fig. 1a) may be a result of improved drug distribution in tumors (pharmacokinetic modulation). We used the autofluorescent drug DOX for this purpose, as we expect that tissue distribution would be similar to that of DOX based on the similar physicochemical properties of these agents, including molecular size (DOX, 543 Da; GEM, 263 Da) and hydrophilicity. Following PTX pretreatment, significant improvement was observed in tissue distribution (Fig. 3) as well as efficacy of DOX (Fig. 2) without changes in vessel density or distribution (CD31‐stained region). These data implied that giving PTX improved antitumor efficacy of DOX as well as GEM by increased drug delivery into tumor tissue by a non‐vasculature‐related mechanism. Nonetheless, vascular involvement may not be ruled out completely because functional improvement of perfusion in the tumor was not determined in the present study.36
We also evaluated the effect of PTX pretreatment on a liposomal formulation (LP‐DOX) that is often considered to provide an advantage in tumoral drug delivery. PTX improved to a similar extent the efficacy and tissue distribution of DOX given either in solution or liposomes (Figs 2 and 3), indicating no additional improvement by liposomal encapsulation of DOX. We speculated that PTX rather improved the distribution of drug released from liposomes, but not the distribution of the liposome itself. The therapeutic advantage of adding antifibrotic drugs to liposomal formulations, however, warrants further evaluation as DOX and LP‐DOX was given as a single dose to exhibit a transient inhibition of tumor growth.
Desmoplasia induced by overproduction of ECM or deregulation of its composition and structure is commonly found in many types of solid tumors. Collagen content and fibrous network creating solid stress in tumors establish a barrier for interstitial drug penetration. ECM deregulation is also involved in creating abnormally high IFP, which acts as a stromal barrier to convective movement of drug molecules out of the vasculature into avascular regions of the tumor parenchyma.3, 8 PTX pretreatment resulted in a significant reduction in type I collagen expression, as shown by measurement of both protein and mRNA levels (Fig. 4), supporting that PTX alleviated ECM‐mediated solid stress to mediate the synergistic interaction with GEM (Fig. 1a). High tumor cell density (tumor hyperplasia) may also create an interstitial barrier for penetration of drug molecules.35 Cell killing by GEM itself may therefore have augmented the reduction in solid stress, synergistically improving drug (GEM) delivery. Other antifibrotic agents, such as halofuginone, losartan, and the hedgehog inhibitor IPI‐926, also demonstrated that their synergistic interactions with various chemotherapeutic agents involved modulation of drug distribution.12, 13, 31, 32, 33 Interestingly, CAF are known to contribute to ECM production significantly more than cancer cells; for example, in a previous study, approximately 76% and 24% of type I collagen content showed host (mouse) and human origin, respectively, in Panc‐2 xenograft tumors.31 Although α‐SMA‐positive CAF were present in a relatively small area of the tumor (as small as 10% of tumor area) (Fig. 5a), reduced α‐SMA expression (decreased activity of CAF) could be associated with a significant decrease in type I collagen content (Fig. 4). The reduced expression of α‐SMA was associated with decreased profibrotic activity of CAF rather than a direct cell kill effect. It can be supported by lack of growth inhibition during PTX treatment (Figs 1 and 2) and minimal cytotoxicity (cell density) seen on the paraffin sections of tumors (Figs 5a and 6a).
TGF‐β1 is generally considered a primary main regulator of the desmoplastic reaction in PDAC.37 A previous report demonstrated that losartan inhibited type I collagen synthesis by blocking TGF‐β signaling in CAF, resulting in improved distribution and efficacy of Doxil in desmoplastic tumors, such as human breast cancer, pancreatic cancer, and murine skin tumors.13 Similar to losartan, PTX also inhibits TGF‐β1 signaling both in vitro and in vivo.( 28, 29, 30, 36 ) In the present study, however, changes in TGF‐β1 expression following PTX treatment were observed only at the mRNA level, not at the protein level. Instead, significant changes in CTGF protein and mRNA expression were induced following PTX treatment in Capan‐1 xenograft tumors (Fig. 5). CTGF derived from tumor cells has also been shown to be a critical regulator of tumor growth. Moreover, CTGF is another potent contributor to the desmoplastic reaction and mediates many of the downstream activities of TGF‐β1 towards overproduction of ECM.38 Recent evidence also suggests that PTX has inhibitory effects on CTGF expression by a Smad3/4‐dependent pathway in renal fibroblasts.39 In PDAC, not only tumor cells but also CAF are suggested to be a major source of CTGF based on studies using in vitro models as well as human tumor samples.40, 41 Moreover, overall, a growing body of evidence has demonstrated that CTGF derived from CAF plays important roles in tumor development, progression, and metastasis,38, 42 and our data showing that PTX may target CTGF‐producing CAF provide a basis for further studies of the potential benefits of the use of PTX in combination with chemotherapies against desmoplastic human PDAC.
Besides improved drug delivery, impairment of the collagen network may also contribute to the increased efficacy of GEM by other mechanisms involved in GEM resistance. Collagen I and IV are known to promote progression and metastasis in PDAC.14, 43, 44 Type I collagen has been shown to be associated with GEM resistance in PDAC models through upregulation of MT1‐MMP on the membrane.11, 45 In the collagen‐rich microenvironment, MT1‐MMP activates TGF‐β1 bound to ECM, promoting TGF‐β1‐mediated cell proliferation by both ERK 1/2 signaling and high mobility group A2 (HMGA2) expression. In the present study, no changes were observed in MT1‐MMP levels after PTX treatment (Fig. 6), thus suggesting that this protein was not involved in the synergistic interaction between PTX and GEM. Further studies are needed to investigate the existence of other pharmacodynamic mechanisms for PTX synergy.
In the present study, we demonstrated that giving PTX induced a synergistic interaction with GEM in a pancreatic tumor xenograft model. This synergy was attributed to decreased collagen expression and improved drug distribution in tumor tissues. Reduced collagen levels were found to be associated with reduced numbers of activated fibroblasts in tumor tissue (Fig. 7). Targeting ECM accumulation and/or ECM‐producing components in tumors may provide a promising strategy to modulate chemotherapeutic efficacy by improving drug distribution in tumor tissues in highly desmoplastic tumors, such as PDAC.46, 47 Evaluation of antifibrotic drugs for their synergistic interaction should be further validated in genetically engineered mouse models before clinical evaluation.48 Tumor models in vitro, such as 3D culture of cancer cells with myofibroblast creating desmoplastic tumor tissue, may serve as an efficient tool for screening of antifibrotic agents, which warrants further studies.49
Figure 7.
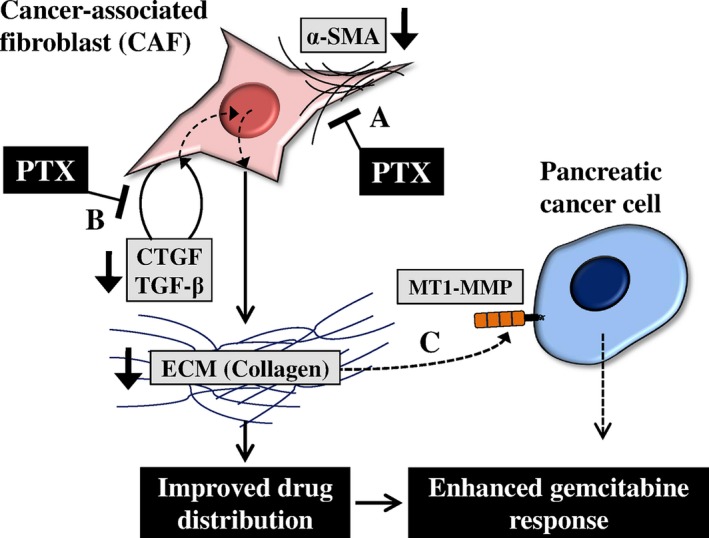
Schematic representation of the effects of pentoxifylline (PTX) on the gemcitabine (GEM) response. In pancreatic ductal adenocarcinoma (PDAC), cancer‐associated fibroblasts (CAF) produce a large amount of collagen, one of the key ECM components. Dense ECM in tumors impairs drug distribution in tissues resulting in attenuation of the GEM response. Our results showed that (a) PTX may inhibit the proliferation or recruitment of CAF, as demonstrated by decreased alpha‐smooth muscle actin (α‐SMA), a marker of CAF (Fig. 5). (b) PTX also inhibited the expression of connective tissue growth factor (CTGF) and transforming growth factor beta (TGF‐β) from CAF, thereby blocking collagen deposition (Fig. 5). Therefore, PTX decreased collagen content and improved drug distribution in tumors, resulting in enhancement of the GEM response. However, (c) PTX did not affect the expression of membrane type I matrix metalloproteinase (MT1‐MMP), a GEM resistance marker induced by collagen in tumors (Fig. 6).
Disclosure Statement
Authors declare no conflicts of interest for this article.
Abbreviations
- CAF
cancer‐associated fibroblast
- CTGF
connective tissue growth factor
- DOX
doxorubicin
- EMDR
environment‐mediated drug resistance
- GEM
gemcitabine
- IFP
interstitial fluid pressure
- IHC
immunohistochemistry
- MT1‐MMP
membrane type I matrix metalloproteinase
- PDAC
pancreatic ductal adenocarcinoma
- PTX
pentoxifylline
- α‐SMA
alpha‐smooth muscle actin
- TGF‐β
transforming growth factor beta
Acknowledgments
The authors thank Dr Wonhee Hur and Dr Mi‐La Cho for helpful discussions. This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korea government (grant nos. 2016R1A2B2011832, 2012R1A5A2047939).
Cancer Sci 108 (2017) 2470–2477
Funding Information
National Research Foundation of Korea (2016R1A2B2011832, 2012R1A5A2047939).
References
- 1. Hariharan D, Saied A, Kocher HM. Analysis of mortality rates for pancreatic cancer across the world. HPB 2008; 10: 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burris HA III, Moore MJ, Andersen J et al Improvements in survival and clinical benefit with gemcitabine as first‐line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997; 15: 2403–13. [DOI] [PubMed] [Google Scholar]
- 3. Sultana A, Ghaneh P, Cunningham D, Starling N, Neoptolemos JP, Smith CT. Gemcitabine based combination chemotherapy in advanced pancreatic cancer‐indirect comparison. BMC Cancer 2008; 8: 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu X, Zhang Z. Understanding the genetic mechanisms of cancer drug resistance using genomic approaches. Trends Genet 2016; 32: 127–37. [DOI] [PubMed] [Google Scholar]
- 5. Holle AW, Young JL, Spatz JP. In vitro cancer cell‐ECM interactions inform in vivo cancer treatment. Adv Drug Deliv Rev 2016; 97: 270–9. [DOI] [PubMed] [Google Scholar]
- 6. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer 2006; 6: 583–92. [DOI] [PubMed] [Google Scholar]
- 7. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007; 99: 1441–54. [DOI] [PubMed] [Google Scholar]
- 8. Khawar IA, Kim JH, Kuh HJ. Improving drug delivery to solid tumors: priming the tumor microenvironment. J Control Release 2015; 201: 78–89. [DOI] [PubMed] [Google Scholar]
- 9. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res 2012; 18: 4266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res 2000; 60: 2497–503. [PubMed] [Google Scholar]
- 11. Dangi‐Garimella S, Krantz SB, Barron MR et al Three‐dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1‐MMP‐mediated expression of HMGA2. Cancer Res 2011; 71: 1019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Olive KP, Jacobetz MA, Davidson CJ et al Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324: 1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diop‐Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci USA 2011; 108: 2909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Neesse A, Frese KK, Bapiro TE et al CTGF antagonism with mAb FG‐3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc Natl Acad Sci USA 2013; 110: 12325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Magzoub M, Jin S, Verkman AS. Enhanced macromolecule diffusion deep in tumors after enzymatic digestion of extracellular matrix collagen and its associated proteoglycan decorin. FASEB J 2008; 22: 276–84. [DOI] [PubMed] [Google Scholar]
- 16. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 21: 418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jacobetz MA, Chan DS, Neesse A et al Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013; 62: 112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nieder C, Zimmermann FB, Adam M, Molls M. The role of pentoxifylline as a modifier of radiation therapy. Cancer Treat Rev 2005; 31: 448–55. [DOI] [PubMed] [Google Scholar]
- 19. Delanian S, Porcher R, Balla‐Mekias S, Lefaix JL. Randomized, placebo‐controlled trial of combined pentoxifylline and tocopherol for regression of superficial radiation‐induced fibrosis. J Clin Oncol 2003; 21: 2545–50. [DOI] [PubMed] [Google Scholar]
- 20. Fang CC, Huang JW, Shyu RS et al Fibrin‐induced epithelial‐to‐mesenchymal transition of peritoneal mesothelial cells as a mechanism of peritoneal fibrosis: effects of pentoxifylline. PLoS ONE 2012; 7: e44765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Movassaghi S, Nadia Sharifi Z, Mohammadzadeh F, Soleimani M. Pentoxifylline protects the rat liver against fibrosis and apoptosis induced by acute administration of 3,4‐methylenedioxymethamphetamine (MDMA or ecstasy). Iran J Basic Med Sci 2013; 16: 922–7. [PMC free article] [PubMed] [Google Scholar]
- 22. Han HD, Lee A, Hwang T, Song CK, Seong H, Hyun J, Shin BC. Enhanced circulation time and antitumor activity of doxorubicin by comblike polymer‐incorporated liposomes. J Control Release 2007; 120: 161–8. [DOI] [PubMed] [Google Scholar]
- 23. Schober M, Jesenofsky R, Faissner R et al Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014; 6: 2137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J, Wientjes MG, Au JL. Pancreatic cancer: pathobiology, treatment options, and drug delivery. AAPS J 2010; 12: 223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Strutz F, Heeg M, Kochsiek T, Siemers G, Zeisberg M, Muller GA. Effects of pentoxifylline, pentifylline and gamma‐interferon on proliferation, differentiation, and matrix synthesis of human renal fibroblasts. Nephrol Dial Transplant 2000; 15: 1535–46. [DOI] [PubMed] [Google Scholar]
- 26. Lee IK, Choi YJ, Shim I, Kim K‐S, Choi CJ. Pentoxifylline induces lipolysis and apoptosis of human preadipocytes, keratinocytes and fibroblasts in vitro . Biomol Ther 2010; 18: 56–64. [Google Scholar]
- 27. Fang CC, Yen CJ, Chen YM et al Pentoxifylline inhibits human peritoneal mesothelial cell growth and collagen synthesis: effects on TGF‐beta. Kidney Int 2000; 57: 2626–33. [DOI] [PubMed] [Google Scholar]
- 28. Ng YY, Chen YM, Tsai TJ, Lan XR, Yang WC, Lan HY. Pentoxifylline inhibits transforming growth factor‐beta signaling and renal fibrosis in experimental crescentic glomerulonephritis in rats. Am J Nephrol 2009; 29: 43–53. [DOI] [PubMed] [Google Scholar]
- 29. Raetsch C, Jia JD, Boigk G et al Pentoxifylline downregulates profibrogenic cytokines and procollagen I expression in rat secondary biliary fibrosis. Gut 2002; 50: 241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong LJ, Zhu JF, Luo DD, Zen LL, Cai SQ. Effects of pentoxifylline on the hepatic content of TGF‐beta1 and collagen in Schistosomiasis japonica mice with liver fibrosis. World J Gastroenterol 2003; 9: 152–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spector I, Zilberstein Y, Lavy A, Nagler A, Genin O, Pines M. Involvement of host stroma cells and tissue fibrosis in pancreatic tumor development in transgenic mice. PLoS ONE 2012; 7: e41833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noguchi R, Yoshiji H, Ikenaka Y et al Synergistic inhibitory effect of gemcitabine and angiotensin type‐1 receptor blocker, losartan, on murine pancreatic tumor growth via anti‐angiogenic activities. Oncol Rep 2009; 22: 355–60. [PubMed] [Google Scholar]
- 33. Bahra M, Kamphues C, Boas‐Knoop S et al Combination of hedgehog signaling blockage and chemotherapy leads to tumor reduction in pancreatic adenocarcinomas. Pancreas 2012; 41: 222–9. [DOI] [PubMed] [Google Scholar]
- 34. Golunski G, Borowik A, Wyrzykowski D, Woziwodzka A, Piosik J. Pentoxifylline as a modulator of anticancer drug doxorubicin. Part I: reduction of doxorubicin DNA binding. Chem Biol Interact 2015; 242: 291–8. [DOI] [PubMed] [Google Scholar]
- 35. Golunski G, Borowik A, Derewonko N et al Pentoxifylline as a modulator of anticancer drug doxorubicin. Part II: reduction of doxorubicin DNA binding and alleviation of its biological effects. Biochimie 2016; 123: 95–102. [DOI] [PubMed] [Google Scholar]
- 36. Mendes JB, Campos PP, Rocha MA, Andrade SP. Cilostazol and pentoxifylline decrease angiogenesis, inflammation, and fibrosis in sponge‐induced intraperitoneal adhesion in mice. Life Sci 2009; 84: 537–43. [DOI] [PubMed] [Google Scholar]
- 37. Lohr M, Schmidt C, Ringel J et al Transforming growth factor‐beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res 2001; 61: 550–5. [PubMed] [Google Scholar]
- 38. Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012; 5(Suppl 1): S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin SL, Chen RH, Chen YM et al Pentoxifylline attenuates tubulointerstitial fibrosis by blocking Smad3/4‐activated transcription and profibrogenic effects of connective tissue growth factor. J Am Soc Nephrol 2005; 16: 2702–13. [DOI] [PubMed] [Google Scholar]
- 40. Bennewith KL, Huang X, Ham CM et al The role of tumor cell‐derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res 2009; 69: 775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hartel M, Di Mola FF, Gardini A et al Desmoplastic reaction influences pancreatic cancer growth behavior. World J Surg 2004; 28: 818–25. [DOI] [PubMed] [Google Scholar]
- 42. Chu CY, Chang CC, Prakash E, Kuo ML. Connective tissue growth factor (CTGF) and cancer progression. J Biomed Sci 2008; 15: 675–85. [DOI] [PubMed] [Google Scholar]
- 43. Ohlund D, Franklin O, Lundberg E, Lundin C, Sund M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer 2013; 13: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Armstrong T, Packham G, Murphy LB et al Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin Cancer Res 2004; 10: 7427–37. [DOI] [PubMed] [Google Scholar]
- 45. Shields MA, Dangi‐Garimella S, Redig AJ, Munshi HG. Biochemical role of the collagen‐rich tumour microenvironment in pancreatic cancer progression. Biochem J 2012; 441: 541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meng H, Zhao Y, Dong J et al Two‐wave nanotherapy to target the stroma and optimize gemcitabine delivery to a human pancreatic cancer model in mice. ACS Nano 2013; 7: 10048–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choi JY, Thapa RK, Yong CS, Kim JO. Nanoparticle‐based combination drug delivery systems for synergistic cancer treatment. J Pharm Investig 2016; 46: 325–39. [Google Scholar]
- 48. Guerra C, Barbacid M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol Oncol 2013; 7: 232–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ware MJ, Keshishian V, Law JJ et al Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials 2016; 108: 129–42. [DOI] [PMC free article] [PubMed] [Google Scholar]