

Abstract
The Proteomics Standards Initiative (PSI) of the Human Proteome Organization (HUPO) has now been developing and promoting open community standards and software tools in the field of proteomics for 15 years. Under the guidance of the chair, cochairs, and other leadership positions, the PSI working groups are tasked with the development and maintenance of community standards via special workshops and ongoing work. Among the existing ratified standards, the PSI working groups continue to update PSI-MI XML, MITAB, mzML, mzIdentML, mzQuantML, mzTab, and the MIAPE (Minimum Information About a Proteomics Experiment) guidelines with the advance of new technologies and techniques. Furthermore, new standards are currently either in the final stages of completion (proBed and proBAM for proteogenomics results as well as PEFF) or in early stages of design (a spectral library standard format, a universal spectrum identifier, the qcML quality control format, and the Protein Expression Interface (PROXI) web services Application Programming Interface). In this work we review the current status of all of these aspects of the PSI, describe synergies with other efforts such as the ProteomeXchange Consortium, the Human Proteome Project, and the metabolomics community, and provide a look at future directions of the PSI.
Keywords: data standard, database, mass spectrometry, proteomics, metabolomics, protein identification, protein quantification, molecular interactions, bioinformatics software, quality control
Introduction
Application of proteomics technologies to identify proteins present in biological samples, measure their abundances, understand their functions, and determine their molecular interaction partners has become a common component of studies of complex biological systems in health and disease. As modern data acquisition instruments generate ever larger data sets, computational software becomes increasingly important in extracting knowledge from the data. The community has developed hundreds of software packages for the analysis of the various types of proteomics data, in the form of both commercial software as well as free and open-source software.
However, the burgeoning array of available software has created difficulties in sharing and comparing data and results among collaborations as well as making data publicly available for the greater community. To foster interoperation of software tools and encourage dissemination of data, common data formats and guidelines are needed. The commonly used data formats in proteomics1 can come in the form of proprietary formats defined by a single group or company where use is restricted or open formats where widespread use is unrestricted and generally encouraged. Open formats may often also come in the form of de facto standards, generally developed for one software tool but eventually used by many tools on account of their utility or ratified standards, which are developed in collaboration by many groups with the aim of creating something that will meet the needs of the entire community, foster the sharing of data, and ultimately accelerate progress in the field.
Since its inception in 2002 as an initiative of the Human Proteome Organization2 (HUPO), the Proteomics Standards Initiative (PSI)3,4 has brought together all participants interested in developing community standards for proteomics. The primary deliverables of the PSI are standardized minimum information specifications, data formats, controlled vocabularies5 (CVs), software tools, and community interaction. Wide participation ensures that the standards that are produced are broadly applicable to the wide variety of workflows practiced by the many different groups in the community. The formats and guidelines thus developed are subjected to a formal procedure to ensure that these products are of high quality, called the PSI Document Process,6 which is an iterative process similar to the review of journal articles. At the conclusion of the Document Process, the formats or guidelines are ratified as a formal PSI standard, and widespread implementation in software is promoted and encouraged.
We provide an update on the state of the PSI in its 15th year of operation. This includes details of the operation of the PSI, including a report from the recent 2017 PSI Spring Workshop (Beijing, China, April 2017). We describe the current state of the existing PSI standards, some of which are nearly 10 years old but continue to evolve with the advancement of proteomics technologies. We then briefly report on a series of new formats that are in the final process of ratification or are still in earlier stages of development. We finish with a look to the future activities and directions of the PSI.
Operation of the PSI
The general operation of the PSI has been described in detail in a previous review.4 In brief, the PSI is led by the chair, who is assisted by two cochairs. They are assisted by additional officers such as the editors, secretary, and others. As of April 2017, the leadership has changed such that Andy Jones serves as chair and Sandra Orchard and Eric Deutsch serve as cochairs. The full listing of PSI officers is provided at http://www.psidev.info/roles. The PSI is governed by a charter that is available at http://www.psidev.info/about.
The development and maintenance of the standards is performed within the PSI working groups. Each working group is led by a chair and one or two cochairs, along with other positions to assist them. The combination of all PSI officers plus the working group chairs and cochairs are collectively known as the Steering Group, which meets, discusses, and votes on matters that affect the operation of the PSI.
Each working group refreshes and submits to the Steering Group a new or refreshed charter each year. These charters describe the leadership and planned activities for the next year. Working groups that do not muster a charter are declared inactive and removed from the table of PSI working groups until such a time when there is sufficient interest to submit a new charter. New working groups may form at any time by submitting a charter to the Steering Group. Since the last review of the PSI in 2014, the Protein Modifications Working Group and the Separations Working Group have become inactive. However, the new Quality Control Working Group has formed, focusing on standards that promote the dissemination of quality control assessment information, including the formalization of the qcML format,7 described further below. The Molecular Interactions Working Group, Mass Spectrometry Working Group, and Proteomics Informatics (PI) Working Group continue active operations. A full listing of all working groups and their leadership is provided at http://www.psidev.info/roles.
The PSI defines standards via a formal, systematic, and transparent workflow called the PSI Document Process6 (http://www.psidev.info/psi-doc-process). A submission to the document process is handled by the PSI Editors. The defined process path depends on the type of document. In the past, mainly two types of PSI documents have been created: (1) “MIAPE documents” (Minimum Information About a Proteomics Experiment), containing the minimal set of information that should be captured about an experiment or analysis to enable its results to be clearly interpreted and validated, and (2) “Recommendation Documents”, which actually specify a technical standard format and its use. For both types of documents a short review phase by the PSI Steering Group is performed to check the document structure and appropriateness of the standard proposed. Then, the actual comprehensive community review phase is performed, requesting comments from the community and from two to three invited reviewers. A manuscript for a journal article is often submitted in parallel, and the comments from the journal review are also taken into account. After a potential revision based on the comments received, a final open community review period is held, during which some additional implementations are sought. After any final comments are addressed, the specification is officially ratified as a formal standard with a version number.
Although activities among the working groups continue all year via e-mail, conference calls, meet-ups at conferences, and collaborative software development environments (http://github.com/HUPO-PSI and https://github.com/MICommunity), the PSI Spring Workshop is the major yearly event that brings the Steering Group and many members together face-to-face. The typically three-day Spring Workshop includes plenary sessions as well as parallel working group tracks that focus on extended discussions of particular topics, making consensus design decisions and dividing the documentation tasks among those present. These workshops typically also provide fresh momentum for progress and greater participation. These workshops are held in a different region each year, and an effort is made to promote extensive local participation with the aim of gathering new long-term membership.
2017 PSI Spring Workshop
The 2017 PSI Spring Workshop was held in Beijing, China at the National Center for Protein Sciences, Beijing (Phoenix Center). The workshop was chaired by Eric Deutsch and the local organizer, Henning Hermjakob. The opening session featured presentations by researchers who have implemented the standards and formats developed by the PSI to further their research work or tool development. Henry Lam (Hong Kong University of Science and Technology) described his work developing applications using spectral libraries in proteomics. In his talk, Lam argued that the process of spectral library building can be improved by first clustering by similarity and then selecting high-quality and confidently identified clusters to be included in the library. Although clustering spectra by similarity is more computationally intensive than simply grouping spectra by identifications, this process is a more robust mechanism for quality control and the detection of errors in spectral libraries. Finally, given the important role of spectral libraries and archives as an information hub and shared community resource, he challenged the PSI to develop a standardized data interchange format, which is currently sorely lacking. Such a standardized format will greatly simplify software development and enable better integration and interoperability of workflows involving spectral libraries and archives.
Jun Qin, Director of the Phoenix Center, updated the attendees on the status of proteomic sciences in China. The Phoenix Center groups have achieved fast proteome sequencing, although there is still an issue with low abundance proteins, which first need to be enriched in a sample. The institute is developing Firmiana, a one-stop proteomic cloud platform for data analysis and processing.8 The aim is to reduce the cost of medicine, with the China Human Proteome Project (CNHPP) focusing on the 10 cancers most prevalent in China.
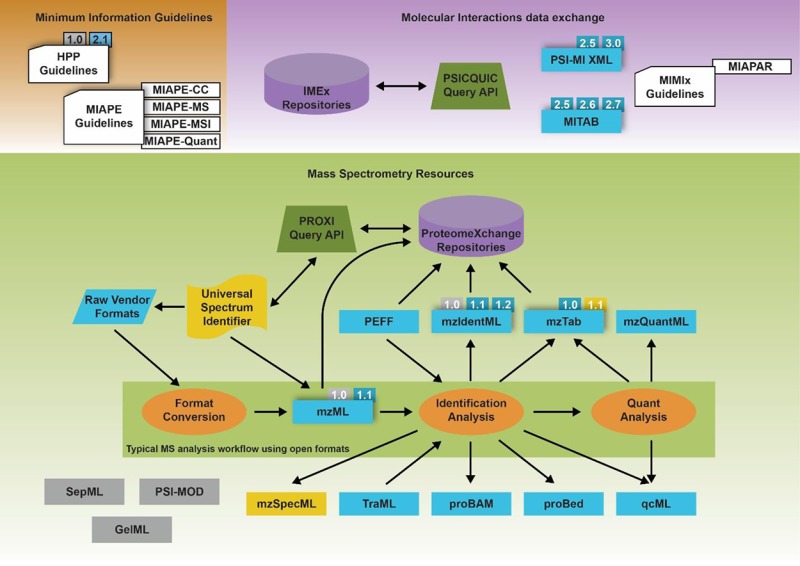
The chairs from the different working groups gave an update on their activities, including nonpurely PSI activities such as ProteomeXchange and the status of metabolomics data standards. The workshop then split into parallel work-tracks for each of the developing formats and standards for the next 2 days, combining presentations with discussion and hands-on development work. A final plenary session reviewed the results of the working groups’ activities at the end of the workshop. In the following sections we provide an overview of the current state of existing standards, new standards, and other related resources, as summarized in Figure 1. The current state of available PSI standards is listed in Table 1. We also provide a historical timeline of PSI developments in Figure 2.
Figure 1.

Overview of the relationship between PSI standard formats (blue rectangles), analysis steps (orange ovals), data repositories (purple cylinders), Application Programming Interfaces (green trapezoids), guidelines (white clipped rectangles), and other related resources described in this article. Resources with multiple prominent versions are decorated with those version numbers (gray for deprecated; blue for active; yellow for in development). Yellow elements are still early in the development process. Mass spectrometry formats are displayed relative to a typical MS workflow using standard formats.
Table 1. Summary of PSI Working Groups, Reporting Guidelines, Data Formats, and Controlled Vocabularies (as of August 2017).
| working groups | guidelines | version | formats | version | controlled vocabularies | version |
|---|---|---|---|---|---|---|
| molecular | MIMIx | 1.1.2 | PSI-MI XML | 2.5.4 | PSI-MI CV | 2.5.0 |
| interactions | MIABE | 1.0.0 | PSI-MI XML (public review) | 3.0 | ||
| MIAPAR | 1.0.0 | MITAB | 2.7 | |||
| mass spectroscopy | MIAPE-MS | 2.98 | mzML | 1.1.0 | PSI-MS | 4.0.13 |
| TraML | 1.0.0 | |||||
| PEFF (nearly final) | 1.0.0-rc | |||||
| proteomics | MIAPE-MSI | 1.1 | mzidentML | 1.2.0 | PSI-MS | 4.0.13 |
| informatics | MIAPE-Quant | 1.0 | mzQuantML | 1.0.1 | XLMOD | 1.0 |
| mzTab | 1.0.0 | |||||
| proBed | 1.0.0 | |||||
| proBam (public review) | 1.0.0-rc | |||||
| quality control | qcML | Beta |
Figure 2.

Overview of the timeline of when PSI standards were published or released. Molecular interactions standards are depicted in the top half, while mass spectrometry standards are depicted in the bottom half. Shapes and colors are the same as described in the Figure 1 legend.
Existing Standards
In this section we briefly describe the current state of the standards previously developed by the PSI (as of August 2017), many of which have received or are in the process of receiving updates since the original release to maintain their usefulness.
Molecular Interactions
The work of the Molecular Interactions Working Group has been to develop the formats and standards required to capture and describe data on interactions between biomolecules. PSI-MI XML 2.5 captures detailed information about interactions between molecules of all different types,9 including metabolites, DNA, proteins, and protein complexes and the experimental details from which the data were derived. However, new use cases that move beyond the scope of PSI-MI XML 2.5 now exist. For example, there is a need to describe dynamic interactions, allosteric interactions, and abstracted data derived from multiple experiments. The format has therefore been extended to version 3.0 to allow these more specialized use cases to be encoded. The IntAct molecular interaction database has implemented a download of this format, and all IMEx (International Molecular Exchange) Consortium data10 are available in PSI-MI XML 3.0. The format will soon be submitted to the PSI Document Process for ratification. The simpler, tab-delimited representation, MITAB, has also grown in complexity in response to user requests, and MITAB 2.5, 2.6, and 2.7 are now all available, with 2.7 being fully backwardly compatible with the earlier versions The versions 2.5, 2.6, and 2.7 of MITAB capture increasing levels of detail; each of these three versions is still considered active, and users may select the version to use based on the level of detail that they wish to encode. The possibility of extending the format to include additional columns to describe statements of the causality of an interaction was discussed at the meeting in Beijing. The required extensions to the PSI-MI CV have already been made.
To support the multiple PSI formats and other applications such as XGMML, the JAMI Java library has been developed recently. JAMI can import and export molecular interaction data in a variety of formats and versions and facilitates data import, integration, and analysis. The library simplifies software development by offering a single API (Application Programming Interface). An officially recognized MI-JSON protocol has also been developed for serving MI data to web pages and visualization tools.
The need to update the PSICQUIC web service11 has been discussed as the user community has grown considerably, and there have been issues with the speed of the service experienced by both data suppliers and data users. The possibility of the service operating in a cloud-based environment will be investigated, as this would enable fault tolerance and ease distribution of the data. A prototype service will be built at EMBL-EBI for testing by other service providers before a final decision is made.
All formats and CVs are available at https://github.com/HUPO-PSI and accompanying tools at https://github.com/MICommunity. The work of the PSI-MI is described at http://www.psidev.info/groups/molecular-interactions.
Mass Spectrometry
Many standards have been developed over the years for mass spectrometry (MS) workflows. The most prominent and widely used format is mzML,12 designed to encode the spectra and chromatograms generated by mass spectrometers. The mzML format is based on Extensible Markup Language (XML) but has an optional indexing scheme that allows random access to spectra inside the XML document. The history of mzML has already been described,13 including the deprecation of the old mzData format, and mzML has been stable at version 1.1 since June 2009. However, there are currently a few pending updates to support ion mobility MS and a few necessary fixes to problems that hinder proper validation in the schema.
One criticism that has been leveled at PSI is that mzML files tend to be larger than vendor raw files, in some cases up to two times as large. There has also been much discussion over the decision to adopt XML over a pure binary format, text-based format, or rich binary format, such as HDF5 (https://support.hdfgroup.org/HDF5/). A lengthy discussion over the pros and cons of the underlying format is beyond the scope of this article. In brief, PSI has tended to use XML as a good compromise, having wide and completely open support in libraries but while using a text-based binary encoding for spectra and encouraging the additional use of internal compression (by a zip-type algorithm) within files to keep file sizes manageable. The PSI has, in addition, been developing more advanced compression for mzML via the MS-Numpress14 technique, which is already implemented in several software packages although is not formally part of the format. Version 1.2 of mzML is anticipated to be completed and released this year. Current format and general working group information is available at http://psidev.info/groups/mass-spectrometry. mzML-specific information is available at http://www.psidev.info/mzml.
Proteomics Informatics
The mzIdentML standard was designed to report peptide and protein identification data, for example, from search engines, and has been released as a stable 1.1 version in 2011.15 The format is intended to allow different software tools to communicate with each other, for example, connecting a search engine to downstream software for protein grouping, statistics, or genome mapping (proteogenomics) as well as to support repository deposition. Since its initial stable release, the adoption of mzIdentML 1.1 has increased enormously. Most notably, several popular proteomics software packages now export mzIdentML natively (http://www.psidev.info/tools-implementing-mzidentml), and this list is growing regularly. In the repository context, mzIdentML is now supported as the primary format for upload of identification data to PRIDE,16−18 jPOST,19 and MassIVE databases within the ProteomeXchange consortium.20,21 An update to the format, mzIdentML version 1.2, has just been released.22 The mzIdentML 1.2 version is backward compatible in that most reading software designed for v 1.1 will be able to read most files without adaptation, but new features have been added to support some special cases that were not considered in the mzIdentML 1.1 release. The newly supported features include the ability to add scores or statistics to modification localization positions at the level of nonredundant peptides (rather than peptide-spectrum matches), cross-linking searches for structural proteomics, and genome-level coordinates for proteogenomics. To directly and unambiguously reference specific cross-linking reagents, we have defined a new CV called XLMOD.
In many proteomics pipelines, different workflows and software are used for quantification, and the needs for data storage are rather different from identification data. The mzQuantML format23 (stable version 1.0) was designed to store the outputs of quantification software from a variety of popular discovery-based workflows including MS1- and MS2-based label-free, MS1 label-based (SILAC or dimethyl), or MS2 tag-based (TMT or iTRAQ). The format was later updated to store results from targeted techniques such as SRM/PRM (selected/parallel reaction monitoring). The design of mzQuantML stores 2D tables of data in the file, where columns are typed as assays (measurement made from one sample by MS) or study variables (groupings of assays for which measurements have been taken) and rows are typed as protein groups, proteins, peptides, or LC–MS features. The format is sufficiently rich to be used for data exchange between software packages or to support LC–MS feature-level visualization.
Both mzIdentML and mzQuantML are represented in XML and can contain substantial levels of detail, requiring some software or coding ability to process data. The PSI–PI Working Group acknowledged that for some uses the formats were challenging to implement, particularly for those only interested in the end results of an analysis and not how they were generated. It was decided that there was a need for a simpler, text-based format to present both identification and quantification results, which resulted in the design of mzTab.24 mzTab follows a tabular design similar to mzQuantML but stores data in a tab-separated text format, making it straightforward to load into a spreadsheet or statistical software. mzTab is now supported as part of the PRIDE, MassIVE, and jPOST submission pipelines, and several popular analysis tools in the field are working toward supporting it (e.g., MaxQuant). In the initial version 1.0 design, an extension for MS metabolomics data was already included, although without the ability to represent detailed results about how metabolites and their adducts were measured by MS. Since 2014, the PSI–PI has partnered with metabolomics standards organizations, working toward mzTab version 1.1, with full support for metabolomics approaches. The mzTab 1.1 update is currently in progress and should be released within the coming year.
A few formats developed have fallen into disuse in the community. The TraML format25,26 was designed for SRM transitions and inclusion lists. Although implemented in a few software packages, it was never implemented in Skyline,27 which came to dominate the field of SRM transition list design and SRM data analysis. The PSI Separations group designed formats in the past to represent data derived from gel-based workflows28 and generically describe separations performed in proteomics workflows, called sepML—both built on top of the FuGE framework.29 GelML and sepML have not been widely implemented, however, and as such they have become deprecated by PSI.
PSI Guidelines
The PSI has also developed several guidelines describing what information should be provided when making a data set public. The MIAPE Guidelines30,31 were developed as a modular set of guidelines specifying what information should be supplied about each component of an experiment, the separation, the chromatography32 (MIAPE-CC), the mass spectrometry33 (MIAPE-MS), the subsequent informatics analysis34 (MIAPE-MSI), and finally the quantitative components31 (MIAPE-Quant). These components may be applied as needed depending on the experiment. These MIAPE components were implemented in the ProteoRed database35 and used as a guide in other software but are not otherwise widely used, most likely due to a lack of implementing software that makes it sufficiently easy to record all of the information needed. Entering all of the information at the time of repository submission is generally seen as disagreeably time-consuming, and the only solution may be more advanced software that can more easily and mostly automatically capture the information as the experiment is performed.
More recently, in conjunction with the Human Proteome Project (HPP), a set of 15 MS data analysis guidelines36 was developed to be applied to data sets contributed as part of the HPP. The guidelines require deposition to a ProteomeXchange data repository, minimum standards for setting and description of false discovery rates, and a minimum of two distinct non-nested peptides of length ≥9 residues for detection claims of proteins or other translation products with no or insufficient explicit evidence of translation yet in neXtProt37 (i.e., not yet PE (Protein Existence) level 1). These guidelines apply to all manuscripts submitted for this special issue and for other HPP-related manuscripts. We aim to recommend their consideration for implementation in other leading journals.
Controlled Vocabularies
An important aspect of all of the PSI standards is the use of standardized, well-defined terminology to complete values in data files via the PSI-controlled vocabularies.5 The PSI-MS CV38 contains 2692 terms in the latest release (4.0.13) for MS instrument and software names and parameters as well as other terms needed to annotate and describe an MS workflow. The molecular interactions formats use the PSI-MI CV. The CVs are available via the Ontology Lookup Service39 and BioPortal,40 updated regularly (every week or two depending on demand for new terms), and new terms can easily be requested by anyone, such as new implementers of exporters for one of the standards.
New Standards
There are also PSI standards that are in the final stages of ratification or are under development, for which further community involvement is actively encouraged. Such involvement includes the development of additional implementing software, creation of additional examples, contribution to the design process, and writing of further documentation.
Two new data file formats were recently devised to aid proteogenomics applications: proBed and proBAM.41 The goal is that the existence of these two formats will increase data sharing and integration between the genomics and proteomics communities. The proBed format is currently at its first mature public release version 1.0.0. The proBAM format is in public review phase after a first successful round of internal revision and will be publicly released soon after completion of the full PSI Document Process. Both formats map MS-based proteomics identifications to genome coordinates and are built as extensions to their widely used genomics counterparts BED (Browser Extensible Data; http://genome.ucsc.edu/FAQ/FAQformat.html#format1) and SAM/BAM42 (Sequence Alignment/Map format and its binary compressed version). Both are tab-delimited and hold mandatory fields from the original formats, containing genomic mapping information. They also accommodate specific proteomics information, either at the peptide-spectrum-match (PSM) or peptide level. (See specification documents for full details at http://www.psidev.info/proBed and http://www.psidev.info/proBAM.) proBAM and proBed serve different purposes: proBed stores high-level track information to present validated MS-based proteomics identification results in a genome-centric way using existing stand-alone or web-based genome browsers: Ensembl,43 UCSC (University of California Santa Cruz) Browser,44 Integrative Genomics Viewer45 (IGV), or JBrowse.46 proBAM also serves this purpose but can also contain more detail describing a full MS proteomics result set (e.g., unmapped, decoy, and lower ranked PSMs), whereupon further analysis can be performed.
Many software implementations are set in place to write/convert, manipulate, analyze, and validate these novel formats. A key point is that proBed and proBAM are intentionally designed as extensions to their genomics format in a way not to break the original format. As a result, existing popular genomics tools as SAMtools and Bedtools42,47 can be used to manipulate them. To convert existing MS-based proteomics identifications results in mzIdentML and mzTab format, among other formats, several tools have also recently been developed. ms-data-core-api48 and PGConverter (https://github.com/PRIDE-Toolsuite/PGConverter) write to proBed, proBAMr writes to proBAM, and proBAMconvert49 (http://probam.biobix.be) writes to both formats. PGConverter also contains a validation module. proBAMtools50 is available for further downstream analysis of proBAM files.
A systematic approach to quality control (QC) is an essential requirement to have confidence in the results of a MS experiment, and as such this has increasingly received attention over the past few years. Although several tools have been developed to compute QC metrics, these initiatives lack a unifying frame of reference, hindering the long-term maturation of MS QC. To this end the Quality Control Working Group, the PSI’s most recently established working group, is undertaking efforts to establish a QC community standard.51 This standard will consist of a formalized version of the qcML file format,7 an associated CV, and a MIAPE-QC specification, along with corresponding resources to generate and aid interpretation of the QC information. The previously proposed qcML file format is currently undergoing substantial changes based on community feedback, as it will be incorporated as an official PSI standard. The group is prioritizing the applicability of QC metrics in diverse MS methods, not limiting qcML to use in data-dependent or “shotgun” LC–MS/MS. Incorporating efforts to apply QC in the context of MALDI-TOF, SRM/PRM, and data-independent acquisition approaches such as UDMSE and SWATH-MS will broaden the applicability of the qcML framework in both proteomics and in metabolomics. Ideally, future developments in instrumentation and bioinformatics software will be able to benefit from qcML-aware statistical frameworks by defining relevant metrics that map to the associated CV. We expect that quantitative proteomics approaches, in particular, will benefit from integrating QC information.
PEFF (PSI Extended FASTA Format, http://www.psidev.info/peff) is a format in the final stages of development designed to encode protein and nucleotide sequence databases. The format is based on the ubiquitous FASTA format used by most proteomics search engines. PEFF extends the FASTA format to enable a standard mechanism by which metadata about the whole collection can be encoded and by which metadata about each entry can be encoded. A header section of metadata describes relevant information about the database(s) from which the sequences have been obtained. For each entry, information about sequence variants and post-translational modifications and much more can be encoded. The format can also be used to fully specify exact proteoforms.52 A CV defines the terms to be used in the instance documents. Currently, although version 1.0 is not yet released, a number of tools (readers and writers) are already supporting it. As examples, neXtProt provides an export functionality, the CompOmics and php-ms projects have written a PEFF viewer, and beta versions of the search engines Comet53 and ProteinProspector54 can read PEFF as a sequence database format. The format has passed a first reviewing round as part of the PSI document process. A number of adaptations were discussed and defined in the meeting in Beijing. A resubmission of the updated format is under preparation.
Because the fragmentation pattern of each peptide ion, captured in a tandem mass spectrum, is largely reproducible across shotgun proteomics experiments employing similar workflows, such a spectrum can function as a fingerprint for the peptide ion. Spectral libraries are simply collections of these fingerprints, which can be used to aid future identifications by spectral matching. Currently, spectral libraries are built by dedicated efforts, such as those undertaken by the U.S. National Institute of Standards and Technology (NIST) and PeptideAtlas. Data generated all over the world and collected in proteomics data repositories are reanalyzed using multiple search engines and state-of-the-art validation methods, condensed by merging replicate observations, annotated and indexed, and finally distributed freely to the community. Open-source software tools also exist for individual researchers to build their own custom-made spectral libraries. Previously, studies have shown that spectral library searching is better suited to detect previously observed peptides than sequence searching, and spectral libraries have become indispensable in targeted quantitative proteomic workflows, such as SRM and DIA. More recently, with the rapid growth in data volume, it has been proposed that tandem mass spectra should be grouped by spectral similarity rather than by peptide identification (if any), in so-called spectral archives. Essentially, spectral archives extend the idea of spectral libraries to include unidentified spectra, which can also function as fingerprints of the yet-to-be identified molecules. As such, spectral archives are especially useful for data repositories to organize and condense vast amount of data while preserving all experimental observations for future discoveries.
Currently, spectral libraries and archives are distributed in different formats by different databases and library builders,55,56 making it difficult to share libraries and compare spectral library searching tools (as highlighted in the Beijing meeting by H. Lam, see above). Also, the lack of a common file format hinders the deposition of spectral searching results in public databases (e.g., ProteomeXchange partners). The PSI is in the early phase of designing a PSI spectral library format (tentatively dubbed mzSpecML). The major shortcoming of the existing formats is a standardized mechanism for encoding extensive metadata about the origins of the library and about each of the entries therein. The format must be flexible enough to fit all of the potential use cases of spectral libraries and yet retain sufficient structure for it to be a practically useful standard. The first implementation of the spectral library file format would be based on the well known and most supported file format MSP. The file format will be enriched with more metadata to describe the method that was used to build the library, and all metadata fields will be represented by CV terms. A new repository (https://github.com/HUPO-PSI/SpectralLibraryFormat) has been created to guide the development process of the standard including the specification, examples, and tools.
Also, early in the requirements gathering and design phase is an effort to define a Universal Spectrum Identifier. Such an identifier would enable authors to reference key spectra that support their finding in their manuscripts or the corresponding Supporting Information and allow reviewers to examine the spectra via their own spectrum viewers interactively rather than rely on screenshot PDFs or other representations of the spectra. It would facilitate discussions over the interpretation of spectra that seem to implicate detections of translation products not yet in the primary reference databases; these identifiers would be particularly helpful in the context of satisfying the HPP Guidelines.36 It would permit referencing of specific spectra within the spectral library format described above and enable software of any kind to refer to permanent, unique identifiers to specific spectra. There are many details yet to be worked out, but an early draft specification and prototype software implementations to facilitate further discussion will unfold in 2017.
Recently, the ProteomeXchange community20,21 has developed a standard representation format (PX XML) to exchange information on public proteomics data sets, which has been implemented by other resources such as OmicsDI,57 a “multi-omics” resource which contains data sets coming from other omics approaches, such as genomics, transcriptomics, and metabolomics.
To provide an easy way of exchanging data among proteomics resources, PSI has started the early design and prototyping of the Protein Expression Interface (PROXI). PROXI is neither a format nor a guideline but rather a standard web services API that will enable users as well as automated software to query, access, and exchange information related to data sets, proteins, protein abundances, peptides, spectra, and peptide-spectrum matches (PSMs). It is envisioned that PROXI will be implemented at all of the major proteomics data repositories PRIDE,18 PeptideAtlas,58−60 MassIVE, and jPOST19 as well as the ProteomeCentral site (http://proteomecentral.proteomexchange.org/) of ProteomeXchange and other willing participants, allowing information access across all sites in a uniform manner. This effort will rely substantially on the Universal Spectrum Identifier and PSI Spectrum Library Format standards described above. Additional participants and funding is still being sought to implement this vision.
Synergies with other Efforts
The efforts and successes of the PSI are entwined with other community efforts that aim to accelerate the progress of biomedical research. One prominent example already mentioned is the ProteomeXchange Consortium20 of proteomics data repositories, which is actively fostering a culture of open data deposition and sharing.21 Crucial to this effort is the existence and widespread implementation of open standards. All of the ProteomeXchange Consortium members participate actively in the PSI and rely on the products of the PSI to streamline the deposition and dissemination of data sets. Similarly, the IMEx Consortium of interaction databases9 has grown from, and actively contributes to, the work of the MI group of the PSI.
For the past several years, the PSI has been actively reaching out to metabolomics initiatives, such as the COordination of Standards in MetabOlomicS (COSMOS),61 the Metabolomics Society, and the Metabolomics Standards Initiative (MSI; http://www.metabolomics-msi.org/), particularly via interaction with the Data Standards Task Group (http://metabolomicssociety.org/board/scientific-task-groups/data-standards-task-group). The COSMOS initiative participated actively in the 2014 PSI Annual Spring Workshop and held a joint meeting there.62 The proteomics and metabolomics communities share underlying computational MS challenges (e.g., http://compms.org/), and it greatly benefits both communities to work together. The Metabolomics Data Standards Task group has members of both MS and PSI communities and regularly holds a workshop during the Annual Metabolomics Society meetings, promoting usage and adoption of data standards. Additionally, the weight of both communities could help with better engagement with the instrument vendors and tools developers to promote adoption and usage of data standards. Currently, the metabolomics community is directly involved with developments of mzTab and qcML. In addition, mzML can already be used to represent metabolomics MS data.
The PSI is primarily an initiative of HUPO and therefore plays an active role in the many activities of HUPO. Typically, at each HUPO World Congress, the PSI hosts a session to communicate its ongoing activities, solicit feedback, and promote involvement with the HUPO membership. PSI members also play an active role in the planning and execution of the HUPO Bioinformatics Hub, a gathering place at the congress where HUPO members can find and ask questions to participating computational researchers, where computational researchers can find each other to discuss current topics and where special educational and discussion sessions can be held to further the advancement of computational proteomics. Finally, the PSI plays an important role in the Human Proteome Project,63 the flagship project of HUPO, by helping to set standards for HPP contributions, such as the HPP MS Data Interpretation Guidelines, as already mentioned36 (http://hupo.org/Guidelines).
Conclusions
We have reviewed the operations of the PSI, the current status of the main existing standards, the plans for upcoming standards, and the synergies between the PSI, MSI, and other groups. These activities demonstrate the commitment of the PSI to accelerating the pace of biomedical research by facilitating the dissemination and reuse of data, interoperability of software, and collaboration between researchers. However, with a full set of standard formats and minimum information guidelines for most data types relevant to proteomics already developed or soon emerging, what is left for the PSI to accomplish?
Although maintenance of and enhancements to existing standards are, of course, necessary as technologies advance to maintain relevance and usability of the standards, future innovations for the PSI will be in the software that supports those standards and the APIs by which they communicate. In the end, most users do not wish to dwell on which standards they use but rather have software that seamlessly implements those standards so that all of the tools that are used can freely interoperate with other software and online services. The future of software is not only better algorithms but also better automation and autonomy. Therefore, future efforts of the PSI must focus on developing standard APIs by which software and web services will communicate, allowing users to gather and integrate information from multiple resources easily and efficiently and allowing data to be shared among collaborators, deposited to repositories, and accessed from repositories without care for the underlying transport mechanisms.
Acknowledgments
We thank C. Sevilla for assistance with the figures. This work (including funding for the organization of the PSI workshops) has been funded in part by the National Institutes of Health under NIGMS grant number R01GM087221 and R24GM127667, NIBIB grant number U54EB020406, the National Science Foundation under grant number IIS-1636903, the Wellcome Trust [grant number WT101477MA], MRC UK MEDical BIOinformatics partnership [grant number MR/L01632X/1], BBSRC grant numbers [BB/M027635/1, BB/K01997X/1, BB/L024128/1, BB/N022440/1, BB/L024225/1, BB/L005239/1, BB/N022432/1], EMBL core funding and the EU FP7 grants ProteomeXchange [grant number 260558] and PRIME-XS [grant number 262067]. G.M. is funded by the BMBF grant de.NBI - German Network for Bioinformatics Infrastructure (FKZ 031 A 534A). M.E. was funded in part by PURE and VALIBIO, projects of Northrhine-Westphalia. The work of standards development in the field of molecular interactions is funded in part by BBSRC MIDAS grant [BB/L024179/1]. D.L.T. was funded in part by a Strategic Health Innovation Partnership (SHIP) grant from the South African (SA) Department of Science and Technology (DST) and SA Medical Research Council (SAMRC) to Gerhard Walzl.
The authors declare no competing financial interest.
References
- Deutsch E. W. File formats commonly used in mass spectrometry proteomics. Mol. Cell. Proteomics 2012, 11 (12), 1612–1621. 10.1074/mcp.R112.019695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanash S.; Celis J. E. The Human Proteome Organization: a mission to advance proteome knowledge. Mol. Cell. Proteomics 2002, 1 (6), 413–414. 10.1074/mcp.R200002-MCP200. [DOI] [PubMed] [Google Scholar]
- Orchard S.; Hermjakob H.; Apweiler R. The proteomics standards initiative. Proteomics 2003, 3 (7), 1374–1376. 10.1002/pmic.200300496. [DOI] [PubMed] [Google Scholar]
- Deutsch E. W.; Albar J. P.; Binz P.-A.; Eisenacher M.; Jones A. R.; Mayer G.; Omenn G. S.; Orchard S.; Vizcaíno J. A.; Hermjakob H. Development of data representation standards by the human proteome organization proteomics standards initiative. J. Am. Med. Inform. Assoc. JAMIA 2015, 22 (3), 495–506. 10.1093/jamia/ocv001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G.; Jones A. R.; Binz P.-A.; Deutsch E. W.; Orchard S.; Montecchi-Palazzi L.; Vizcaíno J. A.; Hermjakob H.; Oveillero D.; Julian R.; et al. Controlled vocabularies and ontologies in proteomics: overview, principles and practice. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844 (1 Pt A), 98–107. 10.1016/j.bbapap.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Martens L.; Hermjakob H.; Julian R. K.; Paton N. W. The PSI formal document process and its implementation on the PSI website. Proteomics 2007, 7 (14), 2355–2357. 10.1002/pmic.200700064. [DOI] [PubMed] [Google Scholar]
- Walzer M.; Pernas L. E.; Nasso S.; Bittremieux W.; Nahnsen S.; Kelchtermans P.; Pichler P.; van den Toorn H. W. P.; Staes A.; Vandenbussche J.; et al. qcML: an exchange format for quality control metrics from mass spectrometry experiments. Mol. Cell. Proteomics 2014, 13 (8), 1905–1913. 10.1074/mcp.M113.035907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Ding C.; Qiu N.; Ni X.; Zhan D.; Liu W.; Xia X.; Li P.; Lu B.; Zhao Q.; et al. Firmiana: towards a one-stop proteomic cloud platform for data processing and analysis. Nat. Biotechnol. 2017, 35 (5), 409–412. 10.1038/nbt.3825. [DOI] [PubMed] [Google Scholar]
- Kerrien S.; Orchard S.; Montecchi-Palazzi L.; Aranda B.; Quinn A. F.; Vinod N.; Bader G. D.; Xenarios I.; Wojcik J.; Sherman D.; et al. Broadening the horizon--level 2.5 of the HUPO-PSI format for molecular interactions. BMC Biol. 2007, 5, 44. 10.1186/1741-7007-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S.; Kerrien S.; Abbani S.; Aranda B.; Bhate J.; Bidwell S.; Bridge A.; Briganti L.; Brinkman F. S. L.; Brinkman F.; et al. Protein interaction data curation: the International Molecular Exchange (IMEx) consortium. Nat. Methods 2012, 9 (4), 345–350. 10.1038/nmeth.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda B.; Blankenburg H.; Kerrien S.; Brinkman F. S. L.; Ceol A.; Chautard E.; Dana J. M.; De Las Rivas J.; Dumousseau M.; Galeota E.; et al. PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat. Methods 2011, 8 (7), 528–529. 10.1038/nmeth.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens L.; Chambers M.; Sturm M.; Kessner D.; Levander F.; Shofstahl J.; Tang W. H.; Römpp A.; Neumann S.; Pizarro A. D.; et al. mzML--a community standard for mass spectrometry data. Mol. Cell. Proteomics 2011, 10 (1), R110.000133. 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E. W. Mass spectrometer output file format mzML. Methods Mol. Biol. 2010, 604, 319–331. 10.1007/978-1-60761-444-9_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teleman J.; Dowsey A. W.; Gonzalez-Galarza F. F.; Perkins S.; Pratt B.; Röst H. L.; Malmström L.; Malmström J.; Jones A. R.; Deutsch E. W.; et al. Numerical compression schemes for proteomics mass spectrometry data. Mol. Cell. Proteomics 2014, 13 (6), 1537–1542. 10.1074/mcp.O114.037879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. R.; Eisenacher M.; Mayer G.; Kohlbacher O.; Siepen J.; Hubbard S. J.; Selley J. N.; Searle B. C.; Shofstahl J.; Seymour S. L.; et al. The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics 2012, 11 (7), M111.014381. 10.1074/mcp.M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens L.; Hermjakob H.; Jones P.; Adamski M.; Taylor C.; States D.; Gevaert K.; Vandekerckhove J.; Apweiler R. PRIDE: the proteomics identifications database. Proteomics 2005, 5 (13), 3537–3545. 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Côté R. G.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Birim M.; Contell J.; et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013, 41 (D1), D1063–1069. 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; Del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44 (D1), D447–456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S.; Watanabe Y.; Moriya Y.; Kawano S.; Yamamoto T.; Matsumoto M.; Takami T.; Kobayashi D.; Araki N.; Yoshizawa A. C.; et al. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. 2017, 45 (D1), D1107–D1111. 10.1093/nar/gkw1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Deutsch E. W.; Wang R.; Csordas A.; Reisinger F.; Ríos D.; Dianes J. A.; Sun Z.; Farrah T.; Bandeira N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32 (3), 223–226. 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E. W.; Csordas A.; Sun Z.; Jarnuczak A.; Perez-Riverol Y.; Ternent T.; Campbell D. S.; Bernal-Llinares M.; Okuda S.; Kawano S.; et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45 (D1), D1100–D1106. 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Mayer G.; Perkins S.; Barsnes H.; Vaudel M.; Perez-Riverol Y.; Ternent T.; Uszkoreit J.; Eisenacher M.; Fischer L.; et al. The mzIdentML Data Standard Version 1.2, Supporting Advances in Proteome Informatics. Mol. Cell. Proteomics 2017, 16 (7), 1275–1285. 10.1074/mcp.M117.068429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzer M.; Qi D.; Mayer G.; Uszkoreit J.; Eisenacher M.; Sachsenberg T.; Gonzalez-Galarza F. F.; Fan J.; Bessant C.; Deutsch E. W.; et al. The mzQuantML data standard for mass spectrometry-based quantitative studies in proteomics. Mol. Cell. Proteomics 2013, 12 (8), 2332–2340. 10.1074/mcp.O113.028506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss J.; Jones A. R.; Sachsenberg T.; Walzer M.; Gatto L.; Hartler J.; Thallinger G. G.; Salek R. M.; Steinbeck C.; Neuhauser N.; et al. The mzTab data exchange format: communicating mass-spectrometry-based proteomics and metabolomics experimental results to a wider audience. Mol. Cell. Proteomics 2014, 13 (10), 2765–2775. 10.1074/mcp.O113.036681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E. W.; Chambers M.; Neumann S.; Levander F.; Binz P.-A.; Shofstahl J.; Campbell D. S.; Mendoza L.; Ovelleiro D.; Helsens K.; et al. TraML--a standard format for exchange of selected reaction monitoring transition lists. Mol. Cell. Proteomics 2012, 11 (4), R111.015040. 10.1074/mcp.R111.015040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsens K.; Brusniak M.-Y.; Deutsch E.; Moritz R. L.; Martens L. jTraML: an open source Java API for TraML, the PSI standard for sharing SRM transitions. J. Proteome Res. 2011, 10 (11), 5260–5263. 10.1021/pr200664h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson F.; Hoogland C.; Martinez-Bartolomé S.; Medina-Aunon J. A.; Albar J. P.; Babnigg G.; Wipat A.; Hermjakob H.; Almeida J. S.; Stanislaus R.; et al. The gel electrophoresis markup language (GelML) from the Proteomics Standards Initiative. Proteomics 2010, 10 (17), 3073–3081. 10.1002/pmic.201000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. R.; Miller M.; Aebersold R.; Apweiler R.; Ball C. A.; Brazma A.; Degreef J.; Hardy N.; Hermjakob H.; Hubbard S. J.; et al. The Functional Genomics Experiment model (FuGE): an extensible framework for standards in functional genomics. Nat. Biotechnol. 2007, 25 (10), 1127–1133. 10.1038/nbt1347. [DOI] [PubMed] [Google Scholar]
- Taylor C. F.; Paton N. W.; Lilley K. S.; Binz P.-A.; Julian R. K.; Jones A. R.; Zhu W.; Apweiler R.; Aebersold R.; Deutsch E. W.; et al. The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 2007, 25 (8), 887–893. 10.1038/nbt1329. [DOI] [PubMed] [Google Scholar]
- Martínez-Bartolomé S.; Binz P.-A.; Albar J. P. The Minimal Information about a Proteomics Experiment (MIAPE) from the Proteomics Standards Initiative. Methods Mol. Biol. 2014, 1072, 765–780. 10.1007/978-1-62703-631-3_53. [DOI] [PubMed] [Google Scholar]
- Jones A. R.; Carroll K.; Knight D.; Maclellan K.; Domann P. J.; Legido-Quigley C.; Huang L.; Smallshaw L.; Mirzaei H.; Shofstahl J.; et al. Guidelines for reporting the use of column chromatography in proteomics. Nat. Biotechnol. 2010, 28 (7), 654. 10.1038/nbt0710-654a. [DOI] [PubMed] [Google Scholar]
- Taylor C. F.; Binz P.-A.; Aebersold R.; Affolter M.; Barkovich R.; Deutsch E. W.; Horn D. M.; Hühmer A.; Kussmann M.; Lilley K.; et al. Guidelines for reporting the use of mass spectrometry in proteomics. Nat. Biotechnol. 2008, 26 (8), 860–861. 10.1038/nbt0808-860. [DOI] [PubMed] [Google Scholar]
- Binz P.-A.; Barkovich R.; Beavis R. C.; Creasy D.; Horn D. M.; Julian R. K.; Seymour S. L.; Taylor C. F.; Vandenbrouck Y. Guidelines for reporting the use of mass spectrometry informatics in proteomics. Nat. Biotechnol. 2008, 26 (8), 862. 10.1038/nbt0808-862. [DOI] [PubMed] [Google Scholar]
- Medina-Aunon J. A.; Martínez-Bartolomé S.; López-García M. A.; Salazar E.; Navajas R.; Jones A. R.; Paradela A.; Albar J. P. The ProteoRed MIAPE web toolkit: a user-friendly framework to connect and share proteomics standards. Mol. Cell. Proteomics 2011, 10 (10), M111.008334. 10.1074/mcp.M111.008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E. W.; Overall C. M.; Van Eyk J. E.; Baker M. S.; Paik Y.-K.; Weintraub S. T.; Lane L.; Martens L.; Vandenbrouck Y.; Kusebauch U.; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J. Proteome Res. 2016, 15 (11), 3961–3970. 10.1021/acs.jproteome.6b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet P.; Michel P.-A.; Zahn-Zabal M.; Cusin I.; Duek P. D.; Evalet O.; Gateau A.; Gleizes A.; Pereira M.; Teixeira D.; et al. The neXtProt knowledgebase on human proteins: current status. Nucleic Acids Res. 2015, 43 (Database issue), D764–770. 10.1093/nar/gku1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G.; Jones A. R.; Binz P.-A.; Deutsch E. W.; Orchard S.; Montecchi-Palazzi L.; Vizcaíno J. A.; Hermjakob H.; Oveillero D.; Julian R.; et al. Controlled vocabularies and ontologies in proteomics: overview, principles and practice. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844 (1), 98–107. 10.1016/j.bbapap.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté R.; Reisinger F.; Martens L.; Barsnes H.; Vizcaino J. A.; Hermjakob H. The Ontology Lookup Service: bigger and better. Nucleic Acids Res. 2010, 38 (Web Server), W155–160. 10.1093/nar/gkq331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy N. F.; Shah N. H.; Whetzel P. L.; Dai B.; Dorf M.; Griffith N.; Jonquet C.; Rubin D. L.; Storey M.-A.; Chute C. G.; et al. BioPortal: ontologies and integrated data resources at the click of a mouse. Nucleic Acids Res. 2009, 37 (Web Server), W170–173. 10.1093/nar/gkp440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menschaert G.; Wang X.; Jones A. R.; Ghali F.; Fenyo D.; Olexiouk V.; Zhang B.; Deutsch E. W.; Ternent T.; Vizcaino J. A.. The proBAM and proBed standard formats: enabling a seamless integration of genomics and proteomics data. bioRxiv 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Handsaker B.; Wysoker A.; Fennell T.; Ruan J.; Homer N.; Marth G.; Abecasis G.; Durbin R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25 (16), 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aken B. L.; Achuthan P.; Akanni W.; Amode M. R.; Bernsdorff F.; Bhai J.; Billis K.; Carvalho-Silva D.; Cummins C.; Clapham P.; et al. Ensembl 2017. Nucleic Acids Res. 2017, 45 (D1), D635–D642. 10.1093/nar/gkw1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner C.; Barber G. P.; Casper J.; Clawson H.; Diekhans M.; Eisenhart C.; Fischer C. M.; Gibson D.; Gonzalez J. N.; Guruvadoo L.; et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 2017, 45 (D1), D626–D634. 10.1093/nar/gkw1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J. T.; Thorvaldsdóttir H.; Winckler W.; Guttman M.; Lander E. S.; Getz G.; Mesirov J. P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29 (1), 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M. E.; Uzilov A. V.; Stein L. D.; Mungall C. J.; Holmes I. H. JBrowse: a next-generation genome browser. Genome Res. 2009, 19 (9), 1630–1638. 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A. R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinforma. 2014, 47 (11.12.1), 11.12.34–34. 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Uszkoreit J.; Sanchez A.; Ternent T.; Del Toro N.; Hermjakob H.; Vizcaíno J. A.; Wang R. ms-data-core-api: an open-source, metadata-oriented library for computational proteomics. Bioinformatics 2015, 31 (17), 2903–2905. 10.1093/bioinformatics/btv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olexiouk V.; Menschaert G. proBAMconvert: A Conversion Tool for proBAM/proBed. J. Proteome Res. 2017, 16 (7), 2639–2644. 10.1021/acs.jproteome.7b00132. [DOI] [PubMed] [Google Scholar]
- Wang X.; Slebos R. J. C.; Chambers M. C.; Tabb D. L.; Liebler D. C.; Zhang B. proBAMsuite, a Bioinformatics Framework for Genome-Based Representation and Analysis of Proteomics Data. Mol. Cell. Proteomics 2016, 15 (3), 1164–1175. 10.1074/mcp.M115.052860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittremieux W.; Walzer M.; Tenzer S.; Zhu W.; Salek R. M.; Eisenacher M.; Tabb D. L. The Human Proteome Organization-Proteomics Standards Initiative Quality Control Working Group: Making Quality Control More Accessible for Biological Mass Spectrometry. Anal. Chem. 2017, 89 (8), 4474–4479. 10.1021/acs.analchem.6b04310. [DOI] [PubMed] [Google Scholar]
- Smith L. M.; Kelleher N. L.; et al. Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10 (3), 186–187. 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J. K.; Jahan T. A.; Hoopmann M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13 (1), 22–24. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Chalkley R. J.; Baker P. R.; Huang L.; Hansen K. C.; Allen N. P.; Rexach M.; Burlingame A. L. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: II. New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol. Cell. Proteomics 2005, 4 (8), 1194–1204. 10.1074/mcp.D500002-MCP200. [DOI] [PubMed] [Google Scholar]
- Lam H.; Deutsch E. W.; Eddes J. S.; Eng J. K.; Stein S. E.; Aebersold R. Building consensus spectral libraries for peptide identification in proteomics. Nat. Methods 2008, 5 (10), 873–875. 10.1038/nmeth.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss J.; Perez-Riverol Y.; Lewis S.; Tabb D. L.; Dianes J. A.; Del-Toro N.; Rurik M.; Walzer M. W.; Kohlbacher O.; Hermjakob H.; et al. Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets. Nat. Methods 2016, 13 (8), 651–656. 10.1038/nmeth.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Bai M.; da Veiga Leprevost F.; Squizzato S.; Park Y. M.; Haug K.; Carroll A. J.; Spalding D.; Paschall J.; Wang M.; et al. Discovering and linking public omics data sets using the Omics Discovery Index. Nat. Biotechnol. 2017, 35 (5), 406–409. 10.1038/nbt.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desiere F.; Deutsch E. W.; Nesvizhskii A. I.; Mallick P.; King N. L.; Eng J. K.; Aderem A.; Boyle R.; Brunner E.; Donohoe S.; et al. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2004, 6 (1), R9. 10.1186/gb-2004-6-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desiere F.; Deutsch E. W.; King N. L.; Nesvizhskii A. I.; Mallick P.; Eng J.; Chen S.; Eddes J.; Loevenich S. N.; Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006, 34 (90001), D655–658. 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E. W.; Sun Z.; Campbell D.; Kusebauch U.; Chu C. S.; Mendoza L.; Shteynberg D.; Omenn G. S.; Moritz R. L. State of the Human Proteome in 2014/2015 As Viewed through PeptideAtlas: Enhancing Accuracy and Coverage through the AtlasProphet. J. Proteome Res. 2015, 14 (9), 3461–3473. 10.1021/acs.jproteome.5b00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salek R. M.; Neumann S.; Schober D.; Hummel J.; Billiau K.; Kopka J.; Correa E.; Reijmers T.; Rosato A.; Tenori L.; et al. COordination of Standards in MetabOlomicS (COSMOS): facilitating integrated metabolomics data access. Metabolomics 2015, 11 (6), 1587–1597. 10.1007/s11306-015-0810-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S.; Albar J. P.; Binz P.-A.; Kettner C.; Jones A. R.; Salek R. M.; Vizcaino J. A.; Deutsch E. W.; Hermjakob H. Meeting new challenges: The 2014 HUPO-PSI/COSMOS Workshop. Proteomics 2014, 14 (21–22), 2363–2368. 10.1002/pmic.201470164. [DOI] [PubMed] [Google Scholar]
- Legrain P.; Aebersold R.; Archakov A.; Bairoch A.; Bala K.; Beretta L.; Bergeron J.; Borchers C. H.; Corthals G. L.; Costello C. E.; et al. The human proteome project: current state and future direction. Mol. Cell. Proteomics 2011, 10 (7), M111.009993. 10.1074/mcp.M111.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]