

Abstract
The pentasaccharide fragment α-d-Man-(1 → 5)-[α-d-Kdo-(2 → 4)-]α-d-Kdo-(2 → 6)-β-d-GlcNAc-(1 → 6)-α-d-GlcNAc equipped with a 3-aminopropyl spacer moiety was prepared by a sequential assembly of monosaccharide building blocks. The glucosamine disaccharide—as a backbone surrogate of the bacterial lipid A region—was synthesized using an 1,3-oxazoline donor, which was followed by coupling with an isopropylidene-protected Kdo-fluoride donor to afford a protected tetrasaccharide intermediate. Eventually, an orthogonally protected manno-configured trichloroacetimidate donor was used to achieve the sterically demanding glycosylation of the 5-OH group of Kdo in good yield. The resulting pentasaccharide is suitably protected for further chain elongation at positions 3, 4, and 6 of the terminal mannose. Global deprotection afforded the target pentasaccharide to be used for the conversion into neoglycoconjugates and “clickable” ligands.
Introduction
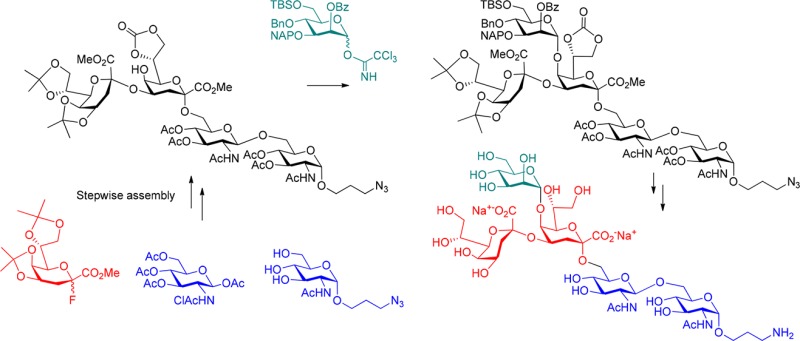
The outer membrane of the Gram-negative bacterial cell wall is covered to a large extent by lipopolysaccharide (LPS) molecules, which are involved in a multitude of major immune reactions in the context of bacterial infections.1,2 In addition to their biomedical relevance, LPS structures are also engaged in important bacteria-plant interactions, e.g., leading to the formation of nodules in leguminous roots when colonized by Rhizobia.3 In structural terms, these glycolipids comprise the endotoxically active lipid anchor (lipid A) and an extended oligosaccharide fraction, which harbors a core region of limited structural variability and the strain-specific, highly diverse O-antigenic polysaccharides.4,5 This core domain may be further divided into an outer and an inner core region, wherein a 3-deoxy-d-manno-2-octulosonic acid (Kdo) usually forms the link to the lipid A part. In many bacterial genera, this Kdo unit is then extended by a lateral α-(2 → 4)-linked Kdo moiety and further elongated at position 5 by additional inner core sugars such as l-glycero-d-manno-heptose (l,d-Hep), d-glucose, d-galacturonic acid or d-mannose.6 Specifically, 5-O-mannosylated Kdo has been found in bacteria such as Francisella tularensis, Brucella melitensis, Agrobacterium tumefaciens, Sinorhizobium meliloti and Rhizobiaceae.2,7 In the latter strains, defects in the mannosyl transferase LpsC impairs their nodulation activity.8,9 Recently, the structure of an oligomannose linked to O-5 of Kdo was reported for the inner core of the lipooligosaccharide (LOS) of Rhizobium radiobacter Rv3, also termed Agrobacterium radiobacter and Agrobacterium tumefaciens (Figure 1).10,11 Of note is the structural resemblance of the α-(1 → 2)-linked oligomannose part to the D1 arm of mammalian high-mannose glycans, which are particularly abundant on HIV-1 gp120.12Rhizobium radiobacter Rv3 LOS and heat-killed cells are bound by the oligomannose-specific HIV-neutralizing antibody 2G12 and a crystal structure of the carbohydrate backbone of Rhizobium radiobacter strain Rv3 complexed to the 2G12 antibody has recently been determined.13 In order to further exploit the natural mimicry of high-mannose carbohydrate epitopes of HIV-1 gp120 seen with this bacterial LOS scaffold, we have set out to extend the bacterial oligosaccharides in order to elicit HIV neutralizing antibodies. In a first step we have prepared the basic branched core oligosaccharide in a sufficient scale to allow for regioselective mannosylation at O-3 and O-6 of the central mannose unit, respectively, as well as for global deprotection to give the pentasaccharide core fragment equipped with a functional spacer group to allow for conversion into neoglycoconjugates.
Figure 1.
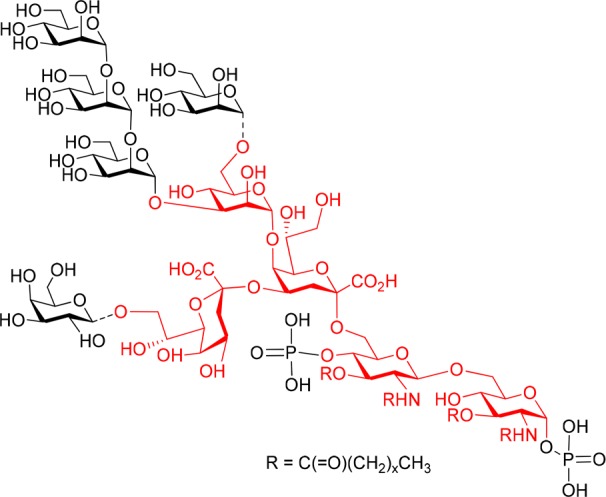
Chemical structure of the inner core and lipid A region of R. radiobacter Rv3 LOS.10 Dotted lines indicate substoichiometric substitution. The targeted pentasaccharide is marked in red.
Results and Discussion
The pentasaccharide was assembled starting from the β-(1 → 6)-linked diglucosamine backbone to be subsequently elongated by the Kdo residues. To simplify deprotection at the final stages, both 2-amino-2-deoxy-groups were protected as N-acetyl derivatives keeping in mind difficulties encountered in several reported glycosylation reactions describing the formation of the respective imidates.14 Although these unwanted reactions were known, the variety of protecting groups had to be limited in order to minimize loss of material in the multistep synthesis. The reducing terminus was equipped with an α-configured 3-aminopropyl spacer glycoside amenable to eventual coupling to proteins as well as suitable for conversion into the corresponding 3-azidopropyl derivatives to be used as substrate for ensuing “click”-chemistry.15
Starting from known16 3-azidopropyl glycoside 1, which was prepared according to an improved procedure in a multigram scale,17 6-O-tritylation was achieved smoothly in 90% yield by treatment of 1 with trityl chloride in pyridine and catalytic 4-dimethylaminopyridine (DMAP). Subsequent introduction of the 1,3-tetraisopropyldisiloxane-1,3-diyl group by reaction with TIPDSCl2 in CH2Cl2 and imidazole gave the crystalline 3,4-O-silyl ether 3 in 87% yield (Scheme 1). Removal of the trityl group could be achieved by treatment with silica-supported NaHSO4. In the process partial migration of the TIPDS-group to give the 4,6-O-substituted derivative was observed which then required cumbersome chromatography to separate the two resulting compounds.18 Hence, as an alternative, the mild trityl deprotection using BCl3 in CH2Cl2 at −20 °C and quenching of the reaction with triethylamine was carried out and afforded compound 4 in a gratifying 93% yield.19
Scheme 1. Synthesis of the β-(1 → 6)-Linked Diglucosamine Acceptor 11 via Two Different Routes.

Next, assembly of the β-(1 → 6)-linked diglucosamine backbone was performed on a multigram-scale. In the presence of FeCl3 the 2-chloroacetamido-2-deoxy-β-1-O-acetyl derivative 5 generates an 1,3-oxazoline intermediate and thus secures the formation of the β-anomeric product.20 This way, the crystalline β-(1 → 6)-linked disaccharide 6 was isolated in 77% yield; the β-anomeric configuration was confirmed by the value of the homonuclear J1,2 coupling constant (8.0 Hz). Subsequent conversion of the chloroacetamido group into the N-acetylated form was challenging. Reaction of 6 with thiourea afforded the corresponding N-acetyl derivative; the procedure, however, was difficult to reproduce and consistently led to thio-containing byproducts which were difficult to separate from the product.21 As an alternative, which concomitantly allowed an early stage removal of the silyl protecting group, hydrolysis of all protecting groups followed by ensuing O- and N-acetylation was performed. The five-step sequence included reaction of the 2-chloroacetamido group with pyridine22 at 90 °C, followed by ester and amide hydrolysis with aqueous NaOH. The crude product was subsequently O,N-acetylated followed by removal of the TIPDS group with TBAF in THF and O-acetylation to afford the penta-O-acetyl derivative 8 in 66% yield (over 5 steps). Having established this reaction sequence, it was decided to further simplify it by exploiting the higher reactivity of the primary hydroxyl group in N-acetyl glucosamine glycosyl acceptor derivatives.23 Direct glycosylation of the triol glycoside 1 with donor 5 in nitromethane/DCM in the presence of FeCl3 afforded the disaccharide 7 in acceptable yields (45%). Conversion of the chloroacetamido group into the N-acetyl group was performed similar to compound 6—without the need to remove the silyl ether group—and eventually gave disaccharide 8 in 81% yield. Overall yields (Route A 37%; Route B 36%) for both pathways were comparable, but route B saved four steps in protecting group manipulation.
In order to selectively protect the primary hydroxy group of the distal glucosamine unit, compound 8 was subjected to Zemplén transesterification affording 9 in near theoretical yield. Next, the 6′-OH group was selectively protected with a TBDPS-group using TBDPSCl and Huenig’s base in dry DMF, followed by a one-pot protection of the remaining OH-groups by acetylation with acetic anhydride in pyridine to deliver 10 in 85% yield. Silyl ether cleavage was carried out using HF-pyridine as reagent at 0 °C. This way, 4 → 6 O-acetyl migration could be minimized and the resulting glycosyl acceptor 11 was obtained in 77% yield by chromatography. The crude product, however, was of sufficient purity and could be directly subjected to glycosylation reaction with Kdo donors.
Glycosylation reactions with glycosyl donors of Kdo suffer from lack of anomeric selectivity, low reactivity due the electron-withdrawing ester group and facile formation of elimination products such as glycal ester 16.24 A limited number of groups have successfully accomplished the challenging glycosylation of the unreactive axial 5-OH group at the proximal unit of the central bacterial Kdo disaccharide α-d-Kdo-(2 → 4)-α-d-Kdo.25 The synthesis of the resulting 4,5-disubstituted oligosaccharides is further complicated by the facile formation of 1′ → 5 interlinked Kdo lactone derivatives under acidic conditions, which would then block glycoside formation. Recently, the per-O-acetylated 3-iodo-Kdo fluoride donor 17 has successfully been employed for the assembly of oligomeric Kdo derivatives.26 Reactions of 17 with disaccharide acceptor 11 promoted either by BF3·Et2O or TMSO-triflate in dichloromethane, however, were not successful and mainly unreacted educts were recovered from the reaction mixtures. Hence, the known 4,5;7,8-di-O-isopropylidene donor 15 -envisaged to be sufficiently α-selective- was selected for the glycosylation steps.27 Donor 15 was prepared via a route different from the previously published procedure, which had been based on direct fluorination of a 4,5;7,8-di-O-isopropylidene protected Kdo hemiketal.28 First, treatment of the peracetylated Kdo methyl ester 12 with HF-pyridine afforded known fluoride 13,29 which was then subjected to Zemplén transesterification to give the tetraol fluoride 14, suitable for further modification and regioselective introduction of protecting groups. Compound 14 is rather unstable and was therefore immediately transformed into the bench stable donor 15 in 64% yield (over two steps from 13) using camphorsulfonic acid and 2-methoxypropene (Scheme 2).
Scheme 2. Synthesis of the Kdo-(2 → 6)-Linked Trisaccharide Acceptor 22.

Coupling of Kdo fluoride donor 15 (2 equiv) to disaccharide acceptor 11 was accomplished using 2.2 equiv of BF3·Et2O as promoter in dichloromethane and in the presence of molecular sieves 4 Å. Trisaccharide 18 was obtained in 64% yield (2 steps, based on 10) after separation from minor byproducts, unreacted educt as well as glycal ester 16 by silica gel chromatography. Next the acetonide groups were hydrolyzed without affecting the acid-labile ketosidic linkage of Kdo by treatment with p-toluenesulfonic acid monohydrate (PTSA) in dry MeOH in 93% crude yield. Unfortunately silica gel chromatography did not lead to separation of the product from PTSA, and the use of basic ion-exchange resin (HCO3– form) led to partial saponification of the acetate groups.
Thus, the milder CeCl3-hydrate/oxalic acid had to be used, affording the tetraol 19 in 76% yield after column chromatography.30 Cleavage of the isopropylidene groups released the steric strain of the skew-boat Kdo conformation in compound 18 and regenerated the 5C2 conformation in compound 19, which then allowed to confirm the α-anomeric configuration of the Kdo unit on the basis of 1H NMR chemical shifts of the 3-deoxy protons as well as 13C NMR data of C-4 and C-6, respectively.31 In order to keep the number of protecting groups at a minimum, the side-chain diol system of Kdo was then protected by a carbonate ester group, based on previous experience in the synthesis of Kdo oligomers.23 Conversion of 19 into the 7,8-O-carbonyl protected trisaccharide 20 had to be performed under carefully controlled conditions. A solution of diphosgene in THF was slowly added at −50 °C to −40 °C to a solution of 19 and sym-collidine in THF. In this way compound 20 could be isolated in yields from 60 to 80% with minor formation of the 4,5;7,8-dicarbonyl derivative (∼10%) and recovery of unreacted 19 (∼10%). Whereas the introduction of the first Kdo unit could be accomplished in good yields, coupling of the lateral Kdo residue to trisaccharide acceptor 20 was less straightforward (Scheme 3, Table 1).
Scheme 3. Synthesis of the Kdo2GlcNAc2 Tetrasaccharide Acceptor 22.

Table 1. Optimization of Kdo-(2 → 4)-Kdo Glycoside Formation.
| isolated
yields (%) |
|||||||
|---|---|---|---|---|---|---|---|
| entry | donor [equiv] | BF3·Et2O [equiv] | T [°C] | time [min] | 21 | 22 | 20 |
| 1 | 2 | 2.1 | 0 | 120 | 40 | 16 | 20 |
| 2 | 1.5 | 1.6 | 0 | 100 | 52 | 25 | 12 |
| 3 | 1.5 | 1.6 | –20 | 120 | 40 | 42 | traces |
| 4a | 1.5 | 1.6 | 0 | 25 | 22 | 6 | 50 |
| 5b | 1.5 | 1.6 | 0 | 100 | 11 | 5 | 65 |
| 6c | 1.5 | 1.6 | 0 | 100 | 23 | 12 | 30 |
BF3·Et2O was slowly added during 20 min using a syringe pump.
Reaction was carried out with 1 equiv of NEt3.
Reaction was carried out with 1 equiv of DIPEA.
Promotion of the glycosylation step in the presence of BF3·Et2O and molecular sieves 4 Å in dichloromethane afforded modest yields of tetrasaccharide 21. The main byproduct of the reaction was identified as the 4,5-O-substituted monoisopropylidene trisaccharide 22 arising from isopropylidene cleavage and transfer to the diol 20. Variation of the reaction time and temperature (see Table 1, entries 1–4) did not lead to any reduction of byproduct formation and even the addition of base (Table 1, entry 4, 5) just slowed down the reaction rate without affecting the product distribution.
Fortunately, byproduct 22 can be reconverted into acceptor 20 by treatment with a 1:1 mixture of DCM/90% aq TFA. Thus, despite the progress in Kdo glycosylation chemistry, generally applicable, selective and high-yielding methods still need to be developed for synthesizing complex Kdo glycosides.
The orthogonally protected mannosyl donor 26, suitable for selective deprotection of positions 3 and 6 was elaborated from known thioglycoside 23 following published procedures (Scheme 4).32 First, compound 23 was subjected to 6-O-silylation using TBS-chloride and DMAP in pyridine which gave the silyl ether 24 in 93% yield. Next, the thioglycoside aglycon was removed by treatment of 24 with NIS/TFA to produce 25 in 92% yield, followed by subsequent introduction of the anomeric trichloroacetimidate according to R. Schmidt33 which provided donor 26 in 88% yield.
Scheme 4. Synthesis of the Mannosyl Trichloroacetimidate Donor 26.

Proceeding toward the branched pentasaccharide 27, donor 26 and acceptor 22 were first reacted for 3 h with 0.1 equiv TMSOTf as promotor in the presence of molecular sieves 4 Å in dry DCM at 0 °C resulting in a mixture of several isomers. One byproduct was isolated by HPLC purification and was identified as the imidate-connected pseudopentasaccharide. The structure of the imidate was proven by NMR data. One NH proton signal was missing and a low-field shifted signal was observed at 6.53 ppm which gave an HMBC-correlation to a 13C NMR signal at 162.0 ppm being characteristic of imidate groups.34 The nucleophilic properties of the amide oxygen of glucosamines had previously been described and higher amounts of promoter as well as higher temperatures were suggested to suppress these side reactions.14b Hence, the mannosylation reaction was repeated at room temperature in the presence of 0.3 equiv TMSOTf affording the desired product 27 in a good yield of 70% (Scheme 5). Unreacted glycosyl acceptor 21 could be recovered in 20% yield. No β-product was isolated and the α-anomeric configuration of the mannosyl residue was proven by the value of the heteronuclear coupling constant JC-1,H-1 (171 Hz).35 Deprotection of the two isopropylidene groups had to be carefully established in order to avoid concomitant hydrolysis of the acid-sensitive Kdo-linkages. Treatment of 27 with CeCl3/oxalic acid selectively removed the 7,8-O-ketal without affecting the 4,5-attached acetonide and thus gave the pentasaccharide 7,8-diol 28 in 67% yield. Compound 28 is a suitable precursor en route to 8-O-glycosylated Kdo core components of rhizobial LPS. Full hydrolysis of both isopropylidene groups was achieved using 90% aqueous TFA in DCM to give 29 in 69% yield. Global deprotection of the remaining protecting groups from 29 was sequentially performed via Zemplén deacylation, hydrogenolysis of benzyl and NAP-group (using H-cube equipment) with concomitant reduction of the terminal azide and alkaline hydrolysis of the methyl ester groups with aqueous sodium hydroxide to afford the target pentasaccharide 30 in 69% yield after final purification on LH-20.
Scheme 5. Synthesis of the Pentasaccharide 27 and Global Deprotection.

13C NMR data of compound 30 (Table 2) could be fully assigned and were in perfect agreement (when correcting for different referencing of chemical shifts) with published data of a related pentasaccharide fragment from Agrobacterium tumefaciens A1.7c Significant low-field shifts were observed for C-4 and C-5 of the branched Kdo residue, whereas the data of the lateral Kdo unit were consistent with similar NMR characteristics of α-(2 → 4)-linked Kdo disaccharide fragments.36
Table 2. 13C NMR Chemical Shifts (δ) of Compound 30.
| carbon | → 6)-α-GlcpN | → 6)-β-GlcpN | → 4,5)-α-Kdop- | α-Kdop-(2 → 4) | α-Manp-(1 → 5) | spacer group |
|---|---|---|---|---|---|---|
| 1 | 97.28 | 102.32 | 175.81b | 175.45b | 101.39 | 65.88 |
| 2 | 54.37 | 56.43 | 100.57 | 102.14 | 70.99 | 27.67 |
| 3 | 71.81 | 74.80 | 35.21 | 35.35 | 71.20 | 38.04 |
| 4 | 70.72 | 71.44 | 71.27 | 66.78 | 67.08 | |
| 5 | 71.75 | 75.24 | 73.99 | 67.50 | 73.51 | |
| 6 | 69.92 | 63.01 | 73.08 | 72.78 | 61.37 | |
| 7 | 70.18 | 71.27 | ||||
| 8 | 64.40 | 63.86 | ||||
| CH3 | 23.06a | 22.67a | ||||
| C=O | 175.17b | 175.17b |
Assignments may be reversed.
Assignments may be reversed.
In conclusion, a central fragment of rhizobial and agrobacterial inner core LPS has been synthesized in a limited number of steps relying mainly on ester, benzyl-type and isopropylidene protecting groups. The latter protecting group was not fully compatible with glycosylation conditions although the bis-isopropylidene protected Kdo fluoride donor was sufficiently reactive and α-selective in the glycosylation steps. Extension of this work toward the oligomannosidic fragments and neoglycoconjugates will be published in due course.
Experimental Section
All purchased chemicals were used without further purification unless stated otherwise. Solvents were dried over activated 4 Å (CH2Cl2, pyridine, THF, DMF, CH3NO2) and 3 Å (CH3CN) molecular sieves. Cation exchange resin DOWEX 50 H+ was regenerated by consecutive washing with HCl (3 M), water and dry MeOH. Aqueous solutions of salts were saturated unless stated otherwise. Concentration of organic solutions was performed under reduced pressure <40 °C. Optical rotations were measured with a PerkinElmer 243 B or Anton Paar MCP100 Polarimeter. Thin layer chromatography was performed on Merck precoated plates: generally on 5 × 10 cm, layer thickness 0.25 mm, Silica Gel 60F254; alternatively on HPTLC plates with 2.5 cm concentration zone (Merck). Spots were detected by dipping reagent (anisaldehyde-H2SO4).37 For column chromatography silica gel (0.040–0.063 mm) was used. HP-column chromatography was performed on prepacked columns (YMC-Pack SIL-06, 0.005 mm, 25 × 1 cm and 25 × 2 cm). NMR spectra were recorded with a Bruker Avance III 600 instrument (600.22 MHz for 1H, 150.93 MHz for 13C) using standard Bruker NMR software. 1H spectra were referenced to 7.26 (CDCl3), 3.34 (MeOD) and 0.00 (D2O, external calibration to 2,2-dimethyl-2-silapentane-5-sulfonic acid) ppm unless stated otherwise. 13C spectra were referenced to 77.00 (CDCl3), 49.00 (MeOD) and 67.40 (D2O, external calibration to 1,4-dioxane) ppm. Assignments were based on COSY, HSQC, HMBC and TOCSY data. ESI-MS data were obtained on a Micromass Q-TOF Ultima Global instrument.
3-Azido-1-propyl 2-acetamido-2-deoxy-6-O-triphenylmethyl-α-d-glucopyranoside (2)
Compound 1 (6.23 g, 20.5 mmol) was dried in vacuo, purged with dry argon and dissolved in anhydrous pyridine (150 mL). DMAP (250 mg, 2.05 mmol) and trityl chloride (12.6 g, 45.2 mmol) were added and the solution was stirred at 55 °C for 20 h. The reaction was quenched by adding MeOH, followed by concentration and coevaporation of the remainder with toluene (2 × 10 mL). The product was purified by flash chromatography (EtOAc/Hex 1:1 → 1:0, then EtOAc/MeOH 50:1 → 20:1) to yield 10.90 g (90%) of 2 as colorless prisms, mp 97–98 °C (dec; EtOAc); [α]D20 + 35 (c 1.0 CHCl3); Rf 0.56 (EtOAc/MeOH 9:1); 1H NMR (600 MHz, CDCl3) δ = 7.45, 7.29, and 7.22 (3 m, 15 H, arom. H), 6.00 (d, 1 H, JNH,2 8.4 Hz, NH), 4.83 (d, 1 H, J1,2 3.9 Hz, H-1), 4.09 (ddd, 1 H, J2,3 10.4 Hz, H-2), 3.87 (dt, 1 H, J 6.0, J 10.1 Hz, OCH2a), 3.68 (ddd, 1 H, J5,6a 3.7, J5,6b 5.5, J5,4 9.6 Hz, H-5), 3.64 (br t, 1 H, J4,3 9.2 Hz, H-4), 3.57–3.51 (m, 2 H, OCH2b, H-3), 3.48–3.34 (m, 5 H, H-6a, CH2N3, H-6b, OH), 2.69 (br s, 1 H, OH), 2.05 (s, 3 H, NAc), 1.92 (dt, 2 H, J 6.3 Hz, CH2); 13C NMR (125 MHz, CDCl3) δ = 172.0 (C=O), 143.9 (C–Ar), 128.8, 128.0, and 127.2 (C–Ar), 97.4 (C-1), 87.0 [C(Ph3)], 74.1 (C-4), 72.7 (C-3), 70.8 (C-5), 65.0 (OCH2), 63.9 (C-6), 53.8 (C-2), 49.0 (CH2N3), 28.8 (CH2), 23.4 (CH3CO); HRMS (ESI-TOF) m/z [M + H]+ Calcd for C30H35N4O6 547.2557, found 547.2563. Anal. Calcd for C30H34N4O6: C, 65.92; H, 6.27; N, 10.25. Found: C, 65.74; H, 6.54; N, 9.75.
3-Azido-1-propyl 2-acetamido-2-deoxy-3,4-O-(tetraisopropyldisiloxane-1,3-diyl)-6-O-triphenylmethyl-α-d-glucopyranoside (3)
1H-Imidazole (2.95 g, 43.4 mmol) followed by TIPDSCl2 (5.47 mL, 17.4 mmol) were added at 0 °C to a solution of alcohol 2 (4.74 g, 8.67 mmol) in dry CH2Cl2 (70 mL). The reaction mixture was stirred overnight at 40 °C. The reaction mixture was quenched by adding aq satd NH4Cl solution (50 mL) and diluted with CH2Cl2 (150 mL). After phase separation, the aqueous layer was extracted with CH2Cl2 (100 mL). The combined organic layers were washed with water (50 mL), brine (30 mL), dried (Na2SO4) and concentrated. The residue was dissolved in the minimal amount of EtOAc (approximately 4 mL) followed by addition of n-hexane (approximately 200 mL). Crystallization was allowed to proceed for 1–2 h at rt followed by storage overnight at 4 °C, which gave 6.0 g (87%) of 3 as colorless prisms, mp 182–183 °C (EtOAc/hexane); [α]D23 + 44.2 (c 1.1, CHCl3); Rf 0.64 (hexane/EtOAc 1:1). 1H NMR (600 MHz, CDCl3) δ = 7.47–7.44 (m, 6 H, arom H), 7.28–7.25 (m, 6 H, arom H), 7.23–7.19 (m, 3 H, arom H), 5.59 (d, 1 H, J2,NH 9.5 Hz, NH), 4.90 (d, 1 H, J1,2 3.8 Hz, H-1), 4.23 (ddd, 1 H, J1,2 3.7, JNH,2 9.5, J3,2 10.0 Hz, H-2), 4.05 (dt, 1 H, J 6.1, 10.3 Hz, OCH2a), 3.84 (ddd, 1 H, J6a,5 1.7, J6b,5 7.6, J4,5 8.9 Hz, H-5), 3.71 (dd, 1 H, J4,3 8.1, J2,3 10.2 Hz, H-3), 3.66 (dt, 1 H, J 6.1, 10.3 Hz, OCH2b), 3.51–3.40 (m, 3 H, H-4, CH2N3), 3.36 (dd, 1 H, J5,6a 1.7, J6b,6a 9.7 Hz, H-6a), 3.17 (dd, 1 H, J5,6b 7.6, J6a,6b 9.7 Hz, H-6b), 2.02 (dt, 2 H, J 1.1, 6.4 Hz, CH2), 1.99 (s, 3 H, NAc), 1.09–0.68 [m, 28 H, 4 × (CH3)2CH]; 13C NMR (150 MHz, CDCl3) δ = 169.7 (C=O), 144.2 (C–Ar), 129.9, 127.9, and 127.1 (C–Ar), 97.6 (C-1), 86.7 [C(Ph3)], 75.8 (C-3), 74.1 (C-4), 72.2 (C-5), 65.2 (OCH2), 64.3 (C-6), 53.3 (C-2), 49.2 (CH2N3), 28.8 (CH2), 23.5 (CH3CO), 17.7, 17.5, 17.4, 17.3, and 17.2 [4(CH3)2CH], 12.8, 12.7, 12.5, and 12.4 [4 × (CH3)2CH]; HRMS (ESI-TOF) m/z [M + Na]+ Calcd for C42H60N4NaO7Si2 811.3898, found 811.3897. Anal. Calcd for C42H60N4O7Si2: C, 63.93; H, 7.66; N, 7.10. Found: C, 63.89; H, 7.83; N, 6.95.
3-Azido-1-propyl 2-acetamido-2-deoxy-3,4-O-(tetraisopropyldisiloxane-1,3-diyl)-α-d-glucopyranoside (4)
A 1.0 M solution of BCl3 in CH2Cl2 (19.14 mL) was added dropwise to a solution of 3 (10.79 g, 13.67 mmol) in dry CH2Cl2 (200 mL) under Argon and stirred for 90 min at −20 °C. The reaction was quenched with MeOH (10 mL) and triethylamine (7 mL) and diluted with CH2Cl2 (100 mL). The organic layer was washed with satd aq NaHCO3 (150 mL) and brine (100 mL) and the water phase was twice re-extracted with CH2Cl2. The combined organic phase was dried (MgSO4) and concentrated. Purification of the residue by silica gel chromatography (EtOAc/hexane 1:1 → 2.5:1) gave 4 (6.94 g, 93%) as colorless crystals; 90–92 °C (EtOAc/hexane); [α]D20 +70.6 (c 1.0, CHCl3); Rf 0.17 (n-hexane/EtOAc 1:1); 1H NMR (600 MHz, CDCl3) δ = 5.59 (d, 1 H, J2,NH 9.2 Hz, NH), 4.80 (d, 1 H, J1,2 3.7 Hz, H-1), 4.18 (ddd, 1 H, J1,2 3.4, JNH,2 9.2, J3,2 9.7 Hz, H-2), 3.85 (dd, 1 H, J5,6a 3.0, J6b,6a 11.6 Hz, H-6a), 3.81 (dt, 1 H, J 5.9, 10.1 Hz, OCH2a), 3.77–3.72 (m, 2 H, H-6b, H-3), 3.69 (dd, 1 H, J3,4 8.5, J5,4 9.2 Hz, H-4), 3.60 (ddd, 1 H, J6a,5 3.0, J6b,5 4.9, J4,5 9.2 Hz, H-5), 3.50 (dt, 1 H, J 6.0, 10.2 Hz, OCH2b), 3.45–3.35 (m, 2 H, CH2N3), 1.97 (s, 3 H, CH3CO), 1.93–1.88 (m, 2 H, CH2), 1.10–0.91 [m, 28 H, 4 × (CH3)2CH]; 13C NMR (150 MHz, CDCl3) δ = 169.7 (C=O), 97.9 (C-1), 75.4 (C-3), 73.6 (C-4), 72.5 (C-5), 65.5 (OCH2), 62.3 (C-6), 53.4 (C-2), 49.0 (CH2N3), 28.7 (CH2), 23.4 (CH3CO), 17.7, 17.5, 17.4, 17.3, and 17.3 [4 × (CH3)2CH], 13.0, 12.9, 12.5, and 12.3 [4 × (CH3)2CH]; HRMS (ESI-TOF) m/z [M + Na]+ Calcd for C23H46N4NaO7Si2 569.2803, found 569.2802. Anal. Calcd for C23H46N4O7Si2: C, 50.52; H, 8.48; N, 10.25. Found: C, 50.71; H, 8.85; N, 9.92.
3-Azido-1-propyl 3,4,6-tri-O-acetyl-2-chloroacetamido-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-2-deoxy-3,4-O-(tetraisopropyldisiloxane-1,3-diyl)-α-d-glucopyranoside (6)
A suspension of 4 (7.3 g, 13.35 mmol) in dry CH2Cl2 (150 mL), 5 (7.92 g, 18.69 mmol) and 4 Å molecular sieves (10 g) was stirred for 30 min at rt. Then FeCl3 (2.81 g, 17.35 mmol) was added and stirring was continued for 42 h. The mixture was diluted with CH2Cl2 (400 mL) and filtered over a layer of Celite. The filtrate was washed with water, then with satd aq NaHCO3 (300 mL) and brine (200 mL). The water phase was twice extracted with CH2Cl2 (2 × 100 mL), the organic phases were combined, dried (MgSO4) and concentrated. The crude was purified by column chromatography (n-hexane/EtOAc 1:1 → 1:3.5) to yield 1.90 g of unreacted donor 5 and 9.40 g (77%) of 6 as colorless needles; mp 187–189 °C; [α]D20 +34.5 (c 1.0, CHCl3); Rf 0.19 (EtOAc/n-hexane 3:1); 1H NMR (600 MHz, CDCl3) δ = 6.63 (d, 1 H, J2′,NH′ 8.8 Hz, NH′), 5.56 (d, 1 H, J2,NH 9.3 Hz, NH), 5.32 (dd, 1 H, J4′,3′ 9.1, J2′,3′ 10.4 Hz, H-3′), 5.06 (t, 1 H, J3′,4′ 9.2, J5′,4′ 9.2 Hz, H-4′), 4.77 (d, 1 H, J1,2 8.0 Hz, H-1′), 4.72 (d, 1 H, J1,2 3.2 Hz, H-1), 4.25 (dd, 1 H, J5′,6a′ 5.1, J6b′,6a′ 12.2 Hz, H-6a′), 4.20 (dd, 1 H, J5,6a 1.7, J6b,6a 10.4 Hz, H-6a), 4.16 (ddd, 1 H, J1,2 3.3, JNH,2 9.3, J3,2 9.6 Hz, H-2), 4.11 (dd, 1 H, J5′,6b′ 2.5, J6a′,6b′ 12.2 Hz, H-6b′), 3.94 (s, 2 H, CH2Cl), 3.90 (ddd, 1 H, J1′,2′ 8.0, JNH′,2′ 8.8, J3′,2′ 10.4 Hz, H-2′), 3.79–3.75 (m, 2 H, OCH2a, H-5), 3.74–3.69 (m, 2 H, H-3, H-5′), 3.55 (dd, 1 H, J5,6b 8.1, J6a,6b 10.4 Hz, H-6b), 3.48–3.36 (m, 4 H, OCH2b, H-4, CH2N3), 2.06, 2.02, 2.01 (3 s, each 3 H, 3 × CH3CO), 1.96 (s, 3 H, NHAc), 1.92–1.87 (m, 2 H, CH2), 1.09–0.90 [m, 28 H, 4 × (CH3)2CH]; 13C NMR (150 MHz, CDCl3) δ = 170.7, 170.7, 169.7, and 169.5 (C=O), 166.3 (ClCH2C=O), 101.0 (C-1′), 97.6 (C-1), 75.6 (C-3), 74.3 (C-4), 72.1 (C-5′), 71.9 (C-5), 71.7 (C-3′), 69.8 (C-6), 68.7 (C-4′), 65.4 (OCH2), 62.4 (C-6′), 55.2 (C-2′), 53.3 (C-2), 49.1 (CH2N3), 42.6 (CH2Cl), 28.6 (CH2), 23.4 (CH3, HNCOCH3), 20.8 and 20.7 (CH3CO), 17.6, 17.5, 17.3 and 17.2, [4 × (CH3)2CH], 12.9, 12.8, 12.6, and 12.4 [4(CH3)2CH]; HRMS (ESI-TOF) m/z [M + H]+ Calcd for C37H65ClN5O15Si2 910.3704, found 910.3712. Anal. Calcd for C37H64ClN5O15Si2: C, 48.81; H, 7.08; N, 7.69. Found: C, 48.60; H, 6.87; N, 7.48.
3-Azido-1-propyl 3,4,6-tri-O-acetyl-2-chloroacetamido-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-2-deoxy-α-d-glucopyranoside (7)
A suspension of 1 (6.3 g, 20.70 mmol), 5 (7.92 g, 18.69 mmol) and 4 Å molecular sieves (27 g) in a mixture of dry CH2Cl2 and dry CH3NO2 (180 mL; 10:1), was stirred for 2 h at rt. Then FeCl3 (5.04 g, 31.06 mmol) was added and stirring was continued for 72 h. The reaction was quenched by addition of satd aq NaHCO3 and the suspension was filtered over a layer of Celite. The phases were separated and the aqu phase was extracted with DCM. The organic phase was dried (Na2SO4) and concentrated. The crude was purified by column chromatography (DCM/MeOH 20:1 → 10:1) to yield 5.5 g (45%) of 7 as colorless amorphous solid; [α]D20 +45.2 (c 0.9, MeOH); Rf 0.48 (DCM/MeOH 10:1); 1H NMR (600 MHz, CDCl3) δ = 5.31 (dd, 1 H, J3′,2′ 10.7, J3′,4′ 9.3 Hz, H-3′), 5.03 (dd, 1 H, J4′,3′ 9.3, J4′,5′ 9.8 Hz, H-4′), 4.76 (d, 1 H, J1,2 3.5 Hz, H-1), 4.76 (d, 1 H, J1′,2′ 8.3 Hz, H-1′), 4.33 (dd, 1 H, J6a′,6b′ 12.3, J6a′,5′ 4.7 Hz, H-6a′), 4.19 (dd, 1 H, J6b,6a 11.0, J6b,5 1.8 Hz, H-6b), 4.17 (dd, 1 H, J6b′,6a′ 12.3, J6b′,5′ 2.5 Hz, H-6b′), 4.03 (d, 2 H J 1.3 Hz, CH2Cl), 3.97 (d, 1 H, J2′,3′ 10.5, J2′,1′ 8.3 Hz, H-2′), 3.90 (dd, 1 H, J2,3 10.7, J2,1 3.7 Hz, H-2), 3.85 (ddd, 1 H, J5′,6b 11.0, J5′,6a′ 4.7, J5′,6b′ 2.5 Hz, H-5′), 3.80–3.72 (m, 2 H, OCH2CH2, H-5), 3.68–3.63 (m, 2 H, H-3, H-6a), 3.53–3.43 (m, 3 H, CH2N3, H-3), 3.26 (dd, 1 H, J4,3 10.1, J4,5 8.7 Hz, H-4), 2.09 (s, 3 H, CH3), 2.04 (s, 3 H, CH3), 2.00 (s, 6 H, CH3), 1.93–1.86 (m, 2 H, OCH2CH2); 13C NMR (150 MHz, CDCl3) δ = 173.6, 172.4, 171.9, 171.3 (C=O), 169.5 (COCH2Cl), 102.5 (C-1′), 98.5 (C-1), 73.8 (C-3′), 73.0 (C-5′), 72.8 (C-3), 72.7 (C-4), 72.5 (C-5), 71.1 (C-6), 70.3 (C-4′), 65.8 (OCH2), 63.3 (C-6′), 55.9 (C-2′), 55.4 (C-2), 49.3 (CH2N3), 43.3 (CH2Cl), 29.9 (OCH2CH2), 22.6 (CH3CO), 20.7 (CH3CO) and 20.6 (2 C, CH3CO). HRMS (ESI-TOF) m/z [M + H]+ Calcd for C25H39ClN5O14 668.2177, found 668.2179. Anal. Calcd for C25H38ClN5O14: C, 44.95; H, 5.73; N, 10.48. Found: C, 44.99; H, 5.72; N, 9.89.
3-Azido-1-propyl 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (8 from 6)
Compound 6 (5.65 g; 6.205 mmol) was dissolved in pyridine (50 mL) under Argon, warmed to 90 °C and stirred for 2 h at this temperature. Then the solution was allowed to cool down to rt and a 1:1 mixture of 5% aq NaOH and MeOH (50 mL) was added. The reaction mixture was stirred for 5 h and subsequently the solvent was removed in vacuo. The crude was dissolved in pyridine (50 mL) under Argon followed by the addition of DMAP (0.076 g; 0.62 mmol). Then Ac2O (11.73 mL; 124.1 mmol) was added dropwise and the reaction mixture was stirred at rt for 40 h. The suspension was quenched by the addition of aq satd NaHCO3 and the aqueous phase was extracted 3 times with CH2Cl2. The combined organic phases were dried (Na2SO4), the solvent was removed in vacuo and the residue was coevaporated with toluene. The residue was dissolved in dry THF under Argon and 1 M TBAF in THF (24 mL; 24 mmol) was added dropwise at 0 °C. The reaction mixture was then warmed to rt and stirred for 2.5 h. In order to remove TBAF, MeOH (20 mL), CaCO3 (5.8 g) and dry DOWEX H+ ion-exchange resin (17 g) were added. The suspension was stirred for 1 h at rt, then filtered over Celite and the solvent was removed in vacuo. The crude intermediate was acetylated according to the same procedure described above, using pyridine (50 mL), Ac2O (5.865 mL; 62.05 mmol) and DMAP (0.076 g; 0.62 mmol). Workup as described followed by silica gel chromatography (DCM/MeOH 50:1 → 10:1) afforded 8 (2.95 g; 66%); colorless crystals, mp 208–209 °C (EtOAc/n-hexane); [α]D20 +25.8 (c 1.1, CHCl3); Rf 0.53 (DCM/MeOH 20:1); 1H NMR (600 MHz, CDCl3) δ = 5.97 (d, 1 H, J2′,NH′ 8.5 Hz, NH′), 5.74 (d, 1 H, J2,NH 9.2 Hz, NH), 5.21 (dd, 1 H, J2′,3′ 10.4, J4′,3′ 9.4 Hz, H-3′), 5.20 (dd, 1 H, J4,3 9.5, J2,3 10.7 Hz, H-3), 5.09 and 5.08 (br t, each 1 H, H-4, H-4′), 4.83 (d, 1 H, J1,2 3.8 Hz, H-1), 4.50 (d, 1 H, J1′,2′ 8.5 Hz, H-1′), 4.29 (ddd, 1 H, J2,3 10.8 Hz, H-2), 4.25 (dd, 1 H, J6′a,6′b 12.3, J6′a,5′ 4.7 Hz, H-6′a), 4.13 (dd, 1 H, J6′a,6′b 12.3, J6′a,5′ 2.5 Hz, H-6′b), 4.04 (dd, 1 H, J6a,6b 11.0, J6a,5 2.0 Hz, H-6a), 3.99 (dt, 1 H, J2′,3′ 10.5 Hz, H-2′), 3.85 (ddd, 1 H, J5,4 10.0, J5,6b 4.6 Hz, H-5), 3.81 (dt, 1 H, J 10.2, 6.2 Hz, OCH2CH2CH2N3), 3.68 (ddd, 1 H, J5′,4′ 9.9 Hz, H-5′), 3.49 (dt, 1 H, J 10.2, 6.2 Hz, OCH2CH2CH2N3), 3.47–3.39 (m, 3 H, H-6b, CH2N3), 2.08, 2.07, 2.03, 2.02, 2.01, 1.97, and 1.94 (7 s, each 3 H, 7 × CH3CO), 1.93–1.89 (m, 2 H, OCH2CH2); 13C NMR (150 MHz, CDCl3) δ = 171.3, 170.8, 170.7, 170.3, 169.9, 169.8, 169.2 (CH3C=O), 101.3 (C-1′), 96.1 (C-1), 72.7 (C-3′), 71.9 (C-5′), 71.4 (C-3), 68.5 (2 C, C-4′, C-5), 68.2 (C-4), 67.7 (C-6), 65.2 (OCH2), 62.0 (C-6′), 54.3 (C-2′), 51.8 (C-2), 48.5 (CH2N3), 28.5 (OCH2CH2), 23.1, 20.7, 20.6, 20.5 (7 C, CH3CO). HRMS (ESI-TOF) m/z [M + H]+ Calcd for C29H44N5O16 718.2778, found 718.2775. Anal. calcd. for C29H43N5O16: C, 48.53; H, 6.04; N, 9.76. Found: C, 48.54; H, 6.27; N, 9.79.
3-Azido-1-propyl 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (8 from 7)
Compound 7 (0.906 g; 1.36 mmol) was dissolved in pyridine (10 mL) under Argon, warmed to 90 °C and stirred for 2 h at this temperature. Then the solution was allowed to cool down to rt and a 1:1 mixture of 10% aq NaOH and MeOH (10 mL) was added. The reaction mixture was stirred for 5 h and subsequently the solvent was removed in vacuo. The crude intermediate was dissolved in pyridine (10 mL) under Argon followed by the addition of DMAP (0.017 g; 0.14 mmol). Then Ac2O (2.564 mL; 27.12 mmol) was added dropwise and the reaction mixture was stirred at rt for 16 h. The suspension was quenched by the addition of aq satd NaHCO3 and the aqueous phase was extracted 3 times with CH2Cl2. The combined organic phases were dried (Na2SO4) and the solvent was removed in vacuo. Silica chromatography (DCM/MeOH 50:1 → 10:1) afforded 7 (0.787 g; 81%). Analytical data were identical to 8 obtained from 6.
3-Azido-1-propyl 2-acetamido-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-2-deoxy-α-d-glucopyranoside (9)
Compound 8 (2.90 g; 4.04 mmol) was dissolved in dry MeOH (25 mL) under Argon and a 0.1 M solution of NaOMe (11.5 mL) was added. The reaction mixture was stirred at rt for 24 h and subsequently made neutral by the addition of DOWEX AG1X resin (H+-form). The resin was filtered off and the filtrate was concentrated to afford 9 (2.05 g; ∼100%), colorless amorphous solid; [α]D20 +59.4 (c 1.1, MeOH); Rf 0.19 (CHCl3/MeOH/H2O 4:1:0.1) 1H NMR (600 MHz, MeOD) δ = 4.76 (d, 1 H, J1,2 3.6 Hz H-1), 4.46 (d, 1 H, J1′,2′ 8.1 Hz, H-1′), 4.20 (dd, 1 H, J6a,6b 10.7, J6a,5 2.1 Hz, H-6a), 3.92 (dd, 1 H, J6′a,6′b 11.9, J6′a,5′ 2.3 Hz, H-6′a), 3.91 (dd, 1 H, J2,3 10.7 Hz, H-2), 3.80 (dt, 1 H, J 10.3, 5.9 Hz, OCH2), 3.73–3.69 (m, 3 H, H-2′, H-6b′, H-5), 3.66 (dd, 1 H, J2,3 10.7, J4,3 8.8 Hz, H-3), 3.64 (dd, 1 H, J6b,5 6.5 Hz, H-6b), 3.54–3.43 (m, 4 H, H-3′, OCH2, CH2N3), 3.35 (br t, 1 H, J4′,5′ 9.7 Hz, H-4′), 3.32 (dd, 1 H, J4,5 9.9, J4,3 8.6 Hz, H-4), 3.30 (ddd, 1 H, J6′b,5′ 5.9 Hz, H-5′), 2.01 (2 × s, 6 H, CH3), 1.90 (dt, 2 H, J 12.7, 6.4 Hz, OCH2CH2); 13C NMR (150 MHz, CDCl3) δ = 173.8 and 173.6 (CH3C=O), 103.3 (C-1′), 98.5 (C-1), 78.0 (C-5′), 76.2 (C-3′), 72.8 (C-3), 72.7 (C-5), 72.6 (C-4), 72.2 (C-4′), 70.4 (C-6), 65.6 (OCH2CH2), 62.8 (C-6′), 57.4 (C-2′), 55.4 (C-2), 49.5 (CH2N3), 29.8 (CH2CH2N3), 23.1 and 22.6 (CH3CO). HRMS (ESI-TOF) m/z [M + H]+ Calcd for C19H34N5O11 508.2249, found 508.2262.
3-Azido-1-propyl 2-acetamido-3,4-di-O-acetyl-6-O-tert-butyldiphenylsilyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (10)
A solution of 9 (2.80 g; 5.52 mmol), freshly distilled DIPEA (8.45 mL; 49.66 mmol) and TBDPSCl (2.15 mL; 8.28 mmol) in dry DMF (30 mL) was stirred under Argon at rt for 16 h until complete conversion to the monosilylated intermediate. Subsequently dry pyridine (30 mL) and DMAP (64 mg; 0.55 mmol) were added, followed by the dropwise addition of Ac2O (4.25 mL; 44.14 mmol). After 24 h TLC indicated full conversion and the reaction was quenched by the addition of aq satd NaHCO3. The aqueous phase was extracted 3 times with CH2Cl2 and the combined organic phases were dried (Na2SO4). The solution was concentrated and the product was purified by flash silica chromatography (CH2Cl2/MeOH 20:1) to give 10 (4.31 g; 85%) as slightly off-white crystals, mp 193–195 °C (EtOAc/n-hexane); [α]D20 +56.45 (c 1.1, CHCl3); Rf 0.45 (DCM/MeOH 20:1). 1H NMR (600 MHz, CDCl3) δ = 7.68–7.63 (m, 4 H, arom H), 7.43–7.34 (m, 6 H, arom H), 5.83 (d, 1 H, J2′,NH′ 8.5 Hz, NH′), 5.70 (d, 1 H, J2,NH 9.4 Hz, NH), 5.20 (dd, 1 H, J2,3 10.6, J4,3 9.4 Hz, H-3), 5.17 (dd, 1 H, J4′,3′ 9.4, J2′,3′ 10.5 Hz, H-3′), 5.13 (t, 1 H, J4,5 9.6 Hz, H-4), 5.07 (t, 1 H, J4′,5′ 9.5 Hz, H-4′), 4.85 (d, 1 H, J1,2 3.7 Hz, H-1), 4.43 (d, 1 H, J1′,2′ 8.4 Hz, H-1′), 4.30 (ddd, 1 H, H-2), 4.06 (dd, 1 H, J6a,6b 11.1, J6a,5 1.9 Hz, H-6a), 3.99 (ddd, 1 H, H-2′), 3.84 (ddd, 1 H, J6b,5 4.3 Hz, H-5), 3.83 (dt, 1 H, J 10.2, J 6.2 Hz, OCH2), 3.74 (dd, 1 H, J6′a,6′b 11.5, J6′a,5′ 5.4 Hz, H-6′a), 3.70 (dd, 1 H, J6′a,6′b 11.5, J6′a,5′ 2.4 Hz, H-6′b), 3.53 (ddd, 1 H, H-5′), 3.50 (dt, 1 H, J 10.1, 6.1 Hz, OCH2), 3.47–3.39 (m, 2 H, CH2N3), 3.35 (dd, 1 H, H-6b), 2.02 (s, 6 H, 2 × CH3CO), 2.01, 1.97, 1.95, and 1.88 (4s, each 3 H, 4 × CH3CO), 1.94–1.88 (m, 2 H, OCH2CH2), 1.03 [s, 9 H, (CH3)3C]; 13C NMR (150 MHz, CDCl3) δ = 171.3, 171.0, 170.2, 169.8, 169.7, 169.1 (C=O), 135.7, 135.6, 133.3, 133.2, 129.7, 127.7, 127.6 (C–Ar), 101.4 (C-1′), 97.2 (C-1), 75.0 (C-5′), 73.2 (C-3′), 71.5 (C-3), 68.8 (C-4′), 68.6 (C-5), 68.2 (C-4), 67.6 (C-6), 65.2 (OCH2), 62.9 (C-6′), 54.5 (C-2′), 51.9 (C-2), 48.5 (CH2N3), 28.6 (OCH2CH2), 26.7 [(CH3)3CSi], 23.2 (2 C, CH3CO), 20.7 (3 C, CH3CO), 20.6 (CH3CO) and 19.2 [(CH3)3CSi]. HRMS (ESI-TOF) m/z [M + H]+ Calcd for C43H60N5O15Si 914.3850, found 914.3877.
Desilylation of 10
A solution of 10 (0.373 g, 0.41 mmol) in dry THF (10 mL) under Argon was cooled to 0 °C and a solution of HF·pyridine (0.6 mL; 4.2 mmol; 70% HF) was added dropwise. The solution was stirred for 16 h and was subsequently quenched by the addition of satd aqueous NaHCO3. The solution was extracted with CH2Cl2 (3×), the combined organic phases were dried (Na2SO4) and the solvent was removed in vacuo. The crude was purified by silica flash chromatography (DCM/MeOH 100:3 → 100:4) to give 11 (0.211 g; 77%) as colorless amorphous solid. Rf 0.24 (DCM/MeOH 25:1); 1H NMR (600 MHz, CDCl3) δ = 5.93 (d, 1 H, JNH′,2′ 8.5 Hz, NH′), 5.72 (d, 1 H, JNH,2 9.4 Hz, NH), 5.20 (dd, 1 H, J3′,2′ 12.7, J3′,4′ 9.4 Hz, H-3′), 5.19 (dd, 1 H, J3,2 12.1, J3,4 9.9 Hz, H-3), 5.13 (t, 1 H, J4,5∼J4,3 9.9 Hz, H-4), 4.95 (t, 1 H, J4′,3′∼J4′,5′ 9.4 Hz, H-4′), 4.83 (d, 1 H, J1,2 3.6 Hz, H-1), 4.53 (d, 1 H, J1′,2′ 8.8 Hz, H-1′), 4.28 (ddd, 1 H, J2,3 9.9, J2,NH 9.4, J2,1 3.6 Hz, H-2), 4.04–3.99 (m, 2 H, H-2′, H-6a), 3.84–3.77 (m, 2 H, H-5, OCH2), 3.70 (dd, 1 H, J6a′,6b′ 12.7, J6a′,5′ 2.2 Hz, H-6a′), 3.58 (dd, 1 H, J6a′,6b′ 12.6, J6b′,5′ 5.3 Hz, H-6b′), 3.53–3.38 (m, 5 H, H-5′, OCH2, H-6b, CH2N3), 2.06, 2.03, 2.00, 1.96, 1.93 (6 × s, each 3 H, 6 CH3CO), 1.92–1.88 (m, 2 H, OCH2CH2); 13C NMR (150 MHz, CDCl3) δ = 171.5, 171.0, 170.4, 170.3, 170.2, 170.0 (C=O), 101.2 (C-1′), 97.4 (C-1), 74.7 (C-5′), 73.0 (C-3′), 71.6 (C-3), 69.0 (C-4′), 68.6 (2 C, C-4, C-5), 67.2 (C-6), 65.4 (OCH2), 61.6 (C-6′), 54.2 (C-2′), 52.0 (C-2), 48.7 (CH2N3), 28.7 (OCH2CH2), 23.3 (2 C, CH3CONH), 21.0, 20.8 (4 C, CH3CO). HRMS (ESI-TOF) m/z [M – H]− calcd for C27H40N5O15: 674.2526, found 674.2517.
Methyl (3-deoxy-4,5;7,8-di-O-isopropylidene-α-d-manno-oct-2-ulopyranosyl)onate fluoride (15)
A solution of 13 (3.65 g, 8.65 mmol, prepared from 12 according to ref (28)) was dissolved in dry MeOH (90 mL) under Argon and was cooled to 0 °C. Subsequently 0.1 M NaOMe (43 mL; 4.3 mmol) was added and the solution was stirred for 6 h at 0 °C. The reaction was quenched by the addition of DOWEX 50 cation exchange resin (H+-form) to give pH 7.5. The resin was filtered off and the filtrate was concentrated to give unstable intermediate 14, which was dried in vacuo for 1 h. The residue was then dissolved in dry DMF (40 mL) under Ar followed by the addition of CSA (0.703 g; 3.03 mmol) and 2-methoxypropene (8.14 mL; 86.51 mmol). The solution was stirred at rt for 16 h. Aqu satd NaHCO3 was added and the mixture was extracted with CH2Cl2 (3×). The combined organic phases were dried (Na2SO4) the solvent was removed in vacuo and the crude product was purified by silica flash chromatography (n-hexane/EtOAc 5:1 → 2.2:1) to give 15 (1.99 g; 69%) as colorless amorphous solid. Analytical data were in agreement with literature.281H NMR (600 MHz, CDCl3) δ = 4.57 (ddd, 1 H, J4,3a 2.8, J4,3e 3.4, J4,5 7.6 Hz, H-4), 4.41–4.37 (m, 2 H, H-5, H-7), 4.15 (dd, 1 H, J8a,8b 9.1, J8a,7 6.3 Hz, H-8a), 4.08 (dd, 1 H, J8b,7 3.6 Hz, H-8b), 3.81 (s, 3 H, CO2CH3), 3.68 (dd, 1 H, J6,7 8.6, J6,5 2.2 Hz, H-6), 3.07 (dt, 1 H, J3e,F 3.4, J3a,3e 15.5 Hz, 1 H, H-3e), 1.98 (ddd, 1 H, J3a,F 18.6, J3a,3e 15.5, J3a,4 2.8 Hz, H-3a), 1.44, 1.39, 1.38, and 1.32 [4 s, each 3 H, 2 × (CH3)2C]; 13C NMR (150 MHz, CDCl3) δ = 166.1 (CO2Me), 109.7 and 109.6 [(CH3)2C], 108.9 (C-2), 73.5 (C-6), 73.1 (C-7), 71.5 (C-5), 69.4 (C-4), 67.0 (C-8), 53.1 (CO2Me), 30.8 (C-3), 27.0, 25.4, 25.1, and 24.7 [(CH3)2C].
3-Azido-1-propyl [methyl (3-deoxy-4,5;7,8-di-O-isopropylidene-α-d-manno-oct-2-ulopyranosyl)onate]-(2 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (18)
A solution of 10 (1.3 g; 1.42 mmol) in dry THF (14 mL) under Argon was cooled to 0 °C and a solution of HF·pyridine (2.96 mL; 14.2 mmol; 70% HF) was added dropwise. The solution was stirred for 16 h and was subsequently quenched by the addition of 10% aq KF. The solution was extracted with CH2Cl2 (3×), the combined organic phases were dried (Na2SO4) and the solvent was removed in vacuo followed by coevaporation of the crude with toluene (2 × 10 mL) and additional drying in vacuum (1.5 h). The crude product 11 and 15 (0.951 g; 2.84 mmol) were dissolved in dry CH2Cl2 (5 mL) under Argon, molecular sieves 4 Å (0.5 g) were added and the suspension was stirred for 1 h at rt. The reaction mixture was cooled to 0 °C followed by the addition of BF3·Et2O (0.65 mL; 3.13 mmol). After stirring for 90 min at 0 °C, the reaction was quenched by the addition of NEt3 (1.5 mL) and the suspension was filtered over Celite. The filtrate was washed with aqu satd NaHCO3 and the aqueous phase was extracted with CH2Cl2 (2×). The combined organic phases were dried (Na2SO4), the solvent was removed in vacuo and the crude was purified by flash chromatography (EtOAc/MeOH 20:1) to afford 18 (0.854 g; 61% for 2 steps) as colorless amorphous solid. [α]D20 +51.6 (c 1.2, CHCl3); Rf 0.41 (DCM/MeOH 20:1); 1H NMR (600 MHz, CDCl3) δ = 6.01 (d, 1 H, J2′,NH′ 8.8 Hz, NH′), 5.77 (d, 1 H, J2,NH 9.4 Hz, NH), 5.19 (dd, 1 H, J2,3 10.7, J4,3 9.4 Hz, H-3), 5.13 (dd, 1 H, J4′,3′ 9.4, J2′,3′ 10.1 Hz, H-3′), 5.08 (t, 1 H, J4,5 9.8 Hz, H-4), 5.07 (t, 1 H, J4′,5′ 9.7 Hz, H-4′), 4.82 (d, 1 H, J1,2 3.7 Hz, H-1), 4.50 (dt, 1 H, J4″,3″a 3.5, J4″,3″e 4.0, J4″,5″ 7.7 Hz, H-4″), 4.40 (d, 1 H, J1′,2′ 8.3 Hz, H-1′), 4.33 (ddd, 1 H, J7″,8″a 6.3, J7″,8″b 5.2, J7″,6″ 7.1 Hz, H-7″), 4.27 (ddd, 1 H, H-2), 4.25 (dd, 1 H, J5″,6″ 1.9 Hz, H-5″), 4.12 (dd, 1 H, J8″a,8″b 8.8 Hz, H-8″a), 4.03 (dd, 1 H, J6a,6b 11.2, J6a,5 1.9 Hz, H-6a), 3.99 (ddd, 1 H, H-2′), 3.98 (dd, 1 H, H-8″b), 3.83 (ddd, 1 H, J6b,5 4.6 Hz, H-5), 3.80 (dt, 1 H, J 6.2, 10.0 Hz, OCH2), 3.74 (s, 3 H, CO2CH3), 3.71 (dd, 1 H, J6′a,6′b 11.1, J6′a,5′ 4.9 Hz, H-6′a), 3.57 (dd, 1 H, J6″,7″ 7.1, J6″,5″ 1.9 Hz, H-6″), 3.54 (ddd, 1 H, J5′,6′b 2.6 Hz, H-5′), 3.49 (dt, 1 H, J 10.1, 6.1 Hz, OCH2), 3.44 (dt, 2 H, CH2N3), 3.40 (dd, 1 H, H-6′b), 3.37 (dd, 1 H, H-6b), 2.84 (dd, 1 H, J3″a,3″e 15.5 Hz, H-3″e), 2.07 (s, 3 H, CH3CO), 2.02 (s, 6 H, 2 × CH3CO), 2.00, 1.96, and 1.94 (3s, each 3 H, 3 × CH3CO), 1.91 (m, 2 H, OCH2CH2), 1.87 (dd, 1 H, H-3″a), 1.41, 1.38, 1.36, and 1.30 [4 s, each 3 H, (CH3)2C]; 13C NMR (150 MHz, CDCl3) δ = 171.1, 170.7, 170.1, 169.9, 169.7, and 169.3 (C=O), 168.5 (C-1″), 109.3 and 108.8 [(CH3)2C], 101.3 (C-1′), 97.3 (C-2″), 97.0 (C-1), 73.3 (C-7″), 73.1 (C-3′), 72.7 (C-5′), 71.9 (C-5″), 71.4 (C-6″), 71.3 (C-3), 69.9 (C-4″), 68.8 (C-5), 68.5 (C-4′), 68.2 (C-4), 67.6 (C-6), 66.5 (C-8″), 65.0 (OCH2), 61.9 (C-6′), 54.0 (C-2′), 52.1 (OCH3), 51.7 (C-2), 48.4 (CH2N3), 32.3 (C-3″), 28.4 (OCH2CH2), 26.7, 25.3, 25.2, and 24.7 [4(CH3)2C], 23.0, 20.6, and 20.5 (6 C, 6 × CH3CO); HRMS (ESI-TOF) m/z [M + H]+ Calcd for C42H63N5O22 990.4037, found 990.4063.
3-Azido-1-propyl [methyl (3-deoxy-α-d-manno-oct-2-ulopyranosyl)onate]-(2 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (19)
Compound 18 (0.521 g; 0.53 mmol) was dissolved in dry CH3CN (5 mL) under Argon and CeCl3 × 7 H2O (0.392 g; 1.05 mmol) was added followed by the addition of oxalic acid × 2 H2O (0.013 g; 0.11 mmol). The resulting suspension was stirred at rt until complete consumption of the starting material (4.5 h). The suspension was filtered over a thin layer of Celite and the filtrate was concentrated under reduced pressure. Flash chromatography of the crude (CH2Cl2/MeOH 10:1→5:1) afforded 19 (0.363 g; 76%) as colorless amorphous solid. [α]D20 +78.7 (c 1.1, MeOH); Rf 0.16 (CH2Cl2/MeOH 10:1); 1H NMR (600 MHz, CDCl3) δ = 6.21 (d, 1 H, J2′,NH′ 8.6 Hz, NH′), 5.89 (d, 1 H, J2,NH 9.4 Hz, NH), 5.20 (dd, 1 H, J2,3 10.6, J4,3 9.5 Hz, H-3), 5.19 (dd, 1 H, J4′,3′ 9.4, J2′,3′ 10.4 Hz, H-3′), 5.10 (t, 1 H, J4,5 9.7 Hz, H-4), 4.95 (t, 1 H, J4′,5′ 9.6 Hz, H-4′), 4.86 (d, 1 H, J1,2 3.5 Hz, H-1), 4.52 (d, 1 H, J1′,2′ 8.4 Hz, H-1′), 4.27 (ddd, 1 H, H-2), 4.09 (br. s, 1 H, H-5″), 4.07–3.92 (m, 4 H, H-4″, H-7″, H-2′, H-6a), 3.88–3.76 (m, 4 H, H-5, H-8″a, H-8″b, OCH2), 3.77 (s, 3 H, CO2CH3), 3.74 (br d, 1 H, J6″,7″ 8.2 Hz, H-6″), 3.68 (ddd, 1 H, J5′,6′b 2.9, J5′,6′a 7.0 Hz, H-5′), 3.61 (dd, 1 H, J6′a,6′b 10.6, J6′a,5′ 6.9 Hz, H-6′a), 3.52 (dt, 1 H, J 6.2, 10.0 Hz, OCH2), 3.49–3.40 (m, 4 H, H-6′b, H-6b, CH2N3), 3.31–3.24 (br. m, 2 H, OH), 2.21–1.90 (m, 4 H, H-3″e, H-3″a, OCH2CH2), 2.08 (s, 3 H), 2.02 (s, 6 H), 2.01, 1.97, and 1.95 (3 s, each 3 H, 3 × CH3CO); 13C NMR (150 MHz, CDCl3) δ = 171.4, 170.9, 170.8, 170.4, 170.3, and 169.8 (C=O), 168.5 (C-1″), 101.3 (C-1′), 98.8 (C-2″), 97.1 (C-1), 72.8 (C-5′), 72.7 (C-3′), 71.8 (C-6″), 71.3 (C-3), 69.7 (C-7″), 69.6 (C-4′), 68.6 (C-5), 68.4 (C-4), 67.8 (C-6), 66.5 (C-5″), 65.8 (C-4″), 65.4 (OCH2), 63.4 (C-8″), 62.6 (C-6′), 54.3 (C-2′), 52.6 (OCH3), 51.9 (C-2), 48.6 (CH2N3), 34.5 (C-3″), 28.5 (OCH2CH2), 23.1 (d.i. 2 × CH3CONH), 20.8 and 20.7 (4 C, 4 × CH3CO). HRMS (ESI-TOF) m/z [M + NH4]+ Calcd for C36H59N6O22 927.3677 Found: 927.3689.
3-Azido-1-propyl methyl (7,8-O-carbonyl-3-deoxy-α-d-manno-oct-2-ulopyranosyl)onate-(2 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (20)
Compound 19 (0.886 g; 0.97 mmol) was dissolved in dry THF (18 mL) under Argon and sym-collidine (0.65 mL; 4.87 mmol) was added. The reaction mixture was cooled to −50 °C followed by dropwise addition of a 0.63 mM of diphosgene in dry THF solution (1 mL; 0.63 mmol) over 1 h using a syringe pump. After completed addition stirring was continued for 2 h at −50 °C and the reaction was then quenched by the addition of MeOH (0.5 mL). The cooling bath was removed and the reaction mixture was allowed to stir for 10 min at rt before aqueous satd NaHCO3 and CH2Cl2 were added. The aqueous phase was extracted with CH2Cl2 (3×) and the combined organic phases were dried (Na2SO4). The solvent was evaporated in vacuo and the crude product was purified by flash chromatography (DCM/MeOH 20:1 → 5:1) affording 20 (0.728 g; 80%) as colorless amorphous solid; [α]D20 +61.3 (c 1.0, CHCl3); Rf 0.38 (DCM/MeOH 10:1); 1H NMR (600 MHz, CDCl3) δ = 6.01 (d, 1 H, J2′,NH′ 9.0 Hz, NH′), 5.79 (d, 1 H, J2,NH 9.4 Hz, NH), 5.20 (dd, 1 H, J2,3 10.6, J4,3 9.5 Hz, H-3), 5.17–5.12 (m, 2 H, H-3′, H-4′), 5.07 (t, 1 H, J4,5 9.8 Hz, H-4), 4.99 (dt, 1 H, H-7″), 4.82 (d, 1 H, J1,2 3.6 Hz, H-1), 4.78 (dd, 1 H, J8″a,7″ 7.3, J8″a,8″b 8.6 Hz, H-8″a), 4.59 (t, 1 H, J8″b,7″ 8.6 Hz, H-8″b), 4.27 (ddd, 1 H, H-2), 4.45 (d, 1 H, J1′,2′ 8.3 Hz, H-1′), 4.29–4.26 (m, 1 H, H-4″), 4.24 (ddd, 1 H, H-2), 4.09–4.05 (m, 1 H, H-2′), 4.00 (dd, 1 H, J6a,6b 11.3, J6a,5 1.7 Hz, H-6a), 3.97 (br s, 2 H, H-5″, H-6″), 3.85 (ddd, 1 H, J6b,5 4.9 Hz, H-5), 3.81 (dt, 1 H, J 6.0, 10.1 Hz, OCH2), 3.77 (s, 3 H, CO2CH3), 3.64–3.61 (m, 1 H, H-5′), 3.59 (dd, 1 H, J6′a,5′ 2.6, J6′a,6′b 10.9 Hz, H-6′a), 3.54 (dd, 1 H, J6′b,5′ 4.4 Hz, H-6′b), 3.52 (dt, 1 H, OCH2), 3.48–3.40 (m, 3 H, H-6b, CH2N3), 3.26 (br s, 1 H, 4″–OH), 3.09 (br s, 1 H, 5″–OH), 2.16 (dd, 1 H, J3″a,4″ 5.0, J3″a,3e″ 12.8 Hz, H-3″e), 2.08, 2.05, 2.03, and 2.01 (4 s, each 3 H, 4 × CH3CO), 1.98 (t, 1 H, H-3″a), 1.98 and 1.95 (2 s, each 3 H, 2 × CH3CO), 1.98–1.90 (m, 2 H, OCH2CH2); 13C NMR (150 MHz, CDCl3) δ = 171.3, 170.9, 170.5, 170.4, 170.2, and 169.9 (C=O), 167.8 (C-1″), 155.0 (OC=O), 101.8 (C-1′), 99.2 (C-2″), 97.1 (C-1), 76.7 (C-7″), 73.2 (C-3′), 72.5 (C-5′), 71.3 (C-3), 71.1 (C-6″), 68.8 (C-5), 68.5 (C-4′), 68.0 (C-4), 68.0 (C-6), 67.0 (C-5″), 66.2 (C-8″), 65.4 (OCH2), 64.6 (C-4″), 61.6 (C-6′), 54.0 (C-2′), 52.7 (OCH3), 51.9 (C-2), 48.6 (CH2N3), 33.9 (C-3″), 28.5 (OCH2CH2), 23.1 (d.i. 2 × CH3CONH), 20.8, 20.7 (d.i.) and 20.6 (4 C, 4 × CH3CO); HRMS (ESI-TOF) m/z [M + H]+ Calcd for C37H54N5O23 936.3204, found 936.3241. Continued elution of the column afforded unreacted 19 (0.08 g; 10%).
3-Azido-1-propyl methyl (3-deoxy-4,5;7,8-di-O-isopropylidene-α-d-manno-oct-2-ulopyranosyl)onate (2 → 4)-methyl (7,8-O-carbonyl-3-deoxy-α-d-manno-oct-2-ulopyranosyl)onate-(2 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (21)
Compound 20 (0.139 g; 0.223 mmol) and compound 15 (0.074 mg; 0.223 mmol) were dissolved in dry CH2Cl2 (1.5 mL) under Argon and were stirred with molecular sieves 4 Å (150 mg) for 40 min at rt. The mixture was cooled to 0 °C followed by the dropwise addition of BF3·Et2O (0.049 mL; 0.238 mmol). The suspension was stirred at 0 °C for 100 min and the reaction was then quenched by the addition of dry NEt3 (0.1 mL). The mixture was filtered over a thin layer of Celite, aqueous satd NaHCO3 was added to the filtrate and the aqueous phase was extracted two more times with CH2Cl2. The combined organic phases were dried (Na2SO4), the solvent was removed in vacuo and the crude product was purified by silica gel chromatography (CH2Cl2/MeOH 100:3 → CH2Cl2/MeOH 100:5). Impure fractions were combined and rechromatographed using the same solvent composition. Byproduct 22 was obtained first as amorphous solid (40 mg, 25%); Rf 0.35 (CH2Cl2/MeOH 20:1); 1H NMR (600 MHz, CDCl3) for 22 δ = 5.97 (d, 1 H, JNH,2 8.3 Hz, NHGlN′), 5.72 (d, 1 H, JNH,2 9.4 Hz, NHGlN), 5.20–5.15 (m, 2 H, H-3GlN, H-3GlN′), 5.08 (t, 1 H, J3,4∼ J4,5 9.9 Hz, H-4GlN), 5.07 (t, 1 H, J3,4∼ J4,5 9.7 Hz, H-4GlN′), 4.87 (td, 1 H, J7,8a ∼ J7,8a 7.8, J7,6 2.6 Hz, H-7Kdo), 4.83–4.79 (m, 1 H, H-1GlN, H-8aKdo), 4.52 (dt, 1 H, J4,5 7.7 J4,3 3.7 Hz, H-4Kdo), 4.47 (t, 1 H, J8a,8b 8.4 Hz, H-8bKdo), 4.36 (d, 1 H, J1,2 8.5 Hz, H-1GlN′), 4.25 (ddd, 1 H, J2,3 10.7, J2,1 3.6 Hz, H-2GlN), 4.19 (dd, 1 H, J5,4 7.7, J5,6 1.8 Hz, H-5Kdo), 4.06–3.96 (m, 3 H, H-2GlN′, H-6aGlN, H-6Kdo), 3.82–3.73 (m, 2 H, H-5Kdo, OCH2), 3.73 (s, 3 H, OCH3), 3.68 (dd, 1 H, J6a,6b 11.0, J6a,5 3.4 Hz, H-6aGlN′), 3.52 (dt, 1 H, J5,4 9.9, J5,6 3.4 Hz, H-5GlN′), 3.45 (dt, 1 H, J 10.1 J 6.2 Hz, OCH2), 3.45–3.33 (m, 4 H, CH2N3, H-6bGlN′, H-6bGlN), 2.89 (dd, 1 H, J3a,3e 15.6, J3e,4 3.8 Hz, H-3eKdo), 2.05, 2.07, 2.00, 1.98, 1.95, and 1.91 (6 s, each 3 H, 6 × CH3CO), 1.91–1.87 (m, 3 H, 3aKdo, OCH2CH2), 1.34, 1.24 [2 s, 6 H, (CH3)2C].
Continued elution of the column afforded 21 (96 mg; 52%) as colorless amorphous solid; [α]D20 +61.4 (c 1.0; CHCl3); Rf 0.36 (CH2Cl2/MeOH 20:1); 1H NMR (600 MHz, CDCl3) δ = 5.98 (d, 1 H, JNH,2 9.1 Hz, NHGlN′), 5.71 (d, 1 H, JNH,2 9.7 Hz, NHGlN), 5.21 (app t, 1 H, J3,4 ∼ J3,2 9.5 Hz, H-3GlN), 5.17 (t, 1 H, J4,5 9.5 Hz, H-4GlN), 5.12 (dd, 1 H, J3,4 9.4, J3,2 10.4 Hz, H-3GlN′), 4.98 (t, 1 H, J3,4 ∼ J4,5 9.7 Hz, H-4GlN′), 4.86 (td, 1 H, J8a,7 ∼ J8b,7 7.6, J7,6 3.5 Hz, H-7Kdo), 4.82 (d, 1 H, J1,2 3.6 Hz, H-1GlN), 4.89 (dd, 1 H, J8a,8b 8.9 Hz, H-8aKdo), 4.56–4.50 (m, 1 H, H-4Kdo′), 4.51 (t, 1 H, H-8bKdo), 4.37 (d, 1 H, J1,2 8.6 Hz, H-1GlN′), 4.37–4.33 (m, 1 H, H-7Kdo′), 4.31–4.25 (m, 3 H, H-4Kdo, H-2GlN, H-5Kdo′), 4.14 (dd, 1 H, J8a,8b 8.8, J8a,7 6.2 Hz, H-8aKdo′), 4.07–4.02 (m, 3 H, H-6aGlN, H-8bKdo′, H-2GlN′), 3.91 (dd, 1 H, J6,7 3.4, J6,5 1.3 Hz, H-6Kdo), 3.86–3.81 (m, 2 H, H-5GlN, OCH2), 3.81 (s, 3 H, OCH3), 3.76 (s, 3 H, OCH3), 3.69 (dd, 1 H, J6,7 6.2, J6,5 1.7 Hz, H-6Kdo′), 3.64 (bs, 1 H, H-5Kdo), 3.60–3.55 (m, 2 H, H-6aGlN′, H-5GlN′), 3.51 (dt, 1 H, J 10.6, J 6.7 Hz, OCH2), 3.49–3.40 (m, 4 H, H-6bGlN, H-6aGlN′, OCH2, CH2CH2N3), 3.39 (dd, 1 H, J6a,6b 11.2, J5,6b 3.2 Hz, H-6bGlN), 2.91 (dd, 1 H, J3a.3e 15.5, J3a,4 3.5 Hz, H-3eKdo′), 2.32 (br s, 1 H, OH), 2.22 (dd, 1 H, J3a,3e 13.0, J3a,4 5.2 Hz, H-3eKdo), 2.10 (s, 3 H, CH3CO), 2.07 (s, 3 H, CH3CO), 2.02–1.99 (m, 1 H, H-3aKdo), 2.02, 2.00, 1.99, and 1.93 (4 s, each 3 H, 4 × CH3CO), 1.91–1.87 (m, 2 H, OCH2CH2), 1.83 (dd, 1 H, J3a,3e 15.5, J3a,4 2.3 Hz, H-3aKdo′), 1.47, 1.41, 1.39, and 1.30 [4 s, each 3 H, 2 × (CH3)2C]; 13C NMR (150 MHz, CDCl3) δ = 171.3, 170.8, 170.4, 170.2, 169.7, 169.4, 167.3 (8 C, C=O), 154.5 (OC=O), 109.6, 108.9 [2 C, (CH3)2C], 101.7 (C-1GlN′), 98.9 (C-2Kdo), 97.3 (C-2Kdo′), 97.1 (C-1GlN), 76.2 (C-7Kdo), 74.2 (C-7Kdo′), 73.4 (C-3GlN′), 72.5 (C-5GlN′), 72.2 (C-5Kdo′), 71.8 (C-6Kdo′), 71.5 (C-3GlN), 70.4 (C-6Kdo), 69.8 (C-4Kdo′), 69.0 (C-4GlN′), 68.5 (C-5GlN), 68.3 (C-4GlN), 67.9 (C-6GlN), 66.8 (C-4Kdo), 66.3 (C-8Kdo′), 66.1 (C-8Kdo), 65.4 (C-5Kdo), 65.2 (OCH2), 62.5 (C-6GlN′), 54.1 (C-2GlN′), 52.7 (OCH3), 52.6 (OCH3), 51.8 (C-2GlN), 48.5 (CH2N3), 33.3 (C-3Kdo), 32.8 (C-3Kdo′), 28.5 (OCH2CH2), 26.8, 25.3, 25.0, 24.6, 23.1 (6 C, CH3CO), 20.8, 20.7 [4 C, (CH3)2C]; HRMS (ESI-TOF) m/z [M + Na]+ Calcd for C52H75N5NaO30 1272.4389, found 1272.4396.
Continued elution of the column eventually gave unreacted 20 (16 mg 12%); Rf 0.11 (CH2Cl2/MeOH 20:1).
Recovery of Acceptor 20 from 22
Compound 22 (0.13 g; 0.13 mmol) was dissolved in CH2Cl2 (0.7 mL) and 90% aq TFA (0.7 mL) was added. The reaction mixture was stirred at rt for 1 h and was subsequently quenched by addition of aqueous satd NaHCO3. CH2Cl2 was added and the aqueous phase was extracted twice with CH2Cl2. The combined organic fractions were dried (Na2SO4) and the solvent was removed in vacuo to afford 20 (0.118 g; 97%) as a colorless amorphous solid (pure by NMR).
4-Methylphenyl 2-O-benzoyl-4-O-benzyl-6-O-tert-butyldimethylsilyl-3-O-(2-naphthylmethyl)-1-thio-α-d-mannopyranoside (24)
Compound 23(31) (1.80 g; 2.90 mmol) was dissolved in 1:1 dry CH2Cl2/dry pyridine (12 mL) under Argon. DMAP (0.035 g; 0.29 mmol) was added, the solution was cooled to 0 °C and TBSCl (0.656 g; 4.35 mmol; 1.5 equiv) was added. The reaction mixture was stirred at rt for 16 h until full conversion of the substrate. The reaction was quenched by the addition of aqueous satd NaHCO3 and was extracted with CH2Cl2 (3×). The combined organic phases were dried (Na2SO4), the solvent was removed in vacuo and the crude product was purified by silica flash chromatography (n-hexane/EtOAc 30:1) to afford 24 (1.95 g; 93%) as colorless amorphous solid. [α]D20 +38.4 (c 1.2, CHCl3); Rf 0.41 (n-hexane/EtOAc 20:1); 1H NMR (600 MHz, CDCl3) δ = 8.18–8.16 (m, 2 H, arom. H), 7.84–7.31 (m, 17 H, arom. H), 7.14–7.12 (m, 2 H, arom. H), 5.97 (t, 1 H, J2,3 2.1 Hz, H-2), 5.61 (d, 1 H, J1,2 2.1 Hz, H-1), 5.02 (d, 1 H, J 10.9 Hz, OCH2Ph), 5.01 (d, 1 H, J 11.6 Hz, OCH2Ph), 4.81 (d, 1 H, J 11.6 Hz, OCH2Ph), 4.76 (d, 1 H, J 10.9 Hz, OCH2Ph), 4.31–4.26 (m, 1 H, H-5), 4.21–4.16 (m, 2 H, H-4, H-3), 4.09 (dd, 1 H, J6a,6b 11.3, J6a,5 4.1 Hz, H-6a), (dd, 1 H, J6b,6a 11.3 J6b,5 1.7 Hz, H-6b), 2.35 (s, 3 H, CH3Ph), 1.00 [s, 9 H, (CH3)3CSi], 0.13 [2 s, 6 H, 2 × CH3Si]; 13C NMR (150 MHz, CDCl3) δ = 165.7 (PhC=O), 138.6, 137.8, 135.3, 133.3, 133.1, 133, 132.3, 132.0, 130.2, 130.0, 129.9, 129.8, 128.5, 128.3, 128.1, 127.9, 127.6, 126.9, 126, 125.9, 125.8 (28 C, C–Ar), 86.7 (C-1), 78.6 (C-3), 75.2 (OCH2Ph), 74.3 (C-4), 72.8 (C-5), 71.7 (OCH2Ph), 70.9 (C-2), 62.2 (C-6), 25.9 (CH3CSi), 21.2 (CH3Ph), 18.3 (CH3CSi), −5.04 (CH3Si), −5.3 (CH3Si); HRMS (ESI-TOF) m/z [M + NH4]+ Calcd for C44H54NO6SSi 752.3436, found 752.3428.
2-O-Benzoyl-4-O-benzyl-6-O-tert-butyldimethylsilyl-3-O-(2-naphthylmethyl)-α-d-mannopyranose 2,2,2-trichloroacetimidate (26)
Compound 24 (1.90 g; 2.59 mmol) was dissolved in dry CH2Cl2 (25 mL) under Argon and cooled to 0 °C. NIS (0.756 g; 3.36 mmol) was added followed by the dropwise addition of TFA (0.257 mL; 3.36 mmol). The solution was stirred at 0 °C for 1 h before piperidine (0.998 mL; 10.08 mmol) was added and stirring was continued for 20 min at 0 °C. The mixture was allowed to warm to rt and stirred for 30 min. The reaction was quenched by the addition of NEt3 (1 mL) and 5% aqueous Na2S2O3. The aqueous phase was extracted with CH2Cl2 (2×). The combined organic phases were dried (Na2SO4), the solvent was removed in vacuo and the crude product was purified via silica flash chromatography (n-hexane/EtOAc 4:1) to obtain lactol 25 (1.5 g; 92%) as a mixture of isomers (α/β = 1/0.04) 1H NMR (600 MHz, CDCl3) δ = 8.14–8.10 (m, 2 H, arom. H), 7.80–7.27 (m, 15 H, arom. H), 5.69 (dd, 1 H, J2,1 2.1 J2,3 0.6 Hz, H-2), 5.35 (dd, 1 H, J1,OH 3.4 J1,2 2.0 Hz, H-1), 4.95–4.92 (m, 2 H, OCH2Ph), 4.75 (d, 1 H, J 11.6 Hz, OCH2Ph), 4.69 (d, 1 H, J 11.8 Hz, OCH2Ph), 4.21 (dd, 1 H, J3,4 9.8, J3,2 3.7 Hz, H-3), 4.09 (t, 1 H, J3,4 ∼ J4,5 10.0 Hz, H-4), 4.01 (dd, 1 H, J6a,6b 11.5, J6a,5 3.8 Hz, H-6a), 3.95 (ddd, 1 H, J5,4 10.0, J5,6a 3.8, J5,6b 1.1 Hz, H-5), 3.85 (dd, 1 H, J6b,6a 11.5, J6b,5 1.1 Hz, H-6b), 2.66 (d, 1 H, JOH,1 3.4 Hz, OH), 0.94 [s, 9 H, (CH3)3Si], 0.10 (s, 3 H, CH3Si), 0.09 (s, 3 H, CH3Si). Compound 25 was then dissolved in dry CH2Cl2 (18 mL) under Argon, K2CO3 (0.989 g; 7.16 mmol) was added and stirred at rt for 10 min. Then CCl3CN (0.957 mL; 9.54 mmol) was added dropwise and stirring was continued for 20 h at rt. The suspension was filtered over a pad of Celite, the solvent was removed in vacuo and the crude product was purified by silica flash chromatography (n-hexane/EtOAc 10:1) to give 26 (1.63 g; 88%) as colorless oil, containing 5% of the β-anomer; [α]D20 −13.4 (c 1.0, CHCl3); Rf 0.42 (n-hexane/EtOAc 10:1); 1H NMR (600 MHz, CDCl3) δ = 8.64 (s, 1 H, NH=C), 8.17–8.14 (m, 2 H, arom. H), 7.79–7.27 (m, 15 H, arom. H), 6.39 (d, 1 H, J1,2 2.0 Hz, H-1), 5.77 (dd, 1 H, J2,3 3.4 J2,1 2.0 Hz, H-2), 4.96 (d, 1 H, J 10.8 Hz, 1 H, OCH2Ph), 4.95 (d, 1 H, J 11.8 Hz, OCH2Ph), 4.79 (d, 1 H, J 11.8 Hz, OCH2Ph), 4.71 (d, 1 H, J 10.8 Hz, OCH2Ph), 4.25 (t, 1 H, J4,5 9.9 Hz, H-4), 4.18 (dd, 1 H, J3,4 9.9, J3,2 3.4 Hz, H-3), 4.03 (dd, 1 H, J6a,6b 12.0, J6a,5 3.4 Hz, H-6a), 3.91–3.86 (m, 2 H, H-6b, H-5), 0.95 [s, 9 H, (CH3)3CSi], 0.11 (s, 3 H, CH3Si), 0.10 (s, 3 H, CH3Si); 13C NMR (150 MHz, CDCl3) δ = 165.6 (COPh), 160.1 (C=NH), 138.4, 135.1, 133.4, 133.2, 133.1, 130.2, 129.7, 128.6, 128.4, 128.2, 128.1, 127.9, 127.8, 127.6, 127.2, 126.3, 126.0, 125.9 (23 C, CCl3, C–Ar), 95.8 (C-1), 77.3 (C-3), 75.6 (OCH2Ph), 75.5 (C-5), 73.5 (C-4), 72.2 (OCH2Nap), 68.0 (C-2), 61.6 (C-6), 25.9 [(CH3)3CSi], 18.3 [(CH3)3CSi], −5.2 (CH3Si), −5.4 (CH3Si); HRMS (ESI-TOF) m/z [M – OCNHCCl3]+ Calcd for C37H43O6Si 611.2823, found 611.2830.
3-Azido-1-propyl 2-O-benzoyl-4-O-benzyl-6-O-tert-butyldimethylsilyl-3-O-(2-naphthylmethyl)-α-d-mannopyranosyl-(1 → 5)[methyl (3-deoxy-4,5;7,8-di-O-isopropylidene-α-d-manno-oct-2-ulopyranosyl)onate-(2 → 4)]-methyl (7,8-O-carbonyl-3-deoxy-α-d-manno-oct-2-ulopyranosyl)onate-(2 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (27)
Compound 21 (203 mg; 0.162 mmol), 26 (188 mg, 0.24 mmol) and molecular sieves 4 Å (0.3 g) were dissolved in dry CH2Cl2 (2 mL) under Argon and the suspension was stirred at rt for 30 min. Then TMSOTf (9 μL; 0.049 mmol) was added and the suspension was stirred for additional 40 min. Subsequently the reaction was quenched via the addition of NEt3 (10 μL). The suspension was filtered over a pad of Celite and the solvent was removed in vacuo. The crude product was purified using silica flash chromatography (DCM/MeOH 25:1 → 25:2) to afford 27 (211 mg; 70%) as colorless amorphous solid; [α]D20 +50.5 (c 1.3, CHCl3); Rf 0.20 (CH2Cl2/MeOH 100:3); 1H NMR (600 MHz, CDCl3) δ = 8.14–8.08 (m, 2 H, arom. H), 7.80–7.69 (m, 3 H, arom. H), 7.66–7.56 (m, 2 H, arom. H), 7.48–7.27 (m, 9 H, arom. H), 6.11 (d, 1 H, JNH,2 8.4 Hz, NHGlN′), 5.71 (d, 1 H, JNH,2 9.4 Hz, NHGlN), 5.51 (dd, 1 H, J2,3 3.2, J2,1 1.9 Hz, H-2Man), 5.22 (t, 1 H, J3,4 ∼ J4,5 9.4 Hz, H-4GlN), 5.19 (app t, 1 H, J2,3 ∼ J3,4 9.4 Hz, H-3GlN), 5.15 (dd, 1 H, J3,4 9.3, J3,2 10.4 Hz, H-3GlN′), 5.06 (d, 1 H, J1,2 1.9 Hz, H-1Man), 4.97 (d, 1 H, J 10.4 Hz, OCH2Ph), 4.89 (d, 1 H, J 12.1 Hz, OCH2Nap), 4.85 (t, 1 H, J3,4 ∼ J4,5 9.3 Hz, H-4GlN′), 4.83 (d, 1 H, J1,2 3.7 Hz, H-1GlN), 4.78 (d, 1 H, J 12.1 Hz, OCH2Nap), 4.74 (d, 1 H, J 10.4 Hz, OCH2Ph), 4.61 (ddd, 1 H, J7,8a 8.0, J7,8b 7.3, J7,6 6.7 Hz, H-7Kdo), 4.39–4.34 (m, 3 H, H-1GlN′, H-4Kdo′, H-8aKdo), 4.31–4.27 (m, 4 H, H-8bKdo, H-2GlN, H-4Man, H-7Kdo′), 4.24 (ddd, 1 H, J4,3a 11.9, J4,3e 4.8, J4,5 2.5 Hz, H-4Kdo), 4.14 (dd, 1 H, J5,4 7.8, J5,6 1.9 Hz, H-5Kdo′), 4.09 (dd, 1 H, J3,4 9.8, J3,2 3.2 Hz, H-3Man), 4.08–3.98 (m, 6 H, H-8aKdo′, H-8bKdo′, H-6aMan, H-6aGlN, H-5Man, H-2GlN), 3.88 (dd, 1 H, J6b,5 1.4, J6a,6b 11.0 Hz, H-6bMan), 3.86–3.80 (m, 3 H, OCH2, H-5GlN, H-6Kdo), 3.76 (br s, 1 H, H-5Kdo), 3.74 (s, 3 H, OCH3), 3.72 (s, 3 H, OCH3), 3.60 (ddd, 1 H, J5,4 9.4, J5,6b 7.7, J5,6a 2.1 Hz H-5GlN′), 3.57 (dd, 1 H, J6,5 1.8, J6,7 6.4 Hz, H-6Kdo′), 3.56–3.51 (m, 1 H, OCH2), 3.48 (dd, 1 H, J6a,6b 10.8, J6a,5 2.1 Hz, H-6aGlN′), 3.43–3.33 (m, 4 H, H-6bGlN′, H-6bGlN, CH2N3), 2.77 (dd, 1 H, J3a,3e 15.7, J3e,4 3.3 Hz, H-3eKdo′), 2.17–2.03 (m, 2 H, H-3eKdo, H-3aKdo), 2.09, 2.02, 2.01, 2.00, 1.99, 1.91 (6 × s, each 3 H, CH3CO), 1.90–1.82 (m, 2 H, OCH2CH2), 1.70 (dd, 1 H, J3e,3a 15.7 J3e,4 2.4 Hz, H-3aKdo′), 1.42, 1.39, 1.34, 1.27 [4 × s, each 3 H, 2 × (CH3)2C], 0.94 [s, 9 H, (CH3)3CSi], 0.11 (s, 3 H, CH3Si), 0.09 (s, 3 H, CH3Si); 13C NMR (150 MHz, CDCl3) δ = 171.3, 170.8, 170.5, 170.3, 169.8, 169.6, 168.6, 167.1, 166.1 (9 C, C=O), 153.6 (OC=O), 138.8, 135.7, 133.3, 133.2, 132.9, 130.1, 129.7, 128.4, 128.1, 127.9, 127.7, 126.5, 126.5, 125.9 (22 C, C–Ar), 109.5, 108.8 [2 C, (CH3)2C], 101.6 (C-1GlN′), 98.9 (C-2Kdo), 98.0 (C-1Man), 97.2 (C-2Kdo′), 97.1 (C-1GlN), 77.5 (C-3Man), 75.5 (OCH2Ph), 74.2 (C-4Man), 73.8 (C-7Kdo′), 73.3 (C-3GlN′), 73.0 (2 C, C-6Kdo, C-5GlN′), 72.8 (2 C, C-5Man, C-7Kdo), 72.3 (C-5Kdo′), 71.6 (CH2Nap), 71.6 (C-3GlN), 71.4 (C-6Kdo′), 71.3 (C-5Kdo), 69.8 (C-4Kdo′), 69.65 (C-2Man), 69.2 (C-4GlN′), 68.4 (2 C, C-4GlN, C-5GlN), 68.1 (C-6GlN), 67.5 (C-8Kdo), 67.0 (C-4Kdo), 66.3 (C-8Kdo′), 65.3 (OCH2), 63.2 (C-6GlN′), 61.7 (C-6Man), 54.3 (C-2GlN′), 52.6 and 52.4 (2 C, OCH3), 51.7 (C-2GlN), 48.7 (CH2N3), 34.4 (C-3Kdo), 32.2 (C-3Kdo′), 28.5 (CH2CH2N3), 26.9 (CH3CO), 26.0 [(CH3)3CSi], 25.4, 25.1, 24.7, 23.1 (5 C, CH3CO), 20.8, 20.7, 20.6 [4 C, (CH3)2C], 18.3 [(CH3)3CSi], −5.0, −5.3 (2 C, CH3Si); HRMS (ESI-TOF) m/z [M + NH4]+ Calcd for C89H117N5O36Si 1877.7586, found 1877.7641.
Continued elution of the column gave 21 (40 mg; 20%); Rf 0.44 (CH2Cl2/MeOH 20:1).
Selective Isopropylidene Cleavage from 27
Compound 27 (15 mg; 0.008 mmol) was dissolved in dry CH3CN (1 mL) under Argon and CeCl3 × 7 H2O (0.012 g, 0.032 mmol) was added followed by the addition of oxalic acid ×2 H2O (0.3 mg; 0.003 mmol). The resulting suspension was stirred at rt for 16 h. The suspension was filtered over a thin layer of Celite and the filtrate was concentrated under reduced pressure. Flash chromatography of the crude (CH2Cl2/MeOH 100:3 → 100:5) afforded 28 (10 mg; 67%) as colorless amorphous solid. Rf 0.32 (CH2Cl2/MeOH 20:1); 1H NMR (600 MHz, CDCl3) δ 8.13–9.08 (m, 2 H, Ar), 7.80–7.73 (m, 3 H, Ar), 7.65–7.57 (m, 2 H, Ar), 7.48–7.27 (m, 9 H, Ar), 6.09 (d, 1 H, JNH,2 8.5 Hz, NHGlN′), 5.70 (d, 1 H, JNH,2 9.3 Hz, NHGlN), 5.47 (dd, 1 H, J2,3 2.9, J2,1 2.0 Hz, H-2Man), 5.22 (t, 1 H J4,3 ∼ J4,5 9.4 Hz, H-4GlN), 5.21 (t, 1 H J3,4 ∼ J3,2 9.6 Hz, H-3GlN′), 5.19 (t, 1 H J4,3 ∼ J3,2 9.4 Hz, H-3GlN), 5.00 (d, 1 H, J1,2 2.0 Hz, H-1Man), 4.96 (t, 1 H J4,3 ∼ J4,5 9.6 Hz, H-4GlN′), 4.96 (d, 1 H, J 10.4 Hz, OCH2Ph), 4.88 (d, 1 H, J 12.0 Hz, OCH2Nap), 4.84 (d, 1 H, J1,2 3.3 Hz, H-1GlN), 4.78 (d, 1 H, J 12.0 Hz, OCH2Nap), 4.75 (d, 1 H, J 10.4 Hz, OCH2Ph), 4.66 (app dd, 1 H, J7,8b 7.4 Hz, H-7Kdo), 4.53 (d, 1 H, J1,2 8.4 Hz, H-1GlN′), 4.47–4.42 (m, 3 H, H-4Kdo, H-4Kdo′, H-8aKdo), 4.34 (dd, 1 H, J8a,8b 8.5, J8b,7 7.4 Hz, H-8bKdo), 4.32–4.26 (m, 3 H, H-2GlN, H-5Kdo′, H-4Man), 4.08–4.04 (m, 2 H, H-3Man, H-8aKdo′), 4.03 (dd, 1 H, J6a,6b 10.6, J6a,5 1.9 Hz, H-6aMan), 4.01–3.87 (m, 6 H, H-6aGlN, H-6Kdo′, H-2GlN′, H-7Kdo′, H-6bMan, H-8bKdo′), 3.86–3.79 (m, 3 H, H-6Kdo, H-5GlN′, OCH2), 3.76 (s, 3 H, CO2CH3), 3.73 (s, 3 H, CO2CH3), 3.75–3.66 (m, 3 H, H-5Kdo, H-5Man, H-5GlN), 3.54 (dt, 1 H, J 10.4, J 5.9 Hz, OCH2CH2), 3.48–3.46 (m, 2 H, H-6aGlN′, H-6bGlN), 3.43–3.34 (m, 3 H, CH2CH2N3, H-6bGlN′), 3.08 (br d, 1 H, J 5.3 Hz, 7-OH), 2.80 (dd, 1 H, J3a,3e 15.5, J3e,4 3.5 Hz, H-3eKdo′), 2.73 (br s, 1 H, 8-OH), 2.21 (dd, 1 H, J3e,3a 12.7, J3e,4 4.0 Hz, H-3eKdo), 2.08 (s, 3 H, CH3CO), 2.07–20.5 (m, 1 H, H-3aKdo), 2.03, 2.00, 1.99, 1.92 (5 s, each 3 H, 5 CH3CO), 1.90–1.84 (m, 2 H, OCH2CH2), 1.89 (dd, 1 H, J3a,3e 15.5, J3a,4 2.3 Hz, H-3aKdo′), 1.37, 1.30 [2 × s, 6 H, (CH3)2C], 0.95 [s, 9 H, (CH3)3Si], 0.12, 0.11 (2 × s, 6 H, (CH3)2Si); 13C NMR (150 MHz, CDCl3) δ = 171.4, 170.8, 170.4, 170.3, 169.9, 168.8, 167.3, 166.1, (9 C, C=O), 153.7 (OC=O), 138.8, 135.6, 133.3, 133.2, 133.0, 130.0, 129.7, 128.4, 128.0, 127.9, 127.7, 127.6, 126.6, 126.0, 125.9 (22 C, Ar), 109.34 [(CH3)2C], 101.0 (C-1GlN′), 99.2 (C-2Kdo), 98.1 (C-1Man), 97.1 (C-1GlN), 96.4 (C-2Kdo′), 77.5 (C-3Man), 75.5 (CH2Ph), 73.8 (C-4Man), 73.3 (C-6Kdo), 73.2 (2 C, C-7Kdo, C-3GlN′), 72.9 (C-6Kdo′), 72.7 and 72.5 (C-5Kdo, C-5Man), 72.3 (C-5Kdo′), 72.1 (C-5GlN′), 71.8 (CH2Nap), 71.5 (C-3GlN), 70.1 (C-7Kdo′), 70.0 (C-4Kdo′), 69.6 (C-2Man), 68.8 (C-4GlN′), 68.5 (C-5GlN), 68.1 (C-4GlN), 67.7 (C-6GlN), 67.1 (C-8Kdo), 65.9 (C-4Kdo), 65.3 (OCH2CH2), 63.1 (C-8Kdo′), 62.5 (C-6GlN′), 61.5 (C-6Man), 54.4 (C-2GlN′), 52.7 (OCH3), 52.6 (OCH3), 51.8 (C-2GlN), 48.6 (CH2CH2N3), 34.2 (C-3Kdo), 31.8 (C-3Kdo′), 28.5 (OCH2CH2), 25.9 [(CH3)3CSi], 25.5, 24.6 [2 C, (CH3)2C], 23.1, 23.1 20.7 (6 C, CH3CO), 18.3 [(CH3)3CSi], −5.0, −5.3 [2 C, (CH3)2Si].
3-Azido-1-propyl 2-O-benzoyl-4-O-benzyl-3-O-(2-naphthylmethyl)-α-d-mannopyranosyl-(1 → 5)[methyl (3-deoxy-α-d-manno-oct-2-ulopyranosyl)onate-(2 → 4)]-methyl (7,8-O-carbonyl-3-deoxy-α-d-manno-oct-2-ulopyranosyl)onate-(2 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-3,4-di-O-acetyl-2-deoxy-α-d-glucopyranoside (29)
Compound 27 (26 mg; 14 μmol) was dissolved in CH2Cl2 (7 mL) and cooled to 0 °C. Then 90% aqueous TFA (0.3 mL) was added and the solution was stirred at 0 °C for 5 h. Aqueous satd NaHCO3 was then added and the aqueous phase was extracted with EtOAc (4×). The combined organic phases were dried (Na2SO4); the solvent was removed in vacuo and the crude product was purified by flash chromatography (CH2Cl2/MeOH 100:7 → 100:10) to afford 29 (16 mg; 69%) as colorless oil; [α]D20 +54.7 (c 0.9, CHCl3); Rf 0.23 (DCM/MeOH 10:1); 1H NMR (600 MHz, CDCl3) δ = 8.09–8.06 (m, 2 H, arom. H), 7.81–7.71 (m, 3 H, arom. H), 7.67–7.59 (m, 2 H, arom. H), 7.51–7.41 (m, 5 H, arom. H), 7.36–7.28 (m, 4 H, arom. H), 6.15 (d, 1 H, JNH,2 8.3 Hz, NHGlN′), 5.79 (d, 1 H, JNH,2 9.3 Hz, NHGlN), 5.41 (t, 1 H, J1,2 ∼ J3,2 2.3 Hz, H-2Man), 5.25 (dd, 1 H, J2,3 10.6, J3,4 9.4 Hz, H-3GlN′), 5.21 (dd, 1 H, J2,3 10.2, J3,4 9.7 Hz, H-3GlN), 5.17 (t, 1 H, J3,4∼ J4,5 9.7 Hz, H-4GlN), 5.10 (t, 1 H, J3,4∼ J4,5 9.7 Hz, H-4GlN′), 5.07 (d, 1 H, J1,2 1.7 Hz, H-1Man), 4.97 (d, 1 H, J 10.8 Hz, OCH2Ph), 4.89 (d, 1 H, J 12.5 Hz, OCH2Nap), 4.86 (d, 1 H, J1,2 3.5 Hz, H-1GlN), 4.81 (d, 1 H, J 12.5 Hz, OCH2Nap), 4.74 (d, 1 H, J 10.8 Hz, OCH2Ph), 4.64 (ddd, 1 H, J7,8a 5.8, J7,8b 7.8, J7,6 5.8 Hz, H-7Kdo), 4.59 (d, 1 H, J1,2 8.3 Hz, H-1GlN′), 4.37 (dd, 1 H, J8a,8b 8.6, J8a,7 5.8 Hz, H-8aKdo), 4.34 (ddd, 1 H, J4,3a 12.1, J4,3e 3.8, J4,5 2.3 Hz, H-4Kdo), 4.34–4.27 (m, 2 H, H-8bKdo, H-2GlN), 4.11–4.00 (m, 7 H, H-5Kdo′, H-7Kdo, H-5Kdo, H-5Man, H-4Man, H-3Man, H-6aGlN), 3.94–3.73 (m, 9 H, H-6aMan, H-6bMan, H-2GlN′, H-8aKdo′, H-8bKdo′, H-4Kdo′, H-5GlN, OCH2, H-6Kdo), 3.80 (s, 3 H, OCH3), 3.76 (s, 3 H, OCH3), 3.66 (br d, 1 H, J6,7 8.2 Hz, H-6Kdo′), 3.61 (dt, 1 H, J5,6a ∼ J5,6b 2.9 Hz, H-5GlN′), 3.54–3.49 (m, 2 H, H-6aGlN′, OCH2), 3.45–3.37 (m, 3 H, CH2N3, H-6bGlN), 3.37 (dd, 1 H, J5,6b 2.9, J6b,6a 13.5 Hz, H-6bGlN′), 3.09 (br s, 1 H, OH), 2.56 (br s, 1 H, OH), 2.35 (br s, 1 H, OH), 2.29 (dd, J3a,3e 12.9 J3a,4 4.3 Hz, H-3eKdo), 2.15 (dd, 1 H, J3a,3e 12.6, J3a,4 4.7 Hz, 3eKdo′), 2.07 (s, 3 H, CH3CO), 2.07–2.03 (m, 1 H, H-3aKdo), 2.03, 2.02, 2.00, 1.98, and 1.93 (5 s, each 3 H, 5 × CH3CO), 1.97–1.91 (m, 1 H, H-3aKdo′), 1.93–1.82 (m, 2 H, OCH2CH2); 13C NMR (150 MHz, CDCl3) δ = 171.4, 179.9, 170.7, 170.2, 169.3 (CH3CO, ArCO) 167.1, 166.0 (CO2Me), 153.8 (OC=O), 138.0, 133.5, 133.2, 133.0, 130.0, 129.5, 128.6, 128.5, 128.2, 128.0, 127.8, 127.7, 126.7, 126.2, 126.0, 125.8 (22 C, C–Ar), 100.8 (C-1GlN′), 100.0 (C-2Kdo′), 99.3 (C-2Kdo), 98.1 (C-1Man), 97.1 (C-1GlN), 77.2 (C-3Man), 75.4 (OCH2Ph), 74.6 (C-4Man), 74.0 (C-7Kdo), 73.2 (C-5Kdo), 72.7 and 72.6 (each 2 C, C-6Kdo′, C-6Kdo, C-3GlN′, C-5Man), 72.0 (C-5GlN′), 71.8 (OCH2Nap), 71.3 (C-3GlN), 70.8 (C-7Kdo′), 69.7 (C-2Man), 68.6 (C-5GlN), 68.1 (C-4GlN), 68.0 (C-4GlN′), 67.8 (C-4Kdo), 67.7 (C-6GlN), 66.3 (C-8Kdo), 66.2 (C-5Kdo′), 65.7 (C-4Kdo′), 65.3 (OCH2), 62.8 (C-6Man), 62.3 (C-8Kdo′), 60.0 (C-6GlN′), 54.5 (C-2GlN′), 53.0 (OCH3), 52.9 (OCH3), 51.9 (C-2GlN), 48.6 (CH2N3), 34.6 (C-3Kdo′), 33.8 (C-3Kdo), 28.6 (OCH2CH2), 23.1, 20.8, 20.7, 20.6 (6 C, CH3CO); HRMS (ESI-TOF) m/z [M + H]+ Calcd for C77H96N5O36 1666.5830, found 1666.5835.
3-Amino-1-propyl α-d-mannopyranosyl-(1 → 5)[sodium (3-deoxy-α-d-manno-oct-2-ulopyranosyl)onate-(2 → 4)]-sodium (3-deoxy-α-d-manno-oct-2-ulopyranosyl)onate-(2 → 6)-2-acetamido-2-deoxy-β-d-glucopyranosyl-(1 → 6)-2-acetamido-2-deoxy-α-d-glucopyranoside (30)
Compound 29 (8 mg; 5 μmol) was dissolved in dry MeOH and a 0.1 M solution of NaOMe (150 μL; 15 μmol) was added. The reaction mixture was stirred at rt for 24 h and subsequently made neutral by the addition of DOWEX AG1X resin (H+-form). The resin was filtered off and the filtrate was concentrated. The residue was dissolved in a 1:1:0.1 mixture of H2O/MeOH/AcOH (2 mL). The solution was treated in an H-cube (cartridge: 10% Pd/C; mode: full H2: 1 mL/min; circulation time 1 h). The solvent was removed in vacuo and NMR analysis of the resulting intermediate confirmed the absence of aromatic signals. The crude product was dissolved in water (1 mL) and 0.1 M NaOH (0.5 mL) was added and stirred for 3 h at rt. DOWEX AG1X resin (H+-form) was added to give a pH 8. The suspension was filtered and the filtrate was concentrated in vacuo. The crude product was purified by gel chromatography on LH20 using 2:1 H2O/MeOH as eluant to afford 30 as colorless amorphous solid (4 mg; 69%); 1H NMR (600 MHz, D2O) δ = 5.19 (d, 1H, J1,2 1.7 Hz, H-1Man), 4.82 (d, 1 H, J1,2 3.6 Hz, H-1GlcN), 4.48 (d, 1 H, J1,2 8.5 Hz, H-1GlcN′), 4.22 (br s, 1 H, H-5Kdo), 4.15 (dd, 1 H, J6a,6b 11.1, J6a,5 1.6 Hz, H-6aGlcN), 4.10 (ddd, 1 H, J4,3e 4.4, J4,5 2.3 Hz, H-4Kdo), 4.09 (dd, 1 H, J2,3 3.1 Hz, H-2Man), 4.01 (dt, 1 H, J4,5 10.1, J5,6a 3.1 Hz, H-5Man), 3.98 (m, 1 H, H-4Kdo′), 3.97 (br. s, 1 H, H-5Kdo′), 3.96 (m, 1 H, H-7Kdo′), 3.93 (m, 1 H, H-8aKdo′), 3.91 (dd, 1 H, J3,4 9.8 Hz, H-3Man), 3.89 (m, 1 H, H-2GlcN), 3.88 (dd, 1 H, J8a,8b 11.4, J8a,7 2.6 Hz, H-8aKdo), 3.84 (app d, 2 H, H-6aMan, H-6bMan), 3.79 (ddd, 1H, J5,4 9.8, J5,6a 1.6, J5,6b 7.0 Hz, H-5GlcN), 3.76 (t, 1 H, J4,3 9.8 Hz, H-4Man), 3.76–3.57 (m, 10 H, OCH2, H-7Kdo′, H-8bKdo′, H-2GlN′, H-8bKdo, H-3GlN, H-6Kdo, H-6Kdo′, H-6bGlN, H-6aGlN′), 3.58–3.55 (m, 1 H, H-5GlN), 3.54 (dt, 1 H, OCH2), 3.50–3.46 (m, 2 H, H-3GlN, H-6bGlN′), 3.44 (app t, 1 H, J 9.9 and 9.2 Hz, H-4GlN), 3.38 (app t, 1 H, J 9.3 and 9.6 Hz, H-4GlN′), 3.11 (t, 2 H, J 7.5 Hz, CH2N), 2.12 (dd, 1 H, J3a,3e 13.3, J3e,4 5.4 Hz, H-3eKdo′), 2.07 (dd, 1 H, J3a,3e 12.5, J3e,4 4.4 Hz, H-3eKdo), 2.01 (s, 6 H, 2 × CH3CO), 1.98 (t, 1 H, H-3aKdo), 2.02–1.95 (m, 2 H, OCH2CH2) and 1.76 (app t, 1 H, J3a,4 11.8 Hz, H-3aKdo′). For 13C NMR data see Table 2. HRMS (ESI-TOF) m/z [M + H]+ Calcd for C41H70N3O30 1084.4039, found 1084.4048.
Acknowledgments
Financial support of this work by the Austrian Science Fund FWF (grant P 26919-N28) is gratefully acknowledged. The authors thank Simon Krauter for the measurement of the HRMS spectra.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b02172.
1H NMR and 13C NMR spectra for known and new compounds (2–11, 15, 18–22, 24–30) (PDF)
Author Present Address
§ Shire, A-1220, Vienna, Austria.
Author Present Address
∥ Department of Chemistry, University of California at Berkeley, Berkeley, California 94720, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Beutler B.; Rietschel E. T. H. Nat. Rev. Immunol. 2003, 3, 169–176. 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- Raetz C. R. H.; Whitfield C. Annu. Rev. Biochem. 2002, 71, 635–700. 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Carlson R. W.; Reuhs B.; Chen T.-B.; Bhat U. R.; Noel K. D. J. Biol. Chem. 1995, 270, 11783–1178. 10.1074/jbc.270.20.11783. [DOI] [PubMed] [Google Scholar]; b Fraysse N.; Couderc F.; Poinsot V. Eur. J. Biochem. 2003, 270, 1365–1380. 10.1046/j.1432-1033.2003.03492.x. [DOI] [PubMed] [Google Scholar]
- a Holst O. In Bacterial Lipopolysaccharides; Knirel Y., Valvano M. A., Eds.; Springer-Verlag: New York, 2011; pp 21–39. [Google Scholar]; b Holst O. FEMS Microbiol. Lett. 2007, 271, 3–11. 10.1111/j.1574-6968.2007.00708.x. [DOI] [PubMed] [Google Scholar]
- Knirel Y. A. In Bacterial Lipopolysaccharides; Knirel Y. A., Valvano M. A., Eds.; Springer-Verlag: New York, 2011; pp 41–115. [Google Scholar]
- Holst O.; Molinaro A. In Microbial Glycobiology; Moran A. P., Holst O., Brennan P. J., von Itzstein M., Eds.; Elsevier: Amsterdam, 2009; pp 29–55. [Google Scholar]
- a De Castro C.; Molinaro A.; Lanzetta R.; Silipo A.; Parrilli M. Carbohydr. Res. 2008, 343, 1924–1933. 10.1016/j.carres.2008.01.036. [DOI] [PubMed] [Google Scholar]; b Kay W.; Petersen B. O.; Duus J. Ø.; Perry M. B.; Vinogradov E. FEBS J. 2006, 273, 3002–3013. 10.1111/j.1742-4658.2006.05311.x. [DOI] [PubMed] [Google Scholar]; c De Castro C.; Carannante A.; Lanzetta R.; Lindner B.; Nunziata R.; Parrilli M.; Holst O. Chem. - Eur. J. 2006, 12, 4668–4674. 10.1002/chem.200501620. [DOI] [PubMed] [Google Scholar]
- Kanipes M. I.; Kalb S. R.; Cotter R. J.; Hozbor D. F.; Lagares A.; Raetz C. R. H. J. Biol. Chem. 2003, 278, 16365–16371. 10.1074/jbc.M301256200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanipes M. I.; Ribeiro A. A.; Lin S.; Cotter R. J.; Raetz C. R. H. J. Biol. Chem. 2003, 278, 16356–16364. 10.1074/jbc.M301255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark B. E.; Auyeung K.; Fregolino E.; Parrilli M.; Lanzetta R.; De Castro C.; Pantophlet R. Chem. Biol. 2012, 19, 254–263. 10.1016/j.chembiol.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Farrand S. K.; Van Berkum P. B.; Oger P. Int. J. Syst. Evol. Microbiol. 2003, 53, 1681–1687. 10.1099/ijs.0.02445-0. [DOI] [PubMed] [Google Scholar]
- Doores K. J.; Huber M.; Le K. M.; Wang S.-K.; Doyle-Cooper C.; Cooper A.; Pantophlet R.; Wong C.-H.; Nemazee D.; Burton D. R. J. Virol. 2013, 87, 2234–2241. 10.1128/JVI.02820-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield R. L.; De Castro C.; Marzaioli A. M.; Wilson I. A.; Pantophlet R. Glycobiology 2015, 25, 412–419. 10.1093/glycob/cwu123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples, see:; a Crich D.; Dudkin V. J. Am. Chem. Soc. 2001, 123, 6819–6825. 10.1021/ja010086b. [DOI] [PubMed] [Google Scholar]; b Liao L.; Auzanneau I. J. Org. Chem. 2005, 70, 6265–6273. 10.1021/jo050707+. [DOI] [PubMed] [Google Scholar]; c Sekljic H.; Wimmer N.; Hofinger A.; Brade H.; Kosma P. J. Chem. Soc., Perkin Trans. 1 1997, 1973–1982. 10.1039/a700497d. [DOI] [Google Scholar]
- For examples, see:; a Tiwari V. K.; Mishra B. B.; Mishra K. B.; Mishra N.; Singh A. S.; Chen X. Chem. Rev. 2016, 116, 3086–3240. 10.1021/acs.chemrev.5b00408. [DOI] [PubMed] [Google Scholar]; b Kolb H. C.; Finn M. G.; Sharpless B. K. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]; c Chabre Y. M.; Roy R. Adv. Carbohydr. Chem. Biochem. 2010, 63, 165–393. 10.1016/S0065-2318(10)63006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau K.; Thon V.; Yu H.; Ding L.; Chen Y.; Muthana M. M.; Wong D.; Huang R.; Chen X. Chem. Commun. 2010, 46, 6066–6068. 10.1039/c0cc01381a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farcet J.-B.; Herczeg M.; Christler A.; Kosma P. In Carbohydrate Chemistry: Proven Synthetic Methods; Murphy P., Vogel C., Eds.; CRC Press: Boca Raton, FL, 2017; Vol. 4, pp 285–292. [Google Scholar]
- Das B.; Mahender G.; Kumar V. S.; Chowdhury N. Tetrahedron Lett. 2004, 45, 6709–6711. 10.1016/j.tetlet.2004.07.075. [DOI] [Google Scholar]
- Jones G. B.; Hynd G.; Wright J. M.; Sharma A. J. Org. Chem. 2000, 65, 263–265. 10.1021/jo9913255. [DOI] [PubMed] [Google Scholar]
- a Kiso M.; Anderson L. Carbohydr. Res. 1979, 72, C12–C14. 10.1016/S0008-6215(00)83962-2. [DOI] [Google Scholar]; b Kiso M.; Anderson L. Carbohydr. Res. 1985, 136, 309–323. 10.1016/0008-6215(85)85205-8. [DOI] [PubMed] [Google Scholar]; c Kosma P.; Strobl M.; Allmaier G.; Schmid E.; Brade H. Carbohydr. Res. 1994, 254, 105–132. 10.1016/0008-6215(94)84246-9. [DOI] [PubMed] [Google Scholar]
- a Masaki M.; Kitahara T.; Kurita H.; Ohta M. J. Am. Chem. Soc. 1968, 90, 4508–4509. 10.1021/ja01018a088. [DOI] [Google Scholar]; b Akamatsu S.; Ikeda K.; Achiwa K. Chem. Pharm. Bull. 1991, 39, 288–296. 10.1248/cpb.39.288. [DOI] [Google Scholar]
- Ikeda K.; Takahashi T.; Shimizu C.; Nakamoto S.; Achiwa K. Chem. Pharm. Bull. 1987, 35, 1383–1387. 10.1248/cpb.35.1383. [DOI] [PubMed] [Google Scholar]
- Kosma P.; Bahnmüller R.; Schulz G.; Brade H. Carbohydr. Res. 1990, 208, 37–50. 10.1016/0008-6215(90)80083-F. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Pradhan T. K.; Mong K. K. T. Isr. J. Chem. 2015, 55, 285–296. 10.1002/ijch.201400145. [DOI] [Google Scholar]; b Kosma P. Tetrahedron Lett. 2016, 57, 2133–2142. 10.1016/j.tetlet.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent examples, see:; a Kong L.; Vijayakrishnan B.; Kowarik M.; Park J.; Zakharova A. N.; Neiwert L.; Faridmoayer A.; Davis B. G. Nat. Chem. 2016, 8, 242–249. 10.1038/nchem.2432. [DOI] [PubMed] [Google Scholar]; b Huang J.-S.; Huang W.; Meng X.; Wang X.; Gao P.-C.; Yang J.-S. Angew. Chem., Int. Ed. 2015, 54, 10894–10898. 10.1002/anie.201505176. [DOI] [PubMed] [Google Scholar]; c Yi R.; Ogaki A.; Fukunaga M.; Nakajima H.; Ichiyanagi T. Tetrahedron 2014, 70, 3675–3682. 10.1016/j.tet.2014.04.024. [DOI] [Google Scholar]; d Pokorny B.; Kosma P. Carbohydr. Res. 2016, 422, 5–12. 10.1016/j.carres.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Boltje T. J.; Zhong W.; Park J.; Wolfert A. A.; Chen W.; Boons G.-J. J. Am. Chem. Soc. 2012, 134, 14255–14262. 10.1021/ja306274v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorny B.; Kosma P. Org. Lett. 2015, 17, 110–113. 10.1021/ol5033128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples, see:; a Imoto M.; Kusunose N.; Kusumoto S.; Shiba S. Tetrahedron Lett. 1988, 29, 2227–2230. 10.1016/S0040-4039(00)86718-0. [DOI] [Google Scholar]; b Yoshizaki H.; Fukuda N.; Sato K.; Oikawa M.; Fukase K.; Suda Y.; Kusumoto S. Angew. Chem., Int. Ed. 2001, 40, 1475–1480. . [DOI] [PubMed] [Google Scholar]; c Zhang Y.; Gaekwad J.; Wolfert M. A.; Boons G.-J. Chem. - Eur. J. 2008, 14, 558–569. 10.1002/chem.200701165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukase K.; Kamikawa T.; Iwai Y.; Shiba T.; Rietschel E. T.; Kusumoto S. Bull. Chem. Soc. Jpn. 1991, 64, 3267–3273. 10.1246/bcsj.64.3267. [DOI] [Google Scholar]
- Rosenbrook W. Jr.; Riley D. A.; Lartey P. A. Tetrahedron Lett. 1985, 26, 3–4. 10.1016/S0040-4039(00)98450-8. [DOI] [Google Scholar]
- Xiao X.; Bai D. Synlett 2001, 2001, 535–537. 10.1055/s-2001-12340. [DOI] [Google Scholar]
- Unger F. M.; Stix D.; Schulz G. Carbohydr. Res. 1980, 80, 191–195. 10.1016/S0008-6215(00)85325-2. [DOI] [Google Scholar]
- Mong K.-K. T.; Shiau K.-S.; Lin Y. H.; Cheng K.-C.; Lin C.-H. Org. Biomol. Chem. 2015, 13, 11550–11560. 10.1039/C5OB01786F. [DOI] [PubMed] [Google Scholar]
- Schmidt R. R.; Michel J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. 10.1002/anie.198007311. [DOI] [Google Scholar]
- Liao L.; Auzanneau F.-I. Org. Lett. 2003, 5, 2607–2610. 10.1021/ol034669x. [DOI] [PubMed] [Google Scholar]
- Bock K.; Pedersen C. J. Chem. Soc., Perkin Trans. 2 1974, 293–297. 10.1039/p29740000293. [DOI] [Google Scholar]
- Paulsen H.; Stiem M.; Unger F. M. Tetrahedron Lett. 1986, 27, 1135–1138. 10.1016/S0040-4039(00)84197-0. [DOI] [Google Scholar]
- Stahl E.; Kaltenbach U. J. Chromatogr. 1961, 5, 351–355. 10.1016/S0021-9673(01)92868-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.