

Abstract
PqqE is a radical S‐adenosyl‐l‐methionine (SAM) enzyme that catalyzes the initial reaction of pyrroloquinoline quinone (PQQ) biosynthesis. PqqE belongs to the SPASM (subtilosin/PQQ/anaerobic sulfatase/mycofactocin maturating enzymes) subfamily of the radical SAM superfamily and contains multiple Fe–S clusters. To characterize the Fe–S clusters in PqqE from Methylobacterium extorquens AM1, Cys residues conserved in the N‐terminal signature motif (CX 3 CX 2C) and the C‐terminal seven‐cysteine motif (CX 9–15 GX 4 CX n CX 2 CX 5 CX 3 CX nC; n = an unspecified number) were individually or simultaneously mutated into Ser. Biochemical and Mössbauer spectral analyses of as‐purified and reconstituted mutant enzymes confirmed the presence of three Fe–S clusters in PqqE: one [4Fe–4S]2+ cluster at the N‐terminal region that is essential for the reductive homolytic cleavage of SAM into methionine and 5′‐deoxyadenosyl radical, and one each [4Fe–4S]2+ and [2Fe–2S]2+ auxiliary clusters in the C‐terminal SPASM domain, which are assumed to serve for electron transfer between the buried active site and the protein surface. The presence of [2Fe–2S]2+ cluster is a novel finding for radical SAM enzyme belonging to the SPASM subfamily. Moreover, we found uncommon ligation of the auxiliary [4Fe–4S]2+ cluster with sulfur atoms of three Cys residues and a carboxyl oxygen atom of a conserved Asp residue.
Keywords: [4Fe–4S] cluster, Mössbauer spectroscopy, pyrroloquinoline quinone, radical SAM enzyme
Abbreviations
- 5′dA
5′‐deoxyadenosine
- anSME
anaerobic sulfatase maturating enzyme
- anSMEcpe
anSME from Clostridium perfringens
- DTT
dithiothreitol
- PQQ
pyrroloquinoline quinone
- RS
radical SAM
- SAM
S‐adenosyl‐l‐methionine
- SPASM
subtilosin/PQQ/anaerobic sulfatase/mycofactocin maturating enzymes
- WT
wild‐type
PqqE is a radical S‐adenosyl‐l‐methionine (SAM) enzyme that catalyzes the initial reaction of pyrroloquinoline quinone (PQQ) biosynthesis, that is, de novo carbon‐carbon bond formation between glutamate and tyrosine residues of a precursor peptide PqqA 1. The reaction is facilitated by a small protein PqqD 1, which forms a complex with PqqA 2.
As other members of the radical SAM superfamily of enzymes, PqqE contains a characteristic N‐terminal CX3CX2C motif that ligates three of the four iron atoms of [4Fe–4S] cluster. Binding of SAM to the remaining iron atom facilitates reductive cleavage of SAM to methionine and 5′‐deoxyadenosyl radical, which then in most cases generates a substrate radical by abstracting a hydrogen atom from the substrate 3. In the case of PqqE, a carbon radical is formed on the γ‐carbon of conserved glutamate of PqqA peptide. This subsequently reacts with the ring C3 carbon of conserved tyrosine forming a carbon‐carbon bond 1.
In addition to the conserved N‐terminal binding motif, PqqE contains a C‐terminal seven‐cysteine motif (CX9–15GX4CXnCX2CX5CX3CXnC; n = an unspecified number) in the domain designated SPASM (subtilosin/PQQ/anaerobic sulfatase/mycofactocin maturating enzymes) 4. The enzymes with the full SPASM domain generally bind additional two [4Fe–4S] clusters (termed auxiliary clusters) with proposed diverse functions such as substrate binding and positioning or electron transfer 5. A subgroup of proteins belonging to the SPASM subfamily contains a shortened version of the SPASM domain (named a Twitch domain 6), which lacks the CX2CX5CX3C motif of the SPASM domain and binds a single auxiliary Fe–S cluster 5. So far only crystal structure of a SPASM domain‐containing protein, anaerobic sulfatase maturating enzyme (anSME) from Clostridium perfringens (anSMEcpe), shows that both auxiliary clusters are fully ligated by cysteine residues, one of the ligating cysteine residues being present upstream of the SPASM domain 6. Available sequence and crystal structure data on the SPASM/Twitch domain‐containing proteins, however, indicate that some of them have an open coordination site for substrate binding on an auxiliary Fe–S cluster or use another amino acid residue to coordinate the cluster 7.
As we reported previously 8, PqqE possibly contains three Fe–S clusters consisting mainly of [4Fe–4S]2+ and [2Fe–2S]2+ forms. To identify the binding of Fe–S clusters in PqqE, conserved Cys residues in both the N‐terminal and C‐terminal signature motifs have been individually or simultaneously mutated into Ser. Biochemical and spectrophotometric analyses of mutant enzymes confirmed the presence of three Fe–S clusters in PqqE: a radical SAM [4Fe–4S]2+ cluster at the N‐terminal region indispensable for the reductive homolytic cleavage of SAM into methionine and 5′‐deoxyadenosyl radical, and two auxiliary clusters (designated Aux I and Aux II clusters), one each [2Fe–2S]2+ cluster (Aux I cluster) and [4Fe–4S]2+ cluster (Aux II cluster), in the C‐terminal SPASM domain. While mutation of Cys residues predicted to bind Aux II cluster resulted in expected loss of cluster accompanied with moderate decrease in SAM cleavage activity, mutation in the Aux I site resulted in marked reduction in protein stability, but not loss of the [2Fe–2S]2+ cluster and SAM cleavage activity. The structure model of PqqE created by homology alignment‐based structure modeling together with analysis of respective mutant enzyme suggests uncommon ligation of a carboxyl group of an Asp residue to an iron atom of the Aux II cluster.
Results
Dual iron isotope analysis
We have recently reported that the as‐purified and reconstituted enzymes of PqqE from Methylobacterium extorquens AM1 contain both [4Fe–4S]2+ and [2Fe–2S]2+ clusters as the major forms with the former being predominant in the reconstituted enzyme 8. In this study, we first employed a dual iron isotope (56Fe/57Fe) analysis to examine whether these Fe–S clusters are derived from biological cluster insertion by the Escherichia coli intracellular system or from chemical reconstitution or both. Thus, PqqE was initially expressed in E. coli cells in the presence of Mössbauer‐silent 56Fe3+ (natural abundance, 92%) and the 56Fe‐as‐purified enzyme was then reconstituted with 57Fe3+. Alternatively, PqqE was initially expressed in the presence of 57Fe3+ and the 57Fe‐as‐purified enzyme was then reconstituted with 56Fe3+. The 56Fe‐as‐purified PqqE reconstituted with 57Fe exhibited a rather simple Mössbauer spectrum (Fig. 1A) simulated with a single major quadrupole doublet (blue) assignable to a [4Fe–4S]2+ (S total = 0) cluster (for Mössbauer parameters, see Table 1). In contrast, the 57Fe‐as‐purified PqqE reconstituted with 56Fe showed a complex spectrum with two major quadrupole doublets assignable to [4Fe–4S]2+ (S total = 0) and [2Fe–2S]2+ (S total = 0) clusters (Fig. 1B; blue and green, respectively), which resembles the complex spectrum of 57Fe‐as‐purified enzyme reported previously 8. This result indicates that reconstitution did not alter the 57Fe‐labeled cluster composition contained in the as‐purified enzyme. The spectra of 57Fe‐reconstituted enzymes (Fig. 1A,C) are accompanied with a small doublet (red) derived from the noncluster iron bound onto the protein surface with Mössbauer hyperfine parameters characteristic of free high‐spin Fe2+ ions (S = 2) 9. In addition, the two minor unassigned doublets (gray and light blue) observed in all spectra have hyperfine parameters identical with those assigned to the [4Fe–4S]2+–pyruvate complex 10, as reported previously 9. They are assumed to be associated with ligation of the conserved Asp319 with an iron atom of the auxiliary [4Fe–4S]2+ cluster bound at the Aux II site, as described later. Using the values of relative spectral area (RA) of Mössbauer signals (Table 1), the [4Fe–4S]2+/[2Fe–2S]2+ cluster ratio is calculated to be approximately 0.75 [= {(53.1 + 3.4 × 2)/4}/(40.0/2)] and 1.9 [= {(67.1 + 3.6 × 2)/4}/(19.9/2)] in the 57Fe‐as‐purified enzymes reconstituted with 56Fe and 57Fe, respectively, with the minor doublets being included in the RA value of [4Fe–4S]2+. The cluster ratio in the as‐purified wild‐type (WT) enzyme (0.89; Table 2) was estimated to be slightly higher than that in the 57Fe‐as‐purified‐56Fe‐reconstituted enzyme. Thus, it is suggested that the cluster insertion into an expressed protein in E. coli cells may vary from cultivation to cultivation. Collectively, these results indicate that chemical reconstitution provides only the [4Fe–4S]2+ cluster and that the [2Fe–2S]2+ cluster contained in the as‐purified enzyme is not converted into the [4Fe–4S]2+ cluster by subsequent reconstitution. The Fe–S cluster bound at the consensus CX3CX2C motif located close to the N terminus of radical SAM enzymes is believed to be a [4Fe–4S]2+ cluster that is directly involved in the homolytic cleavage of SAM and hence often designated ‘RS cluster’ 3, 11, 12. Therefore, the [2Fe–2S]2+ cluster found in the as‐purified PqqE is probably bound at either the Aux I or Aux II site as defined below.
Figure 1.
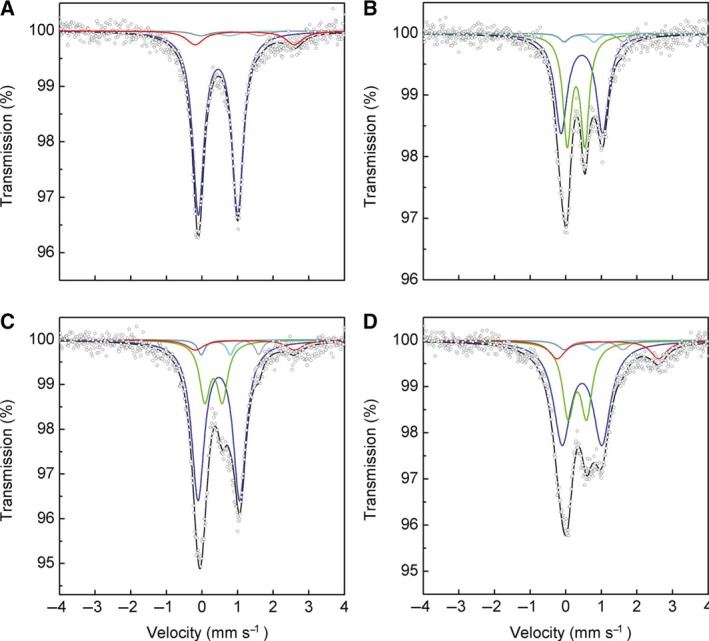
Mössbauer spectra of WT and C32S mutant of PqqE. 57Fe Mössbauer spectra of (A) 56Fe‐as‐purified WT enzyme reconstituted with 57Fe, (B) 57Fe‐as‐purified WT enzyme reconstituted with 56Fe, (C) 57Fe‐as‐purified WT enzyme reconstituted with 57Fe, and (D) 57Fe‐as‐purified C32S mutant reconstituted with 57Fe, all recorded at 5 K. Simulated spectra are shown as follows: blue, [4Fe–4S]2+; green, [2Fe–2S]2+; gray and light blue, ferrous and ferric sites, respectively, of the [4Fe–4S]2+ cluster ligated with the side‐chain carboxyl group of Asp319 at the Aux II site; red, free (noncluster) high‐spin Fe2+; black, the sum of the simulated spectra.
Table 1.
Summary of the Mössbauer hyperfine parametersa
| PqqE variant (Relating Fig.) | Color code for doublet component | δ ± 0.01 (mm·s−1) | ΔE Q ±0.01 (mm·s−1) | RA ± 1 (%) | Cluster assignmentb spin S |
|---|---|---|---|---|---|
| WT | |||||
| 56Fe‐as‐purified‐57Fe‐reconstituted (Fig. 1A) | Blue | 0.45 | 1.10 | 83.6 | [4Fe–4S]2+, S = 0 |
| Light blue | 0.38 | 0.82 | 3.2 | [4Fe–4S]2+ Asp, S = 0 | |
| Gray | 0.78 | 1.62 | 3.2 | [4Fe–4S]2+ Asp, S = 0 | |
| Red | 1.19 | 2.75 | 9.9 | Free Fe2+, S = 2 | |
| 57Fe‐as‐purified‐56Fe‐reconstituted (Fig. 1B) | Green | 0.30 | 0.49 | 40.0 | [2Fe–2S]2+, S = 0 |
| Blue | 0.46 | 1.15 | 53.1 | [4Fe–4S]2+, S = 0 | |
| Light blue | 0.38 | 0.82 | 3.4 | [4Fe–4S]2+ Asp, S = 0 | |
| Gray | 0.78 | 1.62 | 3.4 | [4Fe–4S]2+ Asp, S = 0 | |
| 57Fe‐as‐purified‐57Fe‐reconstituted (Fig. 1C) | Green | 0.32 | 0.49 | 19.9 | [2Fe–2S]2+, S = 0 |
| Blue | 0.47 | 1.17 | 67.1 | [4Fe–4S]2+, S = 0 | |
| Light blue | 0.38 | 0.81 | 3.6 | [4Fe–4S]2+ Asp, S = 0 | |
| Gray | 0.78 | 1.62 | 3.6 | [4Fe–4S]2+ Asp, S = 0 | |
| Red | 1.21 | 2.79 | 5.8 | Free Fe2+, S = 2 | |
| C32S | |||||
| 57Fe‐as‐purified‐57Fe‐reconstituted (Fig. 1D) | Green | 0.33 | 0.51 | 27.1 | [2Fe–2S]2+, S = 0 |
| Blue | 0.46 | 1.10 | 55.9 | [4Fe–4S]2+, S = 0 | |
| Light blue | 0.38 | 0.82 | 3.6 | [4Fe–4S]2+ Asp, S = 0 | |
| Gray | 0.79 | 1.62 | 3.6 | [4Fe–4S]2+ Asp, S = 0 | |
| Red | 1.18 | 2.81 | 9.8 | Free Fe2+, S = 2 | |
| C323S | |||||
| 57Fe‐as‐purified (Fig. 3B) | Green | 0.32 | 0.49 | 57.8 | [2Fe–2S]2+, S = 0 |
| Blue | 0.47 | 1.17 | 27.4 | [4Fe–4S]2+, S = 0 | |
| Light blue | 0.38 | 0.81 | 7.4 | [4Fe–4S]2+ Asp, S = 0 | |
| Gray | 0.78 | 1.62 | 7.4 | [4Fe–4S]2+ Asp, S = 0 | |
| 57Fe‐as‐purified‐57Fe‐reconstituted (Fig. 3C) | Green | 0.32 | 0.53 | 18.3 | [2Fe–2S]2+, S = 0 |
| Blue | 0.44 | 1.12 | 65.5 | [4Fe–4S]2+, S = 0 | |
| Light blue | 0.39 | 0.82 | 2.4 | [4Fe–4S]2+ Asp, S = 0 | |
| Gray | 0.78 | 1.62 | 2.4 | [4Fe–4S]2+ Asp, S = 0 | |
| Red | 1.21 | 2.79 | 11.5 | Free Fe2+, S = 2 | |
| C310S/C313S | |||||
| 57Fe‐as‐purified (Fig. 4B) | Green | 0.28 | 0.54 | 70.7 | [2Fe–2S]2+, S = 0 |
| Blue | 0.46 | 1.10 | 29.3 | [4Fe–4S]2+, S = 0 | |
| 57Fe‐as‐purified‐57Fe‐reconstituted (Fig. 4C) | Green | 0.32 | 0.49 | 19.3 | [2Fe–2S]2+, S = 0 |
| Blue | 0.47 | 1.17 | 65.5 | [4Fe–4S]2+, S = 0 | |
| Purple | 1.42 | 1.32 | 5.0 | Unknown | |
| Red | 1.12 | 2.88 | 10.2 | Free Fe2+, S = 2 | |
| D319S | |||||
| 57Fe‐as‐purified‐57Fe‐reconstituted (Fig. 5B) | Green | 0.27 | 0.56 | 18.6 | [2Fe–2S]2+, S = 0 |
| Blue | 0.46 | 1.12 | 70.8 | [4Fe–4S]2+, S = 0 | |
| Purple | 1.54 | 1.36 | 3.8 | Unknown | |
| Red | 1.25 | 2.72 | 6.7 | Free Fe2+, S = 2 | |
| C28S/C32S/C310S/C313S | |||||
| 57Fe‐as‐purified (Fig. 6B) | Green | 0.28 | 0.56 | 100.0 | [2Fe–2S]2+, S = 0 |
| 57Fe‐as‐purified‐57Fe‐reconstituted (Fig. 6C) | Green | 0.28 | 0.56 | 26.5 | [2Fe–2S]2+, S = 0 |
| Blue | 0.43 | 1.10 | 60.5 | [4Fe–4S]2+, S = 0 | |
| Red | 1.23 | 2.28 | 13.0 | Free Fe2+, S = 2 | |
δ is the isomer shift, ΔE Q is the quadrupole splitting, and RA is the relative spectral area of individual components identified during spectral fitting, assuming an identical recoilless factor for all iron sites within the experimental error of Mössbauer technique.
[4Fe–4S]2+ Asp represents an Fe atom of [4Fe–4S]2+ cluster interacting with the side‐chain carboxyl group of Asp319 at the Aux II site.
Table 2.
Fe and S contents and SAM cleavage activities of the PqqE variants
| PqqE variant | As‐purified | Reconstituted | ||||||
|---|---|---|---|---|---|---|---|---|
| Fe (mol·mol−1) | S (mol·mol−1) | Activity (nmol·min−1·mg−1) | Cluster ratioa | Fe (mol·mol−1) | S (mol·mol−1) | Activity (nmol·min−1·mg−1) | Cluster ratioa | |
| WT | 6.35 ± 0.62 | 10.15 ± 0.65 | 0.442 ± 0.012 | 0.89b | 11.52 ± 1.05 | 16.17 ± 1.21 | 0.858 ± 0.071 | 1.9 |
| C32S | 3.84 ± 0.65b | 5.30 ± 0.33b | ndb, c | 0.78b | 10.50 ± 1.30b | 12.10 ± 1.50b | ndc | 1.2 |
| C268S | 2.12 ± 0.55 | 3.98 ± 0.93 | 0.021 ± 0.006 | NDc | NDc | NDd | NDd | NDd |
| C323S | 4.12 ± 0.57 | 7.29 ± 0.72 | 0.227 ± 0.066 | 0.37 | 10.34 ± 1.16 | 14.18 ± 1.72 | 0.868 ± 0.109 | 1.9 |
| C313S | 4.21 ± 0.54 | 7.52 ± 1.18 | 0.268 ± 0.022 | NDc | 10.36 ± 0.65 | 16.17 ± 0.76 | 0.469 ± 0.019 | 1.7 |
| C310SC313S | 4.09 ± 0.66 | 6.14 ± 0.81 | 0.322 ± 0.012 | 0.21 | 10.08 ± 1.12 | 13.58 ± 1.30 | 0.543 ± 0.042 | 1.7 |
| C310SC313SC341S | 3.72 ± 0.11 | 7.56 ± 0.42 | 0.253 ± 0.046 | NDc | 7.95 ± 0.80 | 12.01 ± 0.68 | 0.428 ± 0.108 | NDd |
| D319S | 6.15 ± 0.53 | 9.57 ± 0.78 | 0.410 ± 0.130 | NDc | 13.20 ± 1.70 | 17.89 ± 0.76 | 0.591 ± 0.031 | 1.9 |
| C32SC310SC313S | 1.69 ± 0.13 | 2.91 ± 0.17 | ndc | 0 | 7.41 ± 0.40 | 10.56 ± 1.46 | ndc | 0.9 |
| C28SC32SC310SC313S | 1.73 ± 0.17 | 2.90 ± 0.18 | ndc | 0 | 6.03 ± 0.28 | 9.51 ± 0.35 | ndc | 1.1 |
| C28SC32SC310SC313SC341S | 1.53 ± 0.12 | 3.10 ± 0.15 | ndc | NDc | 7.47 ± 0.50 | 11.34 ± 1.21 | ndc | NDd |
Cluster molar ratio ([4Fe–4S]2+/[2Fe–2S]2+) was calculated from the relative area (RA) of Mössbauer signals including the RA of signals assigned to an Fe atom of [4Fe–4S]2+ cluster interacting with the side‐chain carboxyl group of Asp319 at the Aux II site (except for Aux II mutants: C313S, C310SC313S, and D319S).
Data adopted from the previous study 8.
Not detected.
Not determined.
Prediction of Fe–S cluster‐binding residues
Amino acid residues putatively involved in Fe–S cluster binding in PqqE have been predicted by homology alignment‐based structure modeling (SWISS‐MODEL) 13. Application of the entire amino acid sequence of M. extorquens AM1 PqqE (UniProt ID: P71517) as a query sequence resulted in building of three models with different template crystal structures: an anaerobic sulfatase maturating enzyme (anSME) from C. perfringens (anSMEcpe) (PDB ID: 4K36) 6, a molybdenum cofactor biosynthetic enzyme MoaA from Staphylococcus aureus (PDB ID: 1TV8) 14, and a maturase HydG involved in the [FeFe]‐hydrogenase H‐cluster assembly from Thermoanaerobacter italicus Ab9 (PDB ID: 4WCX) 15. Although all the enzymes autoselected as the template structures belong to the radical SAM superfamily, the first model with anSMEcpe as a template was employed in this study based on the highest GMQE (global model quality estimate) score (0.50) and overall sequence coverage (0.84). In addition, both PqqE and anSME belong to the SPASM subgroup of the radical SAM superfamily 4 and contain three Fe–S clusters, whereas MoaA and HydG both harbor only two Fe–S clusters. The initial model, however, had to be slightly modified in a partial region of PqqE (Ala247–Lys255) by adopting the coordinates of main chain atoms of the corresponding nonhomologous region of anSMEcpe (Ser254–Thr262), so that the cluster‐ligating Cys residues predicted for the Aux I site are placed at mutually nearby positions (see below). Finally, the coordinates of three [4Fe–4S]2+ clusters found in the anSMEcpe structure were merged into the PqqE model. The coordinates of a [2Fe–2S]2+ cluster taken from the structure of E. coli BioB 16 were also merged, instead of [4Fe–4S]2+, for the Aux I site.
The structure model of PqqE thus constructed (Fig. 2A) is consistent with the previous finding that most, if not all, radical SAM superfamily enzymes would have a common core fold comprising a partial (α/β)6 triosephosphate isomerase (TIM) barrel 17, 18. Three Cys residues (Cys28, Cys32, and Cys35) conserved in the consensus CX3CX2C motif are ideally located to bind the radical SAM [4Fe–4S]2+ cluster (RS cluster) (Fig. 2B) 3, 11, 12. Thus, manual adjustment of the chi angles (N‐CA‐CB‐SG) of Cys residues to ligate the RS cluster led the Cys sulfur‐to‐iron distances to 2.30–2.53 Å that are almost identical with those in the anSMEcpe structure (Fig. 2B). Furthermore, seven Cys residues conserved in the C‐terminal SPASM domain 8, 19 are positioned to bind two auxiliary Fe–S clusters at the sites almost identical with those identified in the anSMEcpe structure 6. The modeled Aux I site consists of four Cys residues (Cys248, Cys268, Cys323, and Cys325) and may be able to accommodate either [4Fe–4S]2+ (Fig. 2C) or [2Fe–2S]2+ (Fig. 2D), as detailed later. Cys248, Cys268, and Cys323 of PqqE correspond to Cys255, Cys276, and Cys330 of anSMEcpe, respectively; a Cys residue corresponding to Cys261 of anSMEcpe is not conserved in PqqE homologs 8 but the conserved Cys325 is instead placed in the Aux I site and may serve as a substitute for Cys261 of anSMEcpe (Fig. 2C,D). However, the Cys sulfur‐to‐iron distances could not be fully optimized even by rotating and/or slightly transferring the introduced Fe–S cluster within the cavity of Aux I site for both [4Fe–4S]2+ and [2Fe–2S]2+, which implies that the Aux I site was not modeled properly. In contrast, the Aux II site was modeled nicely to accommodate a [4Fe–4S]2+ cluster but was found to contain three Cys residues (Cys310, Cys313, and Cys341) (Fig. 2E) unlike in the anSMEcpe structure, in which the Aux II cluster is bound by four Cys residues (Cys317, Cys320, Cys326, and Cys348) 6. Cys310, Cys313, and Cys341 of PqqE correspond exactly to Cys317, Cys320, and Cys348 of anSMEcpe, respectively, with identical Cys sulfur‐to‐iron distances of 2.31–2.44 Å for the introduced [4Fe–4S]2+ cluster (Fig. 2E). Interestingly, Cys326 of anSMEcpe is replaced by an Asp residue (Asp319) in PqqE, which is totally conserved in PqqE homologs 8. As described later, the side‐chain carboxyl group of Asp319 is assumed to ligate an iron atom of the Aux II cluster. The modeled cluster distances (RS–Aux I, Aux I–Aux II, and RS–Aux II) are estimated to be about 16, 14, and 28 Å, respectively (Fig. 2A), which are comparable with those (16.9, 12.9, and 26.7 Å) in the anSMEcpe structure 6.
Figure 2.
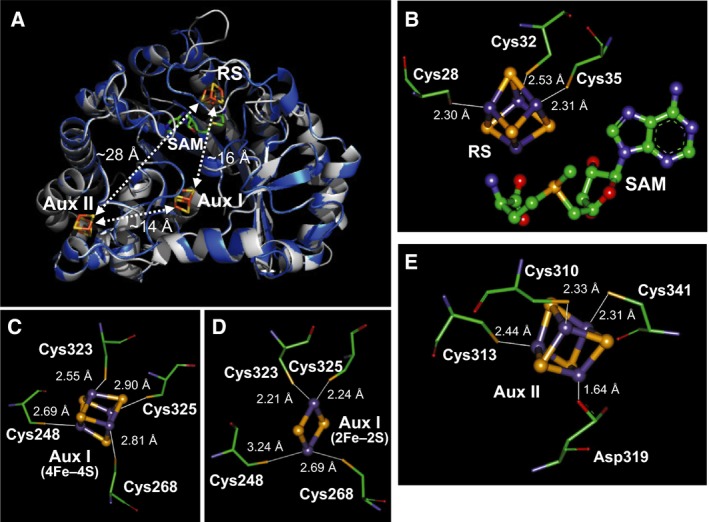
Homology alignment‐based structure model of PqqE. (A) The modeled PqqE structure is shown in ribbon representation (blue) overlaid with the template structure of anSMEcpe (gray) 6. The RS, Aux I, and Aux II clusters and SAM bound in anSMEcpe were merged into the modeled PqqE structure and are shown by stick models with respective atom color codes. Approximate distances between the clusters are also shown. (B–E) Enlarged views around the RS [4Fe–4S]2+ cluster and SAM (B), the Aux I site bound with a [4Fe–4S]2+ (C), or a [2Fe–2S]2+ cluster (D) and the Aux II [4Fe–4S]2+ cluster (E). Amino acid residues are shown in thin stick models and the clusters and SAM in thick ball‐and‐stick models. Chi angles (N‐CA‐CB‐SG) of Cys residues and that (N‐CA‐CB‐CG) of Asp319 were manually adjusted to have shortest possible distances (shown in Å) from Cys sulfur and Asp oxygen atoms to iron atoms (purple) of each cluster. In (C) and (D), the position and orientation of the clusters were also manipulated slightly within the cavity of the Aux I site.
Mutation of residues putatively involved in binding of RS and Aux I clusters
We previously mutated a Cys residue (Cys32) conserved in the consensus CX3CX2C motif and confirmed that the Cys32‐to‐Ser substitution leads to the complete loss of the reductive SAM cleavage activity associated with the RS cluster 8. The zero‐field 57Fe Mössbauer spectra of as‐purified (previous study) and reconstituted (present study, Fig. 1D) C32S mutants provided the [4Fe–4S]2+/[2Fe–2S]2+ cluster ratios of about 0.78 and 1.2 (Table 2), respectively; the cluster ratio (0.6) reported for the as‐purified C32S mutant 8 was recalculated by taking the two minor doublets into account. These results confirm the previous conclusion that the reconstituted C32S mutant contains no [4Fe–4S]2+ cluster at RS site but still retains [4Fe–4S]2+ and [2Fe–2S]2+ clusters at Aux I and II sites. The [2Fe–2S]2+ cluster is the more abundant form in both the as‐purified WT and C32S mutant enzymes 8.
To identify cluster‐binding Cys residues located at the Aux I site, we then mutated Cys268 and Cys323 individually into Ser and investigated the properties of the resultant mutants, C268S and C323S. Unfortunately, however, both C268S and C323S mutants were very unstable during anaerobic purification unlike the WT PqqE that is relatively stable even under semiaerobic conditions 8. Although the addition of 0.1 mm dithiothreitol (DTT) in the affinity purification buffer significantly improved the yield of soluble proteins of these mutants, chemical reconstitution with Fe3+ and S2− in the presence of excess DTT resulted in further production of insoluble proteins, particularly for the C268S mutant. Thus, we could obtain only a small amount (~ 0.3 to 0.8 mg per 1 g of wet cells) of the as‐purified C268S mutant, which was insufficient for subsequent reconstitution and biochemical and spectral characterization. A moderate amount (~ 1.2 to 2.4 mg per 1 g of wet cells) of the as‐purified enzyme was obtained for the C323S mutant that could be used for further analysis.
Both as‐purified C268S and C323S mutants showed considerably lower SAM cleavage activities than the WT PqqE as summarized in Table 2. These results are consistent with the low Fe and S contents in the as‐purified mutant proteins and suggest that the Cys‐to‐Ser mutation at Aux I site also reduces the efficiency of [4Fe–4S]2+ cluster insertion into the RS site in E. coli cells. On the other hand, the reconstituted C323S mutant showed a high SAM cleavage activity comparable with that of the WT PqqE.
As reported previously 8, the WT PqqE shows a UV‐Vis absorption spectrum that is typical for an Fe–S cluster‐containing protein with an absorption peak at 400–420 nm associated with the presence of [4Fe–4S]2+ clusters 20, 21 and shoulders at 300–350 nm and around 450 nm, which are derived from a [2Fe–2S]2+ cluster 20, 22 (Fig. 3A). As‐purified C268S and C323S mutants showed similar UV‐Vis spectra with considerably lower absorption bands throughout the measured wavelength region (250–600 nm) than the WT enzyme (Fig. 3A), in agreement with their low Fe and S contents (Table 2). The shoulder around 450 nm in the spectra of as‐purified C268S and C323S mutants is less visible than in the WT enzyme, presumably reflecting the difference in the contents of [4Fe–4S]2+ and [2Fe–2S]2+ clusters (see below). In contrast, the reconstituted C323S mutant exhibited a UV‐Vis spectrum comparable to the spectrum of the reconstituted WT enzyme (Fig. 3A).
Figure 3.
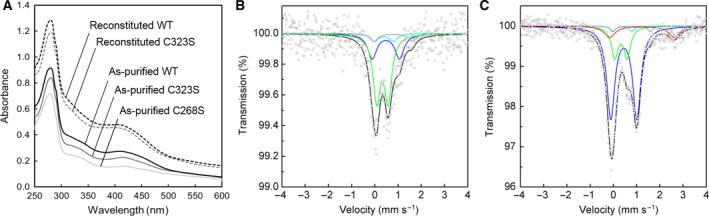
Spectral analyses of Aux I mutants of PqqE. (A) UV‐Vis spectra of the as‐purified and reconstituted C323S and C268S mutants and WT enzyme. (B, C) Mössbauer spectra of the as‐purified (B) and reconstituted (C) C323S mutant, recorded at 5 K. Simulated spectra are shown as follows: blue, [4Fe–4S]2+; green, [2Fe–2S]2+; gray and light blue, ferrous and ferric sites, respectively, of the [4Fe–4S]2+ cluster ligated with the side‐chain carboxyl group of Asp319 at the Aux II site; red, free (noncluster) high‐spin Fe2+; black, the sum of the simulated spectra.
Moreover, Mössbauer spectra of the as‐purified and reconstituted C323S mutants could not be simulated with a single quadrupole doublet (like the spectrum shown in Fig. 1A) but with a mixture of at least two major doublets. Although as‐purified C323S mutant showed a very noisy Mössbauer spectrum due to an inadequate protein amount as well as the low Fe content, the signals may be assignable to [4Fe–4S]2+ (S total = 0) and [2Fe–2S]2+ (S total = 0) clusters (Table 1 and Fig. 3B; blue and green, respectively). Similar parameters were obtained with the reconstituted C323S mutant that showed a less noisy spectrum (Table 1 and Fig. 3C; blue and green for [4Fe–4S]2+ and [2Fe–2S]2+ clusters, respectively). The [4Fe–4S]2+/[2Fe–2S]2+ cluster ratio was estimated to be about 0.37 [= {(27.4 + 7.4 × 2)/4}/(57.8/2)] in the as‐purified C323S mutant and 1.9 [= {(65.5 + 2.4 × 2)/4}/(18.3/2)] in the reconstituted one. Assuming that both RS and Aux II sites would bind only a [4Fe–4S]2+ cluster, as described above and below, we concluded that the C323S mutant can still bind [2Fe–2S]2+ cluster at the mutated Aux I site by utilizing the remaining three Cys residues (Cys248, Cys268, and Cys325; see Fig. 2C) with or without substituted Ser323.
Mutation of residues putatively involved in binding of Aux II cluster
As described above, the predicted Aux II site consists of Cys310, Cys313, Cys341, and Asp319 (Fig. 2E). To investigate the involvement of the three Cys residues in binding of an Fe–S cluster at the Aux II site, we constructed single (C313S), double (C310S/C313S), and triple (C310S/C313S/C341S) Cys‐to‐Ser mutants and studied their properties. We also mutated Asp319 into Ser to examine the role of Asp319 in the Aux II cluster. In contrast to the Aux I mutants described above, all the Aux II mutants were efficiently expressed in E. coli cells and were as stable as the WT PqqE during anaerobic purification and subsequent reconstitution. As summarized in Table 2, iron and sulfur contents of the as‐purified and reconstituted Aux II single, double, and triple Cys‐to‐Ser mutants are similarly lower than those of the WT enzyme, indicating disruption of the Aux II site for binding of an Fe–S cluster. In contrast, even though the RS site of the reconstituted Aux II mutants is assumed to be fully occupied with a [4Fe–4S]2+ cluster, the SAM cleavage activities of all the reconstituted Aux II Cys‐to‐Ser mutants were only about a half (49–63%) of the reconstituted WT enzyme. These results show that the Fe–S cluster bound at the Aux II site ~ 29 Å apart from the RS cluster (Fig. 2A) indirectly affects the reductive SAM cleavage activity of the RS cluster.
As all the Aux II Cys‐to‐Ser mutants exhibited similar UV‐Vis and Mössbauer spectral properties, only those of the double mutant (C310S/C313S) are representatively shown in Fig. 4. As‐purified C310S/C313S mutant showed a UV‐Vis spectrum with a less intensive 400‐ to 420‐nm absorption peak than that of the as‐purified WT enzyme (Fig. 4A) and a Mössbauer spectrum showing a major quadrupole doublet assignable to [2Fe–2S]2+ (Fig. 4B, green). The [4Fe–4S]2+/[2Fe–2S]2+ cluster ratio is calculated to be 0.21 from the RA values shown in Table 1. On the other hand, reconstitution of the Aux II mutant (C310S/C313S) resulted in the production of the mutant enzyme having the cluster ratio of 1.7 (Table 2), which is inconsistent with the loss of Aux II cluster; the cluster ratio should be close to 1.0 (1 [4Fe–4S]2+ in the RS site and 1 [2Fe–2S]2+ in the Aux I site). A possible reason for apparent over‐reconstitution with a [4Fe–4S]2+ cluster is discussed later. The reconstituted Aux II mutant (C310S/C313S) exhibited another minor Mössbauer signal with an isomer shift (δ) of about 1.4 mm·s−1 (Fig. 4C, purple) that remains to be assigned.
Figure 4.
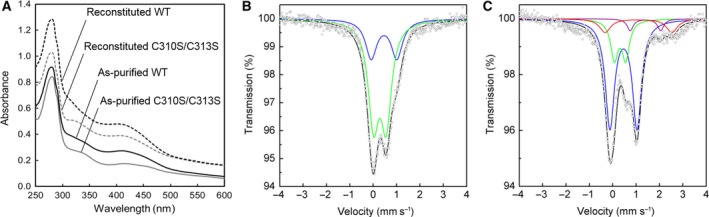
Spectral analyses of Aux II mutant of PqqE. (A) UV‐Vis spectra of the as‐purified and reconstituted C310S/C313S mutant and WT enzyme. (B, C) Mössbauer spectra of the as‐purified (B) and reconstituted (C) C310S/C313S mutant, recorded at 5 K. Simulated spectra are shown as follows: blue, [4Fe–4S]2+; green, [2Fe–2S]2+; red, free (noncluster) high‐spin Fe2+; purple, a doublet simulated for the unassigned signal; black, the sum of the simulated spectra.
As identified in the structure model of PqqE (Fig. 2E), Asp319 has been predicted to ligate an Fe atom of the [4Fe–4S]2+ cluster bound at the Aux II site. Asp319 is conserved in all PqqE homologs 8. We inferred this uncommon ligation to be a source of the two minor unassigned doublets previously identified in the Mössbauer spectra of the reconstituted WT and C32S mutant enzymes 8. The doublets were also found in the reconstituted C323S mutant (Fig. 3C), but not in the Aux II mutant, C310S/C313S (Fig. 4C). Hyperfine parameters of these doublets are almost identical with those found in the [4Fe–4S]2+–pyruvate complex of 4‐demethylwyosine synthase from Pyrococcus abyssi 10, in which one of the four Fe atoms in a [4Fe–4S]2+ cluster has a more charge‐localized character of Fe2+ (showing wider doublets) than the delocalized Fe2.5+ upon binding of a cosubstrate pyruvate to an Fe atom of the [4Fe–4S]2+ cluster 23. The parameters for the ferrous site of a charge‐localized pair, Fe2+–Fe3+ (δ = 0.78 mm·s−1, ΔE Q = 1.62 mm·s−1; Table 1), probably also correspond to those reported for the Asp ligand component (δ = 0.55 mm·s−1, ΔE Q = 1.54 mm·s−1, measured at 77 K) of the oxygen sensing [4Fe–4S]2+ cluster of Bacillus subtilis transcriptional regulator Fnr 24. The reconstituted D319S mutant indeed showed a Mössbauer spectrum lacking the minor unassigned doublets, while giving the [4Fe–4S]2+/[2Fe–2S]2+ cluster ratio of about 1.9 (Fig. 5B), which is identical to that in the reconstituted WT enzyme (Fig. 1C). Its UV‐Vis spectrum (Fig. 5A) and Fe/S contents (Table 2) are also close to those of the WT enzyme. Altogether, these results strongly suggest that the side‐chain carboxyl group of the conserved Asp319 ligates an Fe atom of the [4Fe–4S]2+ cluster bound at the Aux II site but its mutation to Ser does not lead to the release of [4Fe–4S]2+ cluster from the Aux II site. Nevertheless, the reconstituted D319S mutant had a considerably decreased SAM cleavage activity comparable to those of other Aux II mutants. Thus, the ligation of the carboxyl group of Asp319 to the Aux II cluster is important for the indirect participation of the Aux II cluster in the SAM cleavage activity. The significance of Asp ligation for the presumed function of the Aux II cluster is discussed later.
Figure 5.
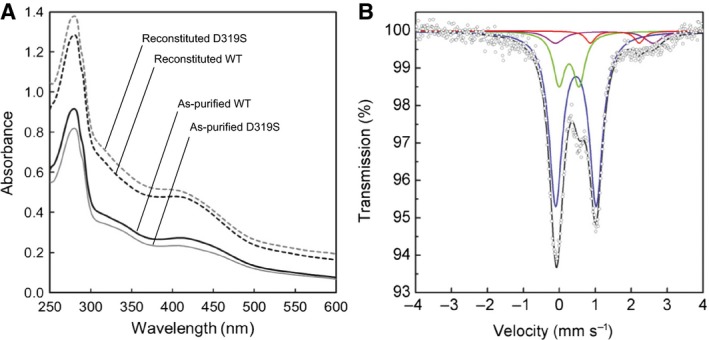
Spectral analyses of D319S mutant of PqqE. (A) UV‐Vis spectra of the as‐purified and reconstituted D319S mutant and WT enzyme. (B) Mössbauer spectra of the reconstituted D319S mutant, recorded at 5 K. Simulated spectra are shown as follows: blue, [4Fe–4S]2+; green, [2Fe–2S]2+; red, free (noncluster) high‐spin Fe2+; purple, a doublet simulated for the unassigned signal; black, the sum of the simulated spectra.
Mutation of multiple residues putatively involved in binding of RS and Aux II clusters
To further characterize the Fe–S cluster bound at the Aux I site, we finally mutated multiple Cys residues located at both RS and Aux II sites simultaneously into Ser (Cys28 and Cys32 at the RS site and Cys310, Cys313, and Cys341 at the Aux II site) and constructed a series of multiple mutants: C32S/C310S/C313S (designated triple mutant), C28S/C32S/C310S/C313S (quadruple mutant), and C28S/C32S/C310S/C313S/C341S (quintuple mutant), which are expected to bind only Aux I cluster. All these mutants were expressed efficiently in E. coli and could be purified in a sufficient amount for characterization. Because of the mutation at the RS site, all the three mutants showed no SAM cleavage activity both in the as‐purified and in reconstituted forms (Table 2). UV‐Vis spectral analyses revealed that all the as‐purified mutant enzymes show very low absorption in the wavelength region above 300 nm (Fig. 6A; only the spectra of quadruple mutant are shown), reflecting the considerable loss of Fe–S clusters. The absorption bands at 410 nm and 450 nm observed at almost the same intensity as well as the low Fe and S contents (Table 2) suggest the presence of only [2Fe–2S]2+ cluster in the as‐purified mutant enzymes, consistent with the Mössbauer spectrum of the as‐purified quadruple mutant (Fig. 6B) that exhibits only a single quadrupole doublet assignable to a [2Fe–2S]2+ cluster.
Figure 6.
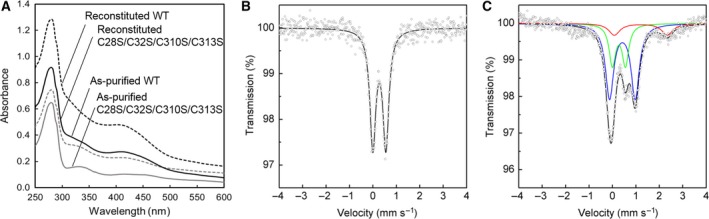
Spectral analyses of multiple RS/Aux II mutant of PqqE. (A) UV‐Vis spectra of the as‐purified and reconstituted C28S/C32S/C310S/C313S mutant and WT enzyme. (B, C) Mössbauer spectra of the as‐purified (B) and reconstituted (C) C28S/C32S/C310S/C313S mutant, recorded at 5 K. Simulated spectra are shown as follows: blue, [4Fe–4S]2+; green, [2Fe–2S]2+; red, free (noncluster) high‐spin Fe2+; black, the sum of the simulated spectra.
Surprisingly, however, chemical reconstitution of the multiply mutated enzymes resulted in marked increases in Fe and S contents (Table 2) and the absorption bands above 300 nm (Fig. 6A); if both the RS and Aux I sites were completely disrupted by the multiple mutation and the Aux I site was fully occupied with a [2Fe–2S]2+ cluster, then chemical reconstitution would not affect the Fe and S contents and UV‐Vis absorption spectra. Mössbauer spectrum of the reconstituted quadruple mutant (Fig. 6C) clearly revealed the presence of another quadrupole doublet (blue) assignable to [4Fe–4S]2+ cluster together with the small doublet of a nonspecifically bound Fe2+ ions (red). The [4Fe–4S]2+/[2Fe–2S]2+ cluster ratio in the reconstituted quadruple mutant is estimated to be about 1.1. Altogether, we conclude that only about 47% of the Aux I site of the as‐purified quadruple mutant is inserted with a [2Fe–2S]2+ cluster by the E. coli system, while the rest of the Aux I site remains vacant and can be reconstituted with a [4Fe–4S]2+ cluster. Thus, the Aux I site of PqqE has unique properties of being able to accommodate either [2Fe–2S]2+ (biologically) or [4Fe–4S]2+ (chemically) cluster. Furthermore, the cluster‐binding ability of the Aux I site is maintained even after the mutation of a single Cys residue contained in this site, as described above (see Fig. 3B,C).
Discussion
The SPASM domain‐containing enzymes constitute the largest subfamily in the RS enzyme superfamily currently compiled in the structure–function linkage database 25; total number of functional domains registered in this subfamily is expanding to 16 026 as of July 2017. Many of these SPASM enzymes are involved in cofactor and peptide maturation processes, using ribosomally translated proteins (peptides), encoded near their genes, as substrate 4, 26. In addition to the RS [4Fe–4S]2+ cluster commonly bound to the N‐terminal signature motif (CX3CX2C), the SPASM enzymes have been reported to bind two auxiliary Fe–S clusters in the seven‐cysteine motif conserved in the C‐terminal half 5, except for those containing a partial SPASM motif (Twitch domain) that binds a single auxiliary cluster 7. As already described, the two auxiliary clusters in anSMEcpe have been both identified as [4Fe–4S]2+ clusters by X‐ray crystallography, in which the four iron atoms are fully ligated with four Cys residues in the seven‐cysteine motif plus one additional cysteine located outside the motif 6. A formylglycine‐generating enzyme AtsB, another member of the SPASM subfamily, contains two auxiliary [4Fe–4S]2+ clusters, one of which may be ligated only with three Cys residues 27. More recently, two other SPASM proteins, QhpD and AlbA involved in maturation of quinohemoprotein amine dehydrogenase 28 and synthesis of a 35‐residue, ribosomally synthesized bacteriocin subtilosin A 19, respectively, have also been predicted to contain two SPASM [4Fe–4S]2+ clusters coordinated by seven Cys residues.
As for PqqE, the enzyme from Klebsiella pneumoniae grown and isolated anaerobically was first reported to contain two [4Fe–4S]2+ clusters in its predominant form (probably one RS and another auxiliary ones) 29. However, we recently demonstrated that PqqE from M. extorquens AM1 contains most likely three Fe–S clusters consisting mainly of [4Fe–4S]2+ and [2Fe–2S]2+ forms 8. The contents of acid‐labile sulfide and iron and the extinction coefficient at 410 nm (about 42 000 m −1·cm−1) of the reconstituted enzyme were consistent with this proposal as estimated from ε410 values reported for [4Fe–4S]2+ (~ 15 000 m −1·cm−1) and [2Fe–2S]2+ (~ 8000–10 000 m −1·cm−1) 22. Subsequently, the presence of two [4Fe–4S]2+ clusters and one in either [2Fe–2S]2+ or [3Fe–4S]+ states was also confirmed for M. extorquens AM1 PqqE by Barr et al. 1. Although the cuboidal [3Fe–4S]+ state has Mössbauer parameters similar to [2Fe–2S]2+ 23 and can be formed by gentle oxidation of [4Fe–4S]2+ (or back‐converted to [4Fe–4S]2+ by reconstitution with iron in the presence of DTT) 30, 31, we conclude that one of the Fe–S clusters contained in PqqE is [2Fe–2S]2+, based on the persistent Mössbauer signals that are not converted to those of [4Fe–4S]2+ by chemical reconstitution as described above. Thus, PqqE is the first example of SPASM proteins containing each one of [4Fe–4S]2+ and [2Fe–2S]2+ clusters at the auxiliary sites.
We then addressed the key issue of the present study: Which of the two predicted auxiliary sites (Aux I and II) binds which of [4Fe–4S]2+ and [2Fe–2S]2+ clusters? Although the final assignment was not as straightforward as we initially anticipated, we concluded that the Aux I site should bind a [2Fe–2S]2+ cluster and the Aux II site should bind a [4Fe–4S]2+ cluster under physiological conditions, based on the following observations. First (and most importantly), simultaneous mutation of multiple Cys residues predicted to be located at the RS and Aux II sites (such as triple and quadruple mutants; see Table 2) yielded the as‐purified mutant enzymes exhibiting only a single quadrupole doublet of Mössbauer signals (Fig. 6B), which is assignable to a [2Fe–2S]2+ cluster that should be bound at the unimpaired Aux I site. Second, all the Aux II mutants showed similar UV‐Vis and Mössbauer properties indicative of the presence of both [4Fe–4S]2+ and [2Fe–2S]2+ clusters (Fig. 4). As the RS site should bind only a [4Fe–4S]2+ cluster, this result strongly suggested that the Aux I site would bind a [2Fe–2S]2+ cluster. Third, three Cys residues (Cys310, Cys313, and Cys341) predicted to be located at the Aux II site by structure modeling are ideally situated to accommodate a cubic [4Fe–4S]2+ cluster (Fig. 2E), but not a rhombic [2Fe–2S]2+ cluster, similar to the three Cys residues (Cys28, Cys32, and Cys35) located at the RS site (Fig. 2B), whereas the Aux I site was modeled with some ambiguity for precise arrangement of the four predicted Cys residues (Cys248, Cys268, Cys323, and Cys325), suggesting a possibility to bind either [4Fe–4S]2+ (Fig. 2C) or [2Fe–2S]2+ (Fig. 2D). Consistent with the presence of four Cys residues at the Aux I site, most [2Fe–2S]2+ clusters have all cysteinyl (Cys)4 ligation with notable exceptions of (Cys)2(His)2 ligation in the Rieske ferredoxins 32, atypical (Cys)3(His)1 ligation in an outer mitochondrial membrane protein MitoNEET 33, 34, 35 and E. coli transcription factor IscR 36 and unique (Cys)3(Arg)1 ligation in biotin synthase (BioB) 16. Although it has been reported that the [2Fe–2S]2+ cluster can be reconstituted chemically into Fe–S proteins such as ferredoxins 37, dual iron isotope analyses indicated that the [2Fe–2S]2+ cluster is inserted into the Aux I site of PqqE only by the E. coli system(s).
Despite the cluster assignment as described above, we obtained contradictory results with the Cys‐to‐Ser mutant of the Aux I site (C323S), which clearly showed that the Aux I mutant did not lose the binding ability for a [2Fe–2S]2+ cluster (Fig. 3), and after reconstitution, it exhibited a high SAM cleavage activity comparable to the WT (Table 2). Consequently, we presumed that the mutated Aux I site may still bind a [2Fe–2S]2+ cluster utilizing the remaining three Cys residues (Cys248, Cys268, and Cys325). Alternatively (and more likely), the Ser residue substituting for Cys323 may participate in binding of a [2Fe–2S]2+ cluster together with the remaining three Cys residues. Such involvement of a Ser residue in ligating an iron atom of the [2Fe–2S]2+ cluster has been previously described for some Cys‐to‐Ser mutants of [2Fe–2S]2+ cluster‐containing proteins, though depending on the Cys residue to be substituted 38. To obtain a mutant without any Fe–S cluster at the Aux I site, mutation of multiple Cys residues may be needed. However, we did not attempt simultaneous mutation of two Cys residues predicted at the Aux I site due to the marked protein lability of even the single mutants (C268S and C323S).
Another unexpected observation relating to the Aux I site was that multiple Cys‐to‐Ser mutants of both the RS and Aux II sites (triple and quadruple mutants; see Table 2) were reconstituted with a [4Fe–4S]2+ cluster while retaining the [2Fe–2S]2+ cluster pre‐existing in the as‐purified enzymes (Fig. 6C), with the content of the [4Fe–4S]2+ cluster accounting for 53% in the reconstituted quadruple mutant. As the Aux I site in these mutants is the sole remaining cluster‐binding site and the [2Fe–2S]2+ cluster is not converted into a [4Fe–4S]2+ cluster by chemical reconstitution, as evidenced by dual iron isotope analysis, this result led us to assume that only about a half of the Aux I site is inserted with a [2Fe–2S]2+ cluster within E. coli cells and the remaining Aux I site is devoid of any cluster, which may be forced to accommodate a [4Fe–4S]2+ cluster by subsequent in vitro reconstitution, as implied by structure modeling (Fig. 2C). The assumption that a fraction of the Aux I site is vacant may also be applicable to the Aux II mutants, in which the [4Fe–4S]2+/[2Fe–2S]2+ cluster ratio significantly exceeds 1.0 after reconstitution; for example, the reconstituted single (C313S) and double (C310S/C313S) Aux II mutants had the cluster ratio of 1.7 (Table 2). It is thus likely that any mutation that leads to the loss of Aux II [4Fe–4S]2+ cluster affects the in vivo [2Fe–2S]2+ cluster insertion into the Aux I site, probably through conformational change in the overall protein folding, and the Aux I site remains consequently vacant in a fraction of the as‐purified mutant protein.
In marked contrast to the RS cluster that plays the well‐established common role for the reductive SAM cleavage, auxiliary clusters found in various SPASM/Twitch domain‐containing enzymes appear to have diverse functions, such as electron transfer, substrate anchoring, and substrate oxidation 5, 7, 39. Based on the crystal structure of anSMEcpe, one of the two auxiliary [4Fe–4S]2+ clusters, designated Aux I and located 16.9 Å from the RS cluster, has been proposed to function as an electron acceptor during substrate oxidation, while the second one (Aux II) located 12.9 Å from Aux I has been suggested to provide a route for the electron from the buried active site to the protein surface 6, 7. Similarly, the Aux I cluster of PqqE predicted to be located about 16 Å from the putative RS cluster (Fig. 2A) may accept an electron from a late radical intermediate derived from substrate PqqA, as proposed recently 1, although the Aux I cluster of PqqE inserted in vivo is not [4Fe–4S]2+ but [2Fe–2S]2+, which generally has higher redox potentials than the former 40. The predicted location of the Aux II cluster of PqqE is about 14 Å from the Aux I cluster and is closest to the protein surface (Fig. 2A), suggesting that the Aux II cluster serves for electron transfer between the Aux I cluster and an external electron acceptor. In view of the significantly reduced SAM cleavage activities of all the reconstituted Aux II mutants (Table 2), efficient electron transfer from the Aux II cluster to the RS cluster via the Aux I cluster may also be required even in the in vitro SAM cleavage reaction using dithionite as an electron donor, as proposed previously for RS enzymes carrying three [4Fe–4S]2+ clusters 19, 41. However, the possibility that the decreased activity of the Aux II mutants is due to the partial replacement of the Aux I [2Fe–2S]2+ cluster by [4Fe–4S]2+ or a consequence of perturbation of the redox or SAM‐binding properties of the RS cluster that are induced by loss of the Aux II cluster also cannot be excluded.
Finally, it is noteworthy that Asp ligation of Fe–S clusters is a rare example of non‐Cys ligands and has been found only for [4Fe–4S]2+ clusters 40, with the first identification in Pyrococcus furiosus ferredoxin 42 followed by Desulfovibrio africanus ferredoxin III 43, the dark‐operative photochlorophyllide reductase complex from Rhodobacter capsulatus 44, and the bacterial transcriptional regulator Fnr 24. In the photochlorophyllide reductase, Asp ligation was proposed to contribute to the low redox potential necessary to reduce photochlorophyllide 45. Although the reconstituted D319S mutant of PqqE did not lose the Aux II [4Fe–4S]2+ cluster, its SAM cleavage activity was considerably decreased (Table 2), suggesting that Asp ligation fine‐tunes the redox potential of the Aux II cluster to suit for the efficient electron transfer to the RS cluster catalyzing reductive SAM cleavage.
Materials and methods
Chemicals
S‐adenosyl‐l‐methionine, DTT, FeCl3, and LB broth were purchased from Sigma Aldrich (Munich, Germany). Li2S, ammonium iron (III) citrate, and 57Fe2O3 (96.64% isotopic enrichment) were purchased from Alfa Aesar (Karlsruhe, Germany), Lach‐Ner (Neratovice, Czech Republic), and ISOFLEX (San Francisco, CA, USA), respectively.
Homology alignment‐based structure modeling
The structure model of PqqE was built by applying the M. extorquens AM1 PqqE amino acid sequence (UniProt ID: P71517) to the web site SWISS‐MODEL 13, and the resultant initial model was modified manually by Coot 46. The coordinates of [4Fe–4S]2+ and [2Fe–2S]2+ clusters were taken from those in the crystal structures of anSMEcpe (PDB ID: 4K36) [AdoMet (RS), Aux I and Aux II clusters] 6 and biotin synthase from E. coli (BioB) (PDB ID: 1R30) 16, respectively, and merged with the coordinates of PqqE model. The cluster–ligand distances were optimized manually using commercial software Discovery Studio (BIOVIA, Tokyo, Japan).
Site‐directed mutagenesis
The C268S and C313S mutants of PqqE were prepared by PCR using Tks Gflex™ DNA polymerase (Takara, Tokyo, Japan) with phosphorylated forward (F) and reverse (R) primers containing a mismatched nucleotide listed in Table S1. The plasmid pET‐pqqE‐N prepared previously 8 was used as a template. The linearized PCR products were self‐ligated with T4 DNA Ligase (NEB, Ipswich, MA, USA) to yield pET‐C268S and pET‐C313S for the C268S and C313S mutants, respectively. The ligation mixtures were directly transformed into E. coli DH5α, and mutations were confirmed by DNA sequencing. For constructions of other PqqE mutants, site‐directed mutagenesis was carried out following a DpnI‐mediated site‐directed mutagenesis method 47. The mutagenesis reactions were performed using a QuickChange Mutagenesis Kit (Agilent Technologies, CA, USA) following the manufacturer's recommended protocol. For constructions of the pET‐C310S, pET‐D319C, pET‐D319S, and pET‐C323S mutant plasmids, pET‐PqqE‐N was used as a template for PCR, and for construction of the pET‐C310S/C313S mutant plasmid, pET‐C313S was used. The primer pairs used in these reactions are listed in Table S1. Construction of pET‐C32S/C310S/C313S was carried out by digestion of pET‐C310S/C313S and pET‐C32S 8 with AflII and BamHI followed by ligation of the smaller fragment from pET‐C310S/C313S with the larger fragment from pET‐C32S. Construction of pET‐C28S/C32S/C310S/C313S was also performed using pET‐C32S/C310S/C313S as a template. The mutant plasmids were transformed into E. coli TOP10, and the mutations were confirmed by DNA sequencing. All mutant proteins thus constructed have an N‐terminal His6‐tag for purification with an affinity column as described below.
Expression and purification of PqqE
Escherichia coli Rosetta 2 (DE3) cells (Novagen, Cambridge, MA, USA) harboring an appropriate plasmid were inoculated from a stock culture into 50 mL of LB medium containing kanamycin (50 μg·mL−1) and chloramphenicol (35 μg·mL−1) and cultivated overnight at 37 °C and 150 rpm. Overnight culture was inoculated (10 mL per 100 mL) into a fresh LB medium containing antibiotics and cultivated for 4 h at the same conditions. Then, 50 mL of this culture was transferred into 500 mL of fresh LB medium supplemented with antibiotics in one‐liter flasks, and the cells were grown at 37 °C and 150 rpm until reaching the OD600 of 0.9. The medium was supplemented with 0.1 mm of 56Fe3+ or 57Fe3+ and expression was induced by 0.1 mm isopropyl‐β‐d‐thiogalactopyranoside. Cells were further cultivated at 18 °C and 120 rpm for 16–18 h, harvested by centrifugation at 5700 g for 5 min, washed with distilled water bubbled with N2, and stored at −20 °C until use.
Purification of all PqqE variants was performed as previously described 8. All purification steps, except centrifugation, were carried out at room temperature (22 °C) in a glove box (SICCO, Grünsfeld, Germany) filled with nitrogen gas (O2 content 1.5 ppm) using buffer solutions that were bubbled with nitrogen for at least 30 min inside the glove box. The oxygen level inside the box was monitored with a GMH 3691 Digital Oximeter (GHM Messtechnik GmbH, Regenstauf, Germany). Cells suspended in the deoxygenated equilibration buffer (50 mm sodium phosphate, pH 8.0, 500 mm NaCl, 10 mm imidazole, and 10% (v/v) glycerol; 4.5 mL per 1 g of cell paste) were disrupted inside the glove box using 30‐minute incubation with BugBuster extraction reagent (0.5 mL per 1 g of cell paste) supplemented with 2.5 μL of benzonase per 1 g of cell paste (Novagen, Cambridge, MA, USA). The cell lysate was clarified by centrifugation at 16 700 g for 15 min at 4 °C in centrifugation tubes that were sealed inside the box. Supernatant was applied onto a Protino® Ni‐NTA Agarose affinity column (Macherey‐Nagel, Düren, Germany) with a column volume of 5 mL, which had been pre‐equilibrated with the deoxygenated equilibration buffer. The column was washed with two volumes of the washing buffer (50 mm sodium phosphate, pH 8.0, 500 mm NaCl, 50 mm imidazole, and 10% (v/v) glycerol) and PqqE was then eluted with the elution buffer (50 mm sodium phosphate, pH 8.0, 500 mm NaCl, 135 mm imidazole, and 10% (v/v) glycerol). The buffer was then exchanged with buffer A (50 mm Tris/HCl, pH 8.0, 150 mm NaCl, 1 mm DTT, and 10% (v/v) glycerol) using a Sephadex G‐25 column (10 mL) and the as‐purified PqqE was immediately used for the subsequent experiments in most cases. Final yields of the as‐purified WT and mutant enzymes of PqqE, except for the mutants of Aux I site (C268S and C323S), were generally 5–6 mg per 1 g of wet cells. For Mössbauer spectral measurement, the purified protein was concentrated to about 0.5 mL in a sealed Amicon Ultra‐15 Centrifugal Filter Unit with 10‐kDa cutoff (Millipore, Darmstadt, Germany), frozen in liquid nitrogen inside the box, and stored in a sealed nitrogen‐filled bottle at −80 °C if not measured immediately. Typical concentration of PqqE for recording Mössbauer spectra was between 1.0 and 1.5 mm.
Protein and activity assay
Soluble protein concentration was measured by the Bradford method 48 with a Bradford Protein Assay Kit (Bio‐Rad, Hercules, CA, USA) and bovine serum albumin as the standard. The values thus obtained were recalculated using Bradford correction factor of 1.025 8.
Assays of PqqE for the reductive homolytic cleavage of SAM were carried out at a room temperature (22 °C) in the anaerobic glove box using buffers bubbled with nitrogen as described 8. The Fe–S clusters of PqqE (2.5 mg·mL−1 in buffer A) were reduced by the addition of sodium dithionite (freshly prepared 1 m solution in buffer A) to the final concentration of 25 mm for 10 min. The enzyme reaction was started by the addition of SAM to the final concentration of 200 μm and was stopped after 30 min by heating at 80 °C for 10 min. The protein precipitates were removed by centrifugation at 17 500 g for 5 min, and the supernatant was analyzed for the formation of 5′‐deoxyadenosine (5′dA) using HPLC with UV detection at 259 nm. Reaction product, 5′dA, was analyzed on a Symmetry C18 column (2.1 × 150 mm, 5 μm; Waters, Milford, CT, USA) connected to an Alliance e2695 high‐performance liquid chromatograph (Waters) and a Waters 2998 photodiode array detector using a linear gradient of 15 mm ammonium formate, pH 4.0, and methanol 8. The concentration of product was calculated by a calibration curve method using authentic standard compound.
Iron and sulfur quantification
Iron and acid‐labile sulfide contents were measured according to the methods of Fish 49 and Beinert 50, respectively.
Reconstitution of PqqE
Reconstitution of Fe–S clusters was carried out under anaerobic conditions inside a glove bag (Sigma Aldrich, Munich, Germany) at 4 °C following the described procedure 8, 51. Briefly, PqqE (140 μm) reduced with 100 equivalents of DTT for 1 h was reconstituted by the addition of 10 equivalents of Fe3+ (in the form of ammonium iron citrate) followed, after five‐minute incubation with gentle stirring, by the addition of 10 equivalents of Li2S. After overnight incubation with gentle stirring and precipitate removal by centrifugation at 17 500 g, the enzyme solution was purified under anaerobic conditions in a glove box on a Sephadex G‐25 column (10 mL) equilibrated with buffer A. For Mössbauer spectra measurement, the purified protein was concentrated and stored as described above.
Spectral analyses of PqqE
UV‐Vis absorption spectra of anaerobic solutions of PqqE variants were measured in a sealed cuvette all at the same protein concentration adjusted to 0.5 mg·mL−1. The spectra were recorded on a Shimadzu UV‐2401 PC spectrophotometer (Shimadzu, Kyoto, Japan). The low‐temperature 57Fe Mössbauer spectra of the samples were measured as described previously 8 and processed using the MossWinn software program 52. The isomer shift values were referred to α‐Fe foil sample at room temperature.
Author contributions
NS, KT, and JF designed and performed experiments, analyzed data, and wrote the manuscript. HU conducted structure modeling. JP and PN measured Mössbauer spectra, analyzed data, and drew simulated spectra. All authors revised the manuscript.
Supporting information
Table S1. PCR primers used for construction of PqqE mutant enzymes
Acknowledgements
We thank Dr Tadashi Nakai for his help in structure modeling and use of Coot. This study was supported by grants LO1204 and LO1305 from the National Program of Sustainability I from the Ministry of Education, Youth and Sports, Czech Republic, and by project of Research Infrastructure NanoEnviCz (LM2015073) from the Ministry of Education, Youth and Sports of the Czech Republic.
References
- 1. Barr I, Latham JA, Iavarone AT, Chantarojsiri T, Hwang JD and Klinman JP (2016) Demonstration that the radical S‐adenosylmethionine (SAM) enzyme PqqE catalyzes de novo carbon‐carbon cross‐linking within a peptide substrate PqqA in the presence of the peptide chaperone PqqD. J Biol Chem 291, 8877–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Latham JA, Iavarone AT, Barr I, Juthani PV and Klinman JP (2015) PqqD is a novel peptide chaperone that forms a ternary complex with the radical S‐adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway. J Biol Chem 290, 12908–12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang J, Woldring RP, Román‐Meléndez GD, McClain AM, Alzua BR and Marsh ENG (2014) Recent advances in radical SAM enzymology: new structures and mechanisms. ACS Chem Biol 9, 1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haft DH and Basu MK (2011) Biological systems discovery in silico: radical S‐adenosylmethionine protein families and their target peptides for posttranslational modification. J Bacteriol 193, 2745–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lanz ND and Booker SJ (2015) Auxiliary iron‐sulfur cofactors in radical SAM enzymes. Biochim Biophys Acta 1853, 1316–1334. [DOI] [PubMed] [Google Scholar]
- 6. Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ and Drennan CL (2013) X‐ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification. Proc Natl Acad Sci USA 110, 8519–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grell TA, Goldman PJ and Drennan CL (2015) SPASM and twitch domains in S‐adenosylmethionine (SAM) radical enzymes. J Biol Chem 290, 3964–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saichana N, Tanizawa K, Pechoušek J, Novák P, Yakushi T, Toyama H and Frébortová J (2016) PqqE from Methylobacterium extorquens AM1: a radical S‐adenosyl‐L‐methionine enzyme with an unusual tolerance to oxygen. J Biochem 159, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mulder DW, Ortillo DO, Gardenghi DJ, Naumov AV, Ruebush SS, Szilagyi RK, Huynh B‐H, Broderick JB and Peters JW (2009) Activation of HydAEFG requires a preformed [4Fe–4S] cluster. Biochemistry 48, 6240–6248. [DOI] [PubMed] [Google Scholar]
- 10. Perche‐Letuvée P, Kathirvelu V, Berggren G, Clemancey M, Latour J‐M, Maurel V, Douki T, Armengaud J, Mulliez E, Fontecave M et al (2012) 4‐Demethylwyosine synthase from Pyrococcus abyssi is a radical‐S‐adenosyl‐L‐methionine enzyme with an additional [4Fe–4S]+2 cluster that interacts with the pyruvate co‐substrate. J Biol Chem 287, 41174–41185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roach PL (2011) Radicals from S‐adenosylmethionine and their application to biosynthesis. Curr Opin Chem Biol 15, 267–275. [DOI] [PubMed] [Google Scholar]
- 12. Broderick JB, Duffus BR, Duschene KS and Shepard EM (2014) Radical S‐adenosylmethionine enzymes. Chem Rev 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L et al (2014) SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42 (Web Server issue), W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hänzelmann P and Schindelin H (2006) Binding of 5′‐GTP to the C‐terminal FeS cluster of the radical S‐adenosylmethionine enzyme MoaA provides insights into its mechanism. Proc Natl Acad Sci USA 103, 6829–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dinis P, Suess DL, Fox SJ, Harmer JE, Driesener RC, De La Paz L, Swartz JR, Essex JW, Britt RD and Roach PL (2015) X‐ray crystallographic and EPR spectroscopic analysis of HydG, a maturase in [FeFe]‐hydrogenase H‐cluster assembly. Proc Natl Acad Sci USA 112, 1362–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berkovitch F, Nicolet Y, Wan JT, Jarrett JT and Drennan CL (2004) Crystal structure of biotin synthase, an S‐adenosylmethionine‐dependent radical enzyme. Science 303, 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nicolet Y and Drennan CL (2004) AdoMet radical proteins–from structure to evolution–alignment of divergent protein sequences reveals strong secondary structure element conservation. Nucleic Acids Res 32, 4015–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dowling DP, Vey JL, Croft AK and Drennan CL (2012) Structural diversity in the AdoMet radical enzyme superfamily. Biochim Biophys Acta 1824, 1178–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benjdia A, Guillot A, Lefranc B, Vaudry H, Leprince J and Berteau O (2016) Thioether bond formation by SPASM domain radical SAM enzymes: Cα H‐atom abstraction in subtilosin A biosynthesis. Chem Commun (Camb) 52, 6249–6252. [DOI] [PubMed] [Google Scholar]
- 20. Agar JN, Krebs C, Frazzon J, Huynh BH, Dean DR and Johnson MK (2000) IscU as a scaffold for iron‐sulfur cluster biosynthesis: sequential assembly of [2Fe–2S] and [4Fe–4S] clusters in IscU. Biochemistry 39, 7856–7862. [DOI] [PubMed] [Google Scholar]
- 21. Hänzelmann P, Hernández HL, Menzel C, García‐Serres R, Huynh BH, Johnson MK, Mendel RR and Schindelin H (2004) Characterization of MOCS1A, an oxygen‐sensitive iron‐sulfur protein involved in human molybdenum cofactor biosynthesis. J Biol Chem 279, 34721–34732. [DOI] [PubMed] [Google Scholar]
- 22. Dailey HA, Finnegan MG and Johnson MK (1994) Human ferrochelatase is an iron‐sulfur protein. Biochemistry 33, 403–407. [DOI] [PubMed] [Google Scholar]
- 23. Pandelia M‐E, Lanz ND, Booker SJ and Krebs C (2015) Mössbauer spectroscopy of Fe/S proteins. Biochim Biophys Acta 1853, 1395–1405. [DOI] [PubMed] [Google Scholar]
- 24. Gruner I, Fradrich C, Bottger LH, Trautwein AX, Jahn D and Hartig E (2011) Aspartate 141 is the fourth ligand of the oxygen‐sensing [4Fe–4S]2+ cluster of Bacillus subtilis transcriptional regulator Fnr. J Biol Chem 286, 2017–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Akiva E, Brown S, Almonacid DE, Barber AE, Custer AF, Hicks MA, Huang CC, Lauck F, Mashiyama ST, Meng EC et al (2014) The Structure–function linkage database. Nucleic Acids Res 42, D521–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haft DH (2011) Bioinformatic evidence for a widely distributed, ribosomally produced electron carrier precursor, its maturation proteins, and its nicotinoprotein redox partners. BMC Genom 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grove TL, Lee K‐H, St. Clair J, Krebs C and Booker SJ (2008) In vitro characterization of AtsB, a radical SAM formylglycine‐generating enzyme that contains three [4Fe–4S] clusters. Biochemistry 47, 7523–7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakai T, Ito H, Kobayashi K, Takahashi Y, Hori H, Tsubaki M, Tanizawa K and Okajima T (2015) The Radical S‐adenosyl‐L‐methionine enzyme QhpD catalyzes sequential formation of intra‐protein sulfur‐to‐methylene carbon thioether bonds. J Biol Chem 290, 11144–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu S‐P, King D, Britt RD and Klinman JP (2009) Pyrroloquinoline quinone biogenesis: demonstration that PqqE from Klebsiella pneumoniae is a radical S‐adenosyl‐L‐methionine enzyme. Biochemistry 48, 10151–10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim A and Gräsland A (2000) Electron paramagnetic resonance evidence for a novel interconversion of [3Fe–4S]+ and [4Fe–4S]+ clusters with endogenous iron and sulfide in anaerobic ribonucleotide reductase activase in vitro. J Biol Chem 275, 12367–12373. [DOI] [PubMed] [Google Scholar]
- 31. Krebs C, Broderick WE, Henshaw TF, Broderick JB and Huynh BH (2002) Coordination of adenosylmethionine to a unique iron site of the [4Fe–4S] cluster of pyruvate formate lyase‐activating enzyme: a Mössbauer spectroscopic study. J Am Chem Soc 124, 912–913. [DOI] [PubMed] [Google Scholar]
- 32. Mason JR and Cammack R (1992) The electron‐transport proteins of hydroxylating bacterial dioxygenases. Annu Rev Microbiol 46, 277–305. [DOI] [PubMed] [Google Scholar]
- 33. Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, Capraro D, Murphy AN, Nechushtai R, Dixon JE et al (2007) MitoNEET is a uniquely folded 2Fe–2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci USA 104, 14342–14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hou XW, Liu RJ, Ross S, Smart EJ, Zhu HN and Gong WM (2007) Crystallographic studies of human MitoNEET. J Biol Chem 282, 33242–33246. [DOI] [PubMed] [Google Scholar]
- 35. Lin JZ, Zhou T, Ye KQ and Wang JF (2007) Crystal structure of human mitoNEET reveals distinct groups of iron‐sulfur proteins. Proc Natl Acad Sci USA 104, 14640–14645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fleischhacker AS, Stubna A, Hsueh KL, Guo Y, Teter SJ, Rose JC, Brunold TC, Markley JL, Münck E and Kiley PJ (2012) Characterization of the [2Fe–2S] cluster of Escherichia coli transcription factor IscR. Biochemistry 51, 4453–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coghlan VM and Vickery LE (1991) Site‐specific mutations in human ferredoxin that affect binding to ferredoxin reductase and cytochrome P450scc. J Biol Chem 266, 18606–18612. [PubMed] [Google Scholar]
- 38. Moulis JM, Davasse V, Golinelli MP, Meyer J and Quinkal I (1996) The coordination sphere of iron‐sulfur clusters: lessons from site‐directed mutagenesis experiments. J Biol Inorg Chem 1, 2–14. [Google Scholar]
- 39. Lanz ND and Booker SJ (2012) Identification and function of auxiliary iron‐sulfur clusters in radical SAM enzymes. Biochim Biophys Acta 1824, 1196–1212. [DOI] [PubMed] [Google Scholar]
- 40. Bak DW and Elliott SJ (2014) Alternative FeS cluster ligands: tuning redox potentials and chemistry. Curr Opin Chem Biol 19, 50–58. [DOI] [PubMed] [Google Scholar]
- 41. Yu L, Blaser M, Andrei PI, Pierik AJ and Selmer T (2006) 4‐Hydroxyphenylacetate decarboxylases: properties of a novel subclass of glycyl radical enzyme systems. Biochemistry 45, 9584–9592. [DOI] [PubMed] [Google Scholar]
- 42. Conover RC, Kowal AT, Fu WG, Park JB, Aono S, Adams MW and Johnson MK (1990) Spectroscopic characterization of the novel iron‐sulfur cluster in Pyrococcus furiosus ferredoxin. J Biol Chem 265, 8533–8541. [PubMed] [Google Scholar]
- 43. Busch JL, Breton JL, Bartlett BM, Armstrong FA, James R and Thomson AJ (1997) [3Fe–4S] ↔ [4Fe–4S] cluster interconversion in Desulfovibrio africanus ferredoxin III: properties of an Asp14→Cys mutant. Biochem J 323, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muraki N, Nomata J, Ebata K, Mizoguchi T, Shiba T, Tamiaki H, Kurisu G and Fujita Y (2010) X‐ray crystal structure of the light‐independent protochlorophyllide reductase. Nature 465, 110–114. [DOI] [PubMed] [Google Scholar]
- 45. Kondo T, Nomata J, Fujita Y and Itoh S (2011) EPR study of 1Asp–3Cys ligated 4Fe–4S iron‐sulfur cluster in NB‐protein (BchN–BchB) of a dark‐operative protochlorophyllide reductase complex. FEBS Lett 585, 214–218. [DOI] [PubMed] [Google Scholar]
- 46. Emsley P, Lohkamp B, Scott WG and Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fisher CL and Pei GK (1997) Modification of a PCR‐based site‐directed mutagenesis method. Biotechniques 23, 570–571, 574. [DOI] [PubMed] [Google Scholar]
- 48. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 49. Fish WW (1988) Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol 158, 357–364. [DOI] [PubMed] [Google Scholar]
- 50. Beinert H (1983) Semi‐micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron‐sulfur proteins. Anal Biochem 131, 373–378. [DOI] [PubMed] [Google Scholar]
- 51. Flühe L, Knappe TA, Gattner MJ, Schäfer A, Burghaus O, Linne U and Marahiel MA (2012) The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat Chem Biol 8, 350–357. [DOI] [PubMed] [Google Scholar]
- 52. Klencsár Z, Kuzmann E and Vértes A (1996) User‐friendly software for Mössbauer spectrum analysis. J Radioanal Nucl Chem 210, 105–118. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. PCR primers used for construction of PqqE mutant enzymes