

Abstract
We have already reported that the inactivated Sendai virus (hemagglutinating virus of Japan; HVJ) envelope (HVJ‐E) has multiple anticancer effects, including induction of cancer‐selective cell death and activation of anticancer immunity. The HVJ‐E stimulates dendritic cells to produce cytokines and chemokines such as β‐interferon, interleukin‐6, chemokine (C‐C motif) ligand 5, and chemokine (C‐X‐C motif) ligand 10, which activate both CD8+ T cells and natural killer (NK) cells and recruit them to the tumor microenvironment. However, the effect of HVJ‐E on modulating the sensitivity of cancer cells to immune cell attack has yet to be investigated. In this study, we found that HVJ‐E induced the production of intercellular adhesion molecule‐1 (ICAM‐1, CD54), a ligand of lymphocyte function‐associated antigen 1, in several cancer cell lines through the activation of nuclear factor‐κB downstream of retinoic acid‐inducible gene I and the mitochondrial antiviral signaling pathway. The upregulation of ICAM‐1 on the surface of cancer cells increased the sensitivity of cancer cells to NK cells. Knocking out expression of ICAM‐1 in MDA‐MB‐231 cells using the CRISPR/Cas9 method significantly reduced the killing effect of NK cells on ICAM‐1‐depleted MDA‐MB‐231 cells. In addition, HVJ‐E suppressed tumor growth in MDA‐MB‐231 tumor‐bearing SCID mice, and the HVJ‐E antitumor effect was impaired when NK cells were depleted by treatment with the anti‐asialo GM1 antibody. Our findings suggest that HVJ‐E enhances NK cell sensitivity against cancer cells by increasing ICAM‐1 expression on the cancer cell surface.
Keywords: Breast cancer, HVJ‐E, ICAM‐1, NK, Sendai virus
Cancer is a leading cause of death worldwide, and its prevalence is increasing as a result of aging and lifestyle alterations.1, 2
Currently, there are many types of cancer therapy, such as surgery, targeted therapy, chemotherapy, radiotherapy, and immunotherapy. Recently, the concept of immune‐checkpoint inhibition has given rise to breakthroughs in cancer immunotherapy. Antibodies against immune‐checkpoint molecules such as PD‐1, PD‐L1, and CTL associated protein‐4 activate CTL against cancers by stopping the inhibitory signal of CD8+ T cells.3, 4, 5, 6 Although antibodies against PD‐1 and PD‐L1 resulted in remission in malignant melanoma, approximately 70% of patients are still resistant to these antibody treatments.7 The insensitivity to immune‐checkpoint inhibitory treatments is a big issue in cancer treatment worldwide. Active β‐catenin signaling in melanoma prevents chemokine CCL4 production, which results in the inhibition of dendritic cell infiltration and subsequent T‐cell activation.8, 9, 10 These reports indicate the importance of the infiltration of antigen‐presenting cells into tumor tissue. The discovery that CD8+ T cells are hardly detected in tumor tissues of non‐responders to the immune‐checkpoint antibody treatment suggests the need for CD8+ T‐cell infiltration into the tumor tissue for the success of immune‐checkpoint blockade therapy. However, even though activated CTLs approach cancer cells, some cancer cells escape from T‐cell attack by suppressing MHC‐class I molecule expression.11 Cells without MHC‐class I molecules are resistant to CTLs, but those cells can be killed by NK cells, which recognize non‐MHC‐class I cells as non‐self.11, 12, 13 Thus, NK‐cell therapy is also very important for cancer immunotherapy. In addition to T‐cell therapy, NK‐cell activation immunotherapy is also carried out by blocking inhibitory receptors on NK cells and by augmenting activating signals in NK cells.14, 15, 16, 17, 18, 19
We have reported the antitumor activity of HVJ‐E, which includes the activation of antitumor immunity and the induction of cancer cell‐selective killing.20, 21, 22, 23, 24, 25, 26 The activity mainly depends on viral RNA fragments that activate RIG‐I and MAVS protein signaling pathway. The pathway activates pro‐apoptotic genes such as TRAIL and Noxa only in cancer cells, such as breast cancer cell line MDA‐MB‐231 and prostate cancer cell line PC3. In immune cells, such as dendritic cells and macrophages, the signaling pathway increases the production of chemokines such as CCL5 and CXCL10 and cytokines such as IFN‐α and ‐β. Both CCL5 and CXCL10 recruit effector T cells and NK cells to the tumor microenvironment. Natural killer cells exposed to type‐I IFNs are activated and secrete IFN‐γ, which activates CD8+ T cells to become CTLs against cancer cells.27 Consequently, both CTL and NK cells are activated by HVJ‐E.24, 25 Apoptotic cell death by HVJ‐E occurred in some human cancer cells such as PC3 cells and MDA‐MB‐231 cells in vitro. In SCID mice transplanted human cancer cells, such as PC3 cells, the elimination of tumors in vivo was very dramatic. We have already shown that such a dramatic tumor suppression in SCID mice was mainly mediated by NK cells and partly by the direct cancer cell killing effect of HVJ‐E.20 However, these effects related to the antitumor immunity of HVJ‐E are caused by the induction of various cytokines and chemokines such as IFN‐β, IL‐6, CXCL10, and CCL5. There is no report showing the modulation of cancer cell responsiveness to host immune reaction by HVJ‐E. Therefore, we examined whether HVJ‐E could augment the sensitivity of cancer cells to NK cells.
We found that HVJ‐E induced ICAM‐1 (CD54) production in several cancer cell lines. Intercellular adhesion molecule‐1 is a transmembrane glycoprotein that is induced by retinoic acid, virus infection, and cytokines such as IL‐1β, tumor necrosis factor‐α, and IFN‐γ.28, 29, 30, 31, 32, 33 The ICAM‐1 protein is expressed on cells and several types of cancer cells including melanoma, prostate cancer, lung cancer, and breast cancer. The function of ICAM‐1 has been reported to be associated with metastatic breast cancer cell line invasion,34, 35 whereas ICAM‐1 has been suggested to suppress M2 macrophage polarization, which induces tumor growth through downregulation of efferocytosis in colon tumors.36 Previous reports have proven that ICAM‐1 can bind with LFA‐1 on CTL and NK cells and induce cell death through these immune cells.37, 38, 39 In our study, we revealed that HVJ‐E enhanced the sensitivity of human cancer cell lines, including MDA‐MB‐231 and PC3 cell lines, previously reported as sensitive to HVJ‐E,22 to NK cells through the upregulation of ICAM‐1. This is the first report to show that virus therapy can enhance NK cell sensitivity in cancer cells. Apoptotic cell death through HVJ‐E occurred in some cancer cells in vitro, and the elimination of those cancer cell‐derived tumor masses in a SCID mouse model in vivo was very dramatic. Therefore, we hypothesized that HVJ‐E might augment the sensitivity of cancer cells to NK cells.
Materials and Methods
Cells
Human breast cancer cell line MDA‐MB‐231, hormone‐resistant human prostate cancer cell line PC3, and normal human prostate epithelial cell line PNT2 were purchased from ATCC (Manassas, VA, USA). The cell lines were maintained in DMEM (Nacalai Tesque, Kyoto, Japan) with 10% FBS (Biowest, Nuaillé, France) and 1% penicillin–streptomycin mixed solution (10 000 U/mL penicillin and 10 000 mg/mL streptomycin in 0.85% NACl; Nacalai Tesque). Human mammary epithelial cells were purchased from Kurabo (Tokyo, Japan) and cultured with a MammaryLife Comp kit (Kurabo) following the manufacturer's instructions.
Reagents and antibodies
The NF‐κB inhibitor Bay11‐7082 was purchased from Wako Pure Chemical Industries (Osaka, Japan). The anti‐CD54/ICAM‐1 antibody (#4915) and anti‐RIG‐I (#4200) antibodies were purchased from Cell Signaling Technology (Tokyo, Japan); the anti‐Fas (sc‐74540) antibody was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The anti‐MAVS antibody (ab25084) was purchased from Abcam (Cambridge, UK), and the anti‐β‐actin (AC‐15) antibody (A5441) was purchased from Sigma‐Aldrich (Tokyo, Japan).
Preparation of HVJ‐E and HVJ‐E RNA extraction
Hemagglutinating virus of Japan (VR‐105 parainfluenza 1 Sendai/52, Z strain; ATCC) was amplified in the chorioallantoic fluid of 10–14‐day‐old chick eggs, and the viral particles were purified by centrifugation and inactivated by UV irradiation (99 mJ/cm2), as previously described.40 The HVJ‐E RNA was isolated by ISOGEN (311‐02501; Nippon Gene, Toyama, Japan).
Small interfering RNA transfection
Human RIG‐I siRNA (DDX58‐HSS177513; Invitrogen, Tokyo, Japan), human MAVS siRNA (Hs_VISA_8707; Sigma‐Aldrich) and scrambled siRNA (46‐2002, negative control low GC duplex; Invitrogen) were transfected into MDA‐MB‐231 cells using Lipofectamine RNAiMAX (Invitrogen, Waltham, MA, USA), and 50 pmol siRNA was used for 3 × 105 cells.
Cancer cell RNA extraction and analysis
Cancer cell RNA was extracted using the RNeasy mini kit (Qiagen, Tokyo, Japan). RNA was converted into cDNA using the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Tokyo, Japan; Thermo Fisher Scientific, Tokyo, Japan) according to the manufacturer's instructions. Quantitative real‐time PCR was carried out on a CFX384 real‐time system (Bio‐Rad, Hercules, CA, USA), using the SYBR qPCR Mix (Toyobo, Osaka, Japan). Primers were ordered from Invitrogen Custom DNA Oligos (Life Technologies, Tokyo, Japan) and Hokkaido System Science (Hokkaido, Japan), and the sequences were as follows: MICA forward, 5′‐GGCATCTTCCCTTTTGCAC‐3′ and reverse, 5′‐AACCCTGACTGCACAGATCC‐3′; MICB forward, 5′‐CCTCTGTGCTCGTGAGTCC‐3′ and reverse, 5′‐CTGACTGCACAGATCCATCC‐3′; ICAM‐1 forward, 5′‐CCTTCCTCACCGTGTACTGG‐3′ and reverse, 5′‐AGCGTAGGGTAAGGTTCTTGC‐3′; Fas forward, 5′‐AATCCTGAAACAGTGGCAATAAA‐3′ and reverse, 5′‐TTTCGAACAAAGCCTTTAACTTG‐3′; ULBP1 forward, 5′‐TGGGGGATTGTAAGATGTGG‐3′ and reverse, 5′‐GGCCAGAGAGGGTGGTTT‐3′; PD‐L1 forward, 5′‐CCATACAGCTGAATTGGTCATC‐3′ and reverse, 5′‐CAGAATTACCAACTGAGTCCTTTCA‐3′; ITGA2 forward, 5′‐GCTAAGATAACCCTAAAGCTCTGCT‐3′ and reverse, 5′‐CGCATCAAGCGTCATGTT‐3′; 18S forward, 5′‐AAACGGCTACCACATCCAAG‐3′ and reverse, 5′‐CCTCCAATGGATCCTCGTTA‐3′; and GAPDH forward, 5′‐GTCTTCACCACCATGGAGAAGGCT‐3′ and reverse, 5′‐CATGCCAGTGAGCTTCCCGTTCA‐3′.
Western blot analysis
The cells were lysed with RIPA buffer containing protease inhibitor (cOmplete protease inhibitor cocktail; Roche, Mannheim, Germany). The lysate was electrophoresed on a 5–20% polyacrylamide gel and transferred onto PVDF membranes (Millipore, Darmstadt, Germany). The membrane was blocked with 5% skim milk and incubated with the primary antibody described above at a 0.1% antibody concentration overnight at 4°C, followed by incubation with an HRP‐linked secondary antibody (GE Healthcare, Tokyo, Japan). Signals were detected using Chemi‐Lumi one (Nacalai Tesque) and ImmunoStar LD (290‐69904; Wako Pure Chemical Industries) by Image Quant LAS 4000 mini software (GE Healthcare, Little Chalfont, UK).
Flow cytometry analysis
Cells were stained in PBS containing 2% FBS with relevant antibodies: FITC anti‐human CD54 antibody, FITC mouse IgG1, κ isotype Ctrl, PE anti‐human CD95 (Fas) and PE mouse IgG1, κ isotype Ctrl (BioLegend, San Diego, CA, USA) for 30 min at 4°C and washed three times with PBS. Flow cytometry was analyzed on a BD FACSCanto II (Becton Dickinson, Franklin Lakes, NJ, USA) with FlowJo software (FlowJo, Ashland, OR, USA).
Mice
Female CB17/Icr‐SCID mice (5–6 weeks old) and female C57BL/6NJcl mice (8–10 weeks old) were purchased from Clea Japan (Tokyo, Japan) and were maintained in a temperature‐controlled, pathogen‐free room. All of the animals were handled according to the approved protocols and guidelines of the Animal Committee of Osaka University (Osaka, Japan).
Calcein release assay
Natural killer cells were isolated from C57BL/6N mice spleens using the mouse NK cell Isolation Kit II (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer's instructions. The cytotoxicity of NK cells was determined in a calcein release assay against cancer cells. The target cells, cancer cells, were labelled with calcein‐AM (#C396; Dojindo, Kumamoto, Japan) at 4 μM per 5 × 105–106 cells in 1 mL PBS for 25 min at 37°C. Labeled cancer cells were incubated with NK cells in a 96‐well plate with 200 μL culture medium for 4 h at 37°C and 5% CO2. After centrifugation, 100 μL supernatant was collected, and the fluorescence was measured with excitation and emission wavelengths at 495/515 nm. The percentage of specific lysis was calculated as follows: 100× (experimental release − spontaneous release)/(maximum release − spontaneous release).
Human breast cancer cell inoculation and treatment
MDA‐MB‐231 cells were washed with cold PBS three times, and 5 × 106 cells in a 100‐μL 1:1 PBS:Matrigel (Corning, Bedford, MA, USA) mixture per mouse were s.c. injected into the backs of the CB17/Icr‐SCID mice. When each tumor had grown to 4–6 mm in diameter, the mice were treated with one intratumor injection of HVJ‐E (1000 HAU in 100 μL per mouse) or 100 μL PBS every 3 days for a total of six injections. Tumor volume was measured in a blinded manner with slide calipers using the following formula: tumor volume (mm3) = length × (width)2/2. To deplete NK cells in vivo, 200 μL anti‐asialo GM1 antibody (1:10 diluted with PBS) was i.p. injected into each mouse on days −1, 0, 1, 2, 4, 6, 9, 12, 15, and 18.
Creation of ICAM‐1 knockout MDA‐MB‐231 cell line
The targeted gRNA oligos were introduced into the pX330 vector (Addgene, Cambridge, MA, USA). Then 1.2 μg each pX330 plasmid DNA with target gRNA sequence and 0.6 μg pPGKpuro (Addgene) were transfected into MDA‐MB‐231 cells (2 × 105 cells) using NEON (Invitrogen) electroporation, and the transfected cells were cultured for 2 days with 1.0 μg/mL puromycin (Nacalai Tesque) in medium for selection. Living cells were diluted in 10‐cm dishes for colony formation. Single colonies were picked and cultured for proliferation. The DNA of each colony was abstracted using the DNeasy Blood & Tissue Kit (Qiagen), and the genomic region containing the CRISPR/Cas9 target site gene was amplified by PCR. The PCR products were purified using QIAquick Gel Extraction Kit (Qiagen) following the manufacturer's protocol and cloned into the pCR‐Blunt II‐TOPO vector (Invitrogen). A number of colonies were selected, and the sequences were analyzed on a 3100 Genetic Analyzer (Applied Biosystems).
Results
Expression of ICAM‐1 in cancer cell lines is increased by HVJ‐E stimulation
To investigate changes in NK cell ligands in cancer cells induced by HVJ‐E, we measured RNA expression levels of a number of NK cell ligands in MDA‐MB‐231 and PC3 cells by quantitative real‐time PCR. RNA expression levels of ICAM‐1 and Fas RNAs were significantly increased in both cell lines stimulated with HVJ‐E for 24 h compared to the expression in cells stimulated with PBS (Fig. 1a,b). The RNA expression level of PD‐L1 was enhanced in PC3 cells, but this enhancement was not observed in MDA‐MB‐231 cells. We further examined the protein expression levels of ICAM‐1 in normal cells (HMECs) and cancer cells by Western blot analysis (Fig. 1c). Hemagglutinating virus of Japan envelope dramatically increased ICAM‐1 expression in human breast cancer cells but not in the normal mammary epithelial cell line, and the HVJ‐E‐induced upregulation of ICAM‐1 in cancer cells was time‐dependent after HVJ‐E treatment. The cancer cell‐specific increase of ICAM‐1 expression by HVJ‐E was also observed in PC3 but not normal prostate epithelial cell line PNT2 (Fig. S1, Appendix S1). Expression of ICAM‐1 on the cell surface was confirmed by flow cytometry analysis (Fig. 1d). Expression of ICAM‐1 on the cell surface was increased with HVJ‐E treatment compared with that in non‐stimulated cells. Although the RNA level of Fas was increased in both cancer cell lines, Western blot analysis showed that there were no significant changes in Fas protein expression in MDA‐MB‐231 or PC3 cells (Fig. S2a, Appendix S1). The results of the flow cytometric assay showed the increase of Fas by comparing the mean fluorescence intensity of cells treated with HVJ‐E or PBS, but the Fas‐positive cell population was not increased by HVJ‐E (Fig. S2b, Appendix S1). Although HVJ‐E might increase the surface expression of Fas in Fas‐positive cells, further analysis is needed. Here, we focused on ICAM‐1 because all the data, including RT‐PCR, Western blot, and FACS analysis, indicate the increase of ICAM‐1 expression by HVJ‐E. We tried to clarify the contribution of ICAM‐1 to NK cell‐mediated cancer suppression triggered by HVJ‐E.
Figure 1.
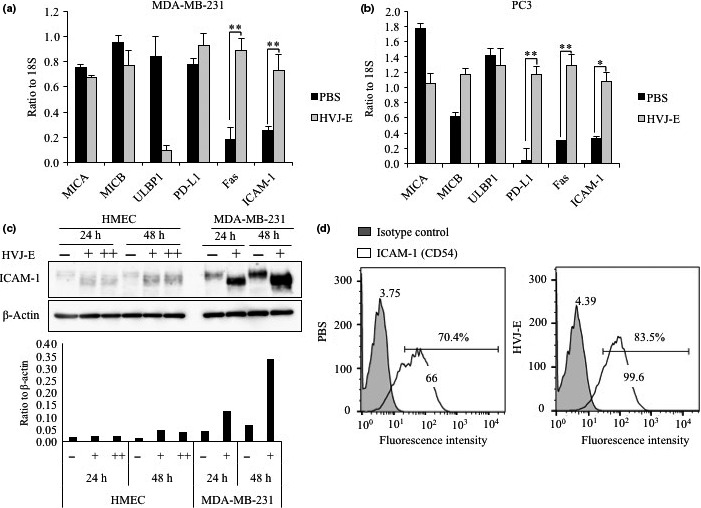
Hemagglutinating virus of Japan envelope (HVJ‐E) induced intercellular adhesion molecule‐1 (ICAM‐1) production in cancer cell lines. (a, b) Quantitative RT‐PCR analysis provided the ratio of RNA levels of natural killer cell ligands to 18S in MDA‐MB‐231 and PC3 cells. Cells were treated with HVJ‐E 1000 MOI or PBS for 24 h before analysis. Mean values ± SE (n = 3). *P < 0.05, **P < 0.01, t‐test. MICA/B, major histocompatibility complex class I polypeptide‐related sequence A/B; PD‐L1, programmed cell death ligand 1; ULBP1, UL16‐binding protein 1. (c) Protein expression levels of ICAM‐1 (CD54) in human mammary epithelial cells (HMEC) and MDA‐MB‐231 cells examined by Western blotting after HVJ‐E (+1000 MOI and ++10 000 MOI) treatment for 24 and 48 h. Bar graph shows the protein expression ratio to β‐actin measured using Image Quant TL Array. (d) Flow cytometry analysis determined the expression of ICAM‐1 on the MDA‐MB‐231 cell surface. Data shown is representative of three independent experiments and mean fluorescence intensity values of the representative experiment are written on each peak.
In the non‐cancerous normal human mammary gland cell line HMEC and prostate epithelial cell line PNT2, HVJ‐E failed to upregulate the expression of ICAM‐1 (Figs 1c, S1, Appendix S1). In addition, we observed that ICAM‐1 became smaller in MDA‐MB‐231 and PC3 cells after treatment with HVJ‐E (Fig. 1c) , and the molecular weight of ICAM‐1 was decreased in a time‐dependent manner (Fig. S3a, Appendix S1). It is known that HVJ‐E introduces its RNA fragments into the cytoplasm when it fuses to a cancer cell.22 To determine whether the HVJ‐E RNA fragments induced ICAM‐1 expression, we isolated the RNAs of HVJ‐E and transfected them into MDA‐MB‐231 cells. The ICAM‐1 protein levels were enhanced by HVJ‐E RNAs in a dose‐dependent manner (Fig. 2a). However, transfection of HVJ‐E‐derived RNA fragments induced ICAM‐1 expression without alteration of the ICAM‐1 protein size (Fig. 2a). This result provides evidence for two points: (i) the signaling pathway of HVJ‐E‐induced ICAM‐1 expression, which is analyzed in the next section; and (ii) the mechanism of ICAM‐1 size reduction by HVJ‐E. The size reduction of ICAM‐1 might result from the fusion of HVJ‐E to the cancer cells. Hemagglutinating virus of Japan has two different glycoproteins, HN and F, on the surface of the viral envelope.41, 42 When HVJ attaches to the host cell surface, the hemagglutinin of HN recognizes the sialic acid on glycoproteins on the host cell surface and cleave the sialic acid with the neuraminidase.43 To determine the mechanism for ICAM‐1 size reduction, we generated HVJ from LLCMK2, a monkey kidney cell line, and depleted the HN protein by HN siRNA transfection (Fig. S3b, Appendix S1). The HVJ derived from LLCMK2 cells is fusion‐incompetent due to an F protein of the F0 form, and trypsin protease was used to cleave F0 into the F1/F2 form.44, 45 In contrast, HVJ from fertilized chick eggs is fusion‐competent because the F protein of egg‐derived HVJ is cleaved into the F1/F2 form by proteolytic activity of Factor Xa in the chorioallantoic fluid of chick eggs. Three types of HVJ, which were egg‐derived, cell‐derived with HN protein expression, and cell‐derived without HN protein expression, were inactivated by UV irradiation to become HVJ‐E and added to cancer cells. Egg‐derived HVJ‐E induced both ICAM‐1 expression and ICAM‐1 size reduction. However, cell‐derived HVJ‐E without the HN protein failed to induce ICAM‐1 expression or ICAM‐1 size reduction. Cell‐derived HVJ‐E with the HN protein induced ICAM‐1 size reduction but did not upregulate ICAM‐1 expression in cancer cells (Fig. S3b, Appendix S1). Additionally, HVJ‐E pretreated with neuraminidase inhibitor failed to induce ICAM‐1 upregulation or size reduction in cancer cells (Fig. S3c, Appendix S1). These data suggest that the neuraminidase activity of the HN protein results in ICAM‐1 size reduction, probably by the digestion of the sialic acid of ICAM‐1 on the cell surface, when HVJ‐E binds to the cell‐surface HVJ receptors, acidic gangliosides.
Figure 2.
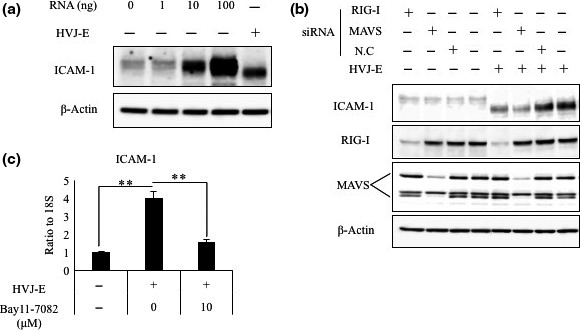
Hemagglutinating virus of Japan envelope (HVJ‐E) RNA‐induced intercellular adhesion molecule‐1 (ICAM‐1) expression was inhibited by knockdown of retinoic acid‐inducible gene I (RIG‐I) or mitochondrial antiviral signaling (MAVS). (a) ICAM‐1 expression in MDA‐MB‐231 cells was analyzed by Western blotting. Cells were transfected with HVJ‐E or 0, 1, 10, or 100 ng HVJ‐E RNA. (b) RIG‐I siRNA, MAVS siRNA, and scrambled siRNA (negative control [N.C]) were transfected into MDA‐MB‐231 cells after 24 h of treatment with HVJ‐E or PBS. ICAM‐1, RIG‐I, and MAVS expression levels in the MDA‐MB‐231 cells were then examined by Western blot analysis. (c) ICAM‐1 RNA levels in MDA‐MB‐231 cells with or without HVJ‐E treatment in the presence of the NF‐κB inhibitor (Bay11‐7082, 0 or 10 μM). Cells were treated with HVJ‐E at 1000 MOI for 24 h. Mean values ± SE (n = 3). *P < 0.05, **P < 0.01, t‐test.
Inactivated Sendai virus RNA‐induced ICAM‐1 expression is mediated by the RIG‐I/MAVS pathway
A previous study identified that RNA fragments of HVJ‐E are able to be recognized by RIG‐I/MAVS and activated transcription factor NK‐κB in cancer cells;20 NF‐κB is one of the nuclear transcription factors that is important for the upregulation of ICAM‐1 expression.46, 47 To further confirm whether HVJ‐E‐induced ICAM‐1 overexpression is dependent on the RIG‐I/MAVS system, we knocked down the RIG‐I or MAVS gene in MDA‐MB‐231 cells using siRNAs and treated the cells with HVJ‐E (Fig. 2b). We found that HVJ‐E‐induced ICAM‐1 expression was reduced in cells transfected with either RIG‐I or MAVS siRNA. In the presence of the NF‐κB inhibitor, the HVJ‐E‐induced enhancement of ICAM‐1 transcription was abolished (Fig. 2c). These results suggest that HVJ‐E induces the production of the ICAM‐1 protein by activating the RIG‐I/MAVS signaling pathway and that NF‐κB acts as a transcription factor for HVJ‐E‐induced ICAM‐1 expression in MDA‐MB‐231 cells.
Both HVJ‐E and HVJ‐E RNA enhance NK cell activity against cancer cells
Intercellular adhesion molecule‐1, which is a ligand of LFA‐1, is required by NK cells to mediate the apoptosis of target cells. We showed that human ICAM‐1 could interact with mouse LFA‐1 on mouse NK cells (Fig. S4, Appendix S1). To determine whether HVJ‐E‐treated cancer cells become more sensitive to NK cells than non‐treated cells, the cytotoxicity of NK cells to HVJ‐E‐treated cancer cells was compared with that of non‐treated cancer cells. Prior to the NK cell‐mediated cytotoxic assay, we determined the effects of the HVJ‐E dose and assay schedule because HVJ‐E itself induces apoptosis in cancer cells, as previously reported.20, 21, 22, 23 As shown in Figure S5 and Appendix S1, HVJ‐E‐mediated cell death was dose‐ and time‐dependent. Based on the results, the survival of MDA‐MB‐231 cells was not significantly affected at 24 h after HVJ‐E treatment at 1000 MOI. Using this condition, NK cells were added to HVJ‐E‐treated MDA‐MB‐231 cells containing calcein. As shown in Figure 3(a), HVJ‐E‐treated MDA‐MB‐231 cells were killed by NK cells more efficiently than non‐treated cells. A similar response was observed in PC3 cells as shown in Figure S6 and Appendix S1. Moreover, the rate of cancer cell death induced by NK cells was significantly higher in MDA‐MB‐231 cells transfected with HVJ‐E RNA than in the cells without HVJ‐E RNA transfection (Fig. 3b). These findings suggest that HVJ‐E increases the sensitivity of cancer cells to NK cells by the introduction of RNA fragments into cancer cells.
Figure 3.
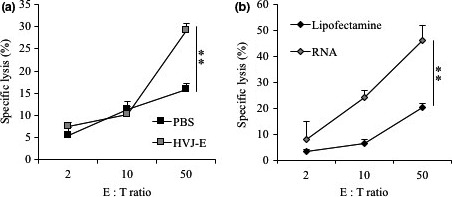
Natural killer (NK) cell cytotoxicity was increased in hemagglutinating virus of Japan envelope (HVJ‐E)‐stimulated MDA‐MB‐231 cells. NK cell cytotoxicity was examined by the calcein release assay at ratios of effector:target (E:T) cells of 2:1, 10:1, and 50:1. (a) MDA‐MB‐231 cells were treated with 1000 MOI HVJ‐E or PBS for 24 h. (b) MDA‐MB‐231 cells were transfected with 100 ng HVJ‐E RNA and incubated for 24 h. Mean values ± SE (n = 4–6). **P < 0.01, t‐test.
Inactivated Sendai virus suppresses MDA‐MB‐231 tumor growth
We inoculated CB17/Icr‐SCID mice with MDA‐MB‐231 cells and treated them with HVJ‐E or PBS. Tumor growth was significantly inhibited by HVJ‐E treatment (Fig. 4a,b). The ICAM‐1 RNA level was enhanced in the HVJ‐E‐treated mouse tumor tissue (Fig. 4c). Expression of ITGA2, a marker for NK cells as well as fibroblasts and platelets, seemed to be increased by HVJ‐E treatment, but the increase was not significant compared with the expression level with PBS treatment (Fig. 4d). To determine if the antitumor effect of HVJ‐E in the MDA‐MB‐231 tumor model is related to NK cells, we depleted NK cells in mice with the anti‐asialo GM1 antibody. Tumor suppression induced by HVJ‐E was attenuated by NK cell depletion (Fig. 4e). Although HVJ‐E treatment seemed to retard tumor progression compared to the progression observed in the PBS treatment group in NK cell‐depleted mice, there was no significant difference between HVJ‐E and PBS treatment groups (Fig. 4e). These data suggest that HVJ‐E suppresses MDA‐MB‐231 tumor development mainly by promoting NK cell activation.
Figure 4.
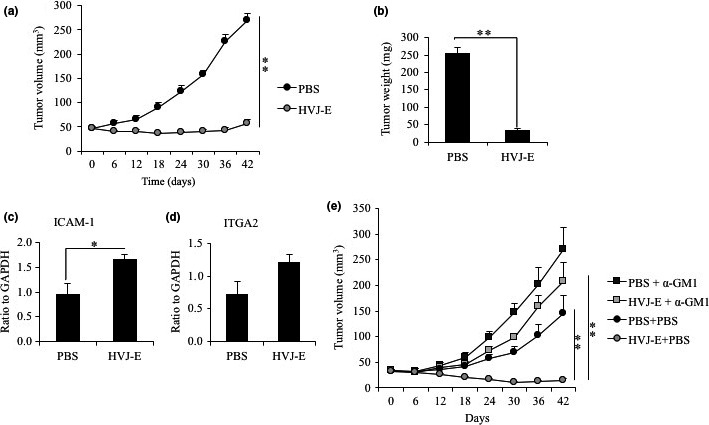
Hemagglutinating virus of Japan envelope (HVJ‐E) treatment inhibited MDA‐MB‐231 tumor growth in vivo. (a) Tumor volume of MDA‐MB‐231 tumor‐bearing mice treated with HVJ‐E (1000 HAU/mouse) or PBS on day 0, 3, 6, 9, 12, and 15. (b) Tumor weight on day 42. Data represent the mean ± SE of seven mice in each group. (c, d) RNA levels of intercellular adhesion molecule‐1 (ICAM‐1) and NK cells in MDA‐MB‐231 tumor tissue of HVJ‐E‐ or PBS‐treated mice were assessed by quantitative RT‐PCR. HVJ‐E (1000 HAU/mouse) or PBS was injected every day for 3 days. Mean values ± SE (n = 3). ITGA2, integrin subunit alpha 2. (e) HVJ‐E‐treated mouse tumor volumes of NK cell‐depleted mice by anti‐asialo GM1 antibody (α‐GM1) treatment. Data represent the mean ± SE (n = 4–6 mice each group). *P < 0.05, **P < 0.01, t‐test.
Antitumor effect of HVJ‐E is associated with tumor cell ICAM‐1 expression
Next, we sought to investigate whether a deficiency of ICAM‐1 abolishes the HVJ‐E‐induced enhancement of NK cell cytotoxicity to cancer cells. We generated an ICAM‐1 knockout MDA‐MB‐231 cell line using the CRISPR/Cas9 system (Fig. 5a), and ICAM‐1 protein expression in the knockout cells was not detected in the Western blot analysis (Fig. 5b). We then compared the cytotoxicity of NK cells to HVJ‐E‐treated cancer cells with or without ICAM‐1 expression. The NK cell‐mediated cancer cell death was reduced in two different ICAM‐1‐deficient cell lines treated with HVJ‐E (Fig. 5c). These data suggest that ICAM‐1 expression on the HVJ‐E‐treated cancer cells is indispensable for the increase in NK cell cytotoxicity.
Figure 5.
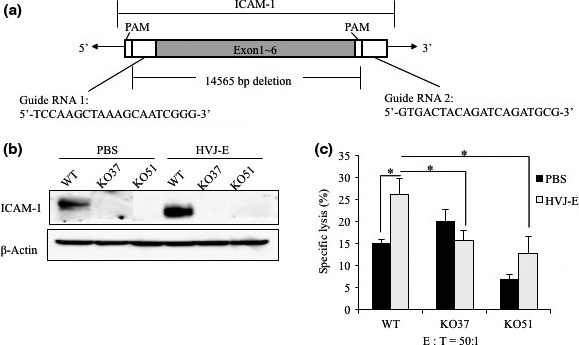
Natural killer cell cytotoxicity was decreased in intercellular adhesion molecule‐1 (ICAM‐1) knockout MDA‐MB‐231 cells. (a) Construction of ICAM‐1 knockout MDA‐MB‐231 cell lines by CRISPR/Cas9. Schematic diagram of ICAM‐1‐targeting gRNA. PAM, protospacer adjacent motif. (b) Examination of ICAM‐1 expression in wild‐type and knockout MDA‐MB‐231 cells treated with or without hemagglutinating virus of Japan envelope (HVJ‐E) for 24 h by Western blot analysis. (c) Natural killer cell cytotoxicity was examined by the calcein release assay at the ratio of effector:target (E:T) cells of 50:1. Mean values ± SE (n = 3). *P < 0.05, t‐test.
Discussion
In this study, we showed that HVJ‐E could enhance the sensitivity of cancer cells to NK cells by upregulation of ICAM‐1.
Inactivated Sendai virus has been shown to have anticancer effects, such as directly killing cancer cells and promoting anticancer immunity.20, 21, 22, 23, 24, 25, 26 We have already reported that HVJ‐E induces anticancer immunity by activating both CD8+ T cells and NK cells.24 However, it has not yet been shown that HVJ‐E can modulate cancer cells to be recognized by immune cells. In this study, we minimized the direct killing effect of HVJ‐E and used the dose of HVJ‐E 1000 HAU per mouse, analyzed in Figure S5 and Appendix S1, for tumor suppression. We showed that HVJ‐E suppressed tumor growth in MDA‐MB‐231 cell‐transplanted SCID mice, and the HVJ‐E tumor suppression was impaired when NK cells were depleted with the anti‐asialo GM1 antibody, as previously reported using PC3‐derived tumors.20 In MDA‐MB‐231‐derived tumors, tumor suppression was greatly abrogated in the HVJ‐E‐treated group by anti‐asialo GM1 antibody. Compared with the PBS‐treated control group, tumor growth was still slightly suppressed by HVJ‐E even in the presence of anti‐asialo GM1 antibody (Fig. 4e). We speculate that this small suppression is likely through direct killing of cancer cells by HVJ‐E. Inactivated Sendai virus recruits and activates NK cells by stimulating dendritic cells to release CXCL10 and type I interferons in the tumor environment.25 Although the result in Figure 4(c) showed no significant increase in NK cells in the tumor environment after HVJ‐E treatment, the sensitivity of cancer cells to NK cells was enhanced. This is probably because of HVJ‐E‐induced ICAM‐1 upregulation, as shown in Figure 3. Moreover, HVJ‐E failed to enhance NK cell sensitivity in ICAM‐1 knockout MDA‐MB‐231 cells. Taken together, HVJ‐E inhibits MDA‐MB‐231 tumor growth by both promoting NK cell activity and upregulating ICAM‐1 expression on MDA‐MB‐231 cells. In the mouse angiosarcoma model, both HVJ‐E and HVJ‐E containing IL‐2 promoted NK cell activity, and NK cell‐mediated cancer cell killing was augmented by the treatment of the mouse angiosarcoma cell line with HVJ‐E.48 This result may be due to the upregulation of ICAM‐1.
The signaling pathway of HVJ‐E‐mediated ICAM‐1 expression is dependent on the RIG‐I/MAVS pathway. This pathway is known to be ubiquitous in various cells. Therefore, the enhancement of NK cell sensitivity by HVJ‐E may occur in all cancer cells with the HVJ receptor. However, it is likely that the increased expression of ICAM‐1 by HVJ‐E is cancer cell‐specific (Figs 1, S1, Appendix S1). We are now analyzing the mechanism of cancer‐specific expression of ICAM‐1 induced by HVJ‐E. The RIG‐I/MAVS signaling pathway has already been reported to contribute to ICAM‐1 expression in Dengue virus‐infected human brain microvascular endothelial cells.49 Other viral RNAs, such as measles virus and mumps virus RNAs, are also known to be recognized by RIG‐I.50 Therefore, virus therapy may generally enhance the sensitivity of cancer cells to NK cells.
Treatment with HVJ‐E induced an increase in ICAM‐1 expression, but it produced a smaller form of the ICAM‐1 protein (Fig. 1c). Neuraminidase treatment of MDA‐MB‐231 cells also gave rise to the smaller ICAM‐1, and the neuraminidase inhibitor blocked the formation of the smaller ICAM‐1 induced by HVJ‐E. Moreover, in HVJ‐E RNA‐transfected cells, ICAM‐1 expression was increased without the reduction in molecular weight. It is likely that HN‐derived neuraminidase removed the sialic acid of ICAM‐1, which resulted in the smaller form of ICAM‐1. However, immunofluorescence analysis of ICAM‐1 showed that cytoplasmic accumulation of ICAM‐1 was detected in both HVJ‐E‐ and PBS‐treated MDA‐MB‐231 cells. To confirm the accumulation of shorter form of ICAM‐1, ICAM‐1 was analyzed in microsomal fractions of MDA‐MB‐231 cells treated with HVJ‐E or PBS. Treatment with HVJ‐E produces shorter form of ICAM‐1 by both removal of sialic acids of ICAM‐1 on the cell surface and increase of unglycosylated form in endoplasmic reticulum (data not shown). This suggests that some stimuli of HVJ‐E might affect the glycosylation condition of ICAM‐1 in endoplasmic reticulum. Although further analysis is required for the analysis of the mechanism of generation of the unglycosylated form of ICAM‐1 by HVJ‐E, it is important to recognize that the smaller ICAM‐1 still retains binding activity with NK cells and contributes to the increase in NK sensitivity in HVJ‐E‐treated cancer cells. Even though ICAM‐1 expression in cancer cells was knocked out by genome editing technology, NK cell sensitivity was not completely abolished in those cancer cells. This remaining sensitivity may be due to the effects of other NK cell ligands expressed on the cancer cell surface, such as Fas and MICB.
In conclusion, these findings suggest that HVJ‐E enhances the NK cell sensitivity of cancer cells by increasing ICAM‐1 expression on the cell surface, which results in the promotion of NK cell anticancer cytotoxicity. This study identified a novel mechanism underlying HVJ‐E antitumor activity. Inactivated Sendai virus can increase the sensitivity of cancers to immunotherapy by modifying the gene expression pattern in cancer cells.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- CCL
chemokine (C‐C motif) ligand
- CXCL
chemokine (C‐X‐C motif) ligand
- F
fusion protein
- HMEC
human mammary epithelial cell
- HN
hemagglutinin–neuraminidase
- HVJ‐E
hemagglutinating virus of Japan envelope
- ICAM‐1
intercellular adhesion molecule‐1
- IFN
interferon
- IL
interleukin
- ITGA2
integrin subunit alpha 2
- LFA‐1
lymphocyte function‐associated antigen 1
- MAVS
mitochondrial antiviral signaling
- MHC
major histocompatibility complex
- MICA/B
MHC class I polypeptide‐related sequence A/B
- NF‐κB
nuclear factor‐κB
- NK
natural killer
- PD
programmed cell death
- PD‐L
programmed cell death ligand
- RIG‐I
retinoic acid‐inducible gene I
- ULBP1
UL16‐binding protein 1
Supporting information
Fig. S1. Hemagglutinating virus of Japan envelope (HVJ‐E)‐induced intercellular adhesion molecule‐1 (ICAM‐1) production was not observed in non‐cancerous prostate epithelial cells.
{kind=link}
Fig. S2. Hemagglutinating virus of Japan envelope (HVJ‐E) induced changes in Fas protein expression level in cancer cells.
{kind=link}
Fig. S3. Hemagglutinating virus of Japan envelope (HVJ‐E)‐induced size reduction of intercellular adhesion molecule‐1 (ICAM‐1) was induced by neuraminidase on the viral surface.
{kind=link}
Fig. S4. Human recombinant intercellular adhesion molecule‐1 (rICAM‐1) binds to lymphocyte function‐associated antigen 1 (LFA‐1) expressed on mouse natural killer cells.
{kind=link}
Fig. S5. Cancer cell survival with or without hemagglutinating virus of Japan envelope (HVJ‐E) treatment.
{kind=link}
Fig. S6. Natural killer cell cytotoxicity was increased in hemagglutinating virus of Japan envelope (HVJ‐E)‐stimulated PC3 cells. E, effector cell; T, target cell.
{kind=link}
Appendix S1. Supplementary material.
Cancer Sci 108 (2017) 2333–2341
Funding InformationPromotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (Project ID: 10‐03) and Grant‐in‐Aid for Scientific Research (B) from Japan Society for the Promotion of Science.
References
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65(2): 87–108. [DOI] [PubMed] [Google Scholar]
- 2. Bae J‐M, Kim EH. Breast density and risk of breast cancer in Asian women: a meta‐analysis of observational studies. J Prev Med Public Health 2016; 49: 367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blank C, Brown I, Peterson AC et al PD‐L1/B7H‐1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res 2004; 64(3): 1140–5. [DOI] [PubMed] [Google Scholar]
- 4. Curran MA, Montalvo W, Yagita H, Allison JP. PD‐1 and CTLA‐4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA 2010; 107: 4275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ott PA, Hodi FS, Robert C. CTLA‐4 and PD‐1/PD‐L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res 2013; 19: 5300–9. [DOI] [PubMed] [Google Scholar]
- 6. Parry RV, Chemnitz JM, Frauwirth KA et al CTLA‐4 and PD‐1 receptors inhibit T‐cell activation by distinct mechanisms. Mol Cell Biol 2005; 25: 9543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Topalian SL, Sznol M, McDermott DF et al Survival, durable tumor remission, and long‐term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014; 32: 1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic β‐catenin signalling prevents anti‐tumour immunity. Nature 2015; 523: 231–5. [DOI] [PubMed] [Google Scholar]
- 9. Harlin H, Meng Y, Peterson AC et al Chemokine expression in melanoma metastases associated with CD8+ T‐cell recruitment. Cancer Res 2009; 69: 3077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ji R‐R, Chasalow SD, Wang L et al An immune‐active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2012; 61: 1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Janeway CA Jr, Travers P, Walport M et al Immunobiology, 5th ed. New York: Garland Science, 2001. [Google Scholar]
- 12. Chadwick BS, Sambhara SR, Sasakura Y, Miller RG. Effect of class I MHC binding peptides on recognition by natural killer cells. J Immunol 1992; 149: 3150–6. [PubMed] [Google Scholar]
- 13. Moretta A, Bottino C, Vitale M et al Receptors for HLA class‐I molecules in human natural killer cells. Annu Rev Immunol 1996; 14(1): 619–48. [DOI] [PubMed] [Google Scholar]
- 14. Li F, Wei H, Wei H et al Blocking the natural killer cell inhibitory receptor NKG2A increases activity of human natural killer cells and clears hepatitis B virus infection in mice. Gastroenterology 2013; 144(2): 392–401. [DOI] [PubMed] [Google Scholar]
- 15. Romagné F, André P, Spee P et al Preclinical characterization of 1‐7F9, a novel human anti–KIR receptor therapeutic antibody that augments natural killer–mediated killing of tumor cells. Blood 2009; 114: 2667–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu L, Zhang C, Zhang J. HMBOX1 negatively regulates NK cell functions by suppressing the NKG2D/DAP10 signaling pathway. Cell Mol Immunol 2011; 8: 433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sutherland CL, Chalupny NJ, Schooley K, VandenBos T, Kubin M, Cosman D. UL16‐binding proteins, novel MHC class I‐related proteins, bind to NKG2D and activate multiple signaling pathways in primary NK cells. J Immunol 2002; 168(2): 671–9. [DOI] [PubMed] [Google Scholar]
- 18. Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 2002; 17(1): 19–29. [DOI] [PubMed] [Google Scholar]
- 19. Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D‐DAP10 triggers human NK cell–mediated killing via a Syk‐independent regulatory pathway. Nat Immunol 2003; 4: 557–64. [DOI] [PubMed] [Google Scholar]
- 20. Kawaguchi Y, Miyamoto Y, Inoue T, Kaneda Y. Efficient eradication of hormone‐resistant human prostate cancers by inactivated Sendai virus particle. Int J Cancer 2009; 124: 2478–87. [DOI] [PubMed] [Google Scholar]
- 21. Tanaka M, Shimbo T, Kikuchi Y, Matsuda M, Kaneda Y. Sterile alpha motif containing domain 9 is involved in death signaling of malignant glioma treated with inactivated Sendai virus particle (HVJ‐E) or type I interferon. Int J Cancer 2010; 126: 1982–91. [DOI] [PubMed] [Google Scholar]
- 22. Matsushima‐Miyagi T, Hatano K, Nomura M et al TRAIL and Noxa are selectively upregulated in prostate cancer cells downstream of the RIG‐I/MAVS signaling pathway by nonreplicating Sendai virus particles. Clin Cancer Res 2012; 18: 6271–83. [DOI] [PubMed] [Google Scholar]
- 23. Jiang Y, Saga K, Miyamoto Y, Kaneda Y. Cytoplasmic calcium increase via fusion with inactivated Sendai virus induces apoptosis in human multiple myeloma cells by downregulation of c‐Myc oncogene. Oncotarget 2016; 7: 36034–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurooka M, Kaneda Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res 2007; 67(1): 227–36. [DOI] [PubMed] [Google Scholar]
- 25. Fujihara A, Kurooka M, Miki T, Kaneda Y. Intratumoral injection of inactivated Sendai virus particles elicits strong antitumor activity by enhancing local CXCL10 expression and systemic NK cell activation. Cancer Immunol Immunother 2008; 57(1): 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nishikawa T, Tung LY, Kaneda Y. Systemic administration of platelets incorporating inactivated Sendai virus eradicates melanoma in mice. Mol Ther 2014; 22: 2046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vankayalapati R, Klucar P, Wizel B et al NK cells regulate CD8+ T cell effector function in response to an intracellular pathogen. J Immunol 2004; 172(1): 130–7. [DOI] [PubMed] [Google Scholar]
- 28. Bassi V, Vitale M, Feliciello A, De Riu S, Rossi G, Fenzi G. Retinoic acid induces intercellular adhesion molecule‐1 hyperexpression in human thyroid carcinoma cell lines. J Clin Endocrinol Metab 1995; 80: 1129–35. [DOI] [PubMed] [Google Scholar]
- 29. Papi A, Johnston SL. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM‐1) via increased NF‐κB‐mediated transcription. J Biol Chem 1999; 274: 9707–20. [DOI] [PubMed] [Google Scholar]
- 30. Matsukura S, Kokubu F, Noda H et al Expression of ICAM‐1 on human bronchial epithelial cells after influenza virus infection. Allergol Int 1996; 45(2): 97–103. [Google Scholar]
- 31. Mulligan MS, Vaporciyan AA, Miyasaka M, Tamatani T, Ward PA. Tumor necrosis factor alpha regulates in vivo intrapulmonary expression of ICAM‐1. Am J Pathol 1993; 142(6): 1739. [PMC free article] [PubMed] [Google Scholar]
- 32. Yang C‐M, Luo S‐F, Hsieh H‐L et al Interleukin‐1β induces ICAM‐1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: involvement of ERK, JNK, AP‐1, and NF‐κB. J Cell Physiol 2010; 224(2): 516–26. [DOI] [PubMed] [Google Scholar]
- 33. Kaiserlian D, Rigal D, Abello J, Revillard J‐P. Expression, function and regulation of the intercellular adhesion molecule‐1 (ICAM‐1) on human intestinal epithelial cell lines. Eur J Immunol 1991; 21: 2415–21. [DOI] [PubMed] [Google Scholar]
- 34. Rosette C, Roth RB, Oeth P et al Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis 2005; 26: 943–50. [DOI] [PubMed] [Google Scholar]
- 35. Guo P, Huang J, Wang L et al ICAM‐1 as a molecular target for triple negative breast cancer. Proc Natl Acad Sci USA 2014; 111: 14710–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang M, Liu J, Piao C, Shao J, Du J. ICAM‐1 suppresses tumor metastasis by inhibiting macrophage M2 polarization through blockade of efferocytosis. Cell Death Dis 2015; 6: e1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jenkinson SR, Williams NA, Morgan DJ. The role of intercellular adhesion molecule‐1/LFA‐1 interactions in the generation of tumor‐specific CD8+ T cell responses. J Immunol 2005; 174: 3401–7. [DOI] [PubMed] [Google Scholar]
- 38. Kikuchi T, Joki T, Akasaki Y, Abe T, Ohno T. Induction of antitumor immunity using Intercellular adhesion molecule 1 (ICAM‐1) transfection in mouse glioma cells. Cancer Lett 1999; 142(2): 201–6. [DOI] [PubMed] [Google Scholar]
- 39. Barber DF, Faure M, Long EO. LFA‐1 contributes an early signal for NK cell cytotoxicity. J Immunol 2004; 173: 3653–9. [DOI] [PubMed] [Google Scholar]
- 40. Kaneda Y, Nakajima T, Nishikawa T et al Hemagglutinating virus of Japan (HVJ) envelope vector as a versatile gene delivery system. Mol Ther 2002; 6(2): 219–26. [DOI] [PubMed] [Google Scholar]
- 41. Curran J, Kolakofsky D. Replication of paramyxoviruses In: Maramorosch K, Murphy FA, Shatkin AJ, eds. Advances in Virus Research [Internet]. Academic Press, 1999; 403–22. [DOI] [PubMed] [Google Scholar]
- 42. Sakaguchi T, Kato A, Kiyotani K, Yoshida T, Nagai Y. Studies on the paramyxovirus accessory genes by reverse genetics in the Sendai virus–mouse system. Proc Jpn Acad Ser B 2008; 84: 439–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takimoto T, Taylor GL, Connaris HC, Crennell SJ, Portner A. Role of the hemagglutinin‐neuraminidase protein in the mechanism of paramyxovirus‐cell membrane fusion. J Virol 2002; 76: 13028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gotoh B, Ogasawara T, Toyoda T, Inocencio NM, Hamaguchi M, Nagai Y. An endoprotease homologous to the blood clotting factor X as a determinant of viral tropism in chick embryo. EMBO J 1990; 9: 4189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kido H, Yokogoshi Y, Sakai K et al Isolation and characterization of a novel trypsin‐like protease found in rat bronchiolar epithelial Clara cells. A possible activator of the viral fusion glycoprotein. J Biol Chem 1992; 267: 13573–9. [PubMed] [Google Scholar]
- 46. Amrani Y, Lazaar AL, Hoffman R, Amin K, Ousmer S, Panettieri RA. Activation of p55 tumor necrosis factor‐α receptor‐1 coupled to tumor necrosis factor receptor‐associated factor 2 stimulates intercellular adhesion molecule‐1 expression by modulating a thapsigargin‐sensitive pathway in human tracheal smooth muscle cells. Mol Pharmacol 2000; 58(1): 237–45. [DOI] [PubMed] [Google Scholar]
- 47. Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule‐1 (CD54) gene expression. J Leukoc Biol 1999; 66: 876–88. [DOI] [PubMed] [Google Scholar]
- 48. Takehara Y, Satoh T, Nishizawa A et al Anti‐tumor effects of inactivated Sendai virus particles with an IL‐2 gene on angiosarcoma. Clin Immunol 2013; 149(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 49. da Conceição TM, Rust NM, Berbel ACER et al Essential role of RIG‐I in the activation of endothelial cells by dengue virus. Virology 2013; 435(2): 281–92. [DOI] [PubMed] [Google Scholar]
- 50. Rehwinkel J, Tan CP, Goubau D et al RIG‐I detects viral genomic RNA during negative‐strand RNA virus infection. Cell 2010; 140(3): 397–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Hemagglutinating virus of Japan envelope (HVJ‐E)‐induced intercellular adhesion molecule‐1 (ICAM‐1) production was not observed in non‐cancerous prostate epithelial cells.
Fig. S2. Hemagglutinating virus of Japan envelope (HVJ‐E) induced changes in Fas protein expression level in cancer cells.
Fig. S3. Hemagglutinating virus of Japan envelope (HVJ‐E)‐induced size reduction of intercellular adhesion molecule‐1 (ICAM‐1) was induced by neuraminidase on the viral surface.
Fig. S4. Human recombinant intercellular adhesion molecule‐1 (rICAM‐1) binds to lymphocyte function‐associated antigen 1 (LFA‐1) expressed on mouse natural killer cells.
Fig. S5. Cancer cell survival with or without hemagglutinating virus of Japan envelope (HVJ‐E) treatment.
Fig. S6. Natural killer cell cytotoxicity was increased in hemagglutinating virus of Japan envelope (HVJ‐E)‐stimulated PC3 cells. E, effector cell; T, target cell.
Appendix S1. Supplementary material.