

Abstract
The aim of this study was to analyze the significance of glucose metabolism‐related enzymes in the proliferation of gastric cancer under hypoxia. Four hypoxia‐resistant gastric cancer cell lines and four parent cell lines were used. Reverse transcription–PCR was used to evaluate the mRNA expression levels of the following metabolism‐related enzymes: pyruvate kinase isozyme M2 (PKM2), glutaminase (GLS), enolase 1 (ENO1), glucose‐6‐phosphate dehydrogenase (G6PDH), and PKM1. The effects of these enzymes on the proliferation of gastric cancer cells were examined using siRNAs, shikonin as a PKM2 inhibitor, or BPTES as a GLS inhibitor, in vitro and in vivo. Levels of both PKM2 and GLS mRNA were significantly high in all hypoxia‐resistant cell lines, compared with those of their parent cells. Knockdown of PKM2 and GLS significantly decreased the proliferation of all hypoxia‐resistant cells. The combination of siPKM2 and siGLS significantly decreased proliferation compared with treatment by siPKM2 or siGLS alone. The knockdown of ENO1, G6PDH, or PKM1 did not decrease the proliferation of all hypoxia‐resistant cells. Combination treatment using shikonin and BPTES inhibited the proliferation of all hypoxia‐resistant cancer cells more than that by either agent alone. The in vivo study indicated that the tumor size treated by the combination of shikonin and BPTES was significantly smaller than that of vehicle‐treated group. These findings suggested that PKM2 and GLS might play important roles in the proliferation of hypoxic gastric cancer cells. A combination of PKM2 and GLS inhibitors could be therapeutically promising for the treatment of gastric cancer.
Keywords: gastric cancer, glucose metabolism‐related enzyme, glutaminase, molecular target, pyruvate kinase isozymes M2
Gastric cancer is one of the most common gastrointestinal malignancies, and is also a leading cause of death from malignant tumors worldwide.1 However, few effective treatments for patients with gastric cancer have been developed.2 Tumor heterogeneity has been suggested to be one of the reasons for the resistance of stomach cancer to cancer therapies. Many kinds of solid tumors, including gastric cancer, have regions of hypoxia.3 Some types of cancer cells survive and proliferate in this hypoxic environment, which might allow them to acquire characteristic malignant potentials such as metastatic or chemoresistant ability. To effectively treat solid gastric tumors, it is important to clarify the molecular mechanisms responsible for the proliferation of cancer cells in hypoxia.
Glycolysis is the primary mode of glucose metabolism, not only in normal cells but also in cancer cells. A number of papers have reported that some glucose metabolism‐related enzymes are key regulators of glycolysis in solid cancer cells.4 The Warburg effect, known as anaerobic glycolysis even in the presence of ample oxygen, might contribute to tumor development in such hypoxic environments.5, 6 It has been suggested that some kinds of glucose metabolism‐related enzymes, including pyruvate kinase isozyme M2 (PKM2), PKM1, enolase 1 (ENO1), and glucose‐6‐phosphate dehydrogenase (G6PDH), might be associated with the anaerobic glycolysis of cancer cells.7, 8 Glutamine is also associated with the proliferation of cancer cells by an alternate source of biosynthesis from the TCA cycle, namely glutaminolysis.9 As glycolysis and glutaminolysis are both reported to play important roles in the proliferation of cancer in hypoxic environments,10 in the present study we evaluated the significance of the enzymes for glycolysis and glutaminolysis in relation to the proliferation of gastric cancer cells in hypoxic environments.
Materials and Methods
Cell lines
Eight gastric cancer cell lines, OCUM12,3 OCUM‐12/hypo,3 OCUM‐2MD3,11 OCUM‐2MD3/hypo,11 NUGC‐3,12 NUGC‐3/hypo, NUGC‐4,12 and NUGC‐4/hypo, were used in this study. NUGC‐3 and NUGC‐4 cells were obtained from the JCRB cell bank (Osaka, Japan). Four hypoxia‐resistant cell lines, OCUM‐12/hypo, OCUM‐2MD3/hypo, NUGC‐3/hypo cells, and NUGC‐4/hypo, were established from the parent cell lines, OCUM12, OCUM‐2MD3, NUGC‐3, and NUGC‐4, respectively, by continuous exposure to 1% oxygen up to 5% oxygen for 4 weeks, as previously reported.3 The culture condition was cultivated in DMEM (Nikken, Kyoto, Japan) with the addition of 10% FBS (Nichirei, Tokyo, Japan), 100 IU/mL penicillin (Wako, Osaka, Japan), 100 mg/mL streptomycin (Wako), and 0.5 mM sodium pyruvate (Wako) at 37°C. All experimental studies by parent cancer cells were carried out at 20% O2, and those by hypoxia‐resistant cells were carried out at 1% O2.
Quantitative real‐time RT‐PCR
The total cellular RNA from cancer cells was extracted using RNeasy Mini Kit (Qiagen, Carlsbad, CA, USA). cDNA was prepared from 2 mg RNA using random primers (Invitrogen, Carlsbad, CA, USA). Real‐time RT‐PCR was undertaken using the ABI Prism 7000 (Applied Biosystems, Foster City, CA, USA). Total cellular RNA was extracted from cell lines with TRIzol (Life Technologies, Carlsbad, CA, USA). Relevant cDNA was amplified by PCR with Taq DNA polymerase (Nippon Gene, Tokyo, Japan) in a thermal cycler. Primer sets were from Applied Biosystems. Reverse transcription–PCR was carried out with commercially available gene expression assays (Applied Biosystems) for PKM2 (assay ID, Hs00761782), GLS (assay ID, Hs00248163), ENO1 (assay ID, Hs00361415), G6PDH (assay ID, Hs01097550), and PKM1 (assay ID, Hs00761782). As an internal control, GAPDH (accession nos. NM‐002046, NM‐001256799; P, 5′‐CCCCTGCAAATGAGCCCCAGCCTTC‐3′; forward, 5′‐CCATCTTCCAGGAGCGAGATC‐3′; and reverse, 5′‐GGCAGAGATGATGACCCTTTTG‐3′) were customized from Sigma‐Aldrich (St. Louis, MO, USA). The threshold cycle (Ct) values were used to calculate the relative expression ratios between control and treated cells. Reverse transcription–PCR was carried out at 95°C for 15 s and 60°C for 60 s for 40 cycles.
Small interfering RNA design
The siRNA and non‐targeting siRNA (negative‐siRNA) were purchased from Ambion (Life Technologies): siPKM2#1 (ID s501106), siPKM2#2 (ID s10575), siGLS#1 (ID s501106), siGLS#2 (ID s5839), siENO1#1 (ID s5838), siENO1#2 (ID s4681), siG6PDH#1 (ID s409), siG6PDH#2 (ID s18369), and siPKM1 (ID S501105). The siRNAs and cancer cells were prepared at 60% confluence in 6‐well dishes. The transfection mixture was prepared by adding 150 μL of Opti‐MEM including 9 μL Lipofectamine RNA iMAX Reagent (Life Technologies) to 150 μL Opti‐MEM including 90 pmol siRNA and incubating for 5 min at room temperature. Finally, the above transfection mixture was added to a 6‐well dish containing 1.7 mL DMEM with 2% FBS. Finally, the above transfection mixture was added to the prepared 6‐well dish. Twenty‐four hours after transfection, RT‐PCR was carried out.
Compounds
Two small compounds, shikonin as a PKM2 inhibitor and BPTES as a GLS inhibitor, were used in this study. Shikonin (≥98%) and BPTES were purchased from Sigma‐Aldrich. Shikonin and BPTES were dissolved in 0.25% methanol and in 0.42% ethanol, respectively, and stored in a light‐shielded container at 4°C. For in vivo experiments, the agent was dissolved in normal saline and i.p. injected. For in vitro experiments, the diluted shikonin and BPTES were mixed at various concentrations with methanol and ethanol.
Proliferation assay
The growth inhibitory effect of siRNAs and their inhibitor on cancer cells were measured by CCK‐8 assay (Dojindo, Kumamoto, Japan). The cells were plated in 96‐well microtiter plates at a density of 1 × 103 cells per well. After incubation for 72 h, cells were treated with 10 μL depsipeptide. Cell viability was assayed 2 h after incubation, measured as absorbance at 450 nm using a microtiter plate reader (PM2004; Wako). The percentage of cell viability was determined as the ratio of the absorbance of the sample versus the control. Survival of gastric cancer cells were presented as a percentage of absorbance with depsipeptide‐treated cells divided by that with cells not exposed to depsipeptide.13
Flow cytometry analysis
Apoptosis was detected using flow cytometry by staining cells with annexin V–FITC and propidium iodide (PI) (BD Pharmingen, San Diego, CA, USA) labeling. OCUM‐12, OCUM‐12/hypo, OCUM‐2MD3, OCUM‐2MD3/hypo, NUGC‐3, NUGC‐3/hypo, NUGC‐4, and NUGC‐4/hypo cells were seeded at a density of 2.0 × 105 cells/mL in a 6‐well plate. With or without the addition of shikonin (0.75 μM) and/or BPTES (7.5 μM) at the concentration of 50 μM, the plates were incubated for 24 h. Cells were stained with annexin V–FITC and/or PI and analyzed by flow cytometry using FACScan (BD LSR II; Becton Dickinson, San Diego, CA, USA).
In vivo tumor model
In vivo experiments were carried out on 4‐week‐old female athymic BALB/c nude mice (CLEA Japan, Tokyo, Japan). Mice were housed in a standard animal laboratory with free access to water and food. They were kept under constant environmental conditions with a 12:12‐h light:dark cycle. OCUM‐2MD3/hypo cells (1 × 107 cells/0.2 mL/site) were injected s.c. into the back upper right, left, and lower right, left regions of mice. The mice were randomly divided into four groups. They were treated daily with normal saline (negative control; n = 4), shikonin (0.8 mg/kg; n = 4), BPTES (6 mg/kg; n = 4), or the combination of shikonin (0.8 mg/kg) and BPTES (6 mg/kg) (n = 4) by i.p. injection on day 5 after inoculation with OCUM‐2MD3/hypo cells. The doses of shikonin and BPTES were based on our previous work that did not show any cytotoxic effect in normal mice. All the mice were killed on day 25 after inoculation. Tumors were measured for largest (a) and smallest (b) diameters, and tumor size was calculated as S = a × b. The selected tissue sections were subjected to immunohistochemical staining. Animal experiments were carried out in compliance with the guidelines of the Osaka City University Ethical Committee (Osaka, Japan).
Immunohistochemical staining
On sections stained with H&E, the total number of mitosis/five fields, under a light microscope at ×200, was counted as the mitotic index. The immunohistochemical determination of proliferation activity of the s.c. xenografted tumor was examined using Ki‐67 antibody (MIB‐1; Dako, Glostrup, Denmark). Ki‐67 expression was analyzed by the Ki‐67 index, the number of MIB‐1‐positive cancer cells in 100 cancer cells. Six randomly chosen fields were counted for each tumor. The mean of six fields was calculated as the sample value. Evaluation was made by two double‐blinded independent observers.
Statistical analysis
Comparisons among the datasets were made by Student's t‐test. Comparisons among datasets were made with the Kruskal–Wallis one‐way anova by ranks followed by the Student–Newman–Keuls multiple comparison test. Data are expressed as mean ± SD. A difference was considered to be statistically significant when the P‐value was <0.05 or less.
Results
Expression levels of PKM2, ENO1, GLS, G6PDH, and PKM1 mRNA in parent cancer cells and hypoxia‐resistant cells
Expression levels of both PKM2 and GLS mRNA were significantly high in hypoxia‐resistant cells of all of four cell lines, compared with those of their parent cells. The ENO1 expression level of hypoxia‐resistant cells was significantly high in all but NUGC‐3/hypo cells. The G6PDH expression level of hypoxia‐resistant cells was significantly high in all but NUGC‐4/hypo cells. In contrast, no significant difference in PKM1 mRNA expression level was found between the hypoxia‐resistant cell lines and their parent cell lines (Fig. 1).
Figure 1.
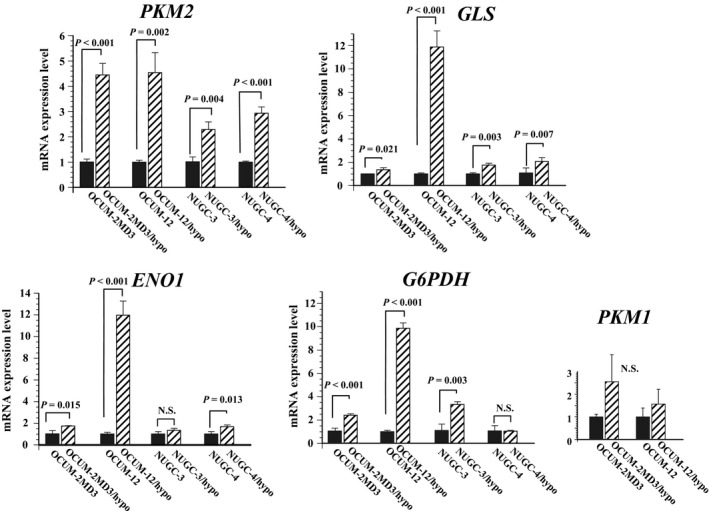
PKM2, ENO1, GLS, G6PDH, and PKM1 expression levels in parent cancer cells and their hypoxia‐resistant daughter cells. PKM2 expression levels in all hypoxia‐resistant cells were significantly (approximately 2.3–4.5 times) higher than in their respective parent cells. GLS expression in all hypoxia‐resistant cells was also significantly (1.3–2.1 times) higher in OCUM‐2MD3/hypo cells, NUGC‐3/hypo cells, and NUGC‐4/hypo cells compared to their respective parent cells, and was especially (11.8 times) higher in OCUM‐212/hypo cells. ENO1 expression levels were significantly higher in hypoxia‐resistant cells compared to parent cells, with the exception of NUGC‐3/hypo cells. G6PDH expression levels were also significantly higher in hypoxia‐resistant cells, again with the exception of NUGC‐4/hypo cells. PKM1 mRNA expression levels did not differ between the hypoxia‐resistant cells and parent cells. Results are presented as means for three independent experiments; bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 versus control. N.S., not significant.
Effect of PKM2, ENO1, GLS, G6PDH, and PKM1 siRNA on proliferation of gastric cancer cells
The siRNA of PKM2, ENO1, GLS, G6PDH, and PKM1 effectively mediated the knockdown of PKM2, ENO1, GLS, G6PDH, and PKM1 (Fig. S1). siPKM2 significantly inhibited the proliferation of cancer cells in all eight cancer cell lines. The growth‐inhibitory effect by siPKM2 was high in hypoxia‐resistant cells, compared with that in their parent cells. Also, the knockdown of GLS significantly decreased the proliferation of three of four hypoxia‐resistant cancer cells and parent NUGC‐3 cells. Combined treatment with siPKM2 and siGLS significantly decreased the proliferation of all four hypoxia‐resistant cell lines, and decreased proliferation significantly more than either siPKM2 or siGLS alone in three of the four hypoxia‐resistant cell lines. In contrast, siENO1 did not significantly affect the proliferation of most cancer cells, except OCUM‐12/hypo cells. Knockdown of G6PDH did not affect the proliferation of most cancer cells. Interestingly, treatment with siPKM1 stimulated the proliferation of all hypoxia‐resistant cancer cells and NUGC‐3 cells (Fig. 2).
Figure 2.
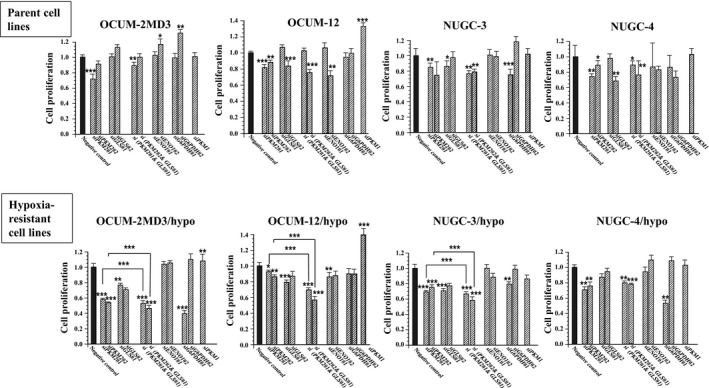
Effect of siRNA on the proliferation of gastric cancer cells. The proliferation of all gastric cancer cell lines was significantly inhibited by siPKM2#1 and siPKM2#2. The knockdown of GLS significantly decreased the proliferation of three of four hypoxia‐resistant cancer cells. The combination of siPKM2 and siGLS significantly decreased the proliferation of all of four hypoxia‐resistant cell lines. Treatment with siENO1 did not affect the proliferation of most cancer cells. Knockdown of G6PDH did not affect the proliferation of most cancer cells. In contrast, siPKM1 stimulated the proliferation of all hypoxia‐resistant cancer cells. Results are presented as means of four independent experiments; bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Effect of KM2 inhibitor and GLS inhibitor on proliferation of cancer cells in vitro
Shikonin, a PKM2 inhibitor, significantly inhibited the proliferation of all of hypoxia‐resistant cancer cell lines. BPTES, a GLS inhibitor, significantly inhibited the proliferation of hypoxia‐resistant cancer cells, except for OCUM‐2MD3/hypo cells. Combined treatment with shikonin and BPTES significantly decreased the proliferation of all hypoxia‐resistant cancer cell lines compared with the addition of shikonin or BPTES alone; the combination of shikonin and BPTES decreased the proliferation of only two of the four parent cancer cell lines. The antitumor effect of the combination of shikonin and BPTES was more remarkable in hypoxia‐resistant cell lines than in parent cell lines (Fig. 3). Figure 4(a) shows representative examples of the apoptosis rate by flow cytometric analysis. Cells staining annexin V‐positive and PI‐negative were considered to be apoptotic. Shikonin significantly increased the apoptosis rate of all of the hypoxia‐resistant cancer cell lines. BPTES did not affect the apoptosis of cancer cells. The combination of shikonin and BPTES significantly increased the apoptosis of cancer cell lines, except for NUGC‐3 cells (Fig. 4b).
Figure 3.
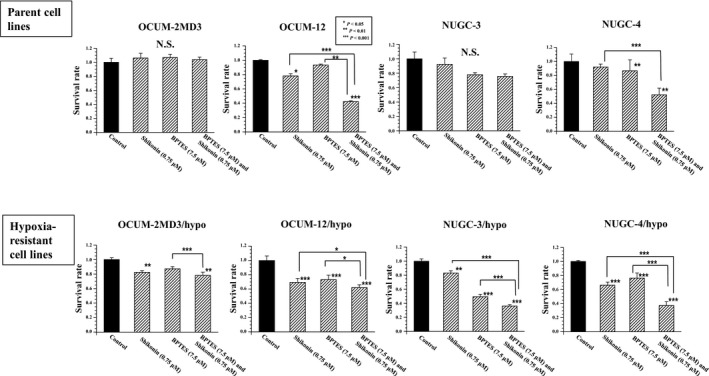
Effect of shikonin and BPTES on the proliferation of gastric cancer cells in vitro. The pyruvate kinase isozyme M2 inhibitor, shikonin, significantly inhibited the proliferation of all hypoxia‐resistant cancer cell lines. The glutaminase inhibitor, BPTES, significantly inhibited the proliferation of hypoxia‐resistant cancer cells except for OCUM‐2MD3/hypo cells. Combined treatment with shikonin and BPTES significantly decreased the proliferation of all hypoxia‐resistant cancer cell lines, compared with treatment with shikonin or BPTES alone. In contrast, combined treatment with shikonin and BPTES significantly decreased proliferation of only two of four parent cancer cell lines. Results are presented as means of four independent experiments; bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001. N.S., not significant.
Figure 4.
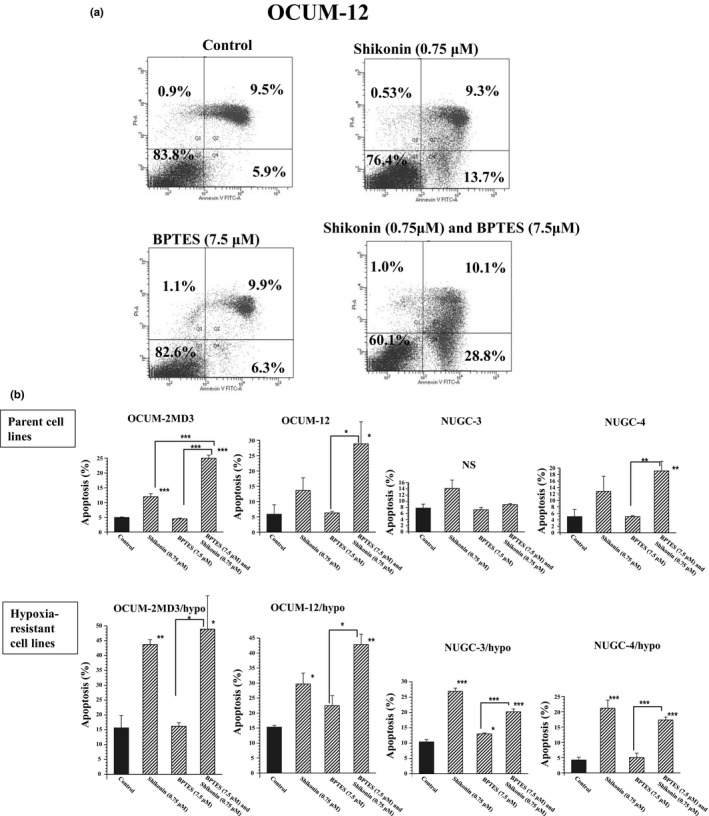
Apoptosis induction by pyruvate kinase isozyme M2 (PKM2) inhibitor (shikonin) and/or glutaminase (GLS) inhibitor (BPTES). (a) Representative examples of flow cytometric analysis. To clarify the induction of apoptosis, double staining of cells with annexin V (FITC) and propidium iodide (PI) was carried out. Cells staining annexin V‐positive and PI‐negative were considered to be apoptotic; cells staining annexin V‐negative and PI‐positive were considered to be viable. (b) Apoptosis rates of cancer cells by PKM2 inhibitor and/or GLS inhibitor. The apoptosis rates of all of hypoxia‐resistant cancer cell lines were significantly increased by shikonin treatment, whereas treatment with BPTES did not affect the apoptosis of cancer cells except NUGC‐3/hypo cells. Combined treatment with shikonin and BPTES significantly increased the apoptosis of cancer cell lines, except NUGC‐3 cells. Results are presented as means of four independent experiments; bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Effect of shikonin and/or BPTES on the growth of OCUM‐2MD3/hypo xenografted tumor in vivo
Subcutaneous inoculation of OCUM‐2MD3/hypo cells (1 × 107 cells/0.2 mL/site) resulted in tumor formation at all sites. Shikonin and/or BPTES were give i.p. five times per week from day 5 to day 25 after inoculation of OCUM‐2MD3/hypo cells. The body weights of mice did not differ among the four treatment groups. The mean volumes of the s.c. tumor of groups treated with vehicle, shikonin, BPTES, and shikonin plus BPTES were 157, 59, 130, and 27 mm2 at day 26 after s.c. inoculation, respectively. The size of tumors in mice receiving a combination of shikonin and BPTES was significantly (P < 0.05) smaller than that in the vehicle‐treated group between days 15 and 26 after inoculation. The tumor size resulting from combined treatment with shikonin plus BPTES was also reduced compared to that from treatment with shikonin or BPTES alone (Fig. 5a). The tumor mitotic indexes of mice treated by Shikonin and/or BPTES were less than that of vehicle‐treated mice. Following the combination treatment of Shikonin and BPTES, the mitotic index was significantly (P < 0.01) decreased compared with that of Shikonin or BPTES alone (Fig. 5b). Expression levels of Ki‐67 in cancer cells were significantly (P < 0.01) decreased by shikonin and/or BPTES compared with vehicle‐treated mice. Ki‐67 index staining in cancer cells was significantly (P < 0.01) decreased by the combination of shikonin and BPTES compared with shikonin or BPTES alone (Fig. 5c).
Figure 5.
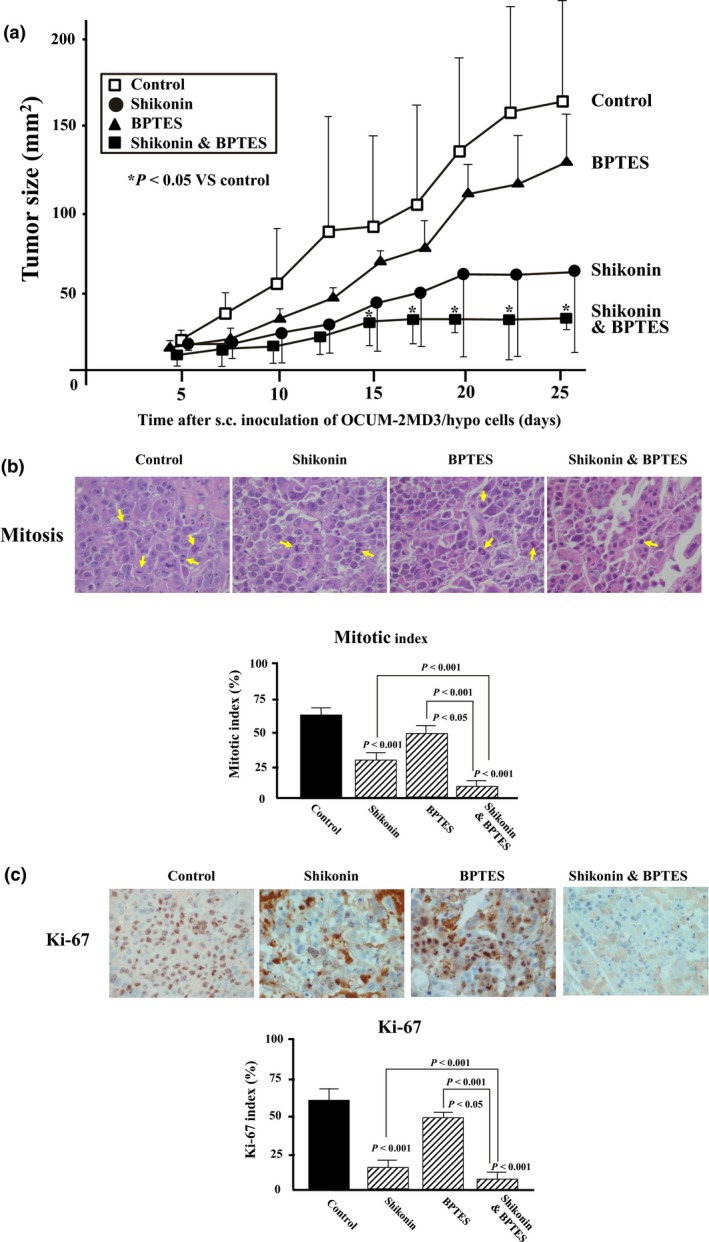
Effect of pyruvate kinase isozyme M2 (PKM2) inhibitor (shikonin) and glutaminase (GLS) inhibitor (BPTES) on the growth of xenografted tumor by OCUM‐2MD3/hypo cells in vivo. (a) Tumor size. Subcutaneous inoculation of OCUM‐2MD3/hypo cells (1 × 107 cells/0.2 mL/site) resulted in tumor formation at all sites. The size of s.c. xenograft tumors treated by the combination of shikonin and BPTES (n = 4) was significantly decreased in comparison with the control (n = 4). Separately, both shikonin (n = 4) and BPTES (n = 4) significantly decreased the tumor size compared with the control. (b) Mitotic indices. The number of tumor cells undergoing mitosis (arrows) was decreased following PKM2 inhibitor and/or GLS inhibitor treatment. (c) Ki‐67 staining of tumor. Expression levels of Ki‐67 in cancer cells of the mice treated by shikonin and/or BPTES were significantly lower than that in vehicle‐treated mice. Six randomly chosen fields were counted for each assay and the mean of six fields was calculated as the sample value. *P < 0.05.
Discussion
In this study, the expression levels of PKM2 and GLS mRNA were significantly high in all hypoxia‐resistant gastric cancer cells compared with those of the parent cells. The knockdown of both PKM2 and GLS significantly inhibited the proliferation of hypoxia‐resistant gastric cancer cells. These findings suggest that PKM2 and GLS might be key molecules for the proliferation of hypoxic cancer cells. Current studies have reported that PKM2 is not essential for all tumor cells, but that PKM2 was active in characteristic populations of tumor cells.14 There is no evidence yet for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis.15 The hypoxia‐resistant cancer cells may exist heterogeneously in a solid tumor, and are a subpopulation of cancer cells. Pyruvate kinase isozyme M2 might be essential for hypoxia‐resistant cancer cells, but not for the main population of cancer cells.
The mRNA level of PKM2 and GLS was significantly high in hypoxia‐resistant cells of all of four cell lines, compared with those of their parent cells. Taken together, siPKM2 significantly inhibited the proliferation of all eight cancer cell lines, and siGLS inhibited the proliferation of most cancer cell lines. In contrast, the growth‐inhibitory effects of siENO1 and siG6PDH were not stable in four hypoxia‐resistant cell lines. Glutaminase is a glutaminolysis enzyme, and PKM2, ENO1, and G6PDH are glycolysis enzymes. Therefore, we selected PKM2 inhibitor as a glycolysis inhibitor, and GLS inhibitor as a glutaminolysis inhibitor. We then evaluated the usefulness of inhibitors of PKM2 and GLS against glycolysis and glutaminolysis in vitro and in vivo, especially in hypoxic environments. Previous studies have shown that shikonin,16, 17 an inhibitor of PKM2, possesses antitumor activity in a variety of solid cancers including gastric cancer. Taken together, BPTES18 appears to be an inhibitor of GLS, which is a key enzyme of glutamine metabolism.19 Proliferation of cancer cells was inhibited by shikonin in all hypoxia‐resistant cancer cells. BPTES decreased the proliferation of hypoxia‐resistant cancer cells except for OCUM‐2MD3/hypo cells. The combination of shikonin and BPTES inhibited the proliferation of all of hypoxia‐resistant cancer cells significantly more than shikonin or BPTES alone. The in vivo study also indicated that the tumor size of mice treated by shikonin plus BPTES was lower than that by shikonin or BPTES alone. These are exciting findings, given that targeting cancer metabolism has emerged as a hot topic in drug discovery.20
Most cancers have a high demand for metabolic inputs, such as glycolysis and glutaminolysis, which aid in their proliferation and survival.19, 21 We here present significant evidence supporting the effective antitumor activity of shikonin and BPTES in gastric cancer in vitro and in vivo. This is the first report to show that the combination of PKM2 and GLS inhibitors is useful for the treatment of gastric cancer.
In this study, we used four cancer cell lines derived from a diffuse‐type of gastric cancer. Shiroki et al. reported that PKM2 expression in gastric cancer was significantly correlated with venous invasion cases.22 Gao et al. reported that the upregulation of PKM2 significantly correlated with both lymph node metastasis and advanced TNM stage.23 Also, Kim et al. reported that GLS expression was significantly associated with lymph node metastasis of breast cancer.24 These findings suggest that PKM2 inhibitor and GLS inhibitor might be useful for the diffuse‐type of gastric cancer with lymph node metastasis.
The expression level of ENO1 was also significantly high in three of four hypoxia‐resistant gastric cancer cell lines, except for NUGC‐3/hypo cells, compared with that of the parent cells. However, siENO1 did not affect the proliferation of most gastric cancer cell lines. It has been reported that ENO1 is involved in pancreatic cancer cell invasion.25 These findings might suggest that the upregulation of ENO1 in hypoxia‐resistant cells might not be associated with proliferation in most gastric cancer cells.
The G6PDH expression level of hypoxia‐resistant cells was significantly high in most hypoxia‐resistant gastric cancer cell lines compared with that of parent cells. Knockdown of G6PDH significantly decreased the proliferation of some gastric cancer cell lines, especially hypoxia‐resistant cell lines. It has been reported that G6PDH participates in the pentose phosphate pathway, and is tightly associated with development and progression of a variety of tumors.26, 27 Glucose‐6‐phosphate dehydrogenase plays a critical role in survival, proliferation, and metastasis of cancer cells.28 These findings might suggest that the development of G6PDH inhibitors could provide new treatments for gastric cancer.
Interestingly, knockdown of PKM1 stimulated the proliferation of hypoxia‐resistant cancer cells. Pyruvate kinase is a glycolytic enzyme that catalyzes the latter step in glycolysis by the conversion of phosphoenolpyruvate to pyruvate. Two alternative splice isoforms of the glycolytic enzyme of PKM, PKM1 and PKM2, have been known to be associated with the Warburg effect in cancer. PKM1 moves the pyruvate into the mitochondria for use in the citric acid cycle, whereas PKM2 favors conversion of pyruvate to lactate.29 Aerobic glycolysis by PKM1 promotes oxidative phosphorylation, which regulates the metabolism of reactive oxygen species and the cytotoxic damage by oxidative stress.30, 31 These findings might suggest that PKM1 suppresses the growth of hypoxia‐resistant cancer cells by promoting oxidative phosphorylation from aerobic glycolysis.
In conclusion, our study showed that PKM2 and GLS might play important roles in the proliferation of gastric cancer cells, especially under hypoxic conditions. Combined treatment with PKM2 and GLS inhibitors could be promising for the treatment of gastric cancer.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Knockdown of PKM2, ENO1, GLS, G6PDH, and PKM1. Reverse transcription–PCR analysis indicated that PKM2, ENO1, GLS, G6PDH, and PKM1 expression were effectively downregulated by siRNA (siPKM2#1, siPKM2#2, siGLS#1, siGLS#2, siENO1#1, siENO1#2, siG6PDH#1, siG6PDH#2, and siPKM1) in all eight cell lines, compared to the negative control siRNA group. Results are presented as the mean for three independent experiments; bars indicate SD.
Acknowledgments
This study was partially funded by KAKENHI Grants‐in‐Aid for Scientific Research, (No. 23390329 to M.Y. and No. 26293307 to K.H. and M.Y.).
Cancer Sci 108 (2017) 2462–2469
Funding Information
Japan Society for the Promotion of Science, KAKENHI Grants‐in‐Aid for Scientific Research (23390329, 26293307).
References
- 1. Hou Y, Xu J, Liu X, Xia X, Li N, Bi X. Shikonin induces apoptosis in the human gastric cancer cells HGC‐27 through mitochondria‐mediated pathway. Pharmacogn Mag 2015; 11: 250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fan Y, Jin S, He J et al Effect of beta, beta‐dimethylacrylshikonin on inhibition of human colorectal cancer cell growth in vitro and in vivo . Int J Mol Sci 2012; 13: 9184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kato Y, Yashiro M, Noda S et al Establishment and characterization of a new hypoxia‐resistant cancer cell line, OCUM‐12/Hypo, derived from a scirrhous gastric carcinoma. Br J Cancer 2010; 102: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov 2013; 12: 829–46. [DOI] [PubMed] [Google Scholar]
- 5. Davidson SM, Papagiannakopoulos T, Olenchock BA et al Environment impacts the metabolic dependencies of ras‐driven non‐small cell lung cancer. Cell Metab 2016; 23: 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vander Heiden MG, Lunt SY, Dayton TL et al Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol 2011; 76: 325–34. [DOI] [PubMed] [Google Scholar]
- 7. Zhu H, Luo H, Zhu X, Hu X, Zheng L, Zhu X. Pyruvate kinase M2 (PKM2) expression correlates with prognosis in solid cancers: a meta‐analysis. Oncotarget 2017; 8: 1628–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu VM, Vander Heiden MG. The role of pyruvate kinase M2 in cancer metabolism. Brain Pathol 2015; 25: 781–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sappington DR, Siegel ER, Hiatt G et al Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim Biophys Acta 2016; 1860: 836–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yashiro M, Chung YS, Nishimura S, Inoue T, Sowa M. Peritoneal metastatic model for human scirrhous gastric carcinoma in nude mice. Clin Exp Metastasis 1996; 14: 43–54. [DOI] [PubMed] [Google Scholar]
- 12. Akiyama S, Amo H, Watanabe T et al Characteristics of three human gastric cancer cell lines, NU‐GC‐2, NU‐GC‐3 and NU‐GC‐4. Jpn J Surg 1988; 18: 438–46. [DOI] [PubMed] [Google Scholar]
- 13. Furuya F, Shimura H, Suzuki H et al Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology 2004; 145: 2865–75. [DOI] [PubMed] [Google Scholar]
- 14. Israelsen WJ, Dayton TL, Davidson SM et al PKM2 isoform‐specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013; 155: 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bluemlein K, Gruning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget 2011; 2: 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gara RK, Srivastava VK, Duggal S et al Shikonin selectively induces apoptosis in human prostate cancer cells through the endoplasmic reticulum stress and mitochondrial apoptotic pathway. J Biomed Sci 2015; 22: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang JT, Li ZL, Wu JY, Lu FJ, Chen CH. An oxidative stress mechanism of shikonin in human glioma cells. PLoS ONE 2014; 9: e94180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elgogary A, Xu Q, Poore B et al Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci USA 2016; 113: E5328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Erickson JW, Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget 2010; 1: 734–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 2015; 11: 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones NP, Schulze A. Targeting cancer metabolism–aiming at a tumour's sweet‐spot. Drug Discov Today 2012; 17: 232–41. [DOI] [PubMed] [Google Scholar]
- 22. Shiroki T, Yokoyama M, Tanuma N et al Enhanced expression of the M2 isoform of pyruvate kinase is involved in gastric cancer development by regulating cancer‐specific metabolism. Cancer Sci 2017; 108: 931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao Y, Xu D, Yu G, Liang J. Overexpression of metabolic markers HK1 and PKM2 contributes to lymphatic metastasis and adverse prognosis in Chinese gastric cancer. Int J Clin Exp Pathol 2015; 8: 9264–71. [PMC free article] [PubMed] [Google Scholar]
- 24. Kim JY, Heo SH, Choi SK et al Glutaminase expression is a poor prognostic factor in node‐positive triple‐negative breast cancer patients with a high level of tumor‐infiltrating lymphocytes. Virchows Arch 2017; 470: 381–9. [DOI] [PubMed] [Google Scholar]
- 25. Principe M, Ceruti P, Shih NY et al Targeting of surface alpha‐enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget 2015; 6: 11098–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu H, Ding X, Yang Y et al Changes in glucose‐6‐phosphate dehydrogenase expression results in altered behavior of HBV‐associated liver cancer cells. Am J Physiol Gastrointest Liver Physiol 2014; 307: G611–22. [DOI] [PubMed] [Google Scholar]
- 27. Wang X, Li X, Zhang X et al Glucose‐6‐phosphate dehydrogenase expression is correlated with poor clinical prognosis in esophageal squamous cell carcinoma. Eur J Surg Oncol 2015; 41: 1293–9. [DOI] [PubMed] [Google Scholar]
- 28. Zhang C, Zhang Z, Zhu Y, Qin S. Glucose‐6‐phosphate dehydrogenase: a biomarker and potential therapeutic target for cancer. Anticancer Agents Med Chem 2014; 14: 280–9. [DOI] [PubMed] [Google Scholar]
- 29. Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014; 71: 2577–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taniguchi K, Sakai M, Sugito N et al PKM1 is involved in resistance to anti‐cancer drugs. Biochem Biophys Res Commun 2016; 473: 174–80. [DOI] [PubMed] [Google Scholar]
- 31. Formentini L, Pereira MP, Sanchez‐Cenizo L et al In vivo inhibition of the mitochondrial H + ‐ATP synthase in neurons promotes metabolic preconditioning. EMBO J 2014; 33: 762–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Knockdown of PKM2, ENO1, GLS, G6PDH, and PKM1. Reverse transcription–PCR analysis indicated that PKM2, ENO1, GLS, G6PDH, and PKM1 expression were effectively downregulated by siRNA (siPKM2#1, siPKM2#2, siGLS#1, siGLS#2, siENO1#1, siENO1#2, siG6PDH#1, siG6PDH#2, and siPKM1) in all eight cell lines, compared to the negative control siRNA group. Results are presented as the mean for three independent experiments; bars indicate SD.