

Abstract
Alzheimer's disease (AD) is associated with neurodegenerative changes resulting clinically in progressive cognitive and functional deficits. The only therapies are the cholinesterase inhibitors donepezil, galantamine and rivastigmine and the N‐methyl‐D‐aspartate‐receptor antagonist memantine. Donepezil acts primarily on the cholinergic system as a symptomatic treatment, but it also has potential for disease modification and may reduce the rate of progression of AD. This review explores the potential for disease modifying effects of donepezil. Several neuroprotective mechanisms that are independent of cholinesterase inhibition, are suggested. Donepezil has demonstrated a range of effects, including protecting against amyloid β, ischaemia and glutamate toxicity; slowing of progression of hippocampal atrophy; and up‐regulation of nicotinic acetylcholine receptors. Clinically, early and continuous treatment with donepezil is considered to preserve cognitive function more effectively than delayed treatment. The possible neuroprotective effects of donepezil and the potential for disease pathway modification highlight the importance of early diagnosis and treatment initiation in AD.
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer's disease
- ADAS
Alzheimer's Disease Assessment Scale
- ADAS‐cog
ADAS–cognitive subscale
- BDNF
brain‐derived neurotrophic factor
- CBF
cerebral blood flow
- ChEI
cholinesterase inhibitor
- CVD
cerebrovascular disease
- GSK‐3
glycogen synthase kinase‐3
- MCI
mild cognitive impairment
- MMSE
Mini‐Mental State Examination
- nAChR
nicotinic ACh receptor
- PP2A
protein phosphatase 2A
Introduction
Alzheimer's disease (AD) is associated with neurodegenerative changes associated with depletion of nicotinic ACh receptors (nAChRs) and loss of cholinergic neurons resulting clinically in progressive cognitive and functional deficits (Krishnan et al., 2003; Takada‐Takatori et al., 2009). The only approved therapies for AD are the cholinesterase inhibitors (ChEIs) donepezil, galantamine and rivastigmine and the N‐methyl‐D‐aspartate‐receptor antagonist memantine. Donepezil acts primarily as symptomatic treatment for AD, by enhancing concentrations of synaptic acetylcholine, thus improving interaction between the neurons of the cholinergic system involved in memory. In addition to this transient symptomatic mechanism, ChEIs have the potential for disease modification (or neuroprotection) such that they may also reduce the rate of progression of AD (Meunier et al., 2006; Leyhe et al., 2008; Takada‐Takatori et al., 2009; Wiendl et al., 2015).
Donepezil is a potent reversible ChEI that provides improvement of cognitive function across the spectrum of mild, moderate and severe AD with minimal side effects (Mohs et al., 2001; Birks and Harvey, 2006; Takada‐Takatori et al., 2009), with the greatest benefit being attained with early treatment initiation (Takada‐Takatori et al., 2009). Clinically, donepezil slows the rate of progression of AD and preserves cognition and function (Dubois et al., 2015; Krishnan et al., 2003), and it has been shown to slow progression of hippocampal atrophy in patients with AD compared with untreated patients (Dubois et al., 2015; Krishnan et al., 2003; Hashimoto et al., 2005). Although donepezil has been considered primarily as an agent for symptomatic treatment, previously published trials of donepezil, including clinical or preclinical/in vitro studies, have suggested a possible disease modifying or neuroprotective role from both cholinergic and non‐cholinergic mechanisms (Figure 1) (Zimmermann, 2013; Francis, 2006; Takada‐Takatori et al., 2009; Meunier et al., 2006; Solntseva et al., 2014; Noh et al., 2009; Noh et al., 2013; Arias et al., 2005; Kimura et al., 2005; Takada‐Takatori et al., 2008; Takada et al., 2003; Akaike, 2006; Shen et al., 2010; Yoshiyama et al., 2010; Akasofu et al., 2008).
Figure 1.
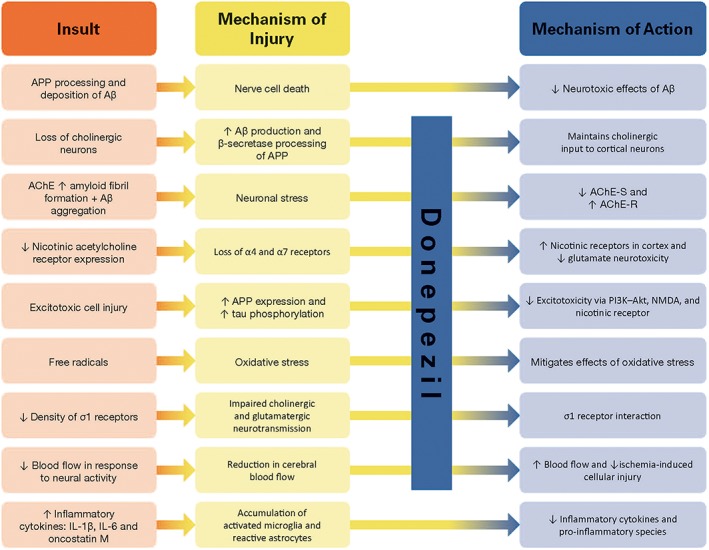
Neuroprotective mechanisms of donepezil. AChE‐R, AChE read‐through form; AChE‐S, AChE synaptic form; APP, amyloid precursor protein.
ChEIs are thought to have pharmacological activity in addition to inhibition of acetylcholinesterase (AChE; Wilkinson et al., 2004). For example, donepezil and galantamine significantly increase nicotinic receptor density, which may be associated with enhanced synaptic strengthening through long‐term potentiation and is related to cognitive function. Galantamine may also potentiate the action of acetylcholine on nicotinic receptors via allosteric modulation, although the effects are concentration‐dependent. Rivastigmine also inhibits butyrylcholinesterase, although it is not clear whether this contributes to its therapeutic effect. In this article, we review the potential for neuroprotective effects of donepezil and the diverse mechanisms of cell death in AD by which these effects might be exerted.
Mechanisms of AD
The pathogenesis of AD progresses in a sequential manner, from preclinical to early and late clinical stages, such that reliable biomarkers including amyloid β (Aβ), neuronal loss (neurofibrillary tangles and degree of hippocampal atrophy) and synaptic loss are seen in a temporally ordered manner (Chan et al., 2003; Vemuri et al., 2009; Jack et al., 2010). There are several mechanisms of cell death in AD, including Aβ accumulation and plaque formation, neurofibrillary tangle formation from hyperphosphorylated tau protein, glutamate excitotoxicity and inflammation (Figure 2) (Jack et al., 2010; Francis, 2006). Biomarkers for AD include CSF Aβ42, increased tau in the CSF, decreased fluorodeoxyglucose uptake on PET, PET amyloid imaging and cerebral atrophy on magnetic resonance imaging (Jack et al., 2010).
Figure 2.
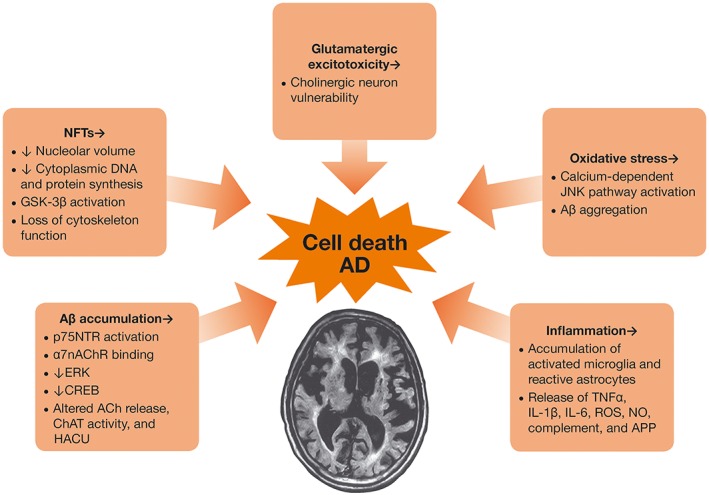
Mechanisms of injury in AD. APP, amyloid precursor protein; ChAT; choline acetylase; CREB, cAMP response element‐binding protein; HACU, high‐affinity choline uptake; p75NTR, p75 neurotrophin receptor.
Aβ has an affinity for the cholinergic system, impairment of which causes loss of learning and memory (Francis, 2006; Meunier et al., 2006). Aβ can be mediated by the p75 neurotrophin receptor, causing neuronal cell death (Perini et al., 2002). Aβ binds to nAChRs, causing desensitization or activation; nAChR activation induces tau phosphorylation, while depletion results in loss of cholinergic neurons (Francis, 2006; Takada‐Takatori et al., 2008; Takada‐Takatori et al., 2009).
Neurofibrillary tangles are caused by tau deposition (Jack et al., 2010). Structural modifications of tau can lead to neuronal dysfunction and cell death in AD. Glycogen synthase kinase‐3 (GSK‐3), a downstream target of the PI3K‐PKB (Akt) pathway, has a role in neuronal cell death (Noh et al., 2009). GSK‐3 activation and decreased protein phosphatase 2A (PP2A) activity also has a role in tau hyperphosphorylation (Noh et al., 2013).
Glutamate excitotoxicity is involved in many neurodegenerative diseases, including AD, in which glutamatergic neurons innervate cholinergic neurons in the basal forebrain (Francis, 2006; Akasofu et al., 2008; Takada‐Takatori et al., 2009). Aβ enhances neuronal vulnerability to glutamate toxicity. Aβ may also be involved in an inflammatory response in AD (Perini et al., 2002). σ1 receptors have been shown to be decreased in patients with AD (Mishina et al., 2008).
Vascular pathology may also contribute to AD, and most patients with AD have cerebral vascular changes (Kandiah et al., 2015; Kuznetsova and Schliebs, 2013; van der Flier, 2012; van Rooden et al., 2014). Microbleeds and microinfarcts correlate with impaired cognition (van der Flier, 2012; van Rooden et al., 2014). There may be a synergistic effect of small vessel cerebrovascular disease (CVD) with amyloid‐tau pathology (Haight et al., 2013; Provenzano et al., 2013; Kandiah et al., 2015). Aβ contributes to vascular injury, and Aβ lesions can be triggered by hypertension and ischaemic brain injury. Cholinergic dysfunction may also result in impaired cerebral blood flow (CBF), and there is an age‐related relationship between Aβ accumulation, cholinergic dysfunction and vascular impairment (Kuznetsova and Schliebs, 2013).
Targets for neuroprotection in AD therefore include Aβ accumulation, glutamate signalling, inflammation, neurofibrillary tangle formation, σ1 receptors and vascular changes (Francis, 2006).
Donepezil and neuroprotection
Preclinical studies in in vivo or in vitro models of AD suggest several neuroprotective mechanisms of donepezil that are independent of cholinesterase inhibition (Takada‐Takatori et al., 2009), including against Aβ toxicity (Arias et al., 2005; Kimura et al., 2005; Meunier et al., 2006; Noh et al., 2009; Noh et al., 2013; Solntseva et al., 2014); glutamate‐induced neurotoxicity (Takada et al., 2003; Akaike, 2006; Takada‐Takatori et al., 2008; Shen et al., 2010); neuroinflammation and tau pathology (Yoshiyama et al., 2010); and vascular pathology (Akasofu et al., 2008). Neuronal AChE splice variants such as AChE read‐through form may also have a role in neuroprotection and thus be a target for donepezil (Zimmermann, 2013). Donepezil has demonstrated a range of activities, including protective effects against Aβ, ischaemia and glutamate toxicity; and slowing of progression of hippocampal atrophy in AD.
Donepezil, galantamine and rivastigmine have shown significant protection against Aβ‐induced apoptosis in a neuroblastoma cell line (Arias et al., 2005). The neuroprotective effects did not appear to be directly related to the primary mechanism of these drugs to inhibit cholinesterase, suggesting that donepezil and galantamine may offer neuroprotection by attenuating Aβ‐induced toxicity via α7nAChRs and the PI3K‐Akt pathway, but not rivastigmine.
Donepezil, galantamine and huperzine A increase neuronal cell viability against Aβ42‐induced toxicity in a concentration‐dependent manner. Donepezil may prevent Aβ‐induced neurotoxicity through the activation of PI3K‐Akt and inhibition of GSK‐3, in addition to up‐regulation of nAChRs (Noh et al., 2009; Noh et al., 2013). Of the three drugs, donepezil provided the most potent inhibition of GSK‐3 activity. A later study by the same group suggested a neuroprotective effect of donepezil via stimulation of PP2A activity (Noh et al., 2013).
In rat septal neurons, donepezil significantly reduced Aβ neurotoxicity, evaluated by LDH efflux, in a concentration‐dependent manner; concentrations of 0.1, 1 and 10 μmol·L−1 attenuated the mean LDH efflux by 9.4, 17.4 and 22.5% respectively (Kimura et al., 2005). These results suggest that donepezil exerts a neuroprotective effect by reducing the amount of Aβ fibrils and that the effect is independent of its cholinergic effects. Both donepezil and tacrine have been found to attenuate Aβ(25–35)‐induced toxicity in rat pheochromocytoma cells (Svensson and Nordberg, 1998). In this study, the neuroprotective effect of tacrine was blocked in the presence of mecamylamine and tubocurarine, suggesting an interaction of tacrine via nicotinic receptors.
In a study of rat cortical neurons exposed to oxygen–glucose deprivation, LDH release was used as a marker of neuronal damage (Akasofu et al., 2003). Cells pretreated with donepezil showed greater morphological preservation after oxygen–glucose deprivation. Donepezil significantly decreased LDH release in a concentration‐dependent manner, while galantamine, tacrine and rivastigmine did not significantly affect LDH release. The neuroprotective effect of donepezil was not antagonized by scopolamine or mecamylamine. The results show that donepezil has a protective effect against oxygen–glucose deprivation‐induced injury and is expected to protect against progressive degeneration of cortical neuronal cells in AD. Both donepezil and huperzine A have protective effects against oxygen–glucose deprivation in rat pheochromocytoma cells shown by reduced markers of cell death (Zhou et al., 2001).
Donepezil may provide protection against glutamate‐induced neurotoxicity by both direct and indirect stimulation of nAChR before the activation points for nitric oxide synthase, nAChR‐PI3K‐Akt and MAP kinase (Takada‐Takatori et al., 2008) in a concentration‐dependent manner (Takada et al., 2003). Exposure of cultured cortical neurons to glutamate markedly reduces the viability of cortical neurons, while cells without drug treatment retain viability. Pretreatment of cultures with donepezil before glutamate exposure significantly retained cell viability, protecting cortical neurons against glutamate neurotoxicity (Takada et al., 2003). Investigation into the mechanism of neuroprotection showed that donepezil activated the PI3K‐Akt pathway and suppressed nitric oxide production and apoptosis (Akaike, 2006). α7nAChR stimulation by donepezil has been shown to decrease glutamate toxicity through down‐regulation of N‐methyl‐D‐aspartate receptors (Shen et al., 2010).
Donepezil administered to a mouse model with a P301S tau mutation was found to ameliorate neuroinflammation, tau pathology, synaptic loss and neuronal loss and decrease tau insolubility and phosphorylation (Yoshiyama et al., 2010). Investigation to confirm the anti‐inflammatory effect of donepezil showed that the drug suppressed IL‐1β and cyclo‐oxygenase‐2 expression in the brain and spleen, suggesting that donepezil directly prevents systemic inflammation.
Donepezil has high affinity for the σ1 receptor. Solntseva et al. (2014) found that the rescue effect of donepezil on hippocampal long‐term potentiation impaired by Aβ was imitated by the σ1‐receptor agonist PRE‐084 and eliminated by the σ1‐receptor antagonist haloperidol. In another study, donepezil showed more potent anti‐amnesic and neuroprotective effects against Aβ‐induced toxicity than PRE‐084, tacrine, rivastigmine and galantamine (Meunier et al., 2006).
Patients with AD have decreased brain‐derived neurotrophic factor (BDNF) in the serum and brain, which may contribute to progressive neurodegeneration. Administration of donepezil results in increased BDNF in the hippocampus and cortex compared with baseline level (Leyhe et al., 2008). Up‐regulation of BDNF might contribute to the neuroprotective effect of donepezil.
Donepezil has been shown to maintain functional brain activity, measured by brain glucose metabolism, within 0.5% of mean baseline levels compared with placebo, which resulted in decline of 10.4% over 24 weeks (Tune et al., 2003). Brain regions in which metabolic activity is enhanced by donepezil compared with placebo are the prefrontal areas, including the middle and superior‐frontal cortices, premotor areas and Broca's area, which are areas that mediate attention, conscious memory and learning.
Early treatment with donepezil is thought to slow disease progression and preserve cognitive function more effectively than delayed treatment, possibly via a neuroprotective mechanism (Winblad et al., 2006). In two studies in patients with mild cognitive impairment (MCI), who are at high risk for progression to AD, donepezil treatment resulted in significant improvement in Alzheimer's Disease Assessment Scale–cognitive subscale (ADAS‐cog) scores (Table 1) (Salloway et al., 2004; Doody et al., 2009). A delayed start study design (Mori et al., 2006) was used to assess the effects of early and late initiation of donepezil in patients with possible or probable AD (Winblad et al., 2006). Patients receiving continuous donepezil therapy for 3 years had less global deterioration (Gottfries‐Bråne‐Steen scale) and better cognitive and functional abilities [Mini‐Mental State Examination (MMSE) and Global Deterioration Scale] than those for whom donepezil initiation was delayed by 1 year (Table 1).
Table 1.
Early treatment with donepezil
| Study | Intervention | Results |
|---|---|---|
| Doody et al. (2009) |
Donepezil 10 mg·day−1
Placebo For 48 weeks |
Between‐treatment differences for modified ADAS‐cog were significant Between‐treatment differences for CDR‐SB were minimal and not significant |
| Salloway et al. (2004) |
Donepezil 10 mg·day−1
Placebo For 24 weeks |
No significant between‐treatment differences for NYU DPR test, except for the fully evaluable population or ADCS CGIC‐MCI scores ADAS‐cog total difference was significant for ≥4 and ≥7 points |
| Winblad et al. (2006) | Donepezil 10 mg for 3 years Placebo for 1 year followed by donepezil 10 mg for 2 years | Trend or significant treatment differences for continuous therapy over delayed start for Gottfries‐Bråne‐Steen scale, MMSE, Global Deterioration Scale and Progressive Deterioration Scale |
| Mohs et al. (2001) | Donepezil 10 mg Placebo For 54 weeks |
More placebo (56%) than donepezil (41%) patients met criteria for clinically evident functional decline Median time to clinically evident functional decline was 357 days for donepezil and 208 days for placebo |
| Wallin et al. (2007) | Donepezil 5–10 mg·day−1 |
Mean decline in MMSE score: 3.8 Mean change in ADAS‐cog (0–70) score: 8.2 CIBIC score at 36 months was improved or unchanged in 30% |
ADCS CGIC‐MCI, Alzheimer Disease Cooperative Study Clinician's Global Impression of Change for MCI; CDR‐SB, Clinical Dementia Rating scale–sum of boxes; CI, confidence interval; CIBIC, Clinician Interview‐Based Impression of Change; NYU DPR, New York University Delayed Paragraph Recall test.
In a study of the effects of donepezil on preservation of function in patients with AD (Mohs et al., 2001), donepezil extended the median time to clinically evident functional decline by 5 months (Table 1) and was associated with a 38% reduction in the risk of functional decline over 1 year compared with placebo. The Swedish Alzheimer Treatment Study, evaluating continuous long‐term donepezil treatment of AD in the clinical setting (Wallin et al., 2007), found that the mean MMSE score increased and remained above baseline for more than 6 months. After 3 years, 38% of patients remained in the study, 30% of whom had unchanged or improved global assessment (measured by Clinician Interview‐Based Impression of Change) (Table 1).
Patients who retain functional ability are better able to maintain activities of daily living (Galasko, 1998). Adequate donepezil dose (≥5 mg·day−1) taken for 9–12 months has been found to delay progression to nursing home placement by up to 2 years compared with untreated patients (Geldmacher et al., 2003). Therefore, use of donepezil may help patients with AD live in the community for longer.
Donepezil, cerebral blood flow and hippocampal atrophy
Donepezil pretreatment has been shown to significantly attenuate cerebral infarction volume after middle cerebral artery occlusion in rats (Fujiki et al., 2005). Thus, donepezil might have a protective effect against progressive neurodegeneration in ischaemic CVD and AD (Akasofu et al., 2008).
A neuroprotective mechanism of donepezil might be preservation of regional CBF, which suggests preservation of functional brain activity (Nakano et al., 2001). Single‐photon emission computed tomography has shown greater preservation of regional CBF in several areas (right and left anterior cingulate gyri, right middle temporal gyrus, right inferior parietal lobules and prefrontal cortex) in patients with mild to moderate AD treated with donepezil for 1 year, than in placebo‐treated patients (Nakano et al., 2001). The anterior cingulate cortex is thought to promote the execution of action, motivation, planning, initiation of action and anticipation (Nakano et al., 2001).
More recently, increased regional CBF in the middle cingulate cortex and posterior cingulate cortex after donepezil treatment has been shown to significantly correlate with behavioural changes measured by ADAS‐cog scores (Table 2) (Li et al., 2012). Intrinsic functional connectivity was significantly enhanced in the medial cholinergic pathway network in the parahippocampal, temporal, parietal and prefrontal cortices. Changes in the medial prefrontal areas correlated with the CBF level in the middle cingulate cortex and posterior cingulate cortex and with the ADAS‐cog scores. Hippocampal functional connectivity has been associated with improvement in ADAS‐cog scores in patients with mild AD treated with donepezil, with greater connectivity recovery correlating with cognitive improvement (Table 2) (Goveas et al., 2011).
Table 2.
Potential neuroprotective effects of donepezil in imaging studies
| Study | Intervention | Results | |
|---|---|---|---|
| Cognitive function | Imaging | ||
| Li et al. (2012) |
Donepezil 5–10 mg·day−1
pCASL and resting‐state functional MRI at baseline and 12 weeks |
ADAS‐cog scores significantly improved |
Regional CBF was significantly increased in MCC and PCC and changes correlated with behavioural changes in ADAS‐cog scores Functional connectivity was enhanced in the medial cholinergic pathway network, and changes in medial prefrontal areas were associated with CBF in MCC and PCC and were correlated with ADAS‐cog scores |
| Goveas et al. (2011) |
Donepezil 5–10 mg·day−1
MRI at baseline and 12 weeks |
ADAS‐cog scores significantly improved | Improvement in ADAS‐cog scores correlated with HFC changes in left dorsolateral prefrontal cortex and middle frontal gyrus |
| Dubois et al. (2015) |
Donepezil 10 mg·day−1
MRI at baseline and at 6–12 months) |
No significant differences between groups in neuropsychological tests | Difference in annualized percentage change in hippocampal atrophy was 45% |
| Ishiwata et al. (2014) |
Donepezil 5–10 mg MRI |
MMSE score was a factor that affects the Z‐score(degree of hippocampal atrophy) |
Decline of cognitive function was correlated with hippocampal volume in donepezil (‐) patients; MMSE score was lower when Z‐score was higher No correlation between cognitive function and hippocampal volume in donepezil (+) patients |
| Hashimoto et al. (2005) |
Donepezil 5 mg·day−1
MRI at baseline and at 1 year |
Mean annual decline in ADAS score:Donepezil+: 0.8 points Donepezil–: 3.6 points | Difference in mean annual rate of hippocampal volume loss was 24% |
HFC, hippocampal functional connectivity; MCC, middle cingulate cortex; pCASL, pseudocontinuous arterial spin labelling; PCC, posterior cingulate cortex.
Measurement of hippocampal volume is important in research into AD, as the level of hippocampal atrophy correlates with the severity of AD and is a reliable marker of disease progression (Jack et al., 1997; Jack et al., 2002). An association between brain atrophy and cognitive decline has been shown by greater hippocampal atrophy rates in patients with AD than in age‐matched controls (Barnes et al., 2009). Increasing rates of hippocampal atrophy have been correlated with progression of AD according to cognitive rating scale scores (MMSE and global and sum‐of‐boxes Clinical Dementia Rating scale) (Dubois et al., 2015; Morra et al., 2009; Jack et al., 2004; Henneman et al., 2009; Vos et al., 2013). Hippocampal atrophy and whole brain atrophy rates have been shown to distinguish between patients with AD, those with MCI and healthy controls (Henneman et al., 2009). Therefore, although hippocampal atrophy is not specific for a diagnosis of AD, it might be a sensitive surrogate marker of pathological stage and progression (Jack et al., 2002; Henneman et al., 2009; Vos et al., 2013).
Donepezil slowed progression of hippocampal atrophy, compared with untreated patients (Dubois et al., 2015; Krishnan et al., 2003; Hashimoto et al., 2005). In patients with suspected prodromal AD, donepezil appeared to significantly slow progression of hippocampal atrophy compared with placebo (Table 2) (Dubois et al., 2015). In a study of patients with probable AD who were or were not receiving donepezil and healthy controls, lower cognitive function was significantly associated with hippocampal atrophy in the group who did not receive donepezil (Table 2) (Ishiwata et al., 2014). The lack of association between cognitive function and hippocampal volume in patients who received donepezil suggests that donepezil affects this relationship, possibly by acting as a disease modifier.
The rate of hippocampal atrophy has been used as a surrogate marker of disease progression in a study of patients with AD receiving donepezil and treatment‐naïve controls (Hashimoto et al., 2005). The mean annual decline in ADAS score and hippocampal volume were significantly greater in the control group than in the treated group (Table 2). The effect of donepezil on hippocampal atrophy remained significant after correcting for age, sex, disease duration, education, magnetic resonance imaging interval, apolipoprotein E genotype and baseline ADAS score. The reduced anatomical progression of AD in the treated group suggests the possibility of a neuroprotective effect of donepezil.
Significantly higher ADAS‐cog scores and greater hippocampal volume have been found in donepezil‐treated patients with AD compared with placebo, as well as higher N‐acetylaspartate concentrations (reflecting reversible neuronal dysfunction) (Krishnan et al., 2003). Cognition improved significantly in the donepezil group and declined in the placebo group, while total hippocampal volume decreased by 0.4% in the donepezil group and by 8.2% in the placebo group. The increase in N‐acetylaspartate concentration also correlated with improvement in cognitive function in the donepezil treatment group, suggesting a neuroprotective effect of donepezil.
Conclusion
Preclinical evidence has demonstrated potential neuroprotective effects of donepezil in several models relevant to AD (Aβ‐induced cell death, oxygen–glucose deprivation, glutamate‐induced cell death). In clinical practice, these neuroprotective effects of donepezil may have a protective effect against progressive degeneration of brain neuronal cells, may allow the retention of functional brain activity and may attenuate hippocampal atrophy, thus slowing conversion of MCI to AD or deterioration in early AD. While this potential for disease modification highlights the importance of early diagnosis of AD and supports the initiation of therapy early in the disease course, further research into early treatment using surrogate markers of progression is warranted.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c,d).
Conflict of interest
S.H.K., J.‐L.H. and C.S. declare no conflicts of interest. N.K. has received honoraria and CME sponsorship from Eisai, Lundbeck and Novartis, and has received research funding from the Biomedical Research Council of Singapore, Media Development Authority of Singapore, National Medical Research Council of Singapore and the SingHealth Foundation. C.U. has received honoraria and meeting support from Eisai (Thailand) Co Ltd. A.D. is an employee of Eisai Singapore Pte. Ltd., Singapore.
Acknowledgements
This manuscript was developed with editorial assistance provided by Mary Smith through an unrestricted grant supported by Eisai Co. Ltd.
Kim, S. H. , Kandiah, N. , Hsu, J.‐L. , Suthisisang, C. , Udommongkol, C. , and Dash, A. (2017) Beyond symptomatic effects: potential of donepezil as a neuroprotective agent and disease modifier in Alzheimer's disease. British Journal of Pharmacology, 174: 4224–4232. doi: 10.1111/bph.14030.
References
- Akaike A (2006). Preclinical evidence of neuroprotection by cholinesterase inhibitors. Alzheimer Dis Assoc Disord 20: S8–11. [DOI] [PubMed] [Google Scholar]
- Akasofu S, Kosasa T, Kimura M, Kubota A (2003). Protective effect of donepezil in a primary culture of rat cortical neurons exposed to oxygen‐glucose deprivation. Eur J Pharmacol 472: 57–63. [DOI] [PubMed] [Google Scholar]
- Akasofu S, Kimura M, Kosasa T, Sawada K, Ogura H (2008). Study of neuroprotection of donepezil, a therapy for Alzheimer's disease. Chem Biol Interact 175: 222–226. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E, Gallego‐Sandín S, Villarroya M, García AG, López MG (2005). Unequal neuroprotection afforded by the acetylcholinesterase inhibitors galantamine, donepezil, and rivastigmine in SH‐SY5Y neuroblastoma cells: role of nicotinic receptors. J Pharmacol Exp Ther 315: 1346–1353. [DOI] [PubMed] [Google Scholar]
- Barnes J, Bartlett JW, van de Pol LA, Loy CT, Scahill RI, Frost C et al (2009). A meta‐analysis of hippocampal atrophy rates in Alzheimer's disease. Neurobiol Aging 30: 1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birks J, Harvey RJ (2006). Donepezil for dementia due to Alzheimer's disease. Cochrane Database Syst Rev 1: CD001190. [DOI] [PubMed] [Google Scholar]
- Chan D, Janssen JC, Whitwell JL, Watt HC, Jenkins R, Frost C et al (2003). Change in rates of cerebral atrophy over time in early‐onset Alzheimer's disease: longitudinal MRI study. Lancet 362: 1121–1122. [DOI] [PubMed] [Google Scholar]
- Doody RS, Ferris SH, Salloway S, Sun Y, Goldman R, Watkins WE et al (2009). Donepezil treatment of patients with MCI: a 48‐week randomized, placebo‐controlled trial. Neurology 72: 1555–1561. [DOI] [PubMed] [Google Scholar]
- Dubois B, Chupin M, Hampel H, Lista S, Cavedo E, Croisile B et al (2015). Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer's disease. Alzheimers Dement 11: 1041–1049. [DOI] [PubMed] [Google Scholar]
- Francis P (2006). Targeting cell death in dementia. Alzheimer Dis Assoc Disord 20: S3–S7. [DOI] [PubMed] [Google Scholar]
- Fujiki M, Kobayashi H, Uchida S, Inoue R, Ishii K (2005). Neuroprotective effect of donepezil, a nicotinic acetylcholine‐receptor activator, on cerebral infarction in rats. Brain Res 1043: 236–241. [DOI] [PubMed] [Google Scholar]
- Galasko D (1998). An integrated approach to the management of Alzheimer's disease: assessing cognition, function and behaviour. Eur J Neurol 5: S9–17. [Google Scholar]
- Geldmacher DS, Provenzano G, McRae T, Mastey V, Ieni JR (2003). Donepezil is associated with delayed nursing home placement in patients with Alzheimer's disease. J Am Geriatr Soc 51: 937–944. [DOI] [PubMed] [Google Scholar]
- Goveas JS, Xie C, Ward BD, Wu Z, Li W, Franczak M et al (2011). Recovery of hippocampal network connectivity correlates with cognitive improvement in mild Alzheimer's disease patients treated with donepezil assessed by resting‐state fMRI. J Magn Reson Imaging 34: 764–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haight TJ, Landau SM, Carmichael O, Schwarz C, DeCarli C, Jagust WJ (2013). Dissociable effects of Alzheimer disease and white matter hyperintensities on brain metabolism. JAMA Neurol 70: 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Kazui H, Matsumoto K, Nakano Y, Yasuda M, Mori E (2005). Does donepezil treatment slow the progression of hippocampal atrophy in patients with Alzheimer's disease? Am J Psychiatry 162: 676–682. [DOI] [PubMed] [Google Scholar]
- Henneman WJ, Sluimer JD, Barnes J, van der Flier WM, Sluimer IC, Fox NC et al (2009). Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology 72: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiwata A, Mizumura S, Mishina M, Yamazaki M, Katayama Y (2014). The potentially protective effect of donepezil in Alzheimer's disease. Dement Geriatr Cogn Disord 38: 170–177. [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Petersen RC, Xu YC, Waring SC, O'Brien PC, Tangalos EG et al (1997). Medial temporal atrophy on MRI in normal aging and very mild Alzheimer's disease. Neurology 49: 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Dickson DW, Parisi JE, Xu YC, Cha RH, O'Brien PC et al (2002). Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology 58: 750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Shiung MM, Gunter JL, O'Brien PC, Weigand SD, Knopman DS et al (2004). Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 62: 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW et al (2010). Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 9: 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandiah N, Chander RJ, Ng A, Wen MC, Cenina AR, Assam PN (2015). Association between white matter hyperintensity and medial temporal atrophy at various stages of Alzheimer's disease. Eur J Neurol 22: 150–155. [DOI] [PubMed] [Google Scholar]
- Kimura M, Akasofu S, Ogura H, Sawada K (2005). Protective effect of donepezil against Abeta(1‐40) neurotoxicity in rat septal neurons. Brain Res 1047: 72–84. [DOI] [PubMed] [Google Scholar]
- Krishnan KR, Charles HC, Doraiswamy PM, Mintzer J, Weisler R, Yu X et al (2003). Randomized, placebo‐controlled trial of the effects of donepezil on neuronal markers and hippocampal volumes in Alzheimer's disease. Am J Psychiatry 160: 2003–2011. [DOI] [PubMed] [Google Scholar]
- Kuznetsova E, Schliebs R (2013). β‐Amyloid, cholinergic transmission, and cerebrovascular system – a developmental study in a mouse model of Alzheimer's disease. Curr Pharm Des 19: 6749–6765. [DOI] [PubMed] [Google Scholar]
- Leyhe T, Stransky E, Eschweiler GW, Buchkremer G, Laske C (2008). Increase of BDNF serum concentration during donepezil treatment of patients with early Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci 258: 124–128. [DOI] [PubMed] [Google Scholar]
- Li W, Antuono PG, Xie C, Chen G, Jones JL, Ward BD et al (2012). Changes in regional cerebral blood flow and functional connectivity in the cholinergic pathway associated with cognitive performance in subjects with mild Alzheimer's disease after 12‐week donepezil treatment. Neuroimage 60: 1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Ieni J, Maurice T (2006). The anti‐amnesic and neuroprotective effects of donepezil against amyloid beta25‐35 peptide‐induced toxicity in mice involve an interaction with the sigma1 receptor. Br J Pharmacol 149: 998–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina M, Ohyama M, Ishii K, Kitamura S, Kimura Y, Oda K et al (2008). Low density of sigma1 receptors in early Alzheimer's disease. Ann Nucl Med 22: 151–156. [DOI] [PubMed] [Google Scholar]
- Mohs RC, Doody RS, Morris JC, Ieni JR, Rogers SL, Perdomo CA et al (2001). A 1‐year, placebo‐controlled preservation of function survival study of donepezil in AD patients. Neurology 57: 481–488. [DOI] [PubMed] [Google Scholar]
- Mori E, Hashimoto M, Krishnan KR, Doraiswamy PM (2006). What constitutes clinical evidence for neuroprotection in Alzheimer disease: support for the cholinesterase inhibitors? Alzheimer Dis Assoc Disord 20: S19–S26. [DOI] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK et al (2009). Automated mapping of hippocampal atrophy in 1‐year repeat MRI data from 490 subjects with Alzheimer's disease, mild cognitive impairment, and elderly controls. Neuroimage 45: S3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Asada T, Matsuda H, Uno M, Takasaki M (2001). Donepezil hydrochloride preserves regional cerebral blood flow in patients with Alzheimer's disease. J Nucl Med 42: 1441–1445. [PubMed] [Google Scholar]
- Noh MY, Koh SH, Kim SM, Maurice T, Ku SK, Kim SH (2013). Neuroprotective effects of donepezil against A_42‐induced neuronal toxicity are mediated through not only enhancing PP2A activity but also regulating GSK‐3β and nAChRs activity. J Neurochem 127: 562–574. [DOI] [PubMed] [Google Scholar]
- Noh MY, Koh SH, Kim Y, Kim HY, Cho GW, Kim SH (2009). Neuroprotective effects of donepezil through inhibition of GSK‐3 activity in amyloid‐beta‐induced neuronal cell death. J Neurochem 108: 1116–1125. [DOI] [PubMed] [Google Scholar]
- Perini G, Della‐Bianca V, Politi V, Della Valle G, Dal‐Pra I, Rossi F et al (2002). Role of p75 neurotrophin receptor in the neurotoxicity by beta‐amyloid peptides and synergistic effect of inflammatory cytokines. J Exp Med 195: 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano FA, Muraskin J, Tosto G, Narkhede A, Wasserman BT, Griffith EY et al (2013). White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA Neurol 70: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, Ferris S, Kluger A, Goldman R, Griesing T, Kumar D et al (2004). Efficacy of donepezil in mild cognitive impairment. Neurology 63: 651–657. [DOI] [PubMed] [Google Scholar]
- Shen H, Kihara T, Hongo H, Wu X, Kem WR, Shimohama S et al (2010). Neuroprotection by donepezil against glutamate excitotoxicity involves stimulation of alpha7 nicotinic receptors and internalization of NMDA receptors. Br J Pharmacol 161: 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solntseva EI, Kapai NA, Popova OV, Rogozin PD, Skrebitsky VG (2014). The involvement of sigma1 receptors in donepezil‐induced rescue of hippocampal LTP impaired by beta‐amyloid peptide. Brain Res Bull 106: 56–61. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson AL, Nordberg A (1998). Tacrine and donepezil attenuate the neurotoxic effect of A beta(25‐35) in rat PC12 cells. Neuroreport 9: 1519–1522. [DOI] [PubMed] [Google Scholar]
- Takada Y, Yonezawa A, Kume T, Katsuki H, Kaneko S, Sugimoto H et al (2003). Nicotinic acetylcholine receptor‐mediated neuroprotection by donepezil against glutamate neurotoxicity in rat cortical neurons. J Pharmacol Exp Ther 306: 772–777. [DOI] [PubMed] [Google Scholar]
- Takada‐Takatori Y, Kume T, Izumi Y, Ohgi Y, Niidome T, Fujii T et al (2009). Roles of nicotinic receptors in acetylcholinesterase inhibitor‐induced neuroprotection and nicotinic receptor up‐regulation. Biol Pharm Bull 32: 318–324. [DOI] [PubMed] [Google Scholar]
- Takada‐Takatori Y, Kume T, Ohgi Y, Izumi Y, Niidome T, Fujii T et al (2008). Mechanism of neuroprotection by donepezil pretreatment in rat cortical neurons chronically treated with donepezil. J Neurosci Res 86: 3575–3583. [DOI] [PubMed] [Google Scholar]
- Tune L, Tiseo PJ, Ieni J, Perdomo C, Pratt RD, Votaw JR et al (2003). Donepezil HCl (E2020) maintains functional brain activity in patients with Alzheimer disease: results of a 24‐week, double‐blind, placebo‐controlled study. Am J Geriatr Psychiatry 11: 169–177. [PubMed] [Google Scholar]
- van der Flier WM (2012). Clinical aspects of microbleeds in Alzheimer's disease. J Neurol Sci 322: 56–58. [DOI] [PubMed] [Google Scholar]
- van Rooden S, Goos JD, van Opstal AM, Versluis MJ, Webb AG, Blauw GJ et al (2014). Increased number of microinfarcts in Alzheimer disease at 7‐T MR imaging. Radiology 270: 205–211. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW et al (2009). MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology 73: 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SJ, van Rossum IA, Verhey F, Knol DL, Soininen H, Wahlund LO et al (2013). Prediction of Alzheimer disease in subjects with amnestic and nonamnestic MCI. Neurology 80: 1124–1132. [DOI] [PubMed] [Google Scholar]
- Wallin AK, Andreasen N, Eriksson S, Båtsman S, Nasman B, Ekdahl A et al (2007). Donepezil in Alzheimer's disease: what to expect after 3 years of treatment in a routine clinical setting. Dement Geriatr Cogn Disord 23: 150–160. [DOI] [PubMed] [Google Scholar]
- Wiendl H, Elger C, Förstl H, Hartung HP, Oertel W, Reichmann H et al (2015). Gaps between aims and achievements in therapeutic modification of neuronal damage (‘neuroprotection’). Neurotherapeutics 12: 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DG, Francis PT, Schwam E, Payne‐Parrish J (2004). Cholinesterase inhibitors used in the treatment of Alzheimer's disease: the relationship between pharmacological effects and clinical efficacy. Drugs Aging 21: 453–478. [DOI] [PubMed] [Google Scholar]
- Winblad B, Wimo A, Engedal K, Soininen H, Verhey F, Waldemar G et al (2006). 3‐year study of donepezil therapy in Alzheimer's disease: effects of early and continuous therapy. Dement Geriatr Cogn Disord 21: 353–363. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Kojima A, Ishikawa C, Arai K (2010). Anti‐inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J Alzheimers Dis 22: 295–306. [DOI] [PubMed] [Google Scholar]
- Zhou J, Fu Y, Tang XC (2001). Huperzine A and donepezil protect rat pheochromocytoma cells against oxygen‐glucose deprivation. Neurosci Lett 306: 53–56. [DOI] [PubMed] [Google Scholar]
- Zimmermann M (2013). Neuronal AChE splice variants and their non‐hydrolytic functions: redefining a target of AChE inhibitors? Br J Pharmacol 170: 953–967. [DOI] [PMC free article] [PubMed] [Google Scholar]