

Abstract
The eye is a highly specialized organ that is subject to a huge range of pathology. Both local and systemic disease may affect different anatomical regions of the eye. The least invasive routes for ocular drug administration are topical (e.g. eye drops) and systemic (e.g. tablets) formulations. Barriers that subserve as protection against pathogen entry also restrict drug permeation. Topically administered drugs often display limited bioavailability due to many physical and biochemical barriers including the pre‐corneal tear film, the structure and biophysiological properties of the cornea, the limited volume that can be accommodated by the cul‐de‐sac, the lacrimal drainage system and reflex tearing. The tissue layers of the cornea and conjunctiva are further key factors that act to restrict drug delivery. Using carriers that enhance viscosity or bind to the ocular surface increases bioavailability. Matching the pH and polarity of drug molecules to the tissue layers allows greater penetration. Drug delivery to the posterior segment is a greater challenge and, currently, the standard route is via intravitreal injection, notwithstanding the risks of endophthalmitis and retinal detachment with frequent injections. Intraocular implants that allow sustained drug release are at different stages of development. Novel exciting therapeutic approaches include methods for promoting transscleral delivery, sustained release devices, nanotechnology and gene therapy.
Abbreviations
- AAV
adeno‐associated‐virus
- ADAs
anti‐drug antibodies
- AMD
age‐related macular degeneration
- BAC
benzalkonium chloride
- BRB
blood–retinal barrier
- CMV
cytomegalovirus
- CNTF
ciliary neurotrophic factor
- CNV
choroidal neovascularization
- DR
diabetic retinopathy
- ECM
extracellular matrix
- EVA
ethylene vinyl acetate
- GFS
glaucoma filtration surgery
- HA
hyaluronic acid
- IOP
intraocular pressure
- i.v.t.
intravitreal
- MEMS
microelectromechanical system
- PAMAM
poly amidoamine
- pNIPAAM
poly(N‐isopropylacrylamide)
- PVA
polyvinyl alcohol
- RP
retinitis pigmentosa
- RPE
retinal pigment epithelium
- RVO
retinal vein occlusion
- TA
triamcinolone acetonide
Introduction
Vision is often considered to be the most important of senses and the one that most people fear losing. Vision is often thought to be the key enabling sense for a person to work and function independently. Considerable efforts to combat vision loss continue to be made as blinding ocular diseases are more prevalent with an increasingly ageing population. All measurements for quality of life, such as disability‐adjusted life‐years, confirm that visual impairment is a highly ranked burden in all countries (Chiang et al., 2006). The leading causes of visual impairment and irreversible blindness are posterior segment‐related diseases (Pascolini and Mariotti, 2012), which include glaucoma, age‐related macular degeneration (AMD), macular oedema secondary to retinal vein occlusion (RVO), cytomegalovirus (CMV) retinitis, posterior uveitis, diabetic retinopathy (DR) and retinitis pigmentosa (RP) (Thrimawithana et al., 2011; del Amo et al., 2015; Waite et al., 2017).
Glaucoma is a term used to describe a group of conditions with an optic neuropathy, characteristic morphological changes in the optic nerve head and visual field defects. It remains a leading cause of irreversible blindness throughout the world and one of the most common neuropathies. It is estimated that 60.5 million people suffered from it in 2010 and 7 million are believed to be bilaterally blind (Quigley and Broman, 2006). The number of people with glaucoma is projected to increase to 111.8 million in 2040 (Tham et al., 2014). Over the last century, in fact, the proportion of blindness attributed to glaucoma in developed countries has changed very little (10%) (Taylor, 2009). In developed countries, the main cause of blindness 100 years ago was corneal disease; fifty years ago, it was cataracts and in the present day, it is AMD. No reduction in the proportion of blindness due to glaucoma is not simply because there have been no advances in treatment. A major factor is the ageing population. As a population becomes healthier and people live longer, the incidence of this degenerative optic neuropathy that reaches a clinically detectable threshold will increase. The main treatment involves trying to slow the optic nerve degeneration. The biggest risk factor for accelerated progression is an increased level of intraocular pressure (IOP) and reducing this is the only proven therapy in everyday use.
AMD affects approximately 10 million people worldwide and is one of the leading causes of vision loss in elderly patients over 60 years of age. The prevalence of AMD accounts for 20% of all posterior segment disorders in the 65–74 years age group and 35% in older age groups (Edelhauser et al., 2010) and is predicted to affect 288 million people by 2040 (del Amo et al., 2017). Cataract, DR, glaucoma and AMD will increase to nearly 70 million adults by 2050 (Joseph and Venkatraman, 2017). Loss of vision will increase with the increase in the ageing population, which in turn will pose a challenge for human health and economic growth (Delplace et al., 2015).
Inflammatory, angiogenic and fibrotic processes cause tissue damage and vision loss. These processes are also major contributors to the failure of current treatments for these diseases. Increased knowledge of the molecular mechanisms of blinding diseases (Rodrigues et al., 2009; de Oliveira Dias et al., 2011) has driven the development of therapeutic proteins including antibody‐based medicines (Penedones et al., 2014). Vascular endothelial growth factor (VEGF)‐neutralizing antibodies have considerably improved the treatment of exudative macular degeneration (wet AMD) and DR. A major challenge during both preclinical and clinical development is to determine the ocular clearance rates of new medicines. Even though the anti‐VEGF antibody‐based medicines can be administered once a month and in some cases every other month (Stewart et al., 2012), much research is focused on developing medicines that will require less frequent dosing regimens.
Another factor in the development of longer acting formulations is the cost of the drugs and overall treatment costs, which is a function of dosing frequency. The expenditure to treat blinding ophthalmic diseases is considerable, with $51.4 billion spent in the USA alone each year (2007). For example, ranibizumab (licensed) and bevacizumab (unlicensed) are widely used to treat AMD. Each injection of ranibizumab (Lucentis®) can cost $2000, whereas bevacizumab (Avastin®) can cost only $50 for each intravitreal (i.v.t.) dose (Raftery et al., 2007). Two multi‐centre randomized controlled clinical trials (IVAN and CATT) compared the use of ranibizumab and bevacizumab (Chakravarthy et al., 2012; Martin et al., 2012; Ahfat and Zaidi, 2013). No difference was found in visual acuity outcome during 1 and 2 year treatment periods respectively (Chakravarthy et al., 2012; Martin et al., 2012). Bevacizumab is normally provided as a solution in a glass vial containing 400 mg of the antibody at a concentration of 25 mg·mL−1. For ocular use, bevacizumab is often transferred under aseptic conditions into ready‐to‐use syringes for i.v.t. injection by compounding pharmacies for local distribution. Many health systems cannot cope with the increasing demands for i.v.t. injection, so bevacizumab is widely used (Elshout et al., 2014; Kwong and Mohamed, 2014).
The eye – a complex organ
The anatomy of the eye and complex physiology of the retina mean that the development of efficacious medicines is challenging. The eye is a specialized organ that enables light to be focused and processed into nerve impulses for interpretation by our brain. A huge range of pathology affects the eye, with either local triggers or manifestations of systemic disease such as diabetes mellitus. Furthermore, different parts of the eye can be affected to different degrees, with implications for drug delivery. The eye is broadly divided into two compartments called the anterior (front of the eye) and posterior (back of the eye) segment. Contact lens‐related infections occur in the cornea of the anterior segment, whereas macular degeneration is in the central retina of the posterior segment. Endophthalmitis (infection within the eye) can cause blindness within a matter of hours. Optic nerve degeneration in open angle glaucoma may take many years before patients are aware that vision is compromised.
The least invasive routes for ocular drug administration are topical (e.g. eye drops) and systemic (e.g. tablets) formulations. The superficial nature of the eye enables local administration (usually eye drops), the main modality of treatment for anterior segment disease. One advantage of this is to bypass the liver, thus avoiding the need for higher systemic doses required with the oral route. The efficacy of drugs may also be assessed at a local level. Parameters can be measured directly, for example, the IOP in glaucoma, or through visual interpretation for signs of oedema in exudative macular degeneration.
Local applications do pose some disadvantages. Most treatments are self‐administered and rely on a compliant patient, but many patients take their medication sporadically. This can be demonstrated by attaching detectors to bottles during glaucoma treatment used by patients (Kass et al., 1986). The reported rate of application from patients differed significantly to the measured rate (Kass et al., 1986). Consequently, even if drug delivery is optimized, efficacy may not be achieved. Topically administered drugs often display limited bioavailability (< 5%) due to many physical and biochemical barriers including the pre‐corneal tear film, the structure and biophysiological properties of the cornea, the limited volume that can be accommodated by the cul‐de‐sac, the lacrimal drainage system, reflex tearing and the aqueous outflow within the eye (Peyman and Ganiban, 1995; Ranta and Urtti, 2006; Rodríguez Villanueva et al., 2016). Newer methods of drug delivery include sustained release preparations that allow reduced frequency of drug administration (Rodríguez Villanueva et al., 2016; Mehta et al., 2017). Preservative‐free eye drops can be achieved with a new multidose bottle with an Airless Antibacterial Dispensing System (Pfizer Inc, USA) containing a valve system and an airless pump with a silver antibacterial coil (Ghate and Edelhauser, 2008).
The patient population is also often elderly and susceptible to side effects. Despite lower doses needed than oral drugs, systemic toxicity can still be an issue. An elderly patient tends to have a lower body mass index, as well as decreased renal function. Furthermore, these patients are more likely to be on multiple medications that may interact. Even small quantities of drug absorbed systemically may have side effects, such as an increase in falls with topical β‐blocker use (Glynn et al., 1991). Another example is the use of atropine. A 50 μL drop of 1% atropine contains 0.5 mg of drug. Treating both eyes with one dose (1.0 mg) can cause tachycardia (Morton and Thomas, 1958).
Significant ocular barriers that exist to prevent pathogen access also hinder drug delivery (Novack and Robin, 2016; Yellepeddi and Palakurthi, 2016). Transparency is necessary for optical function and, therefore, most of the inside of the eye lacks blood vessels. This is at the expense, however, of the host defence, which is weaker compared to rest of body, and if the barriers are breached, as in endophthalmitis, there are devastating effects. However, these barriers are also key restrictors to delivering a drug to the area of need. Blinking and tear film turnover, designed to wash away foreign material and maintain a smooth clear anterior surface, also limits the residence time of a drug. Access to the deeper structures of the eye is hindered by a closely packed corneal epithelium and stroma with varying lipophilicity.
Delivery of a drug to the posterior segment has its own particular challenges. The vascular conjunctiva, sclera and choroid affect delivery, notwithstanding the tight junctions of the blood aqueous barrier of the non‐pigmented ciliary body epithelium, and the endothelium of blood vessels that constitute the blood–retinal barrier (BRB). The BRB controls fluid and molecular movement between the ocular vascular beds and prevents leakage of macromolecules and harmful molecules into the retina (Kaur et al., 2008). Due to the outer and inner BRB, the influx of a drug into the retina and vitreous regions is limited, requiring the systemic administration of high doses to achieve therapeutic concentrations within the eye. As a result, a drug is distributed and accumulated in all tissues in the body, causing unwanted side effects (Myles et al., 2005). Therefore, i.v.t. injections are required to administer drugs directly to the posterior segment to achieve reproducible and high doses. Although i.v.t. injections are required to treat blinding conditions at the back of the eye, repeated administration poses potential complications including endophthalmitis, retinal detachment, traumatic cataract, intraocular haemorrhage and ocular infections (Jager et al., 2004). Particulate contaminants that may be present in a formulation also increase the potential for harmful effects to the posterior segment (Jager et al., 2004). In addition, only a little is known about vitreous dynamics, partly due to the changes in consistency and anatomy with age (Bishop et al., 2004; Laude et al., 2010; Awwad et al., 2015).
Understanding the nature of ocular barriers and developing methods of bypassing or utilizing them together with knowledge of the fluid dynamics within the eye are keys to establishing successful ophthalmic pharmacotherapy. Experimental models act as platforms for drug development (Awwad et al., 2015; Awwad et al., 2017) and translation to the clinic. This review aims to examine these barriers, ways of negotiating them, and methods of pharmacotherapy employed for different types of drugs. As the pathology and pharmacology differs anatomically, we aim to discuss separately the anterior and posterior segment and look at some of the future strategies for drug development on the horizon.
Anterior segment
Barriers
The anterior segment of the eye consists of the cornea, conjunctiva, iris, ciliary body and the lens. The common chronic anterior segment diseases are cataract, glaucoma and uveitis, with cataract accounting for 51% blindness (according on a 2010 survey; Pascolini and Mariotti, 2012; Joseph and Venkatraman, 2017). The vast majority of pharmacotherapy is through topical application by the patients themselves, although other routes such as subconjunctival injection are also used. Tear film turnover is the main factor that limits topical drug residence time. Human tear volume is approximately 7 μL (Gaudana et al., 2010). The volume of an eye drop of drug is up to 50 μL, and although the cul‐de‐sac may briefly expand to 30 μL (Nagataki and Mishima, 1980; Gaudana et al., 2010), most drug is cleared by nasolacrimal drainage, and then to the systemic circulation directly after instillation. Increasing the drop size will therefore not deliver a greater amount of drug to the eye but, instead, increases the chances of systemic side effects. The optimal size of drop to prevent systemic overflow is zero, which is obviously not achievable. Droppers have been created that consistently produce smaller volumes (Van Santvliet and Ludwig, 2004; Kumar et al., 2011). Simpler strategies include nasolacrimal occlusion or closing the eyelid.
Lacrimation caused by irritant drugs, certain excipients and pH deviation increase tear turnover and drug loss. Increase in tonicity can also have significant implications for increase in tearing. The normal tear turnover in humans is 16%·min−1 (Mishima et al., 1966). Increased lacrimation due to drop instillation results in turnover being increased up to 80%·min−1. Therefore, the half‐life of a drug is only 4 min even with a normal turnover. Blinking also increases drainage as eyelid muscle contraction encourages tear flow down the nasolacrimal duct. A significant increase in the drug residence time can be achieved simply by asking patients to close their eyes after drop instillation and apply pressure nasolacrimal duct (Flach, 2008).
The tear film itself traditionally used to be thought of as a simple tri‐layer structure, consisting of an oily surface layer that reduced evaporative loss, an aqueous middle layer that contained enzymes and other anti‐bacterials (such as lactoferrin) and a mucin layer that provides lubrication and protection of the corneal surface. More recently, that view has altered with the discovery of numerous glycosaminoglycans (GAGs), mucopolysaccharides and other molecules responsible for tear film integrity (Tiffany, 2008). Furthermore, the tear film is dynamic as fluids are ‘reshuffled’ with blinking (Cher, 2012). An unstable tear film is a result of dry eye syndrome and chronic eye irritation with changes in the level of lipid, protein and mucin profiles (Yellepeddi and Palakurthi, 2016).
Understanding the tear film constituents and dynamics has led to the development of methods that increase drug residence time and bioavailability. Inserts provide sustained release of drug. Emulsions enable an increased drug load that may not be achievable in solution. Vehicles or ointments increase viscosity. Carriers of drug exist that bind to the tear film or ocular surface with greater adhesion than drug alone.
Bacterial ulceration of the cornea is a significant risk factor in people who regularly wear contact lenses. Poor hygiene combined and overnight use increases the risk of a bacterial infection (Stapleton et al., 2008). Aggressive dosing is required in the initial stages of therapy, with hourly or even half hourly drops to prevent ocular spread and loss of the eye. It is not uncommon for patients to be admitted to hospital for both day and night treatment (Teo et al., 2011). Ocular inserts (Table 1) are solid implants that sit in the cul‐de‐sac of the eyelids and slowly dissolve, releasing drug over a long period of time. The concept is constantly being explored and reported in literature (Saettone et al., 1994; Kumari et al., 2010; Aburahma and Mahmoud, 2011; Franca et al., 2014; Everaert et al., 2017; Rathod et al., 2017; Sharma et al., 2017). Pilocarpine, an IOP lowering treatment, was one of the first drugs to be formulated as an insert (Ocusert) (Langer, 1983). Ocusert is a cul‐de‐sac sustained release insert composed of two membranes of ethylene vinyl acetate (EVA) copolymer surrounding a reservoir of pilocarpine allowing drug diffusion (40 μg·h−1) for over a period of a week for the treatment of glaucoma (del Amo and Urtti, 2008; Peyman and Hosseini, 2011; Barar et al., 2016). An ocular insert that combined ciprofloxacin with sodium carboxymethylcellulose and polyvinyl alcohol (PVA) has shown significant promise. In vitro studies have shown that this insert can maintain levels within a therapeutic range for up to 48 h (D. Jain et al., 2010). Good tissue compatibility has also been demonstrated in an in vivo rabbit model. Ocular inserts have also been clinically tested for patients with dry eye symptoms, with release of hydroxypropyl cellulose alleviating foreign body sensation significantly with blurring of vision being the most common side effect (in 8.7% patients) (Koffler et al., 2010).
Table 1.
FDA‐approved drugs and those in current clinical trials for treatment of anterior segment diseases
| Name and company | Drug delivery platform | Drug and excipients | Indications | Status |
|---|---|---|---|---|
| Acuvail™ (Allergan) | Eye drops | Solution of ketorolac tromethamine (0.45%) in carboxymethylcellulose (pH 6.8) | Reduction of pain and inflammation post cataract surgery | Approved |
| Azasite® (InSite Vision) | Eye drops | Solution of azithromycin (1.0%) in polycarbophil (Durasite®) | Bacterial conjunctivitis | Approved |
| Azasite Plus™ (InSite Vision) | Eye drops | Solution of azithromycin (1.0%)/dexamethasone (0.1%) (ISV‐502) in Durasite® | Blepharoconjunctivitis | Phase III completed |
| Bromsite™ (InSite Vision) | Eye drops | Solution of bromfenac sodium (0.075%) | Reduction of pain and inflammation post cataract surgery | Approved |
| Betoptic‐S™ (Alcon) | Eye drops | Suspension of betaxolol (0.25%) in polystyrene‐divinylbenzene) sulfonic acid, carbomer and BAC (0.01%) | Open angle glaucoma | Approved |
| Cationorm® (Santen Pharma) | Eye drops | Cationic emulsion | Dry eye | Approved |
| DexaSite™ (InSite Vision) | Eye drops | Solution of dexamethasone (0.1%) (ISV‐305) in Durasite® | Reduction of pain and inflammation post cataract surgery; and non‐bacterial blepharitis | Phase III |
| Durezol™ (Alcon) | Eye drops | Emulsion of difluprednate (0.05%) in castor oil, glycerine, boric acid, polysorbate 80 and sorbic acid (0.1%) | Anterior uveitis | Approved |
| EGP‐437 (Eyegate Pharma) | Iontophoresis | Solution of dexamethasone phosphate in EyeGate® II Drug Delivery System (EGDS) | Anterior uveitis | Phase III |
| Indocollirio® (Bausch & Lomb) | Eye drops | Suspension of indomethacin (0.1%) and hydroxypropyl‐b‐cyclodextrin (IND‐CD) | Mydriasis during cataract surgery or conjunctivitis | Approved |
| Lacrisert® (Aton Pharma) | Ocular insert | Sterile, rod‐shaped HPMC insert | Dry eye | Approved |
| Lumigan® (Allergan) | Eye drops | Solution of bimatoprost (0.03%) in BAC (0.005%) | Glaucoma | Approved |
| Mydriasert (Thea Pharma) | Ocular insert | Tropricamide (0.28 mg) and phenylephrine hydrochloride (5.4 mg) in ammoniomethacrylate copolymer, glycerol and ethylcellulose | Induced pre‐operative mydriasis | Approved |
| Ocusert® (Alza) | Ocular insert | Pilocarpine with EVA and alginic acid | Glaucoma | Approved |
| Prolensa™ (Bausch & Lomb) | Eye drops | Solution of bromfenac (0.07%) | Postoperative inflammation | Approved |
| Propine™ (Allergan) | Eye drops | Solution of dipivefrin‐HCl (0.1%) with BAC (0.02%) | IOP control in open angle glaucoma | Approved |
| Restasis™ (Allergan) | Eye drops | Cationic emulsion of cyclosporine (0.05%) in castor oil, glycerine, polysorbate 80 and carbomer | Dry eye | Approved |
| TobraDex® (Alcon) | Eye drops | Suspension of tobramycin/dexamethasone in xanthan gum | Blepharitis | Approved |
| Timoptic‐XE™ (Aton Pharma) | Eye drops | In situ gel solution of timolol maleate (0.25 or 0.5%) in gellan gum, mannitol, tromethamine and BAC (0.012%) | Glaucoma | Approved |
| Travatan™ (Alcon) | Eye drops | Solution of travaprost (0.004%) in BAC (0.015%) | Glaucoma | Approved |
| Xalatan™ (Pfizer) | Eye drops | Solution of latanoprost (0.005%) in BAC (0.015%) | Glaucoma | Approved |
| Zirgan™ (Bausch & Lomb) | Ophthalmic gel | Ophthalmic gel with ganciclovir (0.15%) with carbopol and BAC (0.0075%) | Acute herpetic keratitis | Approved |
All drug therapies mentioned above were also cross‐checked with www.clinicaltrials.gov.uk and its official company site. BAC, benzalkonim chloride; HPMC, hydroxypropylmethylcellulose.
Suspensions are commonly used to enable application of poorly water‐soluble drugs such as the steroid prednisolone acetate. The suspension requires shaking to disperse drug particles in the bottle. However, drug distribution is still not uniform. Oil‐in‐water (o/w) emulsions are prepared to dissolve the drug in an oil phase and disperse it in the aqueous phase with a surfactant, which has been found to improve consistency (Koffler et al., 2010). Microemulsions, which are emulsions that are thermodynamically stable and form spontaneously, have also shown promise. A low concentration of surfactant maintains the drug suspension stability and may enhance penetrance by interacting with the corneal epithelial cells (Fialho and da Silva‐Cunha, 2004). A recent study found that a microemulsion of dexamethasone had an improved anti‐inflammatory effect in a rabbit uveitic model compared to a marketed formulation of dexamethasone (Kesavan et al., 2013).
An alternative to formulating an emulsion for poorly water‐soluble drugs is to combine it with a cyclodextrin (Kurkov and Loftsson, 2013). Cyclodextrins are oligosaccharides consisting of a hydrophilic outer surface and a hydrophobic core (Loftsson and Stefansson, 1997). They are able to form inclusion complexes with ‘guest’ drug molecules, which sit within the core and the hydrophilic outside aids aqueous solubility. A dynamic equilibrium exists between the cyclodextrin drug complex and disassociated drug, enabling drug release (Kek et al., 2010). Cyclodextrins have been used preclinically to formulate eye drops (Loftsson and Brewster, 2011) with various drugs (Siefert and Keipert, 1997; Bary et al., 2000; Cappello et al., 2001; Okamoto et al., 2010; Bozkir et al., 2012; Loftsson et al., 2012; Jóhannsdóttir et al., 2015; Rodriguez‐Aller et al., 2015). Several cyclodextrin‐based eye drops have been evaluated clinically, for example, latanoprost and dexamethasone (Gonzalez et al., 2007; Tanito et al., 2011; Krag and Hessellund, 2014; Shulman et al., 2015), while others have been registered in Europe for clinical use, for example, chromaphenicol (Clorocil®), diclofenac (Voltaren Ophthalmic®) and indomethacin (Indocid®) (Davis and Brewster, 2004). The percentage of cyclodextrin used for drug solubilization was between 10 and 30% w.v‐1 for most of these formulations (Kearse et al., 2001).
Increasing the viscosity with a carrier increases residence time and, in theory, the drug bioavailability (Bourlais, 1995). Ointments are often made from mineral oil, which can pose a challenge for the formulation of water‐based treatments. The oily base may impair drug dissolution, reducing therapeutic efficacy (McCarthy, 1975). The oil itself also interferes with tear film clarity, leading patients to complain of blurry vision or eyelashes being stuck together. Ointments still have an important therapeutic role in the setting of corneal exposure or thermal burns, where production of tear film constituents has been disrupted or where the ocular surface is damaged (Fish and Davidson, 2010).
Another method of increasing the viscosity with less tear film disruption is to use a mucoadhesive component (polymer or excipient) such as polycarbophil (Agrahari et al., 2016) or carboxymethylcellulose (Reddy and Kim, 2011). These substances are long chain polymers that bind to the mucous layer constituents on the ocular surface. Amongst the salts, lipids and proteins are numerous glycoproteins, such as mucin, that have charged carbohydrate groups and end in sialic acid groups. The polymer chains of mucoadhesives, which consist of both polar and non‐polar groups, swell on contact with water, become entangled and adhere to the mucin molecules (Khutoryanskiy, 2010). The polymer length and the pH affect drug residence time and bioavailability. A longer chain polymer will have more viscosity, entanglement and greater adhesiveness to a certain extent. It will also restrict the ability to swell and result in less adhesion. Furthermore, there may be an increase in irritation and lacrimation. The pH is also important in determining how strongly a polymer binds as it governs whether hydrogen bond formation is able to take place (Grabovac et al., 2005). For many polymers, a more acidic pH results in greater hydrogen bonding between polymer and the mucin on the surface.
Hyaluronic acid (HA) has the additional property of shear thinning unlike other mucoadhesive polymers. With movement, the viscosity of HA decreases. Therefore, with blinking, the polymer rearranges, but with an eye that is open there is greater stability (Rah, 2011). HA is commonly used for maintaining anterior chamber volume during intraocular surgery and for the treatment of dry eye. Its potential for enhancing ocular delivery is still being evaluated, although it has shown promise with drugs such as pilocarpine (Pahuja et al., 2012). HA stabilizes and organizes the extracellular matrix (ECM), regulates cell adhesion and motility and mediates cell proliferation and differentiation (Shu et al., 2004; Guter and Breunig, 2017). The commercially available form, sodium hyaluronate, is widely used in biomedical applications such as a scaffold material for wound healing and tissue engineering due to its biocompatible and biodegradable properties (Ha et al., 2006; Baino, 2011).
Routes of drug absorption
From the tear film, there are two main routes of drug absorption to the anterior segment:
The corneal route. The drug passes through the structural layers of the cornea to reach the anterior chamber, including the corneal epithelium, stroma and endothelium.
The non‐corneal route. Drugs pass through the conjunctiva, Tenon's tissue and sclera to reach the anterior chamber.
Corneal route
The cornea consists of three major layers with variable permeability to drug molecules. The epithelium and endothelium both consist of cells linked by tight junctions, which restrict the passage of large molecules, while the stroma is made up of closely packed collagen. It is the epithelium, however, that provides the most resistance to solute diffusion. Its influence on restricting large molecules may be demonstrated by comparing the absorption of [14C]‐dexamethasone with the epithelium intact to when it is debrided in an animal model (Cox et al., 1972a,b). Only after epithelial debridement is [14C]‐dexamethasone detected in the anterior chamber of rabbits after topical administration. Intercellular spaces with pore sizes of 60 A allow the passage of small ionic and hydrophilic molecules (<350 Da) via the paracellular route (Lee, 1990). The transcellular route is the main mechanism of absorption of ocular drugs and other larger lipophilic molecules.
The lipophilicity of the cornea varies with the different layers. The epithelium and endothelium are relatively hydrophobic, whereas the corneal stroma is hydrophilic. Consequently, there is a parabolic relationship between the corneal permeability and the diffusion coefficient (Yoshida and Topliss, 1996). With the stroma playing a lesser role in resistance, corneal penetrance increases with hydrophobicity with a decrease in penetrance in only the most hydrophobic of compounds. In conjunction with lipophilicity, pH is another key factor in determining the permeability of the cornea to a drug (Pahuja et al., 2012) There is a delicate balance therefore between aiming for a pH appropriate for corneal penetration and the physiological pH (7.4), which will result in the least tearing.
Certain drugs harness the differing lipophilicity of the cornea to their advantage for drug delivery. Latanoprost is one of the most common ocular drugs used for the treatment of glaucoma, a disease in which there is accelerated optic nerve degeneration in association with a raised IOP (Digiuni et al., 2012; Pek et al., 2016). Latanoprost is a prostaglandin analogue that reduces IOP by increasing the uveoscleral outflow, through targeting receptors near the drainage angle. Although not fully understood, it is believed that it may cause ciliary muscle relaxation or remodelling of the tissue matrix within the ciliary muscle.
Latanoprost is formulated as a lipophilic pro‐drug ester, which is absorbed easily into the corneal epithelium. It is activated by esterases within the epithelium to the active hydrophilic moiety and passes through into the anterior chamber. The cornea therefore acts as a depot for the pro‐drug with prolonged release after a single application. The elimination half‐life in rabbit corneas has been found to be over an hour (Sjöquist et al., 1998). In humans, IOP lowering begins between 2 and 4 h after application, with the peak effect reached between 8 and 12 h (Digiuni et al., 2012). This effectively means a once a day treatment, which helps with patient compliance.
The non‐corneal route
Less is known about the permeability of the conjunctiva, Tenon's and sclera and the associated pharmacokinetics. Most studies that have examined these different tissues report that the conjunctiva is more permeable to hydrophilic molecules than the cornea, though it is not clear that it is less permeable to hydrophobic molecules. Using a hollow glass cylinder fixed to the corneal surface to isolate cornea versus conjunctiva tissue, Ahmed and Patton (1985) found that the hydrophilic molecule inulin penetrated conjunctiva and sclera more easily than the cornea.
Other conjunctival properties that increase drug absorption include a greater surface area (17 times that of the cornea in humans; Watsky et al., 1988) and a larger pore size with greater permeability than the cornea (Lawrence and Miller, 2004). However, factors that reduce absorption include mucous produced by goblet cells and the presence of lymphatics and a vasculature that enhance systemic loss of drug.
Changing the epithelium permeability directly may increase drug penetration. Surfactants are ionized substances, which can disrupt the plasma membrane of the epithelium. One surfactant in common use is the preservative benzalkonium chloride (BAC). Its ability to kill bacteria and prolong the shelf life of medicines makes it the preservative of choice in many preparations. BAC can cause membrane disruption and emulsification of the lipid layer of the tear film, where the delivery of drug through the corneal layers may be significantly enhanced. This can be seen in studies with acyclovir ointment, a common treatment for herpes simplex keratitis. Acyclovir penetrance in ex vivo corneas of rabbits was increased by three times with a 0.005% BAC solution and by 10 times with a 0.01% BAC solution (Majumdar et al., 2008). A standard formulation is between 0.004 and 0.02%. However, the effects of BAC are toxic and even at low concentrations the presence of BAC can cause cell death. A concentration of 0.0001% is sufficient to cause apoptosis. Higher concentrations cause necrosis. It is no surprise that patients complain of ocular surface irritation when using treatments that contain BAC. Ensuring ocular medications are pathogen‐free is a mandatory part of formulating an ophthalmic preparation, and much research has been performed to find other preservatives that have effective bactericidal properties but without the toxicity seen with BAC.
Iontophoresis is the application of electrical current to drive ionized substances into tissue (del Amo and Urtti, 2008). It is a method of enhancing ocular drug delivery that has been in and out of vogue over many years since Wirtz reported its use for the treatment of infective corneal ulcers with zinc salts in 1908 (Wirtz, 1908). The apparatus usually consists of a reservoir of drug connected to an electrode with the other electrode attached to the patient. It has been used with some success to enhance delivery of antibiotics such as gentamicin, steroids and antiviral treatments both through the cornea and sclera. Side effects such as tissue necrosis and patient discomfort have precluded its widespread use; however, in some circumstances, it may have a useful role (Güngör et al., 2010). Recent phase I/II trials have shown promise in the treatment of dry eye and uveitis (Patane et al., 2011; Cohen et al., 2012; Kang‐Mieler et al., 2014),
Periocular injections
Drugs that are injected underneath the conjunctiva or Tenon's tissue effectively bypass the superficial tear film and conjunctival barriers. Another advantage of this route is the ability to leave a depot of drug within the potential space between tissue layers. Furthermore, more posterior placement of the drug enables delivery to the posterior segment (discussed below). Periocular injections (Ghate et al., 2007) are particularly pertinent for drugs poorly soluble in water such as steroids. Sub‐conjunctival, sub‐Tenon's, peri‐bulbar injections are commonly used in ophthalmology practice. Sub‐conjunctival steroid is used for the treatment of refractory anterior uveitis. Injections of the antifibrotic agents 5‐flurouracil and mitomycin C are also applied using this route to prevent scarring after trabeculectomy surgery. Sub‐Tenon's and peribulbar injections are often the method of choice for administering local anaesthetic. Less commonly used is the retrobulbar injection, which is associated with the small, albeit life‐threatening risk of brain stem anaesthesia.
Posterior segment
The posterior segment of the eye refers to the posterior two‐thirds of the eye, including the anterior hyaloid membrane and all the structures behind it such as the vitreous, retina choroid and optic nerve. The leading causes of visual impairment and irreversible blindness are posterior segment‐related diseases (Thrimawithana et al., 2011). Inflammation and fibrosis are also symptoms associated with posterior segment disorders and ocular tissue damage. The uniqueness of the cellular composition and anatomical structure of the retina means that these biological processes are devastating and detrimental to vision. Inflammatory responses or hypoxic stimuli cause abnormal blood vessels to leak resulting in retinal thickening and oedema, fibrovascular proliferation and fractional retinal detachment. To preserve the retinal structure and function, it is crucial to prevent any primary vascular abnormality (Friedlander, 2007).
Only a decade ago, ocular treatments for these diseases were limited and were, at best, kept in check with laser therapy. With the advent of anti‐VEGF treatment, there have been significant improvements in stabilizing and sometimes improving disease states and vision (Jain et al., 2012). The delivery of anti‐VEGF medicines to the posterior segment poses a particular challenge. The structures in the anterior segment serve as additional barriers to those discussed above. Furthermore, the direction of aqueous flow in the eye (ciliary body to anterior chamber angle) is against the direction of drug delivery via a topical route. It is estimated that drugs applied in a conventional topical way are diluted by a factor of between 250 000 and 1 000 000 by the time the vitreous is reached (Maurice, 2002). Instead, the only modalities of treatment in everyday use are either periocular or i.v.t. injections. Avoiding all barriers, the i.v.t. route results in the greatest bioavailability and, for large molecules, such as proteins, is the only route in current clinical practice. However, each time an injection is performed, there is a small but significant risk of a blinding complication. The risks of retinal detachment and infective endophthalmitis have been reported as 0.9 and 0.2% per injection respectively (Jager et al., 2004). Frequent (4–8 weeks) anti‐VEGF treatment will result in a cumulative increase. Much research has been performed to improve transscleral delivery (Amrite et al., 2008), which would remove the need to breach the walls of the eye, and to develop sustained release preparations, which would reduce the frequency of repeated i.v.t. injections.
Periocular barriers
A drug must penetrate the tissue layers of the globe to reach the back of the eye at a therapeutic level. Various tissue barriers have varying tissue permeability. Unlike the cornea; however, there is blood flow through the periocular tissues, and considerable amounts of a drug are lost to the systemic circulation via venules and lymphatics. The Tenon's tissue and episclera are highly vascular with a network of capillary plexi. The reported rate of removal of drugs is between 5 and 80% (Edelhauser et al., 2010). This has implications not just for reaching the therapeutic concentration but also for preventing systemic toxicity.
Molecules that evade clearance by the subconjunctival circulation reach the sclera, the outer coating of the globe. The sclera is continuous with the cornea at the limbus and has a larger surface area. The sclera is hydrated, mainly composed of collagen fibres and proteoglycans embedded in an ECM, but with few protein binding sites. Its permeability is comparable to the corneal stroma, and aqueous intercellular media carry molecules through pores between the collagen fibres. Molecular radius appears to play a bigger role in determining drug permeability than lipophilicity (Prausnitz and Noonan, 1998), and molecules of linear shape, for example, dextrans, are less able to permeate than globular proteins (Geroski and Edelhauser, 2001). Electrical charge also contributes to permeation. Positively charged molecules penetrate the sclera poorly, believed to be due to their binding to the negatively charged proteoglycan matrix (Kim et al., 2007).
The choroid is below the sclera and is a vascular network whose high flow rate results in considerable drug loss. The main function of the choroid is to supply the retina with nutrients and oxygen. Due to the retina being the most metabolically active tissue in the body (per unit mass), the choroid has a correspondingly high blood flow through it, again being the highest (per unit mass) in the body (Scheinman et al., 2011). The choroid contains an outer layer of large vessels, a middle layer of small vessels and an innermost capillary layer, the choriocapillaris. The choriocapillaris is fenestrated, which enhances metabolic exchange, but this highly dynamic equilibrium also contributes to drug loss.
The retinal pigment epithelium (RPE) is a single‐layered structure that acts as the metabolic interface between the photoreceptors and choroid. It forms the outer BRB, with tight junctions between cells preventing the passage of large molecules and cells across it. The inner BRB is formed by the endothelium of retinal arterioles, which are also linked with tight junctions. Both the choroid and the RPE are pigmented and rich in melanin. Melanin binds to free radicals and other molecules by electrostatic and Van der Waals forces, or simple charge transfer (Larsson, 1993). Melanin‐bound drug can form a reservoir that is then gradually released to the surrounding cells and, as a result, prolongs drug release (Urtti, 2006; Gaudana et al., 2010; Manzanares et al., 2016). The binding of drugs to melanin is especially important in transscleral drug delivery. The unbound drug is the driving force in drug permeation, and the bound drug acts as a reservoir (Ranta and Urtti, 2006). Significant retention of lipophilic compounds occurs within the Bruch's membrane and the choroid presumably due to the high levels of melanin (Cheruvu and Kompella, 2006). However, the choroid‐RPE is even more impermeable to hydrophilic molecules, being up to 20 times less permeable to hydrophilic β‐blockers than lipophilic ones (Pitkanen, 2005). There does appear to be regional and species‐specific differences within the choroid‐RPE (Durairaj et al., 2012). The peripheral choroid‐RPE contains more melanin than the central choroid‐RPE. In humans, unlike monkeys or rabbits, there is also evidence that the sclera may contain more melanin than the central choroid‐RPE (Durairaj et al., 2012). These differences should be considered when using animal models for drug development as it can cause changes in the drug's pharmacokinetic and clearance profiles.
Injection routes of administration
Sub‐Tenon injections
Sub‐Tenon's injection of certain drugs has been shown to be effective for posterior segment disease. Inflammation is a key part of the pathophysiology of macular oedema following cataract surgery (Irvine Gass syndrome), diabetic macular oedema and posterior uveitis. The steroid triamcinolone acetonide (TA) has been used as treatment for these conditions with variable success. Most commonly, it is administered as an i.v.t. injection but this route of administration is associated with the additional specific complications of cataract and raised IOP. Sub‐Tenon's administration may carry a lower risk of complications. No difference in visual acuity outcomes for uveitic macular oedema was noted between i.v.t. and sub‐Tenon administration of TA (Choudhry and Ghosh, 2007). There is also evidence that sub‐Tenon injection leads to a significant increase in the levels of steroid in vitreous fluid. Patients that received sub‐Tenon's TA (40 mg·mL−1) preoperatively before macular hole surgery were found to have vitreous levels maintained between 17 and 31 ng·mL−1 for 2 months (Kovacs et al., 2012). Systemic toxicity may be an issue with sub‐Tenon's steroid injection. In this study, plasma cortisol levels were also altered, which has important implications for patients with poor glucose control such as diabetics.
Other transscleral delivery systems are at different stages of development. Implants have been designed with an impervious casing for sub‐Tenon's placement and openings directed towards the sclera (Pontes De Carvalho, 2006). Clinical trials evaluating efficacy are currently underway for the treatment of retinoblastoma using this method of delivery for the chemotherapeutic agents, carboplatin and topotecan Intravitreal (i.v.t.) injections
The delivery of most treatments for the posterior segment must be by i.v.t. injection due to transscleral barriers. This is particularly pertinent for the delivery of large molecules such as anti‐VEGF antibodies, although periocular delivery and even topical delivery continue to be explored (Chen et al., 2011). Due to the invasive nature and the associated risks of i.v.t. injections, there is much effort to develop longer lasting therapies, which require a reduced dosing frequency.
Understanding the pharmacokinetics of drugs in the vitreous fluid in humans is a significant challenge. The vitreous of humans is of a different consistency to that of other animals. It also undergoes considerable changes with age (Sebag, 1989). As people grow older, the vitreous loses viscosity, with lakes of fluid appearing within it (Bishop, 2000). Collagen fibril links break down in some parts of the vitreous, and fibrils aggregate in other parts (Bishop, 2000; Bishop et al., 2004). At the weaker points, the attachments can fall away and float around inside the eye. This lack of uniformity will affect drug distribution but may preferentially result for example in anti‐VEGF treatment being located over the macula.
The administration of humanized protein therapeutics (e.g. antibodies) intravitreally results in proteins diffusing more slowly in the vitreous than low molecular weight (MW) molecules. Therapeutic protein clearance times tend to be much longer than that of low MW compounds (days rather than hours) (Hayreh, 1966; Kwak and Amico, 1992; Cunha‐Vaz, 1997; Pitkanen, 2005; Urtti, 2006; Laude et al., 2010; Thrimawithana et al., 2011; Haghjou et al., 2013). An issue with therapeutic proteins is that the use of animals to evaluate pharmacokinetics will result in the development anti‐drug antibodies (ADAs). The presence of ADAs will result in the rapid clearance of the candidate protein and can also cause acute hypersensitivity or infusion reactions. ADAs can also bind to the active region of the therapeutic protein, such as the receptor‐binding site, to neutralize the drug. Therefore, it is difficult to determine the efficacy of a drug. ADAs unpredictively change the pharmacokinetic properties, biological effects and toxicity profile of a drug (Brinch et al., 2009; Tamilvanan et al., 2010; Brinks et al., 2011; Vugmeyster et al., 2012; Wang et al., 2012). ADAs are an intractable problem in the growth of protein therapeutics, especially for the development of new formulations with a prolonged duration of action.
Another factor whose contribution to drug distribution in the vitreous is difficult to quantify is eye movements. Saccades can result in sudden globe rotations of up to 900°·s−1, approximately half the speed of an old washing machine on a spin cycle. These abrupt changes shake up the vitreous and affect drug distribution. As the posterior segment is not a perfect sphere, with the lens causing a concave indentation, vortices are created with eye movements (Stocchino et al., 2007; Repetto et al., 2010).
Drug distribution and clearance is difficult to estimate in the new in vitro models recently developed (Repetto et al., 2005; Awwad et al., 2013, 2015, 2017; Fogli et al., 2014; Patel et al., 2015). Therapeutic proteins do not readily permeate the retina and thus clear via aqueous outflow through the anterior chamber into the conjunctiva. A two‐compartment model of the eye that mimics aqueous outflow mimics the human clearance times of therapeutic proteins (Awwad et al., 2015).
Some controversy exists as regards the frequency of the treatment regimen for certain pathologies. Exudative macular degeneration is a pathology that involves blood vessel growth and leakage from the choroid underneath the macula [choroidal neovascularization (CNV)]. Disruption of the retinal layers results in loss of visual function. A major factor for new blood vessel growth is VEGF that is released in association with oxidative stress. Anti‐VEGF antibodies have revolutionized treatment outcomes, with patients showing stabilization or improvement of vision together with a reduction in leakage. What is still unclear is the frequency and time span of drug administration. Both the ANCHOR and MARINA trials examined visual outcomes after monthly dosing of ranibizumab, a humanized monoclonal antibody fragment antigen‐binding (Brown et al., 2006; Rosenfeld et al., 2006). Other trials such as the PRONTO or SUSTAIN trial have shown significant benefit from monthly injections of ranibizumab for 3 months only, then injections as required (Lalwani et al., 2009; Holz et al., 2011). Comparative data between injections‐as‐required and monthly injections found little difference in outcome at 1 year in the CATT trial. The CATT and IVAN trials also compared the use of ranibizumab and bevacizumab, which is used unlicensed. Bevacizumab is a full IgG1 antibody registered for systemic use in cancer, but its use has been shown by the CATT (CATT Research Group et al., 2011) and IVAN (Chakravarthy et al., 2012) trials to be broadly similar to that of ranibizumab. Issues arise because bevacizumab has not been formulated for ocular use (Liu et al., 2011a; Ng et al., 2012; Palmer et al., 2013). Presentation of bevacizumab is in a vial containing 25 mg·mL−1 of antibody, and the dose was established simply by what was in a 50 mL injection. However, costs are dramatically reduced compared to ranibizumab, so bevacizumab is also widely used to treat conditions related to vascularization (e.g. AMD).
Intravitreal (i.v.t.) implants
A strategy to prolong drug action that has long been used clinically for low MW molecules is the i.v.t. injection of a drug suspension of a poorly soluble drug. Although not yet possible with therapeutic proteins, this approach works with poorly soluble steroids. TA, as discussed earlier, consists of a suspension of steroid particles. An injection of 4.0 mg of TA has been shown to maintain elevated levels of steroid up to 3 months in non‐vitrectomized eyes (Mason et al., 2004). The 4.0 mg dose is too large of a mass to dissolve in the eye, so TA dissolution occurs slowly over time with aqueous flow (Jermak et al., 2007; Yilmaz et al., 2011; Zacharias et al., 2013).
A growing field of research is the use of implants for the treatment of posterior segment disease (Figure 1, Table 2). These allow sustained release of drug, removing the need for multiple injections. Taking advantage of the privileged immune state of the inside the eye, many implants have been used successfully without provoking the significant inflammatory reaction that would normally be found with implants used elsewhere in the body (Ratner, 2002). Chronic diseases or those with a relapsing remitting course are particularly appropriate for sustained release implant therapy. The implants may be formulated from biodegradable or non‐biodegradable substances (Choonara et al., 2010; Lee et al., 2010; Thrimawithana et al., 2011; Christoforidis et al., 2012; Gilger et al., 2013; Cima et al., 2014; Kang‐Mieler et al., 2014; Thakur et al., 2014; Bisht et al., 2017; Prata et al., 2017).
Figure 1.
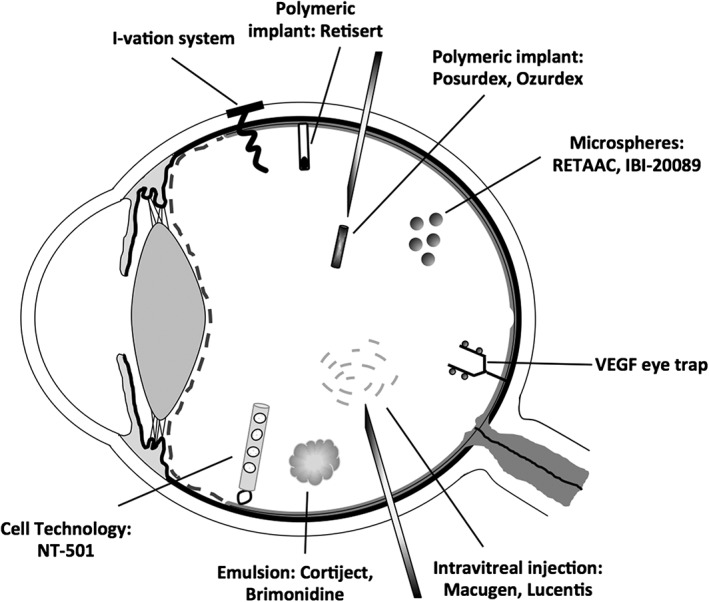
Some of the drug delivery technologies indicated for the treatment of posterior segment diseases
Table 2.
FDA‐approved drugs and those in current clinical trials for treatment of posterior segment diseases
| Name and company | Drug delivery platform | Drug and excipients | Indications and ongoing trials | Status |
|---|---|---|---|---|
| Brimonidine (Allergan) | i.v.t. implant | Brimonidine (0.4 mg) with PLGA | Dry AMD with GA | Phase II |
| Cortiject®/NOVA63035 (Novagali Pharma) | i.v.t. | Dexamethasone emulsion (preservative and solvent‐free) | DME | Phase I/II |
| Durasert™ (pSivida Corp) | i.v.t. implant | Latanoprost with EVA/PVA | Ocular hypertension and glaucoma | Phase I/II |
| I‐vation® (Surmodics Inc) | Reservoir i.v.t. implant | TA (0.925 mg) with PMMA/EVA | DME | Phase IIb |
| IBI‐20089 (Icon Biosciences Inc) | i.v.t. | TA (6.0 mg or 13.8 mg) and benzyl benzoate with lucentis (0.5 mg) | AMD | Phase I |
| CNV | Phase II | |||
| Iluvien™ (Alimera Sciences, Inc.) | 25‐gauge i.v.t. implant | FA (0.19 mg) with polyimide/PVA | DME | Approved |
| AMD with GA | Phase II | |||
| Posterior uveitis | Phase III | |||
| NT‐501/Renexus (Neurotech Pharma) | i.v.t. implant | CNTF with PET and ARPE‐19 cells |
Glaucoma RP |
Phase I |
| NT‐503 (Neurotech Pharma) | i.v.t. implant | Anti‐VEGF receptor protein | Recurrent CNV with wet AMD | Phase I |
| Phase II | ||||
| i.v.t. injection | ||||
| NT‐506 (Neurotech Pharma) | i.v.t. implant | Anti‐PDGF/Anti‐VEGF | Wet AMD | Preclinical |
| Ozurdex® (Allergan) | i.v.t. implant | Dexamethasone (0.7 mg) with PLGA | DME, CRVO, uveitis | Approved |
| Posurdex (Allergan) | i.v.t. implant | Dexamethasone | Posterior uveitis | Phase III |
| DME | Phase III | |||
| Retisert® (Bausch & Lomb) | Reservoir i.v.t. implant | FA (0.59 mg) with silicone/PVA | Non‐infectious uveitis | Approved |
| Vitrasert® (pSivida Corp) | Reservoir i.v.t. implant | Ganciclovir with EVA/ PVA | CMV retinitis | Approved |
All drug therapies mentioned above were also cross‐checked with www.clinicaltrials.gov.uk and its official company site. BRVO, branch retinal vein occlusion; CNTF, ciliary neutrophic factor; CRVO, central retinal vein occlusion; EVA, ethylene‐vinyl acetate; FA, fluocinolone acetonide; GA, geographical atrophy; PET, polyethylene terephthalate; PLGA, poly(lactide‐co‐glycolide); PMMA, nn(methyl methacrylate); PVA, poly(vinyl alcohol).
In immunocompromised patients, such as those with leukaemia or acquired immune deficiency syndrome, the opportunistic infection CMV can cause retinal destruction (Pollard, 1980). Patients may remain asymptomatic until advanced stages of retinal necrosis and haemorrhage. The advent of highly active antiretroviral therapy has reduced the incidence of patients developing CMV retinitis; however, individuals with specific immune deficiency may still develop retinitis when infected with CMV. Effective treatment of CMV retinitis not only reduces the risk of vision loss but is also associated with a decrease in mortality. Standard therapy may include ganciclovir (Pollard, 1996; Martin et al., 1999), foscarnet (Berthe et al., 1994; López‐Cortés et al., 2001) and cidofovir (Ljungman et al., 2001; Biron, 2006).
The ganciclovir slow release implant (Vitrasert) was one of the first approved implants for i.v.t. application (Sepahvandi et al., 2016). It releases ganciclovir at 1 mcg·h−1 from a PVA/EVA polymer‐based system. In a clinical trial, it was found to be twice as effective at slowing retinitis progression as treatment with intravenous ganciclovir (Musch et al., 1997). One of the pitfalls of implant treatment to one eye only, however, is that is that the other eye is not protected, and, in the implant‐treated group of patients, the time to second eye involvement was significantly shorter. Combination therapy with both implant and oral valganciclovir (oral version of ganciclovir) is now advocated for sight threatening retinitis (Jabs, 2008). Controversy exists as to whether implants should be removed from the eye after the drug has been released, as its removal is associated with a risk of retinal detachment, haemorrhage and infection.
Treatment of macular oedema has also been successfully achieved with steroid implants. Macular oedema results from the leakage of fluid from vessels within the retina, which disrupts the retinal architecture, and causes vision loss. It is seen in association with uveitis, RVO and diabetic maculopathy. The i.v.t. injection of the steroid TA has been shown to be beneficial in reducing oedema; however, recurrence is a common feature of all diseases. Work on a dexamethasone pellet coated in PVA/EVA showed promise using a uveitic model of rabbits (Cheng et al., 1995). The Ozurdex® implant has recently been approved by the Food and Drug Administration (FDA) for the treatment of macular oedema in association with non‐infectious uveitis and RVO (Mehta et al., 2015; Barar et al., 2016). It consists of 0.7 mg of dexamethasone integrated into a poly(lactic–co‐glycolic acid) polymer. Pharmacokinetic studies revealed that the steroid continues to be released into the vitreous over a period of 4 months (Chang‐Lin and Attar, 2011). The polymer has the added advantage of being biodegradable, requiring no surgical removal. Other implants are at different stages of development (Table 2).
Future directions
Prolonging the duration of drug action to targets while maintaining drug stability and specificity are key goals of current research. Novel ways of solubilizing drugs by conjugation with carriers, gene therapy and implants that are refillable or able to be directed remotely to the area of treatment are concepts in the process of development.
Dendrimers
Nanotechnology has been used to increase the drug residence time. Dendrimers are nano‐sized, three‐dimensional structures with a central core from which polymeric branches emerge (Patri et al., 2002). The hydrophobic drug is encapsulated in the internal cavity or complexed to the dendrimer. Its tree‐like structure consists of a high surface density of functional groups, which make it possible to attach a wide variety of conjugates. Dendrimers can aid in controlled drug release due to their small size, high chemical versatility, drug loading capacity and ease of preparation (Yavuz et al., 2015; Rodríguez Villanueva et al., 2016). The poly amidoamine (PAMAM) dendrimers include functional carboxylic, hydroxyl and amine groups, and these increase with generation number.
Dendrimer drug complexes have shown potential as an application for prolonging treatment of CNV. A dendrimer complex of anti‐VEGF antisense oligonucleotide was found to inhibit CNV in a laser‐induced rodent model when injected into the vitreous fluid. The effective lifespan of the dendrimer‐bound oligonucleotide was reported to be between 4 and 6 months, with effective protection against nucleases whilst also facilitating its delivery to the target site (Marano et al., 2005).
In a rabbit scarring model after glaucoma filtration surgery (GFS), dendrimer conjugates (dendrimer‐glucosamine as an immunomodulator and dendrimer glucosamine 6‐sulphate as an anti‐angiogenic molecule) were found to be efficacious as an anti‐scarring agent. Histopathological examination of the eyes showed minimal scar tissue formation and no inflammatory or neoangiogenic response, and the success of the GFS was increased from 30 to 80% (Shaunak et al., 2004).
PAMAM dendrimers have also been found to be compatible with topical ocular dosage forms in terms of physicochemical properties (pH, osmolality and viscosity) (Vandamme and Brobeck, 2005). Topical formulations of drugs such as pilocarpine nitrate (Vandamme and Brobeck, 2005), carteolol (Spataro et al., 2010) and dexamethasone (Yavuz et al., 2015) have been reported. The puerarin‐PAMAM dendrimer complex increased the corneal residence time of the drug thereby improving the efficacy and decreasing the frequency of dosing (Yao et al., 2010). This system also improved the availability of puerarin and enhanced the half‐life of the drug in the aqueous humour of rabbits from 0.48 to 1.30 h (Wang et al., 2011).
Hydrogels
Hydrogels have received considerable interest in recent years due to their ability to prolong efficacy whilst being biocompatible and biodegradable. Hydrogels are an alternative to particulate‐associated formulations and have been used for the delivery of large MW molecules (Mitragotri et al., 2014). They are polymeric materials that do not dissolve in water under physiological conditions and swell considerably in aqueous medium. Many hydrogel systems that have been described that do not require organic solvents during preparation (Stile et al., 1999; Vermonden et al., 2012; Shi et al., 2013). Hydrogels are networks of polymer main chains covalently linked together. This is known as cross‐linking, and sometimes, the polymer cross links can be strong non‐covalent interactions. The cross‐linking of polymer chains prevents complete dissolution of the polymer in a good solvent. Hence, hydrogels made of hydrophilic polymers can absorb water into their network structure and swell. The high water content property of hydrogels makes them biocompatible and is being examined for their application for tissue regeneration. However, the high water content of hydrogels is a challenge for developing extended drug release. Hydrogels can be made with polymers that can undergo a phase transition in response to different external conditions such as pH, temperature, electric currents and ionic strength (Simões et al., 2012). Such hydrogels can be made to ‘collapse’ to entrap a drug after administration.
One thermosensitive material that shows gel–sol transition is poly(N‐isopropylacrylamide) (pNIPAAM) (Yıldız et al., 2002; Silva and Oliveira, 2007; Li et al., 2008; Drapala et al., 2011; Klouda, 2015). It has been used in many drug delivery and tissue engineering studies (Li et al., 2008), with no signs of retinal toxicity (Turturro et al., 2011). PNIPAAM has a transition temperature of 32°C, and cross‐linking the hydrogel with poly(ethylene glycol) diacrylate improves the drug release profile by forming homogenous pores (Zhang et al., 2009). This characteristic has been exploited to encapsulate and release protein for ocular delivery to the posterior segment.
Reservoir‐type devices
Another approach being explored recently is the use of refillable reservoir‐type devices. When the existing drug depletes, the reservoir can be refilled with more drug. A microelectromechanical system (MEMS) drug delivery device consisting of a pump, drug reservoir and transscleral cannula showed promise in ex vivo porcine eyes (Li et al., 2008). Another MEMS device described in the literature is formed of layers, by using lithography and master moulds made up of silicon wafers or acrylic. A hard‐core base plate made up of polyetherether ketone is used to control the penetration depth of the needle and prevent the puncture of the entire device by the force of the needle. It is surgically implanted with the reservoir placed beneath the conjunctiva, with the cannula inserted through the sclera and drug dispensing tip, terminating in either the anterior or posterior segment depending on the site of treatment required. When mechanically actuated by force applied by the patient's finger, a pressure gradient occurs and the drug flows into the transscleral cannula. The one‐way valve at the end of the cannula opens and results in the release of a specific dose of the medication. This device was tested in vivo on rabbits using phenylephrine as a model drug (Lo et al., 2009). The Port Delivery System (ForSight VISION4, Inc.) is a refillable drug delivery device currently in phase 1 for preliminary safety for neovascular AMD (www.clinicaltrials.gov: NCT01186432) (Pearce et al., 2015).
Selective i.v.t. drug delivery using a microrobotic device is a novel invention. Controlled electromagnetically, it acts as an i.v.t. implant with a drug reservoir (Weidle et al., 2013). These microrobots can be placed in the lower vessel arcade without obstructing vision. They are non‐biodegradable and are made biocompatible by coating them with polypyrrole. It was demonstrated in vitro and ex vivo that the movement of this device could be wirelessly controlled in the vitreous. The group proposed an algorithm taking into account the complex optics of the eye that helps to localize the device based on 3‐D structure and allows a precise calculation of the gradients and forces acting on the device.
Stem cell technology
Stem cells have been explored as a potential path for the treatment of ocular diseases (Ramsden et al., 2013; Jeon and Oh, 2015; Rao et al., 2017). Stem cells have the ability to grow into many different cell types, that is, specialized cells can arise from their differentiation (Vellonen et al., 2014). Stem cells are critical for the renewal and repair of tissue in the body. Pluripotent stem cells (embryonic stem cells and induced pluripotent stem cells) are a rich source for the regeneration of damaged organs and tissues (Higuchi et al., 2017). Methods have been reported for the differentiation of human pluripotent stem cells (Gao et al., 2013; Burridge et al., 2014; Pagliuca et al., 2014; Rezania et al., 2014; Bao et al., 2015). A key challenge for the clinical use of stem cells is to optimize cell placement and the local host environment so that cell survival, differentiation and integration leads to the long‐term restoration of tissue function (Limb and Daniels, 2008). One of the major barriers to successful stem cell implantation and integration is to control the local inflammatory and fibrotic responses to the transplantation.
Stem cell treatments for corneal injuries are available clinically (Daniels et al., 2006; Daniels et al., 2007). Recent progress in retinal stem cell research has been reported for the treatment of dry AMD. Although no treatment is available for the management of dry AMD, vitamins and anti‐oxidants have been used to slow its progression. Stem cells could possibly help treat the severe retinal degeneration involved in the condition to restore sight (Jeon and Oh, 2015). Several trials are currently underway to treat AMD and retinitis pigmentosa (RP) using human embryonic, fetal and umbilical cord tissue‐derived stem cells and bone marrow‐derived stem cells (Ramsden et al., 2013).
Even though stem cell transplantation for retinal diseases has made considerable progress in the last decade, many issues still need to be resolved before stem cell therapies can be routinely used in the clinic (Rao et al., 2017). Biological risk and technical difficulties associated with the differentiation and culture procedures still need to be resolved (Jeon and Oh, 2015). As our understanding of the retinal regeneration phenomena increases, the likelihood of using cell therapies to regenerate retinal ocular tissues will increase.
Gene therapy
Gene products have been reported to slow or prevent neovascularization (Garoon and Stout, 2016). Gene therapies (Rowe‐Rendleman et al., 2014) require direct cellular delivery (del Amo et al., 2017) using a carrier system (Schön et al., 2015), where a key challenge is to release the gene drug from the carrier within the cell (Karimi et al., 2016). Furthermore, this often requires the gene drug to enter the nucleus. Vector loaded suspensions can be administered via a sub‐retinal injection by pars plana vitrectomy with retinotomy and injection into the subretinal space with a 41‐gauge cannula (Garoon and Stout, 2016). For the gene therapy to be successful, there needs to be no toxicity or immunogenicity after the delivery of highly efficient genes to specific targeted cells (Bloquel et al., 2006). Both viral and non‐viral vectors are available. Examples of viral vectors include adenoviruses, adeno‐associated‐virus (AAV), retroviruses and lentivirus vectors (Chaum and Hatton, 2002; Borrás, 2003; Mohan et al., 2005; Garoon and Stout, 2016). Adenoviruses are the most commonly used vector for targeting a variety of chronic ocular diseases. Some of the AAV‐based systems in clinical trials require a sub‐retinal injection (Bainbridge et al., 2015; Weleber et al., 2016). The administration of adenoviruses allows for stable, long‐term transgene expression in photoreceptors, RPE cells, ganglion cells and Müller cells (Liu et al., 2011b). Viral vectors have been reported to display potential risks, such as mutagenesis and immunogenicity, which hinders the future development of these systems (Bloquel et al., 2006). An alternative solution is non‐viral vectors, which display no toxicity and do not induce significant ocular inflammation (Bloquel et al., 2006). Reviews dedicated to gene therapy for ocular drug delivery can be found elsewhere (Bainbridge et al., 2006; Bloquel et al., 2006; Boye et al., 2013; Garoon and Stout, 2016; Oliveira et al., 2017).
Neuroprotective agents
Ciliary neurotrophic factor (CNTF) is a cytokine belonging to the IL‐6 family. CNTF promotes neurotransmitter synthesis, neurite outgrowth and photoreceptor survival (Liu et al., 2011b). CNTF has been shown to be protective against ganglion cell death in models of oxidative stress and experimental glaucoma (Lipinski et al., 2015). Studies with recombinant adeno‐associated virus‐mediated gene therapy showed sustained expression of CNTF with the protection of photoreceptors (Liang et al., 2001; Bok et al., 2002; Schlichtenbrede et al., 2003). NT‐501 (Neurotech Pharmaceuticals) is an implantable cell technology comprising encapsulated human RPE cells (ARPE‐19) genetically modified to secrete CNTF over an extended period of time for the treatment of glaucoma and retinitis pigmentosa. In a clinical trial, NT‐501 was found to reduce photoreceptor degradation in patients with retinitis pigmentosa and improve their objective visual acuity (Sieving et al., 2006). The implant consists of a scaffold of six strands of polyethylene terephthalate yarn and is sutured onto the sclera through a titanium loop (Thrimawithana et al., 2011). A semipermeable membrane allows the inward and outward diffusion of CNTF and protects the host immune system from being attacked. NT‐501 represents a unique platform for the delivery of protein therapeutics (Sieving et al., 2006). NT‐501 has completed phase II clinical trial for patients with atrophic macular degeneration (NCT00447954), and the implant was reported to be well tolerated (Girmens et al., 2012).
Conclusions
The eye is unique as a therapeutic target in the body. Whilst exhibiting a vast range of pathologies, both local and systemic, its superficial nature enables effective local therapy. The privileged state of the immune system inside the eye means that there is a greater tolerance of non‐host materials, although the risk of infection significantly increases each time the walls are breached. Ocular therapeutic development is therefore geared towards improving the bioavailability of topical and periocular treatments, while efficacious sustained release implants are the cornerstone of development for therapies of the posterior segment.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
We are grateful for the funding from the National Institute of Health Research (NIHR) Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, Moorfields Special Trustees, the Helen Hamlyn Trust (in memory of Paul Hamlyn), Medical Research Council, Fight for Sight, John Nolan, Kard Bos, Moskoutz, Katz Foundation and Sunrise Foundation.
Awwad, S. , Mohamed Ahmed, A. H. A. , Sharma, G. , Heng, J. S. , Khaw, P. T. , Brocchini, S. , and Lockwood, A. (2017) Principles of pharmacology in the eye. British Journal of Pharmacology, 174: 4205–4223. doi: 10.1111/bph.14024.
References
- Aburahma MH, Mahmoud AA (2011). Biodegradable ocular inserts for sustained delivery of brimonidine tartarate: preparation and in vitro/in vivo evaluation. AAPS PharmSciTech 12: 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrahari V, Mandal A, Agrahari V, Trinh HM, Joseph M, Ray A et al (2016). A comprehensive insight on ocular pharmacokinetics. Drug Deliv Transl Res 6: 735–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahfat FG, Zaidi FH (2013). Bevacizumab vs ranibizumab‐an appraisal of the evidence from CATT and IVAN. Eye (Lond) 27: 289–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed I, Patton TF (1985). Importance of the noncorneal absorption route in topical ophthalmic drug delivery. Invest Ophthalmol Vis Sci 26: 584–587. [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Amo EM, Urtti A (2008). Current and future ophthalmic drug delivery systems. A shift to the posterior segment. Drug Discov Today 13: 135–143. [DOI] [PubMed] [Google Scholar]
- del Amo EM, Vellonen K‐S, Kidron H, Urtti A (2015). Intravitreal clearance and volume of distribution of compounds in rabbits: in silico prediction and pharmacokinetic simulations for drug development. Eur J Pharm Biopharm 95: 215–226. [DOI] [PubMed] [Google Scholar]
- del Amo EM, Rimpelä A, Heikkinen E, Kari OK, Ramsay E, Lajunen T et al (2017). Pharmacokinetic aspects of retinal drug delivery. Prog Retin Eye Res 57: 134–185. [DOI] [PubMed] [Google Scholar]
- Amrite AC, Edelhauser HF, Kompella UB (2008). Modeling of corneal and retinal pharmacokinetics after periocular drug administration. Invest Ophthalmol Vis Sci 49: 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awwad S, Lockwood A, Mohamed Ahmed A, Sharma G, Khalili A, Brocchini S et al (2013). Development of an in vitro pharmacokinetic model of the human eye. Invest Ophthalmol Vis Sci 54: 5068. [Google Scholar]
- Awwad S, Lockwood A, Mohamed Ahmed A, Sharma G, Khalili A, Brocchini S et al (2015). The PK‐Eye: a novel in vitro ocular flow model for use in preclinical drug development. J Pharm Sci 104: 3330–3342. [DOI] [PubMed] [Google Scholar]
- Awwad S, Day RM, Khaw PT, Brocchini S, Fadda HM (2017). Sustained release ophthalmic dexamethasone: in vitro in vivo correlations derived from the PK‐Eye. Int J Pharm 522: 119–127. [DOI] [PubMed] [Google Scholar]
- Bainbridge JWB, Tan MH, Ali RR (2006). Gene therapy progress and prospects: the eye. Gene Ther 13: 1191–1197. [DOI] [PubMed] [Google Scholar]
- Bainbridge JWB, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C et al (2015). Long‐term effect of gene therapy on Leber's congenital amaurosis. N Engl J Med 372: 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baino F (2011). Towards an ideal biomaterial for vitreous replacement: historical overview and future trends. Acta Biomater 7: 921–935. [DOI] [PubMed] [Google Scholar]
- Bao X, Lian X, Dunn KK, Shi M, Han T, Qian T et al (2015). Chemically‐defined albumin‐free differentiation of human pluripotent stem cells to endothelial progenitor cells. Stem Cell Res 15: 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barar J, Aghanejad A, Fathi M, Omidi Y (2016). Advanced drug delivery and targeting technologies for the ocular diseases. Bioimpacts 6: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bary AR, Tucker IG, Davies NM (2000). Considerations in the use of hydroxypropyl‐??‐cyclodextrin in the formulation of aqueous ophthalmic solutions of hydrocortisone. Eur J Pharm Biopharm 50: 237–244. [DOI] [PubMed] [Google Scholar]
- Berthe P, Baudouin C, Garraffo R, Hofmann P, Taburet AM, Lapalus P (1994). Toxicologic and pharmacokinetic analysis of intravitreal injections of foscarnet, either alone or in combination with ganciclovir. Invest Ophthalmol Vis Sci 35: 1038–1045. [PubMed] [Google Scholar]
- Biron KK (2006). Antiviral drugs for cytomegalovirus diseases. Antiviral Res 71: 154–163. [DOI] [PubMed] [Google Scholar]
- Bishop PN (2000). Structural macromolecules and supramolecular organisation of the vitreous gel. Prog Retin Eye Res 19: 323–344. [DOI] [PubMed] [Google Scholar]
- Bishop PN, Holmes DF, Kadler KE, McLeod D, Bos KJ (2004). Age‐related changes on the surface of vitreous collagen fibrils. Invest Ophthalmol Vis Sci 45: 1041–1046. [DOI] [PubMed] [Google Scholar]
- Bisht R, Jaiswal JK, Rupenthal ID (2017). Nanoparticle‐loaded biodegradable light‐responsive in situ forming injectable implants for effective peptide delivery to the posterior segment of the eye. Med Hypotheses 103: 5–9. [DOI] [PubMed] [Google Scholar]
- Bloquel C, Bourges JL, Touchard E, Berdugo M, BenEzra D, Behar‐Cohen F (2006). Non‐viral ocular gene therapy: potential ocular therapeutic avenues. Adv Drug Deliv Rev 58: 1224–1242. [DOI] [PubMed] [Google Scholar]
- Bok D, Yasumura D, Matthes MT, Ruiz A, Duncan JL, Chappelow AV et al (2002). Effects of adeno‐associated virus‐vectored ciliary neurotrophic factor on retinal structure and function in mice with a P216L rds/peripherin mutation. Exp Eye Res 74: 719–735. [DOI] [PubMed] [Google Scholar]
- Borrás T (2003). Recent developments in ocular gene therapy. Exp Eye Res 76: 643–652. [DOI] [PubMed] [Google Scholar]
- Bourlais (1995). New ophthalmic drug delivery systems. Drug Dev Ind Pharm 21: 19–59. [Google Scholar]
- Boye SE, Boye SL, Lewin AS, Hauswirth WW (2013). A comprehensive review of retinal gene therapy. Mol Ther 21: 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozkir A, Denli ZF, Basaran B (2012). Effect of hydroxypropyl‐beta‐cyclodextrin on the solubility, stability and in‐vitro release of ciprofloxacin for ocular drug delivery. Acta Pol Pharm 69: 719–724. [PubMed] [Google Scholar]
- Brinch KS, Frimodt‐Møller N, Høiby N, Kristensen H‐H (2009). Influence of antidrug antibodies on plectasin efficacy and pharmacokinetics. Antimicrob Agents Chemother 53: 4794–4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinks V, Jiskoot W, Schellekens H (2011). Immunogenicity of therapeutic proteins: the use of animal models. Pharm Res 28: 2379–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY et al (2006). Ranibizumab versus verteporfin for neovascular age‐related macular degeneration. N Engl J Med 355: 1432–1444. [DOI] [PubMed] [Google Scholar]
- Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD et al (2014). Chemically defined generation of human cardiomyocytes. Nat Methods 11: 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappello B, Carmignani C, Iervolino M, Immacolata La Rotonda M, Fabrizio Saettone M (2001). Solubilization of tropicamide by hydroxypropyl‐beta‐cyclodextrin and water‐soluble polymers: in vitro/in vivo studies. Int J Pharm 213: 75–81. [DOI] [PubMed] [Google Scholar]
- CATT Research Group , Martin DF, Maguire MG, Ying G, Grunwald JE, Fine SL et al (2011). Ranibizumab and bevacizumab for neovascular age‐related macular degeneration. N Engl J Med 364: 1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Wordsworth S et al (2012). Ranibizumab versus bevacizumab to treat neovascular age‐related macular degeneration: one‐year findings from the IVAN randomized trial. Ophthalmology 119: 1399–1411. [DOI] [PubMed] [Google Scholar]
- Chang‐Lin J, Attar M (2011). Pharmacokinetics and pharmacodynamics of a sustained‐release dexamethasone intravitreal implant. Invest Ophthalmol Vis Sci 52: 80–86. [DOI] [PubMed] [Google Scholar]
- Chaum E, Hatton MP (2002). Gene therapy for genetic and acquired retinal diseases. Surv Ophthalmol 47: 449–469. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Ebmeier SE, Sutherland WM, Ghazi NG (2011). Potential penetration of topical ranibizumab (Lucentis) in the rabbit eye. Eye (Lond) 25: 1504–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CK, Berger AS, Pearson PA, Ashton P, Jaffe GJ (1995). Intravitreal sustained‐release dexamethasone device in the treatment of experimental uveitis. Invest Ophthalmol Vis Sci 36: 442–453. [PubMed] [Google Scholar]
- Cher I (2012). Fluids of the ocular surface: concepts, functions and physics. Clin Experiment Ophthalmol 40: 634–643. [DOI] [PubMed] [Google Scholar]
- Cheruvu NPS, Kompella UB (2006). Bovine and porcine transscleral solute transport: influence of lipophilicity and the Choroid–Bruch's layer. Invest Ophthalmol Vis Sci 47: 4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PPC, Keefe JE, Mesurier RTL, Taylor HR (2006). Global burden of disease and visual impairment. Lancet 368: 365. [DOI] [PubMed] [Google Scholar]
- Choonara YE, Pillay V, Danckwerts MP, Carmichael TR, du Toit LC (2010). A review of implantable intravitreal drug delivery technologies for the treatment of posterior segment eye diseases. J Pharm Sci 99: 2219–2239. [DOI] [PubMed] [Google Scholar]
- Choudhry S, Ghosh S (2007). Intravitreal and posterior subtenon triamcinolone acetonide in idiopathic bilateral uveitic macular oedema. Clin Experiment Ophthalmol 35: 713–718. [DOI] [PubMed] [Google Scholar]
- Christoforidis JB, Chang S, Jiang A, Wang J, Cebulla CM (2012). Intravitreal devices for the treatment of vitreous inflammation. Mediators Inflamm 2012: 126463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cima MJ, Lee H, Daniel K, Tanenbaum LM, Mantzavinou A, Spencer KC et al (2014). Single compartment drug delivery. J Control Release 190: 157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AE, Assang C, Patane MA, From S, Korenfeld M, Investigators AS (2012). Evaluation of dexamethasone phosphate delivered by ocular iontophoresis for treating noninfectious anterior uveitis. Ophthalmology 119: 66–73. [DOI] [PubMed] [Google Scholar]
- Cox WV, Kupferman A, Leibowitz HM (1972a). Topically applied steroids in corneal disease. I. The role of inflammation in stromal absorption of dexamethasone. Arch Ophthalmol 88: 308–313. [DOI] [PubMed] [Google Scholar]
- Cox WV, Kupferman A, Leibowitz HM (1972b). Topically applied steroids in corneal disease. II. The role of drug vehicle in stromal absorption of dexamethasone. Arch Ophthalmol 88: 549–552. [DOI] [PubMed] [Google Scholar]
- Cunha‐Vaz JG (1997). The blood‐ocular barriers: past, present, and future. Doc Ophthalmol 93: 149–157. [DOI] [PubMed] [Google Scholar]
- Daniels JT, Secker GA, Shortt AJ, Tuft SJ, Seetharaman S (2006). Stem cell therapy delivery: treading the regulatory tightrope. Regen Med 1: 715–719. [DOI] [PubMed] [Google Scholar]
- Daniels JT, Notara M, Shortt AJ, Secker G, Harris A, Tuft SJ (2007). Limbal epithelial stem cell therapy. Expert Opin Biol Ther 7: 1–3. [DOI] [PubMed] [Google Scholar]
- Davis ME, Brewster ME (2004). Cyclodextrin‐based pharmaceutics: past, present and future. Nat Rev Drug Discov 3: 1023–1035. [DOI] [PubMed] [Google Scholar]
- Delplace V, Payne S, Shoichet M (2015). Delivery strategies for treatment of age‐related ocular diseases: from a biological understanding to biomaterial solutions. J Control Release 219: 652–668. [DOI] [PubMed] [Google Scholar]
- Digiuni M, Fogagnolo P, Rossetti L (2012). A review of the use of latanoprost for glaucoma since its launch. Expert Opin Pharmacother 13: 723–745. [DOI] [PubMed] [Google Scholar]
- Drapala PW, Brey EM, Mieler WF, Venerus DC, Kang Derwent JJ, Pérez‐Luna VH (2011). Role of thermo‐responsiveness and poly(ethylene glycol) diacrylate cross‐link density on protein release from poly(N‐isopropylacrylamide) hydrogels. J Biomater Sci Polym Ed 22: 59–75. [DOI] [PubMed] [Google Scholar]
- Durairaj C, Chastain JE, Kompella UB (2012). Intraocular distribution of melanin in human, monkey, rabbit, minipig and dog eyes. Exp Eye Res 98: 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelhauser HF, Rowe‐Rendleman CL, Robinson MR, Dawson DG, Chader GJ, Grossniklaus HE et al (2010). Ophthalmic drug delivery systems for the treatment of retinal diseases: basic research to clinical applications. Invest Ophthalmol Vis Sci 51: 5403–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshout M, van der Reis MI, Webers CAB, Schouten JSAG (2014). The cost‐utility of aflibercept for the treatment of age‐related macular degeneration compared to bevacizumab and ranibizumab and the influence of model parameters. Graefes Arch Clin Exp Ophthalmol 252: 1911–1920. [DOI] [PubMed] [Google Scholar]
- Everaert A, Wouters Y, Melsbach E, Zakaria N, Ludwig A, Kiekens F et al (2017). Optimisation of HPMC ophthalmic inserts with sustained release properties as a carrier for thermolabile therapeutics. Int J Pharm 528: 395–405. [DOI] [PubMed] [Google Scholar]
- Fialho SL, da Silva‐Cunha A (2004). New vehicle based on a microemulsion for topical ocular administration of dexamethasone. Clin Experiment Ophthalmol 32: 626–632. [DOI] [PubMed] [Google Scholar]
- Fish R, Davidson R (2010). Management of ocular thermal and chemical injuries, including amniotic membrane therapy. Curr Opin Ophthalmol 21: 317–321. [DOI] [PubMed] [Google Scholar]
- Flach AJ (2008). The importance of eyelid closure and nasolacrimal occlusion following the ocular instillation of topical glaucoma medications, and the need for the universal inclusion of one of these techniques in all patient treatments and clinical studies. Trans Am Ophthalmol Soc 106 138‐45–8. [PMC free article] [PubMed] [Google Scholar]
- Fogli G, Orsi G, De Maria C, Montemurro F, Palla M, Rizzo S et al (2014). New eye phantom for ophthalmic surgery. J Biomed Opt 19: 68001. [DOI] [PubMed] [Google Scholar]
- Franca JR, Foureaux G, Fuscaldi LL, Ribeiro TG, Rodrigues LB, Bravo R et al (2014). Bimatoprost‐loaded ocular inserts as sustained release drug delivery systems for glaucoma treatment: in vitro and in vivo evaluation. PLoS One 9: e95461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander M (2007). Fibrosis and diseases of the eye. J Clin Invest 117: 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Kishida T, Ejima A, Gojo S, Mazda O (2013). Myostatin acts as an autocrine/paracrine negative regulator in myoblast differentiation from human induced pluripotent stem cells. Biochem Biophys Res Commun 431: 309–314. [DOI] [PubMed] [Google Scholar]
- Garoon RB, Stout JT (2016). Update on ocular gene therapy and advances in treatment of inherited retinal diseases and exudative macular degeneration. Curr Opin Ophthalmol 27: 1. [DOI] [PubMed] [Google Scholar]
- Gaudana R, Ananthula HK, Parenky A, Mitra AK (2010). Ocular drug delivery. AAPS J 12: 348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geroski DH, Edelhauser HF (2001). Transscleral drug delivery for posterior segment disease. Adv Drug Deliv Rev 52: 37–48. [DOI] [PubMed] [Google Scholar]
- Ghate D, Edelhauser HF (2008). Barriers to glaucoma drug delivery. J Glaucoma 17: 147–156. [DOI] [PubMed] [Google Scholar]
- Ghate D, Brooks W, McCarey BE, Edelhauser HF (2007). Pharmacokinetics of intraocular drug delivery by periocular injections using ocular fluorophotometry. Invest Ophthalmol Vis Sci 48: 2230–2237. [DOI] [PubMed] [Google Scholar]
- Gilger BC, Abarca EM, Salmon JH, Patel S (2013). Treatment of acute posterior uveitis in a porcine model by injection of triamcinolone acetonide into the suprachoroidal space using microneedles. Invest Ophthalmol Vis Sci 54: 2483–2492. [DOI] [PubMed] [Google Scholar]
- Girmens J‐F, Sahel J‐A, Marozova K (2012). Dry age‐related macular degeneration: a currently unmet clinical need. IRDR 1: 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn RJ, Seddon JM, Krug JH, Sahagian CR, Chiavelli ME, Campion EW (1991). Falls in elderly patients with glaucoma. Arch Ophthalmol 109: 205–210. [DOI] [PubMed] [Google Scholar]
- Gonzalez JR, Baiza‐Duran L, Quintana‐Hau J, Tornero‐Montaño R, Castaneda‐Hernandez G, Ortiz M et al (2007). Comparison of the stability, efficacy, and adverse effect profile of the innovator 0.005% latanoprost ophthalmic solution and a novel cyclodextrin‐containing formulation. J Clin Pharmacol 47: 121–126. [DOI] [PubMed] [Google Scholar]
- Grabovac V, Guggi D, Bernkop‐Schnürch A (2005). Comparison of the mucoadhesive properties of various polymers. Adv Drug Deliv Rev 57: 1713–1723. [DOI] [PubMed] [Google Scholar]
- Güngör S, Delgado‐Charro MB, Ruiz‐Perez B, Schubert W, Isom P, Moslemy P et al (2010). Trans‐scleral iontophoretic delivery of low molecular weight therapeutics. J Control Release 147: 225–231. [DOI] [PubMed] [Google Scholar]
- Guter M, Breunig M (2017). Hyaluronan as a promising excipient for ocular drug delivery. Eur J Pharm Biopharm 113: 34–49. [DOI] [PubMed] [Google Scholar]
- Ha DI, Lee SB, Chong MS, Lee YM, Kim SY, Park YH (2006). Preparation of thermo‐responsive and injectable hydrogels based on hyaluronic acid and poly(N‐isopropylacrylamide) and their drug release behaviors. Macromol Res 14: 87–93. [Google Scholar]
- Haghjou N, Abdekhodaie MJ, Cheng Y‐L (2013). Retina‐choroid‐sclera permeability for ophthalmic drugs in the vitreous to blood direction: quantitative assessment. Pharm Res 30: 41–59. [DOI] [PubMed] [Google Scholar]
- Hayreh SS (1966). Posterior drainage of the intraocular fluid from the vitreous. Exp Eye Res 5: 123–144. [Google Scholar]
- Higuchi A, Suresh Kumar S, Ling QD, Alarfaj AA, Munusamy MA, Murugan K et al (2017). Polymeric design of cell culture materials that guide the differentiation of human pluripotent stem cells. Prog Polym Sci 65: 83–126. [Google Scholar]
- Holz FG, Amoaku W, Donate J, Guymer RH, Kellner U, Schlingemann RO et al (2011). Safety and efficacy of a flexible dosing regimen of ranibizumab in neovascular age‐related macular degeneration: the SUSTAIN study. Ophthalmology 118: 663–671. [DOI] [PubMed] [Google Scholar]
- Jabs DA (2008). AIDS and ophthalmology, 2008. Arch Ophthalmol 126: 1143–1146. [DOI] [PubMed] [Google Scholar]
- Jager RD, Aiello LP, Patel SC, Cunningham ET (2004). Risks of intravitreous injection: a comprehensive review. Retina 24: 676–698. [DOI] [PubMed] [Google Scholar]
- Jain D, Carvalho E, Banerjee R (2010). Biodegradable hybrid polymeric membranes for ocular drug delivery. Acta Biomater 6: 1370–1379. [DOI] [PubMed] [Google Scholar]
- Jain GK, Warsi MH, Nirmal J, Garg V, Pathan SA, Ahmad FJ et al (2012). Therapeutic stratagems for vascular degenerative disorders of the posterior eye. Drug Discov Today 17: 1–12. [DOI] [PubMed] [Google Scholar]
- Jeon S, Oh IH (2015). Regeneration of the retina: toward stem cell therapy for degenerative retinal diseases. BMB Rep 48: 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermak CM, Dellacroce JT, Heffez J, Peyman GA (2007). Triamcinolone acetonide in ocular therapeutics. Surv Ophthalmol 52: 503–522. [DOI] [PubMed] [Google Scholar]
- Jóhannsdóttir S, Jansook P, Stefánsson E, Loftsson T (2015). Development of a cyclodextrin‐based aqueous cyclosporin A eye drop formulations. Int J Pharm 493: 86–95. [DOI] [PubMed] [Google Scholar]
- Joseph RR, Venkatraman SS (2017). Drug delivery to the eye: what benefits do nanocarriers offer? Nanomedicine (Lond) 12: 683–702. [DOI] [PubMed] [Google Scholar]
- Kang‐Mieler JJ, Osswald CR, Mieler WF (2014). Advances in ocular drug delivery: emphasis on the posterior segment. Expert Opin Drug Deliv 11: 1647–1660. [DOI] [PubMed] [Google Scholar]
- Karimi M, Ghasemi A, Sahandi Zangabad P, Rahighi R, Moosavi Basri SM, Mirshekari H et al (2016). Smart micro/nanoparticles in stimulus‐responsive drug/gene delivery systems. Chem Soc Rev 45: 1457–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass MA, Meltzer DW, Gordon M, Cooper D, Goldberg J (1986). Compliance with topical pilocarpine treatment. Am J Ophthalmol 101: 515–523. [DOI] [PubMed] [Google Scholar]
- Kaur C, Foulds WS, Ling EA (2008). Blood‐retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res 27: 622–647. [DOI] [PubMed] [Google Scholar]
- Kearse EC, McIntyre OL, Johnson MH, Phillips CI, Lathe R, Adams W et al (2001). Influence of dehydroepiandrosterone on rabbit intraocular pressure. Ophthalmic Res 33: 42–47. [DOI] [PubMed] [Google Scholar]
- Kek WK, Miller J, Rawson‐Lax E, Wilson CG, Uttamchandani D (2010). In situ measurement of spectral changes in the anterior eye following application of ultraviolet‐absorbing compounds. Eur J Pharm Biopharm 75: 200–205. [DOI] [PubMed] [Google Scholar]
- Kesavan K, Kant S, Singh PN, Pandit JK (2013). Mucoadhesive chitosan‐coated cationic microemulsion of dexamethasone for ocular delivery: in vitro and in vivo evaluation. Curr Eye Res 38: 1–11. [DOI] [PubMed] [Google Scholar]
- Khutoryanskiy V (2010). Advances in mucoadhesion and mucoadhesive polymers. Macromol Biosci 11: 748–764. [DOI] [PubMed] [Google Scholar]
- Kim S, Lutz R, Wang N, Robinson M (2007). Transport barriers in transscleral drug delivery for retinal diseases. Ophthalmic Res 39: 244–254. [DOI] [PubMed] [Google Scholar]
- Klouda L (2015). Thermoresponsive hydrogels in biomedical applications A seven‐year update. Eur J Pharm Biopharm 97: 338–349. [DOI] [PubMed] [Google Scholar]
- Koffler B, Mcdonald M, Nelinson D (2010). Improved signs, symptoms, and quality of life associated with dry eye syndrome: hydroxypropyl cellulose ophthalmic insert patient registry. Eye Contact Lens 36: 170–176. [DOI] [PubMed] [Google Scholar]
- Kovacs K, Wagley S, Quirk M, Ceron O, Silva P, Singh R et al (2012). Pharmacokinetic study of vitreous and serum concentrations of triamcinolone acetonide after posterior sub‐Tenon's injection. Am J Ophthalmol 153: 939–948. [DOI] [PubMed] [Google Scholar]
- Krag S, Hessellund A (2014). Topical dexamethasone‐cyclodextrin microparticle eye drops for uveitic macular oedema. Acta Ophthalmol 92: e689–e690. [DOI] [PubMed] [Google Scholar]
- Kumar S, Karki R, Meena M, Prakash T, Rajeswari T, Goli D (2011). Reduction in drop size of ophthalmic topical drop preparations and the impact of treatment. J Adv Pharm Technol Res 2: 192–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari A, Sharma PK, Garg VK, Garg G (2010). Ocular inserts – advancement in therapy of eye diseases. J Adv Pharm Technol Res 1: 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurkov SV, Loftsson T (2013). Cyclodextrins. Int J Pharm 453: 167–180. [DOI] [PubMed] [Google Scholar]
- Kwak HW, Amico DJD (1992). Evaluation of the retinal toxicity and pharmacokinetics of dexamethasone after intravitreal injection. Arch Ophthalmol 110: 259–266. [DOI] [PubMed] [Google Scholar]
- Kwong TQ, Mohamed M (2014). Anti‐vascular endothelial growth factor therapies in ophthalmology: current use, controversies and the future. Br J Clin Pharmacol 78: 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalwani GA, Rosenfeld PJ, Fung AE, Dubovy SR, Michels S, Feuer W et al (2009). A variable‐dosing regimen with intravitreal ranibizumab for neovascular age‐related macular degeneration: year 2 of the PrONTO study. Am J Ophthalmol 148: 43–58.e1. [DOI] [PubMed] [Google Scholar]
- Langer R (1983). Implantable controlled release systems. Pharmacol Ther 21: 35–51. [DOI] [PubMed] [Google Scholar]
- Larsson BS (1993). Interaction between chemicals and melanin. Pigment Cell Res 6: 127–133. [DOI] [PubMed] [Google Scholar]
- Laude A, Tan LE, Wilson CG, Lascaratos G, Elashry M, Aslam T et al (2010). Intravitreal therapy for neovascular age‐related macular degeneration and inter‐individual variations in vitreous pharmacokinetics. Prog Retin Eye Res 29: 466–475. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Miller JW (2004). Ocular tissue permeabilities. Int Ophthalmol Clin 44: 53–61. [DOI] [PubMed] [Google Scholar]
- Lee SS, Hughes P, Ross AD, Robinson MR (2010). Biodegradable implants for sustained drug release in the eye. Pharm Res 27: 2043–2053. [DOI] [PubMed] [Google Scholar]
- Lee VHL (1990). Mechanisms and facilitation of corneal drug penetration. J Control Release 11: 79–90. [Google Scholar]
- Li X, Wu W, Liu W (2008). Synthesis and properties of thermo‐responsive guar gum/poly(N‐isopropylacrylamide) interpenetrating polymer network hydrogels. Carbohydr Polym 71: 394–402. [Google Scholar]
- Liang FQ, Dejneka NS, Cohen DR, Krasnoperova NV, Lem J, Maguire AM et al (2001). AAV‐mediated delivery of ciliary neurotrophic factor prolongs photoreceptor survival in the rhodopsin knockout mouse. Mol Ther 3: 241–248. [DOI] [PubMed] [Google Scholar]
- Limb GA, Daniels JT (2008). Ocular regeneration by stem cells: present status and future prospects. Br Med Bull 85: 47–61. [DOI] [PubMed] [Google Scholar]
- Lipinski DM, Barnard AR, Singh MS, Martin C, Lee EJ, Davies WIL et al (2015). CNTF gene therapy confers lifelong neuroprotection in a mouse model of human retinitis pigmentosa. Mol Ther 23: 1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Ammar DA, Ross LA, Mandava N, Kahook MY, Carpenter JF (2011a). Silicone oil microdroplets and protein aggregates in repackaged bevacizumab and ranibizumab: effects of long‐term storage and product mishandling. Invest Ophthalmol Vis Sci 52: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MM, Tuo J, Chan C‐C (2011b). Gene therapy for ocular diseases. Br J Ophthalmol 95: 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman P, Deliliers GL, Platzbecker U, Matthes‐Martin S, Bacigalupo A, Einsele H et al (2001). Cidofovir for cytomegalovirus infection and disease in allogeneic stem cell transplant recipients. The infectious diseases working party of the European group for blood and marrow transplantation. Blood 97: 388–392. [DOI] [PubMed] [Google Scholar]
- Lo R, Li PY, Saati S, Agrawal RN, Humayun MS, Meng E (2009). A passive MEMS drug delivery pump for treatment of ocular diseases. Biomed Microdevices 11: 959–970. [DOI] [PubMed] [Google Scholar]
- Loftsson T, Brewster ME (2011). Pharmaceutical applications of cyclodextrins: Effects on drug permeation through biological membranes. J Pharm Pharmacol 63: 1119–1135. [DOI] [PubMed] [Google Scholar]
- Loftsson T, Jansook P, Stefánsson E (2012). Topical drug delivery to the eye: dorzolamide. Acta Ophthalmol 90: 603–608. [DOI] [PubMed] [Google Scholar]
- Loftsson T, Stefansson E (1997). Effect of cyclodextrins on topical drug delivery to the eye. Drug Dev Ind Pharm 23: 473–481. [Google Scholar]
- López‐Cortés L, Pastor‐Ramos M, Ruiz‐Valderas R, Cordero E, Uceda‐Montañés A, Claro‐Cala C et al (2001). Intravitreal pharmacokinetics and retinal concentrations of ganciclovir and foscarnet after intravitreal administration in rabbits. Invest Ophthalmol Vis Sci 42: 1024–1028. [PubMed] [Google Scholar]
- Majumdar S, Hippalgaonkar K, Repka MA (2008). Effect of chitosan, benzalkonium chloride and ethylenediaminetetraacetic acid on permeation of acyclovir across isolated rabbit cornea. Int J Pharm 348: 175–178. [DOI] [PubMed] [Google Scholar]
- Manzanares JA, Rimpelä AK, Urtti A (2016). Interpretation of ocular melanin drug binding assays. Alternatives to the model of multiple classes of independent sites. Mol Pharm 13: 1251–1257. [DOI] [PubMed] [Google Scholar]
- Marano RJ, Toth I, Wimmer N, Brankov M, Rakoczy PE (2005). Dendrimer delivery of an anti‐VEGF oligonucleotide into the eye: a long‐term study into inhibition of laser‐induced CNV, distribution, uptake and toxicity. Gene Ther 12: 1544–1550. [DOI] [PubMed] [Google Scholar]
- Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA (1999). Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N Engl J Med 340: 1063–1070. [DOI] [PubMed] [Google Scholar]
- Martin DF, Maguire MG, Fine SL, Ying GS, Jaffe GJ, Grunwald JE et al (2012). Ranibizumab and bevacizumab for treatment of neovascular age‐related macular degeneration: two‐year results. Ophthalmology 119: 1388–1398. [DOI] [PubMed] [Google Scholar]
- Mason JO, Somaiya MD, Singh RJ (2004). Intravitreal concentration and clearance of triamcinolone acetonide in nonvitrectomized human eyes. Retina (Philadelphia, Pa) 24: 900–904. [DOI] [PubMed] [Google Scholar]
- Maurice DM (2002). Drug delivery to the posterior segment from drops. Surv Ophthalmol 47: pp. S41–S52. [DOI] [PubMed] [Google Scholar]
- McCarthy TJ (1975). The effect of vehicle composition on the release of chloramphenicol from creams and eye ointments. S Afr Med J 49: 1259–1262. [PubMed] [Google Scholar]
- Mehta H, Gillies M, Fraser‐Bell S (2015). Perspective on the role of Ozurdex (dexamethasone intravitreal implant) in the management of diabetic macular oedema. Ther Adv Chronic Dis 6: 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P, Haj‐Ahmad R, Al‐Kinani A, Arshad MS, Chang M‐W, Alany RG et al (2017). Approaches in topical ocular drug delivery and developments in the use of contact lenses as drug‐delivery devices. Ther Deliv 8: 521–541. [DOI] [PubMed] [Google Scholar]
- Mishima S, Gasset A, Klyce SD, Baum JL (1966). Determination of tear volume and tear flow. Invest Ophthalmol 5: 264–276. [PubMed] [Google Scholar]
- Mitragotri S, Burke PA, Langer R (2014). Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov 13: 655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan RR, Sharma A, Netto MV, Sinha S, Wilson SE (2005). Gene therapy in the cornea. Prog Retin Eye Res 24: 537–599. [DOI] [PubMed] [Google Scholar]
- Morton HJ, Thomas ET (1958). Effect of atropine on the heart‐rate. Lancet 2: 1313–1315. [DOI] [PubMed] [Google Scholar]
- Musch DC, Martin DF, Gordon JF, Davis MD, Kuppermann BD (1997). Treatment of cytomegalovirus retinitis with a sustained‐release ganciclovir implant. The ganciclovir implant study group. N Engl J Med 337: 83–90. [DOI] [PubMed] [Google Scholar]
- Myles M, Neumann D, Hill J (2005). Recent progress in ocular drug delivery for posterior segment disease: emphasis on transscleral iontophoresis. Adv Drug Deliv Rev 57: 2063–2079. [DOI] [PubMed] [Google Scholar]
- Nagataki S, Mishima S (1980). Pharmacokinetics of instilled drugs in the human eye. Int Ophthalmol Clin 20: 33–49. [DOI] [PubMed] [Google Scholar]
- Ng DS, Chan CW, Kwok AKH, Li WWT (2012). Intravitreal bevacizumab: safety of multiple doses from a single vial for consecutive patients. Hong Kong Med J 18: 488–495. [PubMed] [Google Scholar]
- Novack GD, Robin AL (2016). Ocular pharmacology. J Clin Pharmacol 56: 517–527. [DOI] [PubMed] [Google Scholar]
- Okamoto N, Ito Y, Nagai N, Murao T, Takiguchi Y, Kurimoto T et al (2010). Preparation of ophthalmic formulations containing cilostazol as an anti‐glaucoma agent and improvement in its permeability through the rabbit cornea. J Oleo Sci 59: 423–430. [DOI] [PubMed] [Google Scholar]
- Oliveira AV, Rosa da Costa AM, Silva GA (2017). Non‐viral strategies for ocular gene delivery. Mater Sci Eng C 77: 1275–1289. [DOI] [PubMed] [Google Scholar]
- de Oliveira Dias JR, Rodrigues EB, Maia M, Magalhães O, Penha FM, Farah ME (2011). Cytokines in neovascular age‐related macular degeneration: fundamentals of targeted combination therapy. Br J Ophthalmol 95: 1631–1637. [DOI] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH et al (2014). Generation of functional human pancreatic β cells in vitro. Cell 159: 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahuja P, Arora S, Pawar P (2012). Ocular drug delivery system: a reference to natural polymers. Expert Opin Drug Deliv 9: 837–861. [DOI] [PubMed] [Google Scholar]
- Palmer JM, Amoaku WM, Kamali F (2013). Quality of bevacizumab compounded for intravitreal administration. Eye (Lond) 27: 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolini D, Mariotti SP (2012). Global estimates of visual impairment: 2010. Br J Ophthalmol 96: 614–618. [DOI] [PubMed] [Google Scholar]
- Patane MA, Cohen A, From S, Torkildsen G, Welch D, Ousler GW (2011). Ocular iontophoresis of EGP‐437 (dexamethasone phosphate) in dry eye patients: results of a randomized clinical trial. Clin Ophthalmol 5: 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Müller G, Stracke JO, Altenburger U, Mahler H‐C, Jere D (2015). Evaluation of protein drug stability with vitreous humor in a novel ex‐vivo intraocular model. Eur J Pharm Biopharm 95: 407–417. [DOI] [PubMed] [Google Scholar]
- Patri AK, Majoros IJ, Baker JR (2002). Dendritic polymer macromolecular carriers for drug delivery. Curr Opin Chem Biol 6: 466–471. [DOI] [PubMed] [Google Scholar]
- Pearce W, Hsu J, Yeh S (2015). Advances in drug delivery to the posterior segment. Curr Opin Ophthalmol 26: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pek YS, Wu H, Mohamed ST, Ying JY (2016). Long‐term subconjunctival delivery of brimonidine tartrate for glaucoma treatment using a microspheres/carrier system. Adv Healthc Mater 5: 2823–2831. [DOI] [PubMed] [Google Scholar]
- Penedones A, Mendes D, Alves C, Batel Marques F (2014). Safety monitoring of ophthalmic biologics: a systematic review of pre‐ and postmarketing safety data. J Ocul Pharmacol Ther 30: 729–751. [DOI] [PubMed] [Google Scholar]
- Peyman G, Ganiban G (1995). Delivery systems for intraocular routes. Adv Drug Deliv Rev 16: 107–123. [Google Scholar]
- Peyman GA, Hosseini K (2011). Combination therapies in ophthalmology: Implications for intravitreal delivery. J Ophthalmic Vis Res 6: 36–46. [PMC free article] [PubMed] [Google Scholar]
- Pitkanen L (2005). Permeability of retinal pigment epithelium: effects of permeant molecular weight and lipophilicity. Invest Ophthalmol Vis Sci 46: 641–646. [DOI] [PubMed] [Google Scholar]
- Pollard R (1996). CMV retinitis: ganciclovir/monoclonal antibody. Antiviral Res 29: 73–75. [DOI] [PubMed] [Google Scholar]
- Pollard RB (1980). Cytomegalovirus retinitis in immunosuppressed hosts: I. Natural history and effects of treatment with adenine arabinoside. Ann Intern Med 93: 655–664. [DOI] [PubMed] [Google Scholar]
- Pontes De Carvalho R (2006). Delivery from episcleral exoplants. Invest Ophthalmol Vis Sci 47: 4532–4539. [DOI] [PubMed] [Google Scholar]
- Prata AI, Coimbra P, Pina ME (2017). Preparation of dexamethasone ophthalmic implants: a comparative study of in vitro release profiles. Pharm Dev Technol 1: 1–20. [DOI] [PubMed] [Google Scholar]
- Prausnitz MR, Noonan JS (1998). Permeability of cornea, sclera, and conjunctiva: a literature analysis for drug delivery to the eye. J Pharm Sci 87: 1479–1488. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Broman AT (2006). The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery J, Clegg A, Jones J, Tan SC, Lotery A (2007). Ranibizumab (Lucentis) versus bevacizumab (Avastin): modelling cost effectiveness. Br J Ophthalmol 91: 1244–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rah MJRO (2011). A review of hyaluronan and its ophthalmic applications. Optometry 82: 38–43. [DOI] [PubMed] [Google Scholar]
- Ramsden CM, Powner MB, Carr A‐JF, Smart MJK, da Cruz L, Coffey PJ (2013). Stem cells in retinal regeneration: past, present and future. Development 140: 2576–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranta V, Urtti A (2006). Transscleral drug delivery to the posterior eye: prospects of pharmacokinetic modeling. Adv Drug Deliv Rev 58: 1164–1181. [DOI] [PubMed] [Google Scholar]
- Rao RC, Dedania VS, Johnson MW (2017). Stem cells for retinal disease: a perspective on the promise and perils. Am J Ophthalmol 179: 32–38. [DOI] [PubMed] [Google Scholar]
- Rathod LV, Kapadia R, Sawant KK (2017). A novel nanoparticles impregnated ocular insert for enhanced bioavailability to posterior segment of eye: in vitro, in vivo and stability studies. Mater Sci Eng C 71: 529–540. [DOI] [PubMed] [Google Scholar]
- Ratner BD (2002). Reducing capsular thickness and enhancing angiogenesis around implant drug release systems. J Control Release 78: 211–218. [DOI] [PubMed] [Google Scholar]
- Reddy R, Kim SJ (2011). Critical appraisal of ophthalmic ketorolac in treatment of pain and inflammation following cataract surgery. Clin Ophthalmol (Auckland, NZ) 5: 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repetto R, Siggers JH, Stocchino A (2010). Mathematical model of flow in the vitreous humor induced by saccadic eye rotations: effect of geometry. Biomech Model Mechanobiol 9: 65–76. [DOI] [PubMed] [Google Scholar]
- Repetto R, Stocchino A, Cafferata C (2005). Experimental investigation of vitreous humour motion within a human eye model. Phys Med Biol 50: 4729–4743. [DOI] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A et al (2014). Reversal of diabetes with insulin‐producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32: 1121–1133. [DOI] [PubMed] [Google Scholar]
- Rodrigues EB, Farah ME, Maia M, Penha FM, Regatieri C, Melo GB et al (2009). Therapeutic monoclonal antibodies in ophthalmology. Prog Retin Eye Res 28: 117–144. [DOI] [PubMed] [Google Scholar]
- Rodríguez Villanueva J, Navarro MG, Rodríguez Villanueva L (2016). Dendrimers as a promising tool in ocular therapeutics: latest advances and perspectives. Int J Pharm 511: 359–366. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Aller M, Guinchard S, Guillarme D, Pupier M, Jeannerat D, Rivara‐Minten E et al (2015). New prostaglandin analog formulation for glaucoma treatment containing cyclodextrins for improved stability, solubility and ocular tolerance. Eur J Pharm Biopharm 95: 203–214. [DOI] [PubMed] [Google Scholar]
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY et al (2006). Ranibizumab for neovascular age‐related macular degeneration. N Engl J Med 355: 1419–1431. [DOI] [PubMed] [Google Scholar]
- Rowe‐Rendleman CL, Durazo SA, Kompella UB, Rittenhouse KD, Di Polo A, Weiner AL et al (2014). Drug and gene delivery to the back of the eye: from bench to bedside. Invest Ophthalmol Vis Sci 55: 2714–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saettone MF, Monti D, Torracca MT, Chetoni P (1994). Mucoadhesive ophthalmic vehicles: evaluation of polymeric low‐viscosity formulations. J Ocul Pharmacol 10: 83–92. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Vooturi SK, Kompella UB (2011). Druggable targets and therapeutic agents for disorders of the back of the eye In: Kompella UB, Edelhauser HF. (eds). Drug product development for the back of the eye. Springer: New York, 495–563. [Google Scholar]
- Schlichtenbrede FC, MacNeil a, Bainbridge JWB, Tschernutter M, Thrasher aJ, Smith aJ et al (2003). Intraocular gene delivery of ciliary neurotrophic factor results in significant loss of retinal function in normal mice and in the Prph2Rd2/Rd2 model of retinal degeneration. Gene Ther 10: 523–527. [DOI] [PubMed] [Google Scholar]
- Schön C, Biel M, Michalakis S (2015). Retinal gene delivery by adeno‐associated virus (AAV) vectors: strategies and applications. Eur J Pharm Biopharm 95 (Pt B): 343–352. [DOI] [PubMed] [Google Scholar]
- Sebag J (1989). Development and aging of the vitreous In: The Vitreous. Structure, function and pathobiology. Springer: New York, pp. 73–95. [Google Scholar]
- Sepahvandi A, Eskandari M, Moztarzadeh F (2016). Drug delivery systems to the posterior segment of the eye: implants and nanoparticles. BioNanoScience 6: 276–283. [Google Scholar]
- Sharma R, Das A, Alzhrani RM, Kang D, Bhaduri SB, Boddu SHS (2017). Comparison of electrospun and solvent cast polylactic acid (PLA)/ poly(vinyl alcohol) (PVA) inserts as potential ocular drug delivery vehicles. Mater Sci Eng C 77: 895–903. [DOI] [PubMed] [Google Scholar]
- Shaunak S, Thomas S, Gianasi E, Godwin A, Jones E, Teo I et al (2004). Polyvalent dendrimer glucosamine conjugates prevent scar tissue formation. Nat Biotechnol 22: 977–984. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lv H, Fu Y, Lu Q, Zhong J, Ma D et al (2013). Preparation and characterization of a hydrogel carrier to deliver gatifloxacin and its application as a therapeutic contact lens for bacterial keratitis therapy. Biomed Mater (Bristol, England) 8: 55007. [DOI] [PubMed] [Google Scholar]
- Shu XZ, Liu Y, Palumbo FS, Luo Y, Prestwich GD (2004). In situ crosslinkable hyaluronan hydrogels for tissue engineering. Biomaterials 25: 1339–1348. [DOI] [PubMed] [Google Scholar]
- Shulman S, Johannesson G, Stefansson E, Loewenstein A, Rosenblatt A, Habot‐Wilner Z (2015). Topical dexamethasone‐cyclodextrin nanoparticle eye drops for non‐infectious Uveitic macular oedema and vitritis – a pilot study. Acta Ophthalmol 93: 411–415. [DOI] [PubMed] [Google Scholar]
- Siefert B, Keipert S (1997). Influence of alpha‐cyclodextrin and hydroxyalkylated β‐cyclodextrin derivatives on the in vitro corneal uptake and permeation of aqueous pilocarpine‐HCl solutions. J Pharm Sci 86: 716–720. [DOI] [PubMed] [Google Scholar]
- Sieving PA, Caruso RC, Tao W, Coleman HR, Thompson DJS, Fullmer KR et al (2006). Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci U S A 103: 3896–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R, Oliveira MGD (2007). Effect of the cross‐linking degree on the morphology of poly(NIPAAm‐co‐AAc) hydrogels. Polymer 48: 4114–4122. [Google Scholar]
- Simões S, Figueiras A, Veiga F (2012). Modular hydrogels for drug delivery. J Biomater Nanobiotechnol 3: 185–199. [Google Scholar]
- Sjöquist B, Basu S, Byding P, Bergh K, Stjernschantz J (1998). The pharmacokinetics of a new antiglaucoma drug, latanoprost, in the rabbit. Drug Metab Dispos 26: 745–754. [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spataro G, Malecaze F, Turrin CO, Soler V, Duhayon C, Elena PP et al (2010). Designing dendrimers for ocular drug delivery. Eur J Med Chem 45: 326–334. [DOI] [PubMed] [Google Scholar]
- Stapleton F, Keay L, Edwards K, Naduvilath T, Dart JK, Brian G et al (2008). The incidence of contact lens‐related microbial keratitis in Australia. Ophthalmology 115: 1655–1662. [DOI] [PubMed] [Google Scholar]
- Stewart MW, Rosenfeld PJ, Penha FM, Wang F, Yehoshua Z, Bueno‐Lopez E et al (2012). Pharmacokinetic rationale for dosing every 2 weeks versus 4 weeks with intravitreal ranibizumab, bevacizumab, and aflibercept (vascular endothelial growth factor Trap‐eye). Retina (Philadelphia, Pa) 32: 434–457. [DOI] [PubMed] [Google Scholar]
- Stile RA, Burghardt WR, Healy KE (1999). Synthesis and characterization of injectable poly (N‐isopropylacrylamide)‐based hydrogels that support tissue formation in vitro. Macromolecules 32: 7370–7379. [Google Scholar]
- Stocchino A, Repetto R, Cafferata C (2007). Eye rotation induced dynamics of a Newtonian fluid within the vitreous cavity: the effect of the chamber shape. Phys Med Biol 52: 2021–2034. [DOI] [PubMed] [Google Scholar]
- Tamilvanan S, Raja NL, Sa B, Basu SK (2010). Clinical concerns of immunogenicity produced at cellular levels by biopharmaceuticals following their parenteral administration into human body. J Drug Target 18: 489–498. [DOI] [PubMed] [Google Scholar]
- Tanito M, Hara K, Takai Y, Matsuoka Y, Nishimura N, Jansook P et al (2011). Topical dexamethasone‐cyclodextrin microparticle eye drops for diabetic macular edema. Invest Ophthalmol Vis Sci 52: 7944–7948. [DOI] [PubMed] [Google Scholar]
- Taylor HR (2009). Glaucoma: where to now? Ophthalmology 116: 821–822. [DOI] [PubMed] [Google Scholar]
- Teo L, Lim L, Tan DT, Chan TK, Jap A, Ming LH (2011). A survey of contact lens complications in Singapore. Eye Contact Lens 37: 16–19. [DOI] [PubMed] [Google Scholar]
- Thakur SS, Barnett NL, Donaldson MJ, Parekh HS (2014). Intravitreal drug delivery in retinal disease: are we out of our depth? Expert Opin Drug Deliv 11: 1575–1590. [DOI] [PubMed] [Google Scholar]
- Tham Y‐C, Li X, Wong TY, Quigley HA, Aung T, Cheng C‐Y (2014). Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta‐analysis. Ophthalmology 121: 2081–2090. [DOI] [PubMed] [Google Scholar]
- Thrimawithana T, Young S, Bunt C (2011). Drug delivery to the posterior segment of the eye. Drug Discov Today 16: 270–277. [DOI] [PubMed] [Google Scholar]
- Tiffany JM (2008). The normal tear film. Dev Ophthalmol 41: 1–20. [DOI] [PubMed] [Google Scholar]
- Turturro SB, Guthrie MJ, Appel AA, Drapala PW, Brey EM, Pérez‐luna VH et al (2011). The effects of cross‐linked thermo‐responsive PNIPAAm‐based hydrogel injection on retinal function. Biomaterials 32: 3620–3626. [DOI] [PubMed] [Google Scholar]
- Urtti A (2006). Challenges and obstacles of ocular pharmacokinetics and drug delivery. Adv Drug Deliv Rev 58: 1131–1135. [DOI] [PubMed] [Google Scholar]
- Van Santvliet L, Ludwig A (2004). Determinants of eye drop size. Surv Ophthalmol 49: 197–213. [DOI] [PubMed] [Google Scholar]
- Vandamme TF, Brobeck L (2005). Poly(amidoamine) dendrimers as ophthalmic vehicles for ocular delivery of pilocarpine nitrate and tropicamide. J Control Release 102: 23–38. [DOI] [PubMed] [Google Scholar]
- Vellonen KS, Malinen M, Mannermaa E, Subrizi A, Toropainen E, Lou YR et al (2014). A critical assessment of in vitro tissue models for ADME and drug delivery. J Control Release 190: 94–114. [DOI] [PubMed] [Google Scholar]
- Vermonden T, Censi R, Hennink WE (2012). Hydrogels for protein delivery. Chem Rev 112: 2853–2888. [DOI] [PubMed] [Google Scholar]
- Vugmeyster Y, Xu X, Theil F‐P, Khawli La, Leach MW (2012). Pharmacokinetics and toxicology of therapeutic proteins: Advances and challenges. World J Biol Chem 3: 73–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite D, Wang Y, Jones D, Stitt A, Raj Singh TR (2017). Posterior drug delivery via periocular route: challenges and opportunities. Ther Deliv 8: 685–699. [DOI] [PubMed] [Google Scholar]
- Wang W, Singh SK, Li N, Toler MR, King KR, Nema S (2012). Immunogenicity of protein aggregates‐concerns and realities. Int J Pharm 431: 1–11. [DOI] [PubMed] [Google Scholar]
- Wang WY, Yao C, Shao YF, Mu HJ, Sun KX (2011). Determination of puerarin in rabbit aqueous humor by liquid chromatography tandem mass spectrometry using microdialysis sampling after topical administration of puerarin PAMAM dendrimer complex. J Pharm Biomed Anal 56: 825–829. [DOI] [PubMed] [Google Scholar]
- Watsky MA, Jablonski MM, Edelhauser HF (1988). Comparison of conjunctival and corneal surface areas in rabbit and human. Curr Eye Res 7: 483–486. [DOI] [PubMed] [Google Scholar]
- Weidle UH, Tiefenthaler G, Weiss EH, Georges G, Brinkmann U (2013). The intriguing options of multispecific antibody formats for treatment of cancer. Cancer Genomics Proteomics 10: 1–18. [PubMed] [Google Scholar]
- Weleber RG, Pennesi ME, Wilson DJ, Kaushal S, Erker LR, Jensen L et al (2016). Results at 2 years after gene therapy for RPE65‐deficient leber congenital amaurosis and severe early‐childhood‐onset retinal dystrophy. Ophthalmology 123: 1606–1620. [DOI] [PubMed] [Google Scholar]
- Wirtz R (1908). Die ionentherapie in der Augenheilkunde. Klin Mbl Augenheilk.
- Yao W, Sun K, Mu H, Liang N, Liu Y, Yao C et al (2010). Preparation and characterization of puerarin‐dendrimer complexes as an ocular drug delivery system. Drug Dev Ind Pharm 36: 1027–1035. [DOI] [PubMed] [Google Scholar]
- Yavuz B, Pehlivan SB, Vural İ, Ünlü N (2015). In vitro/in vivo evaluation of dexamethasone – PAMAM dendrimer complexes for retinal drug delivery. J Pharm Sci 104: 3814–3823. [DOI] [PubMed] [Google Scholar]
- Yellepeddi VK, Palakurthi S (2016). Recent advances in topical ocular drug delivery. J Ocul Pharmacol Ther 32: 67–82. [DOI] [PubMed] [Google Scholar]
- Yıldız B, Işık B, Kış M (2002). Synthesis and characterization of thermoresponsive isopropylacrylamide–acrylamide hydrogels. Eur Polym J 38: 1343–1347. [Google Scholar]
- Yilmaz T, Cordero‐Coma M, Federici TJ (2011). Pharmacokinetics of triamcinolone acetonide for the treatment of macular edema. Expert Opin Drug Metab Toxicol 7: 1327–1335. [DOI] [PubMed] [Google Scholar]
- Yoshida F, Topliss JG (1996). Unified model for the corneal permeability of related and diverse compounds with respect to their physicochemical properties. J Pharm Sci 85: 819–823. [DOI] [PubMed] [Google Scholar]
- Zacharias LC, Lin T, Migon R, Ghosn C, Orilla W, Feldmann B et al (2013). Assessment of the differences in pharmacokinetics and pharmacodynamics between four distinct formulations of triamcinolone acetonide. Retina (Philadelphia, Pa) 33: 522–531. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Krishna N, Lettinga MP, Vermant J, Grelet E (2009). Reversible gelation of rod‐like viruses grafted with thermoresponsive polymers. Langmuir 25: 2437–2442. [DOI] [PubMed] [Google Scholar]