

Abstract
Background and Purpose
The tumour suppressor p53 is traditionally recognized as a surveillance molecule to preserve genome integrity. Recent studies have demonstrated that it contributes to metabolic diseases. Here, we investigated the role of p53 in the regulation of bile acid disposition and cholestasis.
Experimental Approach
The bile acid disposition‐related gene expression profile affected by p53 activation was assessed in mouse primary hepatocytes with p53 depletion and in Trp53‐null mice. Dual luciferase reporter assay was used to detect the transcriptional activities of target genes. Anticholestatic effects of p53 activator doxorubicin were investigated in a 0.5% cholic acid‐fed mouse model of cholestasis. Changes in bile acids were evaluated using metabolomics analysis.
Key Results
Doxorubicin‐mediated p53 activation induced Cyp2b10, Sult2a1 and Abcc2/3/4 expression in mice in vitro and in vivo. ABCC3 and CYP2B6 (human orthologue of Cyp2b10) were identified as direct p53 target genes. Doxorubicin attenuated cholic acid‐induced cholestasis in mice, as demonstrated by shrunken gall bladder, decreased serum total bile acid and total bilirubin levels and alkaline phosphatase activity. Targeted metabolomics analysis revealed that doxorubicin enhanced the excretion of bile acid metabolites from serum and liver to intestine and faeces. Up‐regulation of Cyp2b10, Sult2a1 and Abcc2/3/4 expression was further confirmed in cholestatic mice. Cholic acid‐induced cholestatic injury was aggravated in p53‐deficient mice and levels of bile acid in intestine and faeces were decreased.
Conclusions and Implications
Our findings suggest a novel role of p53 in promoting bile acid disposition and alleviating cholestatic syndrome, which provides a potential therapeutic target for cholestasis.
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate transaminase
- BSEP
bile salt export pump
- CCND1
cyclin D1
- CDCA
chenodeoxycholic acid
- CYP
cytochrome P450
- DCA
deoxycholic acid
- H&E
haematoxylin and eosin
- HDCA
hyodeoxycholic acid
- MRP
multidrug resistance‐associated protein
- NTCP
Na+‐dependent taurocholate transporter
- OATP
organic anion transporting polypeptide
- OST
organic solute transporter
- p53RE
p53 response element
- PCA
principal components analysis
- qRT‐PCR
quantitative real‐time PCR
- siRNA
small interference RNA
- SULT
sulfotransferase
- TCA
taurocholic acid
- THDCA
taurohyodeoxycholic acid
- T‐β‐MCA
tauro‐β‐muricholic acid
- WT
wild type
Introduction
Cholestasis is a classic disorder of bile acid homeostasis, which is caused by a disruption of bile flow and accumulation of toxic bile acids in the liver and systemic circulation. Cholestasis can subsequently lead to injury of hepatocytes and biliary epithelial cells, which eventually causes liver failure and increases the risk of hepatocellular or cholangiocellular carcinomas (Trauner et al., 1998). A useful strategy for protecting against cholestasis is to regulate bile acid haemostasis by promoting the elimination of excess bile acids.
Multiple enzymes and transporters regulating bile acids synthesis and disposition are implicated in the response of adaptive defence to cholestasis (Boyer, 2007). Cholesterol 7α‐hydroxylase (CYP7A1) mediates the rate‐limiting step of bile acid synthesis in the classical pathway. The Na+‐dependent taurocholate transporter (NTCP) is the major bile acid uptake transporter in the basolateral membrane of hepatocytes, while organic anion transporting polypeptides (OATPs) mediate the Na+‐independent bile acid uptake (Trauner and Boyer, 2003). In the liver, cytochrome P450 (CYP) 3A4 and CYP2B6 (human orthologue of mouse Cyp3a11 and Cyp2b10, respectively) are the major phase I enzymes that convert bile acids into more hydrophilic and less toxic metabolites under cholestasis (Wagner et al., 2005; Chen et al., 2014a). Bile acids also undergo glucuronidation and sulfonation under catalysis of uridine diphosphate (UDP)‐glucuronosyltransferase and sulfotransferase 2A isoforms (SULT2As) respectively (Weinshilboum et al., 1997; Gall et al., 1999; Belanger et al., 2003; Trottier et al., 2006). The bile salt export pump (BSEP) and multidrug resistance‐associated protein‐2 (ABCC2 also known as MRP2) are responsible for canalicular efflux of bile acids, while MRP3 (ABCC3), MRP4 (ABCC4) and organic solute transporter α and β (OSTα/β) heterodimer are involved in sinusoidal excretion of bile acids into the systemic circulation.
The underlying molecular mechanisms orchestrating these enzymes and transporters are mediated mainly at a transcriptional level via a complex network involving nuclear receptors such as the farnesoid X receptor, pregnane X receptor, constitutive androstane receptor and vitamin D receptor (Makishima et al., 1999; Xie et al., 2001; Makishima et al., 2002). Activation of nuclear receptor‐mediated pathways has currently become a promising therapeutic approach in cholestasis (Gonzalez‐Sanchez et al., 2015). However, whether transcription factors other than nuclear receptors are involved in bile acid metabolism and transport still needs to be investigated, and revealing these potential targets may provide more therapeutic options for cholestasis.
The tumour suppressor protein p53 traditionally acts as a transcription factor in controlling genomic stability and cell growth. Upon stress, activated p53 accumulates in the nucleus, which promotes the transcription of a myriad of genes to launch cell cycle arrest, DNA repair and apoptosis (Levine and Oren, 2009; Goldstein et al., 2011). Recent studies reported that p53 has a role in regulating metabolic pathways, such as glycolysis, oxidative phosphorylation, amino acid metabolism and lipid metabolism (Bensaad et al., 2006; Matoba et al., 2006; Kawauchi et al., 2008; Hu et al., 2010; Suzuki et al., 2010; Goldstein et al., 2012). Most recently, an increase in p53 has also been shown to be associated with bile acid metabolic disorders. Immunostaining of liver sections from patients with primary biliary cirrhosis showed that they exhibited increased levels of p53‐positive biliary epithelial cells (Harada et al., 2001). A markedly increased p53 expression level was also observed in mice with common bile duct ligation and in lithocholic acid‐treated primary human hepatocytes and HuH‐7 cells (Yang et al., 2009). These studies indicate that an up‐regulation of p53 may represent a positive feedback to protect against injuries caused by bile acid overload. However, the underlying mechanism still needs to be elucidated. A recent study demonstrated that activation of p53 inhibits bile acid synthesis by regulating the expression of a small heterodimer partner via p53 response elements (p53REs), which subsequently suppresses the expression of bile acid synthesis enzymes CYP7A1 and CYP8B1 (Kim and Lee, 2011). In addition to synthesis, the metabolism and transport of bile acids also play an important role in maintaining bile acid homeostasis. The maintenance of bile acid homeostasis through promoting metabolism and elimination of excess bile acids is a key strategy for protecting against cholestasis. Therefore, the promotion of the transformation of hydrophilic bile acids and/or secretion of bile acids are key therapeutic strategies for cholestasis and bile acid metabolic disorders. However, whether p53 is involved in bile acid disposition such as its metabolism and transport remains unclear.
In the current study, the effect of p53 on the expression of genes involved in bile acid disposition was examined in primary hepatocytes and mice. We found that the expression of bile acid disposition‐related genes Cyp2b10, Sult2a1 and Abcc2/3/4 (encoding MRP2/3/4) was increased by p53 and p53 directly activated the transcription of CYP2B6 and ABCC3. By using a cholic acid‐induced cholestatic mouse model, we further investigated the effects of p53 activation on cholestasis and liver injury. Furthermore, targeted metabolomic analysis was performed to investigate detailed changes of bile acids constitution and abundance. The results demonstrate a role of p53 in promoting bile acid disposition and alleviating cholestatic syndrome by regulating expression of genes involved in bile acid metabolism and transport, including Cyp2b10, Sult2a1 and Abcc2/3/4. This finding provides a potential new therapeutic target for cholestasis and bile acid metabolic disorders.
Methods
Animals and treatments
Male C57BL/6 mice (8–9 weeks) were purchased from Guangdong Animal Experimental Centre (Guangzhou, China). The Trp53‐null mice were established by NIFDC and Beijing Biocytogen Co., Ltd, and supplied by the National Centre of Laboratory Rodents. The animal study protocols were approved by the Institutional Animal Care and Use Committee at Sun Yat‐sen University (Guangzhou, China). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Mice were housed in stainless polypropylene cage (size: 320 * 215 * 170 mm) with wood shavings as bedding materials. A maximum of six mice was kept in a single cage. Housing rooms were maintained at 22–24°C with a light/dark cycle of 12/12 h and 55–60% relative humidity with feed and water provided ad libitum in a specific pathogen‐free environment. Mice were assigned to groups randomly, and no mice were excluded from statistical analysis. Upon the completion of testing, all mice were killed by inhalation of diethyl ether, followed by cervical dislocation. Blood samples were collected from the retroorbital plexus of anaesthetized mice.
Treatment 1
Doxorubicin (Dox) (Aladdin Company, Shanghai, China) at 10 mg·kg−1 or saline was administered to Trp53‐null and corresponding wild‐type (WT) mice by i.p. injections. Mice were killed 24 h after doxorubicin treatment. Liver tissues were collected and snap‐frozen in liquid nitrogen.
Treatment 2
Mice were fed either a standard rodent chow diet or a chow diet supplemented with 0.5% (wt.wt‐1) cholic acid (Aladdin Company) for 4 days. Doxorubicin (10 mg·kg−1) or saline was i.p. injected to mice on day 1 and day 3. Then 24 h after doxorubicin treatment, the mice were killed and serum, liver, intestine and faeces samples were collected and snap‐frozen in liquid nitrogen.
Treatment 3
The Trp53‐null and corresponding WT mice were fed a chow diet supplemented with 0.5% (wt.wt‐1) cholic acid for 4 days. They were then killed and the samples were collected as above.
Isolation, culture and treatment of mouse primary hepatocytes
Hepatocytes from male C57BL/6 mice (8–9 weeks) were isolated by a two‐step collagenase digestion method as described previously (Klaunig et al., 1981). Hepatocytes were cultured according to our previously published procedure (Chen et al., 2014b). Hepatocytes were seeded in 12‐well plates at a density of 6 × 105 cells per well. Then 100 nmol negative small interference RNA (siRNA) or siRNA targeting Trp53 (RiboBio, Guangzhou, China) were transiently transfected using X‐tremeGENE siRNA Transfection Reagent (Roche Diagnostics, Mannheim, Germany). Cells were treated with 0.5 μM doxorubicin at 12 h after siRNA transfection and incubated for 48 h. The total culture medium volume in each well was not changed during the treatment period, in accord with the instructions of the transfection reagent. Cells were harvested for quantitative real‐time PCR (qRT‐PCR) or Western blot analysis.
Histological and biochemical assessments
Liver tissues were immediately fixed in formaldehyde, embedded in paraffin wax, sectioned and stained for haematoxylin and eosin (H&E). H&E‐stained liver sections were examined using an Olympus BX41 microscope. Cholestasis and liver injury were evaluated by measuring the serum activities of alanine aminotransferase (ALT), aspartate transaminase (AST) and alkaline phosphatase (ALP). Total serum bile acids and bilirubin levels and hepatic total bile acid levels were measured using commercially available kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The volume of the contents obtained from each gall bladder was measured so as to obtain the gall bladder volume.
Dual luciferase activity assay
HEK‐293T cells were plated in 96‐well plates and transfected (Lipofectamine 2000, Invitrogen, Grand Island, NY, USA) with a pGL3‐basic vector harbouring 3.0 kb upstream of the transcription start site (TSS) of the specified gene fragment (120 ng per well), luc‐TK (5 ng per well) and p53 overexpression plasmid (60 ng per well; kindly provided by Dr Jing Huang, National Institutes of Health, Bethesda, MD, USA). Luminescence values were measured at 24 h after transfection, by using the Dual Reporter Assay System and following the manufacturer's instructions (Promega, Madison, WI, USA). Firefly luciferase activity was normalized to renilla activity for each well.
Chromatin immunoprecipitation–qPCR assay
The chromatin immunoprecipitation assay was performed using Pierce Agarose ChIP Kit (Thermo Fisher Scientific, Rockford, IL, USA) in HepG2 cells. Briefly, nuclear proteins were crosslinked to genomic DNA by 1% formaldehyde. Subsequently, cells were collected in cold PBS containing protease inhibitors. Following centrifugation, pellets were resuspended in lysis buffer. Chromatin DNA fragments were precipitated with 5 μg anti‐p53 antibody (Abcam, Cambridge, MA, USA) overnight at 4°C. Protein G‐sepharose beads were added, then sequentially, the resultant immune complexes were washed with a series of wash buffers. After centrifugation, the immune complexes were resuspended in elution buffers. Human genomic DNA was amplified using primers (Supporting Information Table S2) by qRT‐PCR, according to the putative p53 binding sites identified in silico. Input sample and IgG antibody were used as a positive and negative control respectively.
LC/MS and metabolomics analysis
Protein in serum samples was removed by mixing with 67% aqueous acetonitrile, followed by centrifuging at 18 000× g for 20 min. Protein in liver or intestine was removed by homogenization in 50% aqueous acetonitrile, followed by the centrifugation. Twenty milligram faeces in 400 μL PBS was vortexed and centrifuged at 18 000× g for 20 min at 4°C, and 20 μL supernatant was added to the tube with 67% aqueous acetonitrile followed by centrifugation to remove proteins and particulates.
The supernatant was transferred to an UPLC vial, and a 5 μL aliquot of deproteinized samples was injected into an Ultimate 3000 HPLC system (Dionex Corporation, Sunnyvale, CA, USA) interfaced with a Q Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Chromatographic separations were performed on a Waters XTerra® MS C18 5 μm column (100 × 2.1 mm, Thermo Fisher Scientific, Waltham, MA, USA). The mobile phase consisted of solvent A (0.1% formic acid in water) and solvent B (acetonitrile). The gradient programme was as follows: 0 min 0% B, 1 min 0% B, 9 min 70% B, 16 min 83% B, 17 min 90% B, 19 min 100% B, 20 min 100% B, 25 min 0% B and 25 min stop. The flow rate was 0.3 mL·min−1. Electrospray negative ionization mode was used for analysis. The spray voltage was set to 2.8 kV. Capillary and aux gas heater temperatures were set at 325 and 350°C respectively. Nitrogen was used as both sheath gas flow rate 40 arb and aux gas flow rate 10 arb.
The mass spectral data were aligned by using SIEVE 2.2 (Thermo Fisher Scientific, Waltham, MA, USA). The multivariate data matrix was further exported into SIMCA‐P + 13 software (Umetrics, Kinnelon, NJ, USA). Either unsupervised principal components analysis (PCA) or supervised orthogonal partial least squares discriminant analysis models were constructed to analyse the data from the serum, liver, intestine and faeces samples. Ions that were significantly altered were subjected to further analysis, for confirmation, by comparing the retention times and fragmentation patterns with authentic standards of bile acids.
qRT‐PCR analysis
The qRT‐PCR analysis of the expression of target genes in mouse livers and primary hepatocytes was performed as described previously (Chen et al., 2014b; 2015). The gene‐specific primers were obtained from a PrimerBank (Spandidos et al., 2010; Wang et al., 2012), and the sequences are listed in Supporting Information Table S1.
Western blot analysis
Western blot analysis of target proteins in mice livers was carried out as described previously (Chen et al., 2014b). Blots were incubated with primary antibodies against, including CYP3A (L‐14), p21 (F‐8), MRP2 (H‐17), MRP3 (C‐18) and SULT2A1 (FL‐285) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); p53 (pab 240 and ab26) and MRP4 (M4I‐10) (Abcam); GAPDH (14c10) (Cell Signalling Technologies, Danvers, MA, USA); CYP2B10 (AB9916) (Millipore, Darmstadt, Germany); cyclin D1 (CCND1) (AB20509b) and PCNA (AB20014) (Sangon, Shanghai, China).
Statistical analysis
The data are presented as mean ± SEM. One‐way ANOVA followed by Dunnett's multiple comparison post hoc test or unpaired Student's t‐test was used for statistical analysis of data using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA). Only the comparisons indicated above the bars are being made, and difference was considered as significant if the probability (P value) was less than 0.05 (P < 0.05). The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
p53 regulates the expression of genes involved in bile acid disposition in mouse primary hepatocytes
Mouse primary hepatocytes were adopted as the cell model in the current study, since bile acid disposition‐related metabolic pathways in this model were preserved as in vivo. As a p53 signalling pathway activator, doxorubicin markedly up‐regulated the expression of p53, whereas this increase was abolished in p53 knockdown mouse primary hepatocytes (Figure 1A). Furthermore, mRNA levels of Cdkn1a (encoding p21), a well‐established p53 target gene, were regulated correspondingly by doxorubicin treatment and p53 knockdown (Figure 1B).
Figure 1.
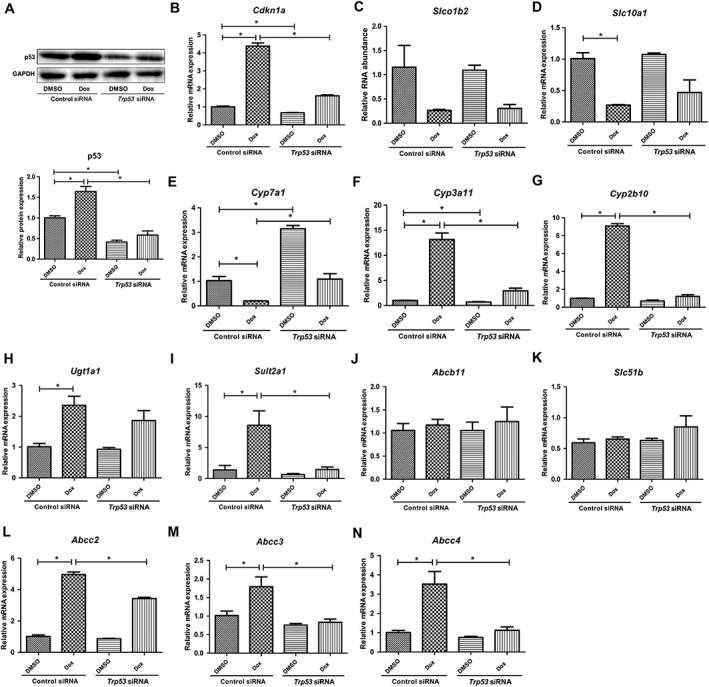
Effects of p53 activation on bile acid disposition‐related gene expression in primary hepatocytes of Trp53‐WT mice. The cells were incubated with Trp53 siRNA for 60 h and treated with 0.5 μM doxorubicin (Dox) for the last 48 h. mRNA and protein were measured by qRT‐PCR and Western blot analysis respectively. (A) Western blots of p53 and GAPDH proteins and the corresponding quantification of p53 relative to GAPDH. (B–N) mRNA expression of Cdkn1a, Slco1b2, Slc10a1, Cyp7a1, Cyp3a11, Cyp2b10, Ugt1a1, Sult2a1, Abcb11, Slc51b, Abcc2, Abcc3 and Abcc4. Data are the mean ± SEM (n = 5). *P < 0.05.
The expression of genes involved in bile acid disposition was detected after doxorubicin treatment in both normal and p53 knockdown mouse primary hepatocytes. The expression of bile acid uptake transporters Slc10a1 (encoding NTCP) and Slco1b2 (encoding OATP1b2) was suppressed by doxorubicin even under p53 knockdown conditions (Figure 1C, D). This indicates that changes in these two genes were independent of p53 activation. Consistent with previous findings (Kim and Lee, 2011), Cyp7a1 expression was significantly down‐regulated by p53 (Figure 1E). The expression of Cyp3a11 and Cyp2b10, two genes encoding enzymes involved in bile acid hydroxylation, was significantly up‐regulated by doxorubicin treatment (Figure 1F, G). Similarly, the expression levels of the gene encoding the enzyme for bile acid sulfomation, Sult2a1, were elevated after doxorubicin treatment (Figure 1I). Although doxorubicin increased the gene expression level of the glucuronidation enzyme Ugt1a1; this effect was not mediated via the p53 pathway, as the increase in Ugt1a1 levels was not abolished by Trp53 silencing (Figure 1H). We found that activation of p53 had no effect on Abcb11 (encoding BSEP) and Slc51b (encoding OSTβ) expression, but induced a significant up‐regulation of Abcc2, Abcc3 and Abcc4 levels (Figure 1J–N). Our results demonstrate that various genes involved in bile acid disposition are regulated by p53 signalling pathway.
p53 regulates bile acid disposition‐related genes in vivo
Trp53‐null mice were used to investigate the regulation of p53 on bile acid disposition‐related gene expression in vivo. After ensuring p21 induction by doxorubicin in Trp53‐WT mice instead of Trp53‐null mice, we measured the mRNA and protein levels of bile acid disposition‐related enzymes or transporters after doxorubicin treatment in both Trp53‐WT and Trp53‐null mice. Activation of p53 induced an up‐regulation of CYP2B10, SULT2A1 and MRP2/3/4 expression and a down‐regulation of Cyp7a1 expression, which were consistent with our observations in vitro (Figure 2B, D–K). Interestingly, CYP3A11 expression was decreased in doxorubicin‐treated Trp53‐WT mice, which is opposite to the result in vitro (Figure 2C, I, J). However, a decrease in CYP3A11 expression was also observed in Trp53‐null mice, which indicates that the role of p53 in regulating CYP3A11 expression is complicated (Figure 2C, I, J).
Figure 2.
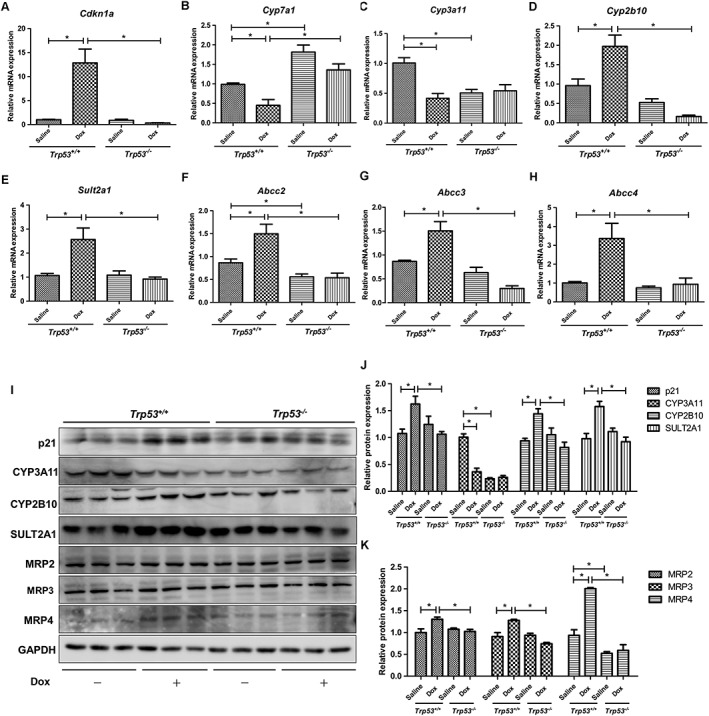
Effects of p53 activator doxorubicin (Dox) on hepatic bile acid disposition‐related gene expression in Trp53‐WT and Trp53‐null mice. Liver tissues were collected at 24 h after saline or 10 mg·kg−1 doxorubicin treatment. mRNA and protein were measured by qRT‐PCR and Western blot analysis respectively. (A–H) mRNA expression of Cdkn1a, Cyp7a1, Cyp3a11, Cyp2b10, Sult2a1, Abcc2, Abcc3 and Abcc4. (I) Western blots of p21, CYP7A1, CYP3A11, CYP2B10, SULT2A1, MRP2, MRP3, MRP4 and GAPDH, and (J, K) the corresponding quantifications relative to GAPDH. Data are the mean ± SEM (n = 5). *P < 0.05.
p53 directly activates the transcription of CYP2B6 and ABCC3
We firstly performed a reporter gene assay to determine if these bile acid disposition‐related genes were transcriptionally activated by p53. A 3.0 kb upstream of each gene's TSS was cloned into a vector containing a luciferase reporter gene, and the luciferase expression was measured following ectopic expression of p53. Luciferase expression was significantly elevated by p53 in constructs harbouring promoters of CYP2B6 and ABCC3, but not in constructs harbouring promoters of SULT2A1, ABCC2 or ABCC4 (Figure 3). These data indicate that p53 activates the transcription of CYP2B6 and ABCC3. Furthermore, we analysed human ABCC3 and CYP2B6 for motifs similar to the consensus p53Res (Smeenk et al., 2008; Schlereth et al., 2013) and identified four putative p53REs in the ABCC3 promoter region and two in the CYP2B6 promoter region. To determine whether these putative p53REs were occupied by p53 in a cellular context, we performed a chromatin immunoprecipitation–qPCR assay in HepG2 cells and confirmed that p53 was able to bind all of the predicted p53REs in promoter regions of ABCC3 and CYP2B6 (Figure 4). Collectively, these data indicate that p53 might initiate the transcription of ABCC3 and CYP2B6 by directly binding to their promoter regions.
Figure 3.
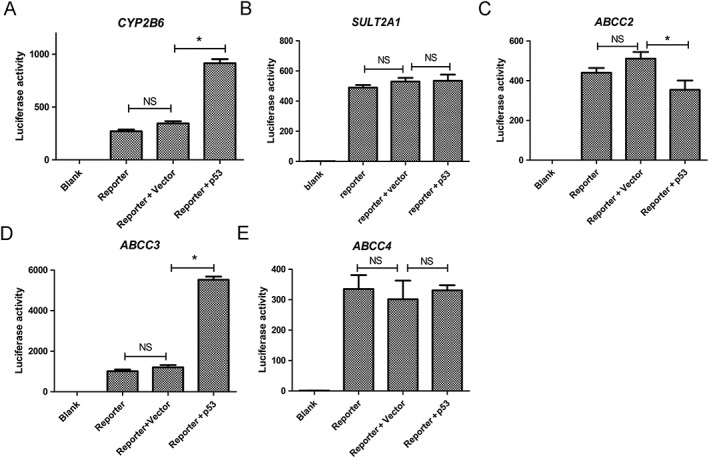
Effects of p53 on transcriptional activation of bile acid disposition‐related genes. (A–E) HEK‐293T cells were transiently transfected with a pGL3‐basic vector harbouring a fragment of 3.0 kb, upstream of TSS, of CYP2B6, SULT2A1, ABCC2, ABCC3 or ABCC4, 5 ng per well of luc‐TK and 60 ng per well of p53 overexpression plasmid. Luminescence values were estimated 24 h later. Data are the mean ± SEM (n = 5). *P < 0.05; NS, not significant.
Figure 4.
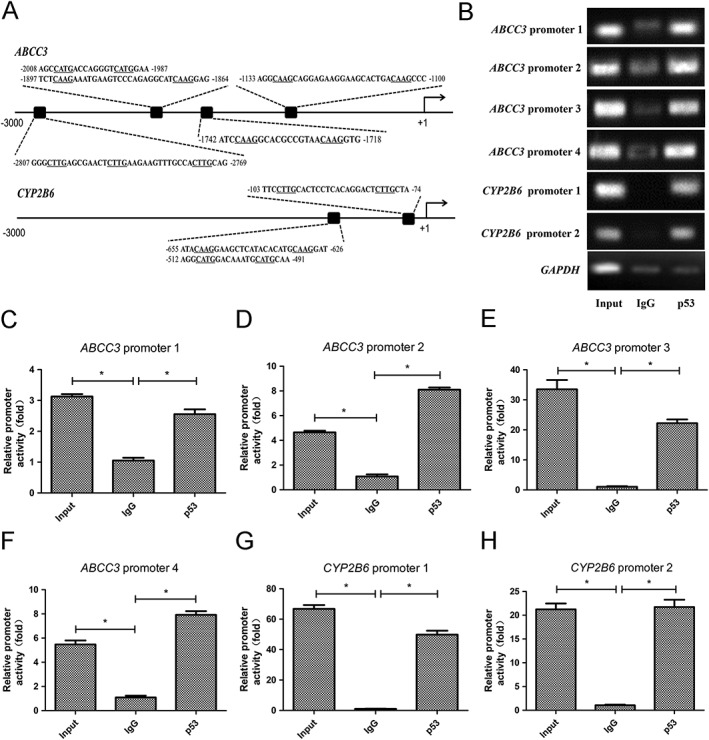
p53 directly activates the transcription of ABCC3 and CYP2B6 by binding to p53REs. (A) Alignments and locations of the indicated p53REs in promoters of ABCC3 and CYP2B6. (B) p53 binds to the genes' p53REs. Chromatin DNA fragments were precipitated with 5 μg anti‐p53 antibody. Human genomic DNA was amplified using primers by qRT‐PCR according to the putative p53 binding sites identified in silico. Input sample and IgG antibody served as positive and negative control respectively. Data are the mean ± SEM (n = 5). *P < 0.05.
Doxorubicin attenuates cholic acid‐induced cholestasis in p53‐WT mice
To further investigate the effects of p53 activation on bile acid homeostasis and cholestasis, a cholestasis model was produced by feeding Trp53‐WT mice with a diet containing 0.5% cholic acid. Doxorubicin‐treated mice exhibited dramatically shrunken gall bladders compared with saline‐treated mice (Figure 5A and Supporting Information Figure S2). Quantification of gall bladder volume is shown in Figure 5B.In addition, doxorubicin treatment diminished the cholic acid‐induced elevation of serum total bile acid and total bilirubin levels (Figure 5C, D). Also the activities of ALP were significantly decreased by doxorubicin treatment in both saline‐ and cholic acid‐treated animals, while serum ALT and AST levels were not changed by doxorubicin in cholic acid‐treated mice and even slightly increased in the saline group (Figure 5E–G). Cholic acid‐induced liver jury was not alleviated by doxorubicin, which is consistent with its lack of effect on ALT and AST levels (Figure 5A).
Figure 5.
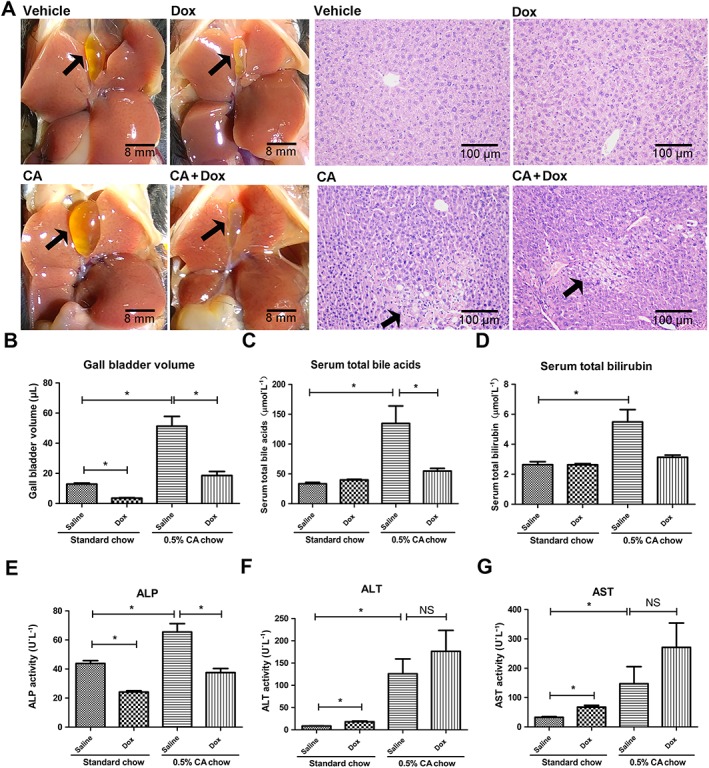
Ameliorating effects of p53 activator doxorubicin on cholic acid (CA)‐induced cholestasis in Trp53‐WT mice. Mice were fed with standard or cholic acid supplemented chow for 4 days. Doxorubicin (10 mg·kg−1) or saline was administered once per 3 days. Mice were killed at 24 h after the final doxorubicin treatment, and samples were collected. (A) Morphology of representative livers. Gall bladders are indicated by black arrows. (B) Representative H&E stained liver sections (10 × 10). Apparent subcapsular necrotic foci in the liver are indicated by black arrows. (C–G) Gall bladder volume, levels of serum total bile acid and total bilirubin, as well as serum ALP, ALT and AST activities. Data are the mean ± SEM (n = 5–6). *P < 0.05; NS, not significant.
Metabolomics analysis was performed to examine dynamic changes in bile acids in the serum, liver, intestine and faeces. PCA analysis revealed that a distribution pattern of cholic acid‐treated mice in the scores scatter plot was clearly separated from doxorubicin co‐treated mice, which indicates there is a significant difference in the endogenous metabolome between these two groups (Figure 6A–D). The above results were in accordance with the tendency observed in serum biochemistry. We further performed target metabolomics analysis on bile acids. The heat map (Figure 6E) and corresponding relative quantitative analysis using log intensity of peak area (Figure 6F–I) showed that the concentrations of all of the individual bile acids in serum and liver were decreased after doxorubicin treatment. In serum, the concentrations of cholic acid, chenodeoxycholic acid (CDCA), hyodeoxycholic acid (HDCA), tauro‐β‐muricholic acid (T‐β‐MCA) and taurocholic acid (TCA) in the doxorubicin‐treated cholic acid group were all significantly lower than those in the vehicle‐treated cholic acid group. In livers, the cholic acid, CDCA, taurodeoxycholic acid, taurohyodeoxycholic acid (THDCA) and T‐β‐MCA in doxorubicin‐treated cholic acid group were also lower than that in the vehicle‐treated cholic acid group. In contrast, increased amounts of these bile acids were observed in the faeces and intestine of the doxorubicin‐treated cholic acid group. The concentrations of cholic acid, CDCA, deoxycholic acid (DCA), taurochenodeoxycholic acid and T‐β‐MCA in intestines and cholic acid, HDCA, DCA,THDCA, T‐β‐MCA and taurocholic acid in faeces were all significantly increased in doxorubicin‐treated cholic acid group, as compared with those in the vehicle‐treated group. Of note, no significant differences were found in the taurocholic acid levels between the two groups. The heat map data indicate that doxorubicin promotes the transportation of bile acids from the serum and liver to the intestine and faeces. These results are in accord with the biochemical parameters observed. Overall, this analysis confirms that doxorubicin attenuates cholestasis by altering the distribution and abundance of individual bile acids.
Figure 6.
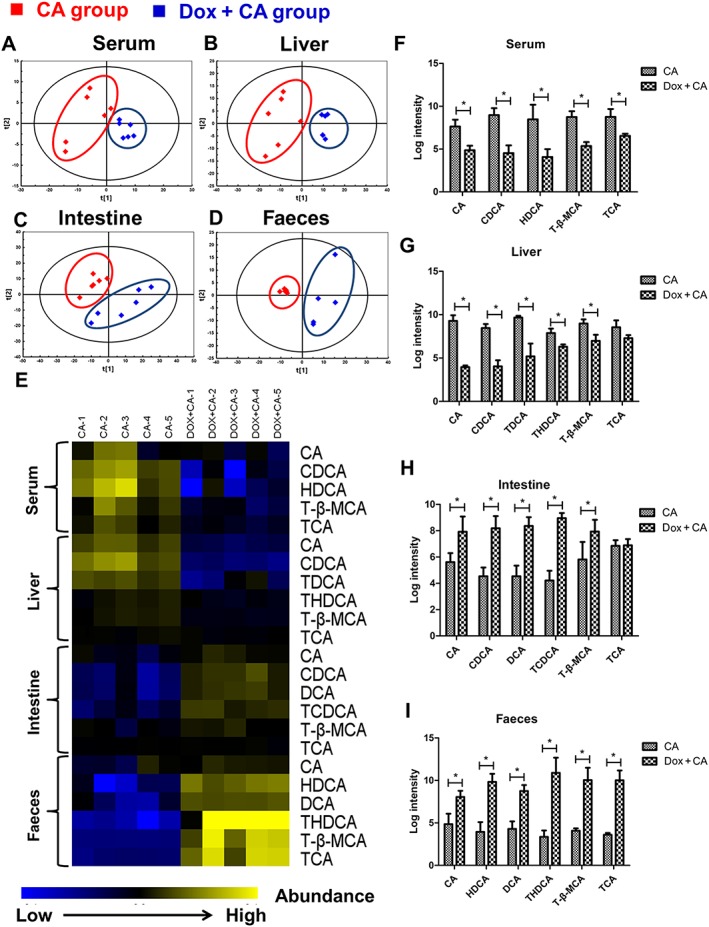
Effects of p53 activator doxorubicin on bile acid metabolome of serum, liver, intestine and faeces in cholic acid (CA)‐fed Trp53‐WT mice. Mice were fed with standard or cholic acid supplemented chow for 4 days. Doxorubicin (10 mg·kg−1) or saline was administered once per 3 days. Mice were killed at 24 h after the final doxorubicin treatment, and samples were collected (n = 5). (A–D) PCA analysis in four samples between cholic acid group (red) and cholic acid/doxorubicin group (blue). (E) Heat maps of bile acid profiles of four samples. (F–I) The relative amount of bile acid was displayed as log intensity of peak area. Data are expressed as the mean ± SEM (n = 5–6). *P < 0.05.
Doxorubicin affects bile acid disposition‐related genes in cholestatic mice
The above data demonstrate that p53 affects the expression of genes involved in bile acid disposition. We further determined whether doxorubicin had an effect on the expression of these gene in cholic acid‐induced cholestatic mice. An increase in Cdkn1a levels after doxorubicin treatment confirmed the activation of the p53 signalling pathway (Figure 7A). Doxorubicin treatment decreased Cyp7a1 expression and increased Sult2a1 and Abcc2/3/4 expression in both the standard and cholic acid diet groups (Figure 7B, F–H). Notably, a decrease in Cyp3a11 and an elevated Cyp2b10 expression was observed only in the cholic acid fed group after doxorubicin treatment (Figure 7C, D). Western blot analysis illustrated that the protein expression of CYP3A11, CYP2B10, SULT2A1 and MRP2/3/4 was consistent with the corresponding mRNA levels (Figure 7I–K). These data indicate that an up‐regulation of CYP2B10, SULT2A1, MRP2, MRP3 and MRP4, as well as a down‐regulation of CYP7A1 induced by p53 activation may contribute to the ameliorative effects of doxorubicin on cholic acid‐induced cholestasis.
Figure 7.
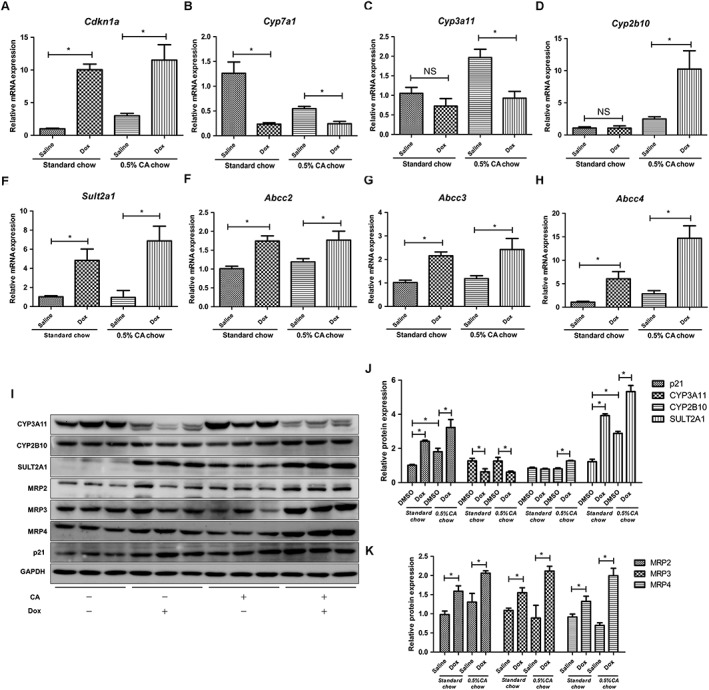
Effects of p53 activator doxorubicin on hepatic bile acid disposition‐related gene expression in cholic acid‐fed Trp53‐WT mice. With the same treatment strategy, mice were killed 24 h after the final doxorubicin treatment, and liver tissues were collected. mRNA and protein were measured by qRT‐PCR and Western blot analysis respectively. (A–H) mRNA expression of Cdkn1a, Cyp7a1, Cyp3a11, Cyp2b10, Sult2a1, Abcc2, Abcc3 and Abcc4. (I) Western blots of p21, CYP7A1, CYP3A11, CYP2B10, SULT2A1, MRP2, MRP3, MRP4 and GAPDH, and (J, K) the corresponding protein levels quantified relative to GAPDH. Data are the mean ± SEM (n = 5–6). *P < 0.05.
The effect of p53 deficiency on cholic acid‐induced cholestasis in mice
To further determine the role of p53 in cholestasis, we compared the cholestatic effects of cholic acid in Trp53‐WT and Trp53‐null mice. No apparent differences in gall bladder morphology were observed between these two groups (Figure 8A). However, gall bladder size tended to be elevated in the Trp53‐null mice (Figure 8C). Furthermore, H&E staining of liver sections illustrated that necrosis foci were more apparent in Trp53‐null mice (Figure 8B). No significant changes in serum total bile acid levels were observed in Trp53‐null mice compared with Trp53‐WT mice, although they tended to be elevated in Trp53‐null mice (Figure 8C), while serum ALP, ALT and AST levels were significantly increased in Trp53‐null mice (Figure 8D–G). These results indicate that Trp53‐null mice are more susceptible to cholestasis and liver injury.
Figure 8.
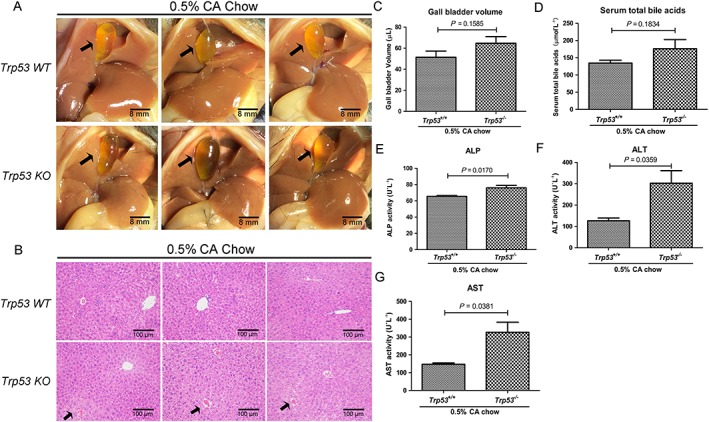
Effects of excessive cholic acid (CA) diet on cholestasis in Trp53‐WT and Trp53‐null mice. Mice were fed a chow diet supplemented with 0.5% (wt.wt‐1) CA for 4 days, and then samples were collected. (A) Morphology of representative livers. Gall bladders are indicated by black arrows. (B) Representative H&E stained liver sections (10 × 10). Apparent subcapsular necrotic foci in the liver are indicated by black arrows. (C–G) Gall bladder volume, levels of serum total bile and serum ALP, ALT and AST activities. Data are the mean ± SEM (n = 5). *P < 0.05. KO, knockout.
PCA analysis demonstrated a separated distribution pattern of intestine and faeces in Trp53‐null mice, while this separation was not observed in serum and liver (Figure 9A–D). As shown in the heat map (Figure 9E) and corresponding relative quantitative analysis using log intensity of peak area (Figure 9F–I), there were no apparent differences in the serum levels of cholic acid, CDCA, THDCA and T‐β‐MCA between cholic acid‐treated Trp53‐WT and Trp53‐null mice. In livers, cholic acid and CDCA were significantly lower in cholic acid‐treated Trp53‐WT mice than in cholic acid‐treated Trp53‐null mice, while THDCA and T‐β‐MCA showed no differences between the two groups. In addition, a decreased amount of these bile acids was observed in faeces and intestines of cholic acid‐treated Trp53‐null mice. The concentrations of cholic acid, CDCA and DCA in intestines and cholic acid, CDCA, DCA and HDCA in faeces were all significantly reduced in cholic acid‐treated Trp53‐null mice, as compared with those in cholic acid‐treated Trp53‐WT mice. However, no significant difference was found in T‐β‐MCA content between the two groups. These data suggest that the p53 deficiency hindered the transit of bile acid into the intestine and faeces.
Figure 9.
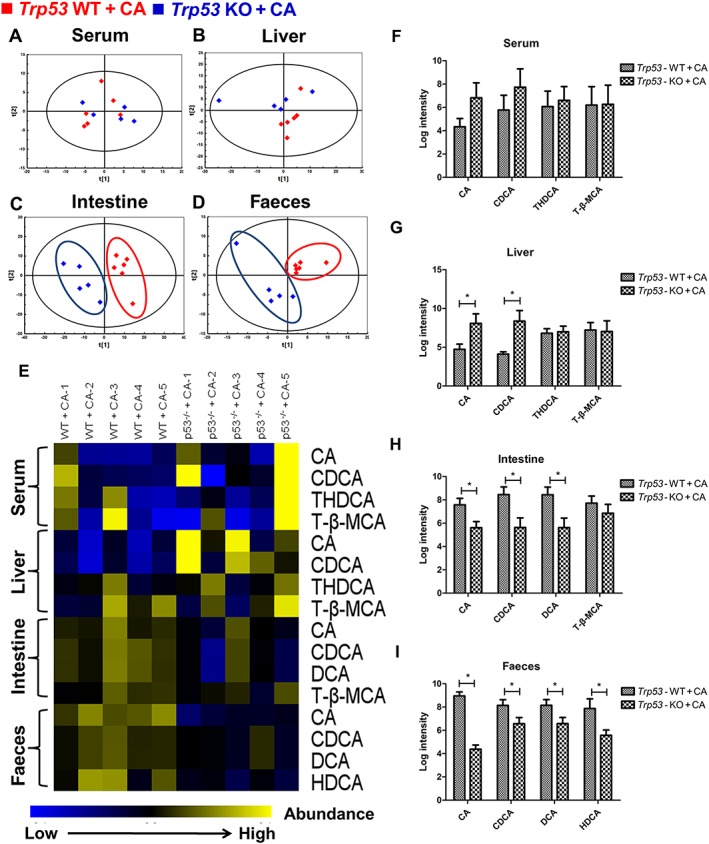
Effects of excessive cholic acid diet on bile acid metabolome of serum, liver, intestine and faeces in cholic acid‐fed Trp53‐WT and Trp53‐null mice. Mice were fed a chow diet supplemented with 0.5% (wt.wt‐1) cholic acid for 4 days, and then four samples were collected. (A–D) PCA analysis in four samples between cholic acid‐fed Trp53‐WT (red) and Trp53‐null mice (blue). (E) Heat maps of bile acid profiles of four samples. (F–I) The relative amount of bile acid was displayed as log intensity of peak area. Data are expressed as the mean ± SEM (n = 5–6). *P < 0.05. KO, knockout.
Discussion
The present study reveals a novel role of p53 in regulating the expression of genes in bile acid disposition. We demonstrated that Cyp2b10, Sult2a1 and Abcc2/3/4 expression was increased by doxorubicin‐mediated p53 activation both in vitro and in vivo. A reporter gene assay further demonstrated that p53 directly activated the transcription of CYP2B6 and ABCC3. In addition, our study showed that doxorubicin attenuated cholic acid‐induced cholestasis in mice by enhancing the excretion of bile acid metabolites from the serum and liver to the intestine and faeces, while cholic acid‐fed Trp53‐null mice exhibited more severe liver and bile duct injuries with suppressed secretion of bile acids. Overall, our study comprehensively investigated the effect of p53 on bile acid haemostasis in cholic acid‐induced cholestasis.
Primary hepatocytes were selected as the cell model in our study, since many of characteristic hepatic metabolic pathways for bile acid disposition are still preserved in these cells (Hylemon et al., 1985). Considering that doxorubicin activates p53 signalling in a stress‐responsive manner following DNA damage, it should be noted that other toxic compounds or anti‐tumour drugs with DNA‐damaging effects might also trigger p53‐mediated changes in bile acid disposition. Nutlin‐3, which activates p53 by disrupting the interaction between p53 and its negative regulator mouse double minute 2 homologue (MDM2), also affected bile acid‐related gene expression (Supporting Information Figure S1) (Vassilev et al., 2004), which indicates that p53 still has a regulatory effect in physiological conditions. The in vivo results further confirmed the regulative role of p53 in bile acid disposition. Interestingly, paradoxical results were observed for CYP3A11. The CYP3A11 level was up‐regulated in mouse primary hepatocytes, which was consistent with the result reported by Goldstein et al. (2013). However, p53 activation suppressed the expression of CYP3A11 in vivo, which may be explained by other signalling pathways triggered by doxorubicin counteracting the inducing effect of p53 on CYP3A11 expression. Further studies are needed to explain the contradictory results on CYP3A11 regulation by p53 found in vitro and in vivo.
Cholic acid, as a primary bile acid in both humans and rodents, is substantially elevated in cholestatic diseases (Bremmelgaard and Alme, 1980; Fischer et al., 1996). Thus, a diet supplemented with excessive cholic acid (0.5%) is a commonly used model to induce cholestasis (Rost et al., 2003; Teng and Piquette‐Miller, 2007). A significant alleviation of cholestasis was observed in doxorubicin‐treated groups. It should be noted that ALP levels were also significantly reduced by doxorubicin, suggesting that doxorubicin mitigates bile duct injury during cholestasis. However, liver injury caused by excessive cholic acid in the diet was not alleviated by doxorubicin, which might be due to the inhibition of liver regeneration mediated by p53/p21 pathway (Yang et al., 2011; Zhang et al., 2015). In fact, we found that doxorubicin significantly reduced the protein expression of PCNA and CCND1 (Supporting Information Figure S3), which hinders the initiation of liver regeneration by reducing DNA synthesis and cell proliferation.
Since cholic acid‐induced cholestasis was attenuated by doxorubicin, we hypothesized that Trp53‐null mice might be more susceptible to cholestatic injury. However, we only detected a significant elevation of ALT, AST and ALP activities in Trp53‐null mice; other cholestasis indicators were not affected. One potential explanation is that the treatment (4 days) is too short to observe effects on cholestatic indicators in Trp53‐null mice. Also, other compensatory pathways that adapt to the p53 deficiency might also play a role in preventing cholestatic injury.
Bile acid homeostasis is tightly maintained under the regulation of multiple enzymes and transporters in the liver. Cholestatic injury or hereditary mutations of transporter genes lead to dysfunctional bile secretory mechanisms in the liver, which subsequently causes cell injury, inflammation, liver fibrosis and even liver failure. The targeting of not only inflammation and liver fibrosis, but also bile acid hydrophilicity and bile flow could provide clinically relevant therapies for cholestatic liver diseases. Our data demonstrated that CYP2B10 and SULT2A1 are regulated by the p53 signalling pathway, indicating that p53 activation, by increasing hydroxylation and sulfonation, is beneficial for the detoxification of bile acids. MRP2 is a crucial canalicular transporter for mediating the efflux of bilirubin and bile acid conjugates. In particular, divalent bile acids with two negative charges such as those sulfated, taurocholate or glycolithocholate are transported by MRP2, whereas monovalent bile acids are not substrates for MRP2 (Trauner and Boyer, 2003). An increase in MRP2 might promote canalicular bile flow and subsequently faecal bile acid excretion via the bile duct, which is in line with the data obtained in the present study. Based on our observations, we speculate that induction of MRP2 by p53 activation leads to decreased amounts of bile acid in serum and liver and an increase in faeces and intestine.
Besides MRP2, bile acids can also be transported by MRP3 and MRP4 into sinusoidal blood as a compensatory excretion pathway. Bile acids in sinusoidal blood are filtered at the glomerulus from plasma into urine or re‐cycled into hepatocytes along the sinusoid (Rius et al., 2006; Keppler, 2017). An increase in MRP3 and MRP4 might enhance the levels of bile acids excreted in urine and via the systemic circulation. Our result alsos showed that the expression of MRP3 and MRP4 was up‐regulated by p53 activation, but whether the induction of these two genes by p53 is associated with an increased disposition of bile acid in urine needs to be further investigated.
The reporter gene assay and ChIP experiments further showed that p53 directly activates the transcription of CYP2B6 and ABCC3, suggesting CYP2B6 and ABCC3 are new target genes of p53. However, SULT2A1 and MRP2 and MRP4 might also be regulated through their own p53REs. The 3 kb upstream region of each candidate target gene of p53 was chosen in the reporter assays and many p53REs are located far from the gene body.
Cyp2b10, Sult2a1 and Abcc2/3/4 were demonstrated to be increased by p53, and we propose that p53 directly regulates the transcription of these genes. However, it should be noted that the p53‐mediated induction of these genes might also occur as a result of crosstalk with nuclear receptors, such as the farnesoid X receptor, pregnane X receptor and constitutive androstane receptor, since these nuclear receptors have long been considered to induce the upregulation of many enzymes and transporters involved in bile acid disposition (Claudel et al., 2011). Thus, we cannot exclude the possibility of crosstalk between p53 and nuclear receptors, and any indirect effects of p53 on bile acid disposition will be investigated in the future.
In the present study, we just focused on investigating the effect of p53 on the levels of genes expressed in liver cells. However, whether similar effects also occur in intestines would be an interesting future study.
We also evaluated gall bladder volume as one of indices for bile acid disposition in our study. However, in a previous study it was found that primary biliary cirrhosis patients exhibited increased intensity of p53‐positive biliary epithelial cells (Harada et al., 2001), indicating that p53 may also mediate water excretion from cholangiocytes and enhance bile flow. Thus, bile flow needs to be measured to fully reveal the mechanism by which p53 affects bile acid disposition.
Currently, the role of p53 in hepatic and systemic bile acid disposition is not fully understood. Our study provides direct evidence that p53 can affect the enzymes and transporters involved in the metabolism of bile acid. More importantly, the regulation by p53 of bile acid homeostasis has also been demonstrated in a cholic acid‐induced cholestasis mice model. Overall, the results of the current study extend our understanding of the pathological mechanisms of cholestasis, indicate that p53 has an important role in maintaining the homeostasis of bile acids and may be a potential target for diagnosis and clinical interventions of cholestasis.
Author contributions
H.B. and M.H. conceived or designed the study; P.C., D.L., Y.C., K.F., L.G., H.Z., Y.J., X.L., X.Z. and X.C. performed the research; P.C. and H.B. analysed the data; P.C., D.L. and Y.C. contributed the new methods or models; and P.C., J.S. and H.B. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effects of p53 activation on bile acid disposition related gene expression in primary hepatocytes of Trp53‐WT mice. The cells were incubated with Trp53 siRNA for 60 hours and treated with 25 μM Nutlin‐3 for the last 48 hours, and then mRNA and protein were measured by qRT‐PCR analysis. (A‐G) mRNA expression of Cdkn1a, Cyp3a11, Cyp2b10, Sult2a1, Abcc2, Abcc3 and Abcc4. Data are the mean ± S.E.M. (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure S2 Effects of p53 activator doxorubicin on morphology of liver and gallbladder in Trp53‐WT mice. Mice were fed either a standard rodent chow diet or a chow diet supplemented with 0.5% (wt/wt) cholic acid for 4 days. doxorubicin (10 mg/kg) or saline was administered to mice once per three days. Mice were sacrificed 24 hours after the final doxorubicin treatment. Gallbladders were marked by black arrows. (n = 6).
Figure S3 Effects of p53 activator doxorubicin on liver regeneration related gene expression in cholic acid‐fed Trp53‐WT mice. Mice were fed either a standard rodent chow diet or a chow diet supplemented with 0.5% (wt/wt) cholic acid for 4 days. doxorubicin (10 mg/kg) or saline was administered to mice once per three days. Mice were sacrificed 24 hours after the final treatment and liver tissues were collected. Protein was measured by Western Blot analysis. (A) Western blots of PCNA, CCND1, GAPDH, and (B) the corresponding PCNA and CCND1 protein quantifications relative to GAPDH. Data are the mean ± S.E.M. (n = 5–6). *P < 0.05, **P < 0.01, ***P < 0.001.
Table S1 Sequences of the gene‐specific primers.
Table S2 Sequences of Primers Used in ChIP Assay.
Acknowledgements
This work was supported by the Natural Science Foundation of China (grants 81573489, 81522047, 81373470, 81503156 and 81320108027), the 111 Project (grant B16047), the National Key R&D Program (grants 2017YFC0909300, 2017YFC0909303, 2016YFC0905000 and 2016YFC0905001) and the Key Laboratory Foundation of Guangdong Province (grant 2011A060901014).
Chen, P. , Li, D. , Chen, Y. , Sun, J. , Fu, K. , Guan, L. , Zhang, H. , Jiang, Y. , Li, X. , Zeng, X. , Chen, X. , Huang, M. , and Bi, H. (2017) p53‐mediated regulation of bile acid disposition attenuates cholic acid‐induced cholestasis in mice. British Journal of Pharmacology, 174: 4345–4361. doi: 10.1111/bph.14035.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger A, Pelletier G, Labrie F, Barbier O, Chouinard S (2003). Inactivation of androgens by UDP‐glucuronosyltransferase enzymes in humans. Trends in endocrinology and metabolism: TEM 14: 473–479. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R et al (2006). TIGAR, a p53‐inducible regulator of glycolysis and apoptosis. Cell 126: 107–120. [DOI] [PubMed] [Google Scholar]
- Boyer JL (2007). New perspectives for the treatment of cholestasis: lessons from basic science applied clinically. J Hepatol 46: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremmelgaard A, Alme B (1980). Analysis of plasma bile acid profiles in patients with liver diseases associated with cholestasis. Scand J Gastroenterol 15: 593–600. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhao KN, Chen C (2014a). The role of CYP3A4 in the biotransformation of bile acids and therapeutic implication for cholestasis. Ann Transl Med 2: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Li J, Fan X, Zeng H, Deng R, Li D et al (2015). Oleanolic acid attenuates obstructive cholestasis in bile duct‐ligated mice, possibly via activation of NRF2‐MRPs and FXR antagonism. Eur J Pharmacol 765: 131–139. [DOI] [PubMed] [Google Scholar]
- Chen P, Zeng H, Wang Y, Fan X, Xu C, Deng R et al (2014b). Low dose of oleanolic acid protects against lithocholic acid‐induced cholestasis in mice: potential involvement of nuclear factor‐E2‐related factor 2‐mediated upregulation of multidrug resistance‐associated proteins. Drug Metab Dispos 42: 844–852. [DOI] [PubMed] [Google Scholar]
- Claudel T, Zollner G, Wagner M, Trauner M (2011). Role of nuclear receptors for bile acid metabolism, bile secretion, cholestasis, and gallstone disease. Biochim Biophys Acta 1812: 867–878. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, Beuers U, Spengler U, Zwiebel FM, Koebe HG (1996). Hepatic levels of bile acids in end‐stage chronic cholestatic liver disease. Clin Chim Acta; international journal of clinical chemistry 251: 173–186. [DOI] [PubMed] [Google Scholar]
- Gall WE, Zawada G, Mojarrabi B, Tephly TR, Green MD, Coffman BL et al (1999). Differential glucuronidation of bile acids, androgens and estrogens by human UGT1A3 and 2B7. J Steroid Biochem Mol Biol 70: 101–108. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Ezra O, Rivlin N, Molchadsky A, Madar S, Goldfinger N et al (2012). p53, a novel regulator of lipid metabolism pathways. J Hepatol 56: 656–662. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Marcel V, Olivier M, Oren M, Rotter V, Hainaut P (2011). Understanding wild‐type and mutant p53 activities in human cancer: new landmarks on the way to targeted therapies. Cancer Gene Ther 18: 2–11. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Rivlin N, Shoshana OY, Ezra O, Madar S, Goldfinger N et al (2013). Chemotherapeutic agents induce the expression and activity of their clearing enzyme CYP3A4 by activating p53. Carcinogenesis 34: 190–198. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Sanchez E, Firrincieli D, Housset C, Chignard N (2015). Nuclear receptors in acute and chronic cholestasis. Dig Dis 33: 357–366. [DOI] [PubMed] [Google Scholar]
- Harada K, Furubo S, Ozaki S, Hiramatsu K, Sudo Y, Nakanuma Y (2001). Increased expression of WAF1 in intrahepatic bile ducts in primary biliary cirrhosis relates to apoptosis. J Hepatol 34: 500–506. [DOI] [PubMed] [Google Scholar]
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z (2010). Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 107: 7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylemon PB, Gurley EC, Kubaska WM, Whitehead TR, Guzelian PS, Vlahcevic ZR (1985). Suitability of primary monolayer cultures of adult rat hepatocytes for studies of cholesterol and bile acid metabolism. J Biol Chem 260: 1015–1019. [PubMed] [Google Scholar]
- Kawauchi K, Araki K, Tobiume K, Tanaka N (2008). p53 regulates glucose metabolism through an IKK‐NF‐kappaB pathway and inhibits cell transformation. Nat Cell Biol 10: 611–618. [DOI] [PubMed] [Google Scholar]
- Keppler D (2017). Progress in the molecular characterization of hepatobiliary transporters. Dig Dis 35: 197–202. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Lee JW (2011). Tumor suppressor p53 regulates bile acid homeostasis via small heterodimer partner. Proc Natl Acad Sci U S A 108: 12266–12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig JE, Goldblatt PJ, Hinton DE, Lipsky MM, Chacko J, Trump BF (1981). Mouse liver cell culture. I. Hepatocyte isolation. In Vitro 17: 913–925. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Oren M (2009). The first 30 years of p53: growing ever more complex. Nature Reviews Cancer 9: 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM et al (2002). Vitamin D receptor as an intestinal bile acid sensor. Science 296: 1313–1316. [DOI] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A et al (1999). Identification of a nuclear receptor for bile acids. Science 284: 1362–1365. [DOI] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O et al (2006). p53 regulates mitochondrial respiration. Science 312: 1650–1653. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius M, Hummel‐Eisenbeiss J, Hofmann AF, Keppler D (2006). Substrate specificity of human ABCC4 (MRP4)‐mediated cotransport of bile acids and reduced glutathione. Am J Physiol Gastrointest Liver Physiol 290: G640–G649. [DOI] [PubMed] [Google Scholar]
- Rost D, Herrmann T, Sauer P, Schmidts HL, Stieger B, Meier PJ et al (2003). Regulation of rat organic anion transporters in bile salt‐induced cholestatic hepatitis: effect of ursodeoxycholate. Hepatology 38: 187–195. [DOI] [PubMed] [Google Scholar]
- Schlereth K, Heyl C, Krampitz AM, Mernberger M, Finkernagel F, Scharfe M et al (2013). Characterization of the p53 cistrome – DNA binding cooperativity dissects p53's tumor suppressor functions. PLoS Genet 9: e1003726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC et al (2008). Characterization of genome‐wide p53‐binding sites upon stress response. Nucleic Acids Res 36: 3639–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Seed B (2010). PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res 38 (Database issue): D792–D799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S et al (2010). Phosphate‐activated glutaminase (GLS2), a p53‐inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 107: 7461–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng S, Piquette‐Miller M (2007). Hepatoprotective role of PXR activation and MRP3 in cholic acid‐induced cholestasis. Br J Pharmacol 151: 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M, Boyer JL (2003). Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev 83: 633–671. [DOI] [PubMed] [Google Scholar]
- Trauner M, Meier PJ, Boyer JL (1998). Molecular pathogenesis of cholestasis. N Engl J Med 339: 1217–1227. [DOI] [PubMed] [Google Scholar]
- Trottier J, Verreault M, Grepper S, Monte D, Belanger J, Kaeding J et al (2006). Human UDP‐glucuronosyltransferase (UGT)1A3 enzyme conjugates chenodeoxycholic acid in the liver. Hepatology 44: 1158–1170. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al (2004). In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science 303: 844–848. [DOI] [PubMed] [Google Scholar]
- Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C et al (2005). CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 42: 420–430. [DOI] [PubMed] [Google Scholar]
- Wang X, Spandidos A, Wang H, Seed B (2012). PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40 (Database issue): D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshilboum RM, Otterness DM, Aksoy IA, Wood TC, Her C, Raftogianis RB (1997). Sulfation and sulfotransferases 1: sulfotransferase molecular biology: cDNAs and genes. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 11: 3–14. [PubMed] [Google Scholar]
- Xie W, Radominska‐Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES et al (2001). An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A 98: 3375–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Li TW, Ko KS, Xia M, Lu SC (2009). Switch from Mnt‐Max to Myc‐Max induces p53 and cyclin D1 expression and apoptosis during cholestasis in mouse and human hepatocytes. Hepatology 49: 860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Li TW, Peng J, Tang X, Ko KS, Xia M et al (2011). A mouse model of cholestasis‐associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology 141: 378–388 388 e371‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Liu L, He Z, Li G, Liu J, Song Z et al (2015). Inhibition of wild‐type p53‐induced phosphatase 1 promotes liver regeneration in mice by direct activation of mammalian target of rapamycin. Hepatology 61: 2030–2041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of p53 activation on bile acid disposition related gene expression in primary hepatocytes of Trp53‐WT mice. The cells were incubated with Trp53 siRNA for 60 hours and treated with 25 μM Nutlin‐3 for the last 48 hours, and then mRNA and protein were measured by qRT‐PCR analysis. (A‐G) mRNA expression of Cdkn1a, Cyp3a11, Cyp2b10, Sult2a1, Abcc2, Abcc3 and Abcc4. Data are the mean ± S.E.M. (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure S2 Effects of p53 activator doxorubicin on morphology of liver and gallbladder in Trp53‐WT mice. Mice were fed either a standard rodent chow diet or a chow diet supplemented with 0.5% (wt/wt) cholic acid for 4 days. doxorubicin (10 mg/kg) or saline was administered to mice once per three days. Mice were sacrificed 24 hours after the final doxorubicin treatment. Gallbladders were marked by black arrows. (n = 6).
Figure S3 Effects of p53 activator doxorubicin on liver regeneration related gene expression in cholic acid‐fed Trp53‐WT mice. Mice were fed either a standard rodent chow diet or a chow diet supplemented with 0.5% (wt/wt) cholic acid for 4 days. doxorubicin (10 mg/kg) or saline was administered to mice once per three days. Mice were sacrificed 24 hours after the final treatment and liver tissues were collected. Protein was measured by Western Blot analysis. (A) Western blots of PCNA, CCND1, GAPDH, and (B) the corresponding PCNA and CCND1 protein quantifications relative to GAPDH. Data are the mean ± S.E.M. (n = 5–6). *P < 0.05, **P < 0.01, ***P < 0.001.
Table S1 Sequences of the gene‐specific primers.
Table S2 Sequences of Primers Used in ChIP Assay.