

Abstract
Background and Purpose
Gabapentin is commonly prescribed for nerve pain but may also cause dizziness, sedation and gait disturbances. Similarly, inhibition of the endogenous cannabinoid enzyme monoacylglycerol lipase (MAGL) has antinociceptive and anti‐inflammatory properties but also induces sedation in mice at high doses. To limit these side effects, the present study investigated the analgesic effects of coadministering a MAGL inhibitor with gabapentin.
Experimental Approach
Mice subjected to the chronic constriction injury model of neuropathic pain were administered the MAGL inhibitor KML29 (1–40 mg·kg−1, i.p.), gabapentin (1–50 mg·kg−1, i.p.) or both compounds. Mice were tested for mechanical and cold allodynia. The function and expression of cannabinoid CB1 receptors in whole brain homogenates and lipid profile of spinal cords were assessed after repeated drug administration.
Key Results
The combination of low‐dose KML29:gabapentin additively attenuated mechanical allodynia and synergistically reduced cold allodynia. The CB1 antagonist, rimonabant, partially reversed the anti‐allodynic effects of KML29:gabapentin in mechanical allodynia but not cold allodynia. The anti‐allodynic effects of KML29:gabapentin did not undergo tolerance in mechanical allodynia after repeated administration but produced mild tolerance in cold allodynia. High dose KML29 alone reduced CB1 receptor expression and function, but KML29:gabapentin reduced the density of CB1 receptors but did not alter their function. KML29:gabapentin influenced additional signalling pathways (including fatty acids) other than the pathways activated by a higher dose of either drug alone.
Conclusion and Implications
These data support the strategy of combining MAGL inhibition with a commonly prescribed analgesic as a therapeutic approach for attenuating neuropathic pain.
Abbreviations
- 2‐AG
2‐arachidonylglycerol
- 2‐LG
2‐linoleoyl glycerol
- anandamide
arachidonoylethanolamine
- CB receptor
cannabinoid receptor
- CCI
chronic constriction injury
- CI
confidence interval
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- NAEs
N‐acylethanolamines
- NAGly
N‐arachidonoyl glycine
- PEA
N‐palmitoyl ethanolamine
- TRPV
transient receptor potential cation channel subfamily V
- Zadd
predicted additive ED50 values
- Zmix
experimentally derived ED50 values
Introduction
Neuropathic pain is characterized by altered nerve function stemming from peripheral nerve injury, autoimmune and other disease states, or toxic insult. Treating neuropathic pain is difficult, because it is often refractory to traditional analgesics (Attal et al., 2006; Rahn and Hohmann, 2009). As compared with the well‐documented side effects and abuse potential of traditional analgesics, such as opioids, anticonvulsants have a relatively strong safety profile with minimal adverse drug interactions and are the recommended first‐line treatment for neuropathic pain (Kukkar et al., 2013). However, anticonvulsants are associated with negative side effects, including drowsiness, dizziness and ataxia (Beal et al., 2012; Stahl et al., 2013). Gabapentin is the most commonly prescribed anticonvulsant for neuropathy and, unlike opioids, repeated administration of gabapentin does not undergo tolerance (Hao et al., 2000; Gottrup et al., 2004), which reduces abuse potential. Gabapentin is a structural analogue of GABA but binds to α2δ subtype 1 of voltage‐gated calcium channels (Sills, 2006; Kukkar et al., 2013). The specific mechanisms through which gabapentin produces analgesia are not completely clear. However, gabapentin is thought to suppress central sensitization by blocking α2δ channels, which are densely expressed on presynaptic dorsal horn neurons (Tuchman et al., 2010; Stahl et al., 2013), thus reducing hyperexcitability of spinal nociceptive neurons.
Despite being routinely prescribed, gabapentin is effective in only half of chronic pain patients (Moore et al., 2014). Using an alternative approach targeting multiple systems with combination therapies could increase the efficacy of treating neuropathic pain (Chaparro et al., 1996; Gilron et al., 2005; Grim et al., 2014; Crowe et al., 2015). Combination therapy combines two or more drugs with different mechanisms of action, thereby increasing potential efficacy as compared with administration of either drug alone (Raffa, 2001; Perez et al., 2013). Using this approach offers multiple advantages, such as allowing for lower doses of each drug, thereby potentially reducing negative side effects of each drug, while maintaining pain relief (Raffa, 2001). For example, gabapentin has been characterized alone (Moore et al., 2014) and in combination with other analgesics, including opioids and antidepressants (Chaparro et al., 1996; Gilron et al., 2005), in neuropathic pain patients.
Preclinical studies have evaluated the effects of endogenous cannabinoid ligands (i.e. endocannabinoids) anandamide (arachidonoylethanolamine) and 2‐arachidonylglycerol (2‐AG) by inhibiting their respective catabolic enzymes, fatty acid amide hydrolase (FAAH) or monoacylglycerol lipase (MAGL), which have analgesic and anti‐inflammatory properties (Klein, 2005; Schlosburg et al., 2009; Kinsey et al., 2011; Crowe et al., 2015). Further, inhibiting FAAH or MAGL increases brain levels of anandamide or 2‐AG, respectively (Lichtman et al., 2004; Kinsey et al., 2009; Long et al., 2009a), thus increasing the bioavailability of the endocannabinoids to bind to cannabinoid CB1 and CB2 receptors. Specifically, inhibiting FAAH reduces neuropathic (Russo et al., 2007; Kinsey et al., 2009), inflammatory (Schlosburg et al., 2009; Booker et al., 2012) and visceral pain (Naidu et al., 2009). However, the FAAH inhibitor PF‐04457845 did not reduce pain in a clinical trial of osteoarthritis (Huggins et al., 2012), raising questions about the translation of preclinical to clinical application of this enzyme. Similarly, the MAGL inhibitors, JZL184 and KML29, attenuate pain in models of neuropathic pain (Kinsey et al., 2010; 2013; Ignatowska‐Jankowska et al., 2014) and inflammatory pain (Guindon et al., 2011; Ghosh et al., 2013). MAGL inhibitors attenuate allodynia (i.e. the painful perception of non‐noxious stimuli) and hyperalgesia (i.e. increased sensitivity to noxious stimuli) after nerve injury through a CB1 receptor‐mediated mechanism of action (Kinsey et al., 2009; 2010; Ignatowska‐Jankowska et al., 2014).
Despite the benefits of MAGL inhibition, chronic administration of high‐dose JZL184 or KML29 induces physical dependence and cross‐tolerance to cannabinoid receptor agonists and down‐regulates and desensitizes CB1 receptors (Ignatowska‐Jankowska et al., 2014; Schlosburg et al., 2014). To circumvent the effects of high doses and maximize analgesia, research has focused on using lower doses of endocannabinoid modulators in combination with other analgesics (Gunduz et al., 2011; Grim et al., 2014; Kazantzis et al., 2016). For example, the combined pharmacological inhibition of MAGL and COX1 and 2 synergistically attenuated mechanical allodynia and additively attenuated cold allodynia (Crowe et al., 2015), indicating that MAGL might be a good candidate for combination therapy. However, repeated administration of MAGL combined therapy has not been studied. In addition, commonly prescribed anticonvulsants have not been evaluated in conjunction with drugs affecting the endocannabinoid system. Therefore, the objective of the present study was to determine the anti‐allodynic effects of acute and chronic administration of the preclinical MAGL inhibitor, KML29, in combination with the clinically available anticonvulsant, gabapentin.
Methods
Animals
All animal care and experimental procedures were in accordance with ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015) and were approved by the Institutional Animal Care and Use Committee at West Virginia University prior to the start of any experiments. A total of 95 male C57BL/6 J mice (Jackson Laboratory, Bar Harbor, ME, USA) approximately 20 weeks old at the start of the experiment were used. Every effort was made to reduce animal suffering and to minimize the number of mice used whenever possible. Mice were housed in polysofone plastic NextGen cages (Allentown, Inc.) with corncob bedding. Mice were housed three to five per cage in a temperature (20–22°C) and humidity‐controlled environment with ad libitum access to food and water in an AAALAC‐accredited facility at West Virginia University. C57BL/6 J mice are widely studied and have been evaluated extensively in pain and behavioural assays (Crowe et al., 2015; Ignatowska‐Jankowska et al., 2015; Deng et al., 2015b). Mice were randomly assigned, and all experiments were carried out by trained technicians who were blinded to treatment conditions.
Chronic constriction injury (CCI)
Surgery was performed as described previously (Russo et al., 2007; Kinsey et al., 2009). Mice were anaesthetized with inhaled isoflurane (Phoenix Pharmaceuticals, Burlingame, CA, USA) with oxygen. Anesthesia was confirmed by toe pinch. The right hind leg was shaved and cleaned with three alternating wipes of Betadine solution, followed by 70% ethanol. An incision was made on the skin lateral to the femur. After separating the muscle, the sciatic nerve was isolated and partially ligated with a single, double‐knotted 5–0 silk suture. The muscle and skin were then closed with 6–0 nylon suture. Mice recovered in clean, heated cages and were observed for ataxia before being returned to the vivarium. Mice were administered ketoprofen (5 mg·kg−1, i.p.) for 3 days, as a postoperative analgesic. The mice were tested repeatedly, starting 7 days post‐surgery, to confirm that allodynia developed. Testing with experimental drug treatments started 4 weeks post‐surgery.
Behaviour assessments
The mice were brought into the testing room, weighed, injected and subjected to behavioural testing. Mice were injected with gabapentin or vehicle 60 min before testing. Pilot data in mice subjected to CCI were administered gabapentin (50 mg·kg−1, i.p.) either 60 or 120 min prior to allodynia testing, and no differences were found between pretreatment times in mechanical (P = 0.70; data not show) and cold allodynia (P = 0.12; data not shown). For experiments using KML29 alone and in combination with gabapentin, mice were injected with drug or vehicle 120 min before testing (Ignatowska‐Jankowska et al., 2014). The mice subjected to CCI were tested for allodynia, starting 7 days after CCI surgery, and placed inside ventilated polycarbonate chambers on an aluminium mesh table and allowed to acclimatize for 60 min before testing. In the CCI antagonist studies, rimonabant (3 mg·kg−1, i.p.), SR144528 (3 mg·kg−1, i.p.) or vehicle was given 15 min before administering the KML29:gabapentin combination (Crowe et al., 2015). A separate group of mice without CCI were subjected to the ‘Billy Martin tetrad battery’ to evaluate potential negative side effects of the drugs used in the present study, including non‐selective motor impairment.
A statistical power analysis was performed for sample size estimation of n = 14 (80% power, α 0.05, effect size 0.40) for acute allodynia testing, n = 8.5 (80% power, α 0.05, effect size 0.40) for repeated allodynia testing and n = 9 (80% power, α 0.05, effect size 0.60) for the tetrad battery. Final sample sizes are reported in the figure legends.
Mechanical allodynia test
Mechanical allodynia was tested using von Frey filaments (North Coast Medical, Morgan Hill, CA, USA) using the ‘up‐down’ method (Chaplan et al., 1994; Crowe et al., 2015). The plantar surface of either hind paw was stimulated with each filament, ranging from 0.16 to 6.0 g, starting with the 0.6 g filament, five times at a frequency of ~2 Hz (Kinsey et al., 2010; 2011; Grim et al., 2014; Crowe et al., 2015). The filaments were tested in ascending order until the mouse lifted its paw after three out of the five stimulations (i.e. a positive response). Once a positive response occurred, the filaments were tested in descending order until a positive response was no longer recorded, thus establishing a sensory threshold.
Acetone‐induced cold allodynia test
Immediately following the von Frey test (i.e. 30 min after starting von Frey test), 10 μL of acetone (99% HPLC grade; Thermo Fisher Scientific, Waltham, MA, USA) was applied via a 100 μL pipette (USA Scientific, Ocala, FL, USA) onto the plantar surface of each hind paw to test cold allodynia (Choi et al., 1994; Decosterd and Woolf, 2000). Acetone was applied from below the testing table via air burst by ‘expressing’ the pipette, thereby avoiding mechanical stimulation of the paw with the pipette tip. Cold allodynia was operationally defined as total time lifting or clutching the hind paw, which included paw lifting when walking or grooming. A maximum cut‐off time of 20 s was used (Decosterd and Woolf, 2000).
In order to assess the effects of repeated administration of the low dose combination of KML29:gabapentin, mice were administered KML29 (40 mg·kg−1, i.p.; Ignatowska‐Jankowska et al., 2014), gabapentin (50 mg·kg−1, i.p.; Kinsey et al., 2010), KML29:gabapentin combination (13.33:4 mg·kg−1, i.p.) or 1:1:18 vehicle for 7 days, with each administration separated by approximately 24 h. The low combination dose was chosen due to its similar effect on both mechanical and cold allodynia. On the first and sixth day, mice were tested for mechanical and cold allodynia, as described above, 2 h after drug administration. Two hours after the final injection on the seventh day, mice were killed via CO2 asphyxia, and brains and lumbar spinal cords were dissected, snap frozen in liquid nitrogen and stored at −80°C until assayed.
Tetrad
Testing was conducted 120 min following administration of KML29 (40 mg·kg−1, i.p.), gabapentin (50 mg·kg−1, i.p.), KML29:gabapentin (13.33:4 mg·kg−1, i.p.) or vehicle in the following order: locomotor activity, bar test (catalepsy), tail immersion test and rectal temperature. Testing was performed according to previously described procedures (Martin et al., 1991; Long et al., 2009b; Schlosburg et al., 2010). Briefly, locomotor activity was video recorded for a 5 min period in a Plexiglas chamber placed within a lighted, sound‐attenuating chamber. ANYmaze (Stoelting, Wood Dale, IL, USA) software was used to determine time spent immobile. Catalepsy was assessed using a horizontal bar (0.75 cm diameter) placed 4.5 cm off the benchtop. The mouse was placed with its front paws on the bar and a timer was started. Time spent immobile on the bar was recorded, for up to 60 s maximum. If the mouse moved off the bar, it was placed back on in the original position up to three times. Nociception was then assessed in the tail immersion assay. The mouse was placed head first into a custom‐fabricated restraint from absorbent under pads (VWR Scientific Products, Radnor, PA, USA) with the tail protruding out. The distal 1 cm of the tail was submerged into a 56°C water bath. The latency for the mouse to withdraw its tail (within a 10 s cut‐off time) was scored. Rectal temperature was assessed by inserting a mouse thermocouple probe 2 cm into the rectum, and temperature was determined by telethermometer (Physitemp Bat‐12).
Homologous competition receptor binding
Increasing concentrations of the non‐radioactive CB1/CB2 full agonist CP‐55,940 (10−10 to 10−7 M) were incubated in triplicate with 0.5 nM of [3H]‐CP‐55,940 in a final volume of 1 mL of binding buffer (50 mM Tris, 0.05% BSA, 5 mM MgCl2, pH 7.4) as described previously (Brents et al., 2011). Each binding assay contained 50 μg of crude whole mouse brain homogenates. To achieve equilibrium, reactions were incubated for 90 min at room temperature and terminated by rapid vacuum filtration through Whatman GF/B glass fibre filters, followed by three washes with ice‐cold binding buffer. Filters were immediately placed into scintillation vials with 4 mL of scintiverseTM BD cocktail scintillation fluid (Fisher Scientific, Fair Lawn, NJ, USA). Samples were incubated overnight in scintillation fluid, vortexed and bound reactivity determined by employing a liquid scintillation spectrophotometer (Tri Carb 2100 TR Liquid Scintillation Analyser, Packard Instrument Company, Meriden, CT, USA).
[35S]‐GTPγS binding
[35S]‐GTPγS assays were conducted as described previously (Brents et al., 2012) in buffer containing 20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 0.05% BSA, 10 μM GDP and 20 U L−1 of adenosine deaminase. Assays were performed in triplicate in a final volume of 1 mL, with all reactions containing 0.1 nM [35S]‐GTPγS and increasing concentrations of the CB1/CB2 full agonist CP‐55,940 (10−9 to 3 × 10−7), and 10 μg of crude whole mouse brain homogenates. To achieve maximal G‐protein activation, reactions were incubated at 30°C for 30 min and terminated by rapid vacuum filtration through Whatman GF/B glass fibre filters and followed by four 1 mL washes with ice‐cold filtration buffer (20 mM HEPES, pH 7.4, 0.05% BSA). Filters were immediately placed into scintillation vials with 4 mL of scintiverseTM BD cocktail scintillation fluid (Fisher Scientific, Fair Lawn, NJ, USA). Samples were incubated overnight in scintillation fluid, vortexed and bound reactivity determined by employing a liquid scintillation spectrophotometer (Tri Carb 2100 TR Liquid Scintillation Analyser, Packard Instrument Company, Meriden, CT, USA).
Lipid extraction, HPLC/MS/MS analysis, lipid quantification
Lipid extraction was performed on frozen spinal cord tissue as previously described (Leishman et al., 2016a). In brief, frozen tissue underwent mechanical, methanolic extraction in the presence of deuterated standards, then partial purification on C18 columns (Agilent Technologies, Santa Clara, CA, USA). Eluent fractions were analysed using HPLC/MS/MS (Shimadzu autosampler and pumps, Columbia, MD, USA; API 3000 triple quadrupole, Applied Biosystems/MDS Sciex; Foster City, CA, USA). Quantification of lipids is through a series of standard curve analyses using Analyst software (Applied Biosystems/MDS Sciex; Foster City, CA, USA) and was previously described in greater detail (Leishman et al., 2016b).
To determine the magnitude change in one treatment group relative to another and, therefore, the number of arrows to assign each significant difference, the mean level of a particular lipid in a selected treatment group was divided by that same lipid's mean level in the comparison group (Table 3). For example, the average level of 2‐AG in the spinal cord of KML29‐treated mice was 6.39 × 10−9 mol·g−1, and the average level of 2‐AG in the spinal cord of the vehicle‐treated mice was 1.90 × 10−9 mol·g−1; 6.39 × 10−9 divided by 1.90 × 10−9 equals 3.36, meaning that 2‐AG levels are over three times as high in the spinal cord of the KML29‐treated mice and assigning it four up arrows in the 2‐AG cell for the ‘Change with KML relative to vehicle’ column because the magnitude of change was between 3 and 10 times higher. For decreases, the process was very similar: the mean level in spinal cord of one group was divided by the same lipid's mean level in the spinal cord of the comparison group; however, the reciprocal of the decimal was taken to express a fold decrease (if the level in the one group is half of the level of another then that is a twofold decrease). As an example, the mean level of PGE2 was 4.90 × 10−11 mol·g−1 in the spinal cord of the KML29‐treated mice and 8.18 × 10−11 mol·g−1 in the corresponding gabapentin‐treated spinal cord. 4.90 × 10−11 divided by 8.18 × 10−11 is 0.599 and the reciprocal of 0.599 is 1.67, meaning that the decrease is between 1.5 and 2 fold and giving it two down arrows on our scale in PGE2 cell of the ‘Change with KML relative to gabapentin’ column.
Data analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All results are presented as means ± SEM. Results were considered significant at P < 0.05. Ipsilateral paw sensitivity data were analysed using a t‐test compared to the contralateral paw. Dose–response data were analysed using one‐way ANOVA, followed by Dunnett's post hoc test. The antagonist studies were analysed by two‐way (combo vs. antagonist) between‐subjects ANOVA followed by Bonferroni post hoc tests. The repeated administration study was analysed by a two‐way (acute vs. chronic) ANOVA followed by paired t‐tests. No animals were excluded from analyses.
For the mechanical allodynia assay, raw paw threshold was expressed as % maximum possible effect (%MPE) using the equation %MPE = (test threshold / max threshold)*100, where ‘max threshold’ was the assay's maximum filament (i.e. 6 g), and ‘test threshold’ was the paw's established filament threshold. For the cold allodynia assay, raw seconds the paw was lifted was expressed as %MPE using the equation %MPE = [(max cut‐off − test time)/(Max Cut‐off)]*100, where ‘max cut‐off’ was the assay's maximum cut‐off point (i.e. 20 s), ‘test time’ was the time (s) the paw was lifted off the testing table. The ED50 values were calculated by interpolation when only two data points were available (one below and one above 50% MPE) or by standard linear regression analysis when at least three data points were available on the linear portion of the dose‐effect curve (Crowe et al., 2015). To determine synergistic, additive or subadditive interactions, the theoretical additive ED50 value of the combined drugs was calculated from the individual dose–response curves. The combination is assumed to equal the sum of the effects of each drug.
Dose‐addition analysis was carried out as previously published (Tallarida, 2006; Naidu et al., 2009; Crowe et al., 2015). For graphical display, the ED50 of gabapentin was plotted on the abscissa (x axis), and the isoeffective dose of KML29 was plotted on the ordinate (y axis) to generate the isobologram. A line connecting the two points represents the theoretical additive effect of KML29 and gabapentin dose combinations. The drug mixture ED50 value was determined by linear regression as the overall mixture dose (KML29 and gabapentin doses were summed). The experimentally derived ED50 values (Zmix) from the dose–response curves of the ratios were compared to the predicted additive ED50 values (Zadd). Zadd values were calculated using the equation: Zadd = fA + (1 − f)B, where Α was the KML29 alone ED50 value, Β was the gabapentin alone ED50 value and ƒ was a fractional multiplier of Α in the computation of the additive total dose. The experiments described in this manuscript tested mixtures that yielded values of ƒ=0.25, ƒ=0.5 and ƒ=0.75, where ƒ is related to the proportion of KML29 in a mixture per the equation ρA=ƒ/Zadd. If the ED50 values of the Zmix are below those of Zadd and the confidence limit (CI) does not overlap, then the interaction is considered synergistic. The statistical difference between the theoretical additivity ED50 value (Zadd) and the experimental ED50 value (Zmix) was analysed using a Fisher's exact test.
For receptor binding assays, curve‐fitting and statistical analyses were conducted utilizing nonlinear regression; the one site homologous competition binding equation was used to determine the affinity (KD) of [3H]‐CP‐55,940 and CB1 receptor density (BMAX) expression in crude whole mouse brain homogenates. Curve fitting of concentration–effect curves via nonlinear regression was also employed to determine the EC50 (a measure of potency, in nM) and EMAX (a measure of efficacy, in pmol·mg−1) for the [35S]‐GTPγS binding experiments. Measures of affinity (KD) and potency (EC50) were converted to pKD or pEC50 values by taking the negative log of each value so that parametric tests could be used for statistical comparisons. Data were analysed for statistical differences by a one‐way ANOVA, followed by Tukey's post hoc comparisons. For the HPLC/MS/MS data, concentrations of each detected lipid in mol·g−1 adjusted for % recovery were analysed using one‐way ANOVA followed by post hoc Fisher's Least Significant Differences Test to determine significant differences between treatment groups. All statistical tests for HPLC/MS/MS data were carried out using SPSS Statistics (IBM, Armonk, NY, USA).
Materials
Gabapentin was purchased from Sigma‐Aldrich (St. Louis, MO, USA). KML29, rimonabant (SR141716; CB1 receptor antagonist) and SR144528 (CB2 receptor antagonist) were purchased from Cayman Chemical (Ann Arbor, MI, USA). All compounds were dissolved in a vehicle consisting of ethanol, Cremophor (Sigma‐Aldrich, St. Louis, MO, USA) and normal saline in a ratio of 1:1:18 parts (Pinto et al., 2010). All solutions were warmed to room temperature and injected i.p. at a volume of 10 μL·g−1 body mass. While both KML29 and gabapentin are orally bioavailable, i.p. and p.o. routes of administration of KML29 have comparable levels of MAGL inhibition (Chang et al., 2012) and gabapentin pharmacokinetics are not influenced by route of administration (Gambelunghe, Mariucci, Tantucci, & Ambrosini, 2005). CP‐55,940 was purchased from Tocris Bioscience (Bristol, UK) and prepared as a stock solution in 100% DMSO at a concentration of 10 mM, divided into aliquots and maintained at −4°C until use. [3H]‐CP‐55950 (168 Ci mmol−1) and [35S]‐GTPγS (1250 Ci mmol−1) were purchased from Perkin Elmer (Boston, MA, USA). All other reagents for binding assays were purchased from Fisher Scientific Inc (Pittsburgh, PA, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
CCI induces mechanical and cold allodynia
Following the CCI surgery, mice were tested repeatedly, in the absence of any drug administration, to establish allodynia development before treatment testing. The mice developed mechanical [t(29) = 9.47, P < 0.05] and cold [t(29) = −9.50, P < 0.05] allodynia in the ipsilateral paw [mean(SEM) = 1.23(0.22)g and 13.27(0.79)s], as compared to the contralateral paw [mean(SEM) = 4.74(0.28)g and 3.98(0.53)s].
Coadministration of KML29 and gabapentin additively attenuated mechanical allodynia and synergistically reduced cold allodynia
Administration of KML29 has been previously shown to significantly attenuate mechanical and cold allodynia at ≥30 mg·kg−1 (Crowe et al., 2015). Gabapentin significantly reduced mechanical and cold allodynia at ≥10 mg·kg−1 (data not shown). To determine the overall ED50 for KML29, the ED50s for mechanical and cold allodynia, 16.62 and 27.26 mg·kg−1, respectively, were averaged, resulting in an overall ED50 of 22 mg·kg−1 (Crowe et al., 2015). For the overall ED50 of gabapentin, the ED50s for mechanical and cold allodynia, 6.68 and 7.65 mg·kg−1, respectively, were averaged, resulting in an overall ED50 of 7 mg·kg−1. Using the overall ED50s for KML29 and gabapentin, the 1:1 ratio reflects 1 part KML29 to 0.3 parts gabapentin, the 3:1 ratio reflects 1 part KML29 to 0.1 parts gabapentin and the 1:3 ratio reflects 1 part KML29 and 0.9 parts gabapentin (Figure 1).
Figure 1.
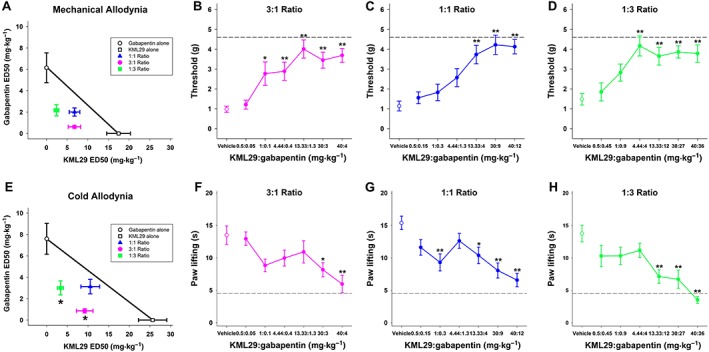
Coadministration of the selective MAGL inhibitor KML29 and gabapentin additively attenuated mechanical allodynia and synergistically reduced cold allodynia in mice subjected to CCI. A separate group of mice was subjected to CCI and then administered KML29, gabapentin or vehicle and tested for mechanical and cold allodynia. Then, KML29 was coadministered with gabapentin and mechanical (A) and cold allodynia (E) were assessed. The isobole of additivity is shown as a solid line connecting the ED50 values of KML29 and gabapentin and depicting the theoretical line of additivity. The experimental points of the collective mechanical allodynia tests (B–D) do not differ significantly from line of additivity, indicating an additive interaction. The experimental points of the collective cold allodynia tests (F–H) significantly lie below the theoretical line of additivity, indicating a synergistic interaction. Data are expressed as mean (±) SEM. *P < 0.05 versus vehicle (n = 15).
The Zmix in the 1:1, 3:1 and 1:3 ratios in the mechanical allodynia test was lower than the Zadd with some CI overlap, indicating that the interaction was additive (Table 1). The Zmix in the 1:1 ratio in the cold allodynia test was less than the Zadd; however, there was some CI overlap, and thus, the interaction was considered additive (Table 1). The Zmix in the 3:1 and 1:3 ratios in the cold allodynia test was less than the Zadd and did not have any CI overlap, indicating the interactions were synergistic (Table 1).
Table 1.
ED50 values of KML29 and gabapentin (GBP) in combination
| Combination (doses in mg·kg−1) | Combination Ratio | ED50 mg·kg−1 (95% confidence interval) KML29:GBP | ||
|---|---|---|---|---|
| Zadd (theoretical) | Zmix (experimental) | |||
| KML29:GBP | 1:1 | Mechanical Allodynia | 11.80 (8.68–14.93) | 11.94 (7.35–19.36) |
| 0.5:0.15 | Cold Allodynia | 16.62 (12.97–20.27) | 13.65 (9.02–20.65) | |
| 1:0.3 | ||||
| 4.44:1.3 | ||||
| 13.33:4 | ||||
| 30:9 | ||||
| 40:12 | ||||
| KML29:GBP | 3:1 | Mechanical Allodynia | 14.63 (10.36–18.90) | 7.38 (4.83–11.27) |
| 0.5:0.05 | Cold Allodynia | 21.13 (16.04–26.23) | 10.06 (6.52–15.52)* | |
| 1:0.1 | ||||
| 4.44:0.4 | ||||
| 13.33:1.3 | ||||
| 30:3 | ||||
| 40:4 | ||||
| KML29:GBP | 1:3 | Mechanical Allodynia | 8.98 (6.49–11.46) | 4.27 (2.76–6.60) |
| 0.5:0.45 | Cold Allodynia | 12.11 (9.40–14.82) | 5.28 (3.43–8.11)* | |
| 1:0.9 | ||||
| 4.44:4 | ||||
| 13.33:12 | ||||
| 30:27 | ||||
| 40:36 | ||||
The Zadd and Zmix values reflect the total amount of both drugs combined, where KML29 and gabapentin were summed for each combination.
Experimentally determined Zmix values and predicted Zadd values (95% CI) for mixtures of KML29 and gabapentin in assays of acetone and von Frey. Asterisks indicate a synergistic interaction as evidence of non‐overlapping 95% CI between Zmix and Zadd values.
P < 0.05 compared with Zadd using the Fisher test.
Because the experimental points of the collective mechanical allodynia tests do not differ significantly from the theoretical line of additivity, the interaction was additive. The experimental points of the collective cold allodynia of the 1:1 ratio did not differ significantly from the theoretical line of additivity, and thus, the interaction was additive (Figure 1A–D). Because the experimental points of the cold allodynia in the 3:1 and 1:3 ratios were significantly different from the line of additivity, the interaction was synergistic (Figure 1E–H).
Anti‐allodynia effects were partially blocked by a CB1 receptor antagonist
To determine the relative contribution of either cannabinoid receptor to the observed anti‐allodynia, the CB1 antagonist, rimonabant (3 mg·kg−1; Crowe et al., 2015), or the CB2 antagonist, SR144528 (3 mg·kg−1; Crowe et al., 2015), was administered prior to the combination of KML29 (13.33 mg·kg−1) and gabapentin (4 mg·kg−1) to assess receptor mechanism. There was an overall main effect of treatment in mechanical allodynia [F(3,52) = 5.15, P < 0.05; Figure 2A] and cold allodynia [F(3,52) = 9.49, P < 0.05; Figure 2B]. In mechanical allodynia, post hoc analyses did not indicate a difference between KML29:gabapentin and rimonabant or SR144528. Similarly, in cold allodynia, post hoc analyses did not indicate a difference between KML29:gabapentin and rimonabant or SR144528.
Figure 2.
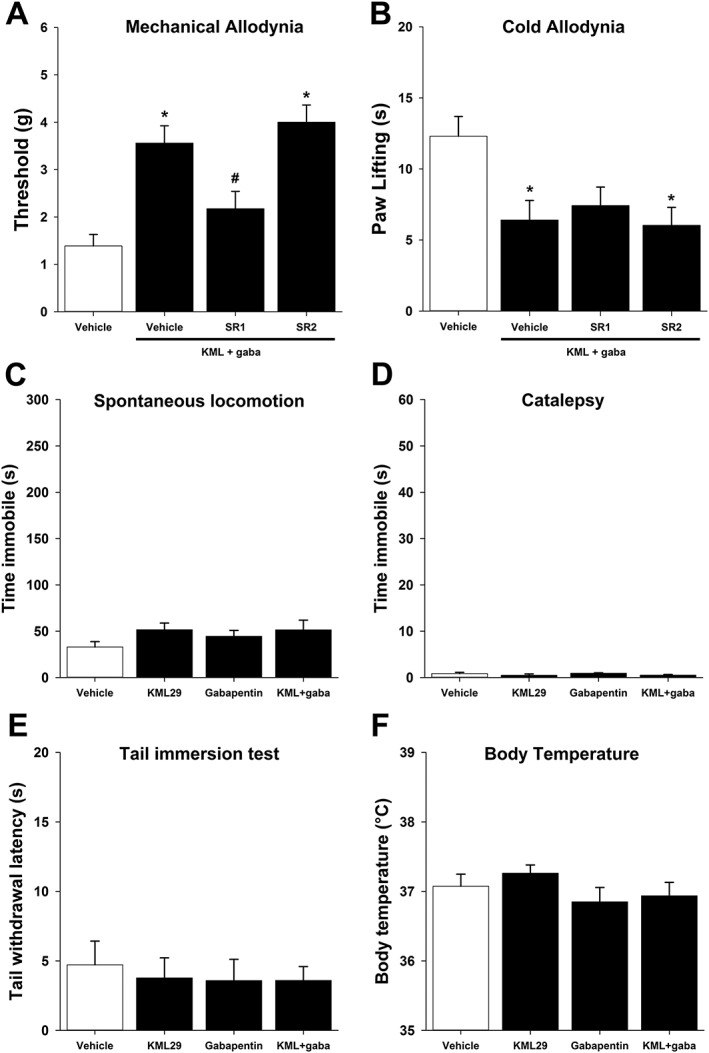
Anti‐allodynic effects of KML29:gabapentin combination treatment are partially mediated by CB1 receptors. Mice subjected to CCI were administered a combination of KML29:gabapentin (13.33:4 mg·kg−1). Pretreatment with rimonabant (SR1; 3 mg·kg−1), but not SR144528 (SR2, 3 mg·kg−1), partially reduces anti‐allodynia in the von Frey mechanical allodynia test (A), but neither had any effect in the acetone‐induced cold allodynia test (B), (n = 14). KML29 (40 mg·kg−1), gabapentin (50 mg·kg−1) or KML29:gabapentin (13.33:4 mg·kg−1) did not elicit any behavioural changes in spontaneous locomotor activity (C), catalepsy (D), tail immersion (E) or body temperature (F), (n = 8). Data are expressed as mean (±) SEM. *P < 0.05 versus vehicle.
Neither drug nor the combination elicited classic cannabimimetic effects
KML29 (40 mg·kg−1), gabapentin (50 mg·kg−1) and KML29:gabapentin (13.33:4 mg·kg−1) were assessed for classic cannabinoid effects using the ‘Billy Martin tetrad battery’. Neither drug, alone or in combination altered spontaneous locomotor activity [F(3,28) = 0.13, P = 0.94; Figure 2C], bar test catalepsy [F(3,28) = 1.11, P = 0.36; Figure 2D], tail immersion analgesia [P = 0.94; Figure 2E] or core body temperature [F(3,28) = 1.04, P = 0.39 Figure 2F].
No tolerance to repeated KML29 and gabapentin combination in mechanical allodynia
Mice were treated repeatedly with KML29 (40 mg·kg−1), gabapentin (50 mg·kg−1), KML29:gabapentin (13.33:4 mg·kg−1) or vehicle for 7 days and were assessed for mechanical and cold allodynia on the first (i.e. acute effect) and sixth day (i.e. chronic effect). On Day 1 of the experiment, KML29, gabapentin and KML29:gabapentin attenuated mechanical allodynia [F(3,27) = 21.88, P < 0.05; Figure 3A], which replicated the acute results reported above. On Day 6, gabapentin differed from vehicle (P < 0.05), but KML29 (P = 0.29) and the KML29:gabapentin combination (P = 0.02) did not differ from chronic vehicle. There was an interaction between the time of treatment (Day 1 or Day 6) and drug group [F(3,54) = 8.35, P < 0.05; Figure 3A]. Post hoc analyses revealed that acute and chronic administration of KML29 (40 mg·kg−1) differed (P < 0.05), indicating that repeated administration of high dose KML29 leads to tolerance. Acute and repeated gabapentin differed from each other (P < 0.05), whereas the acute and chronic combination of KML29:gabapentin group did not differ (P = 0.28), indicating that repeated administration of the low dose combination does not lead to tolerance.
Figure 3.
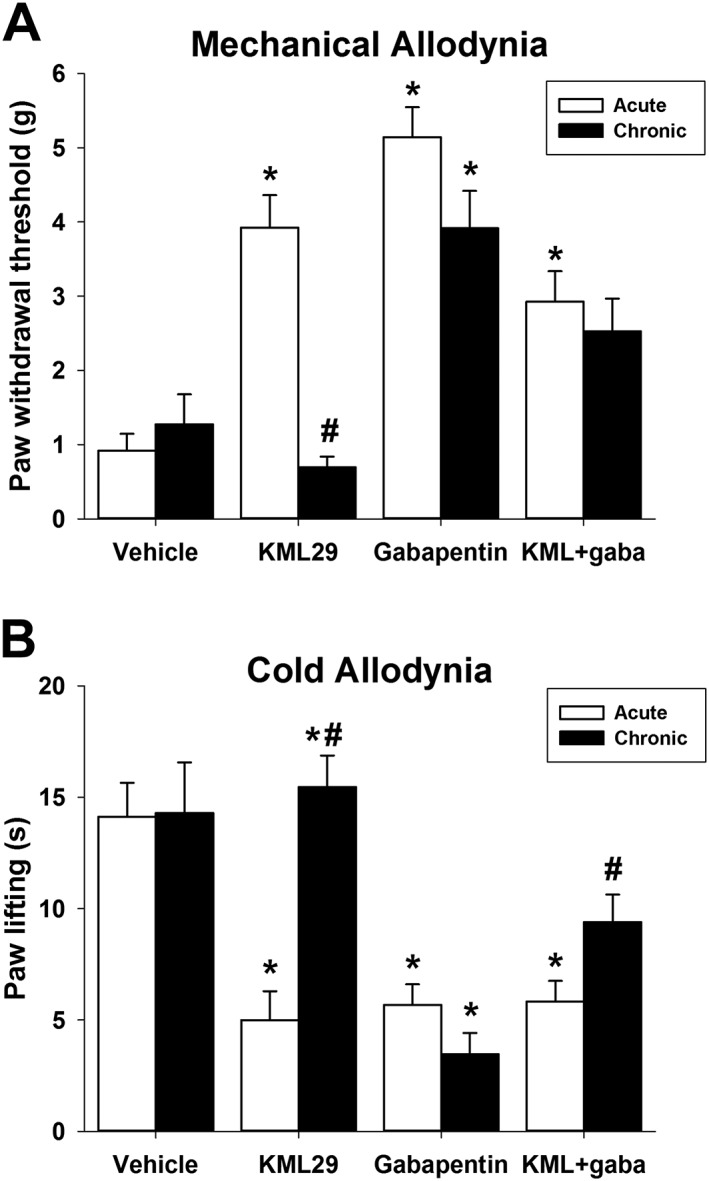
The anti‐allodynic effects of KML29:gabapentin do not undergo tolerance in mechanical allodynia (A) after chronic administration but show partial tolerance in cold allodynia (B). Mice were administered KML29 (40 mg·kg−1), gabapentin (50 mg·kg−1) or KML29:gabapentin (combo; 13.33:4 mg·kg−1) daily for 6 days. Data are expressed as mean (±) SEM. *P < 0.05 versus vehicle, #P < 0.05 versus acute treatment. For vehicle, KML29 and KML29:gabapentin groups n = 8 and for the gabapentin group n = 7. The difference in experimental numbers reflects an odd number of animals evenly distributed in the experimental design.
Similarly, KML29, gabapentin and KML29:gabapentin attenuated cold allodynia [F(3,27) = 13.16, P < 0.05; Figure 3B] on Day 1. On Day 6, gabapentin differed from vehicle (P < 0.05), but KML29 (P = 0.60) and the combination (P = 0.04) were not statistically significantly different from chronic vehicle. There was an interaction between the time of treatment (Day 1 or Day 6) and drug group [F(3,54) = 7.60, P < 0.05; Figure 3B]. Acute and chronic administration of KML29 (40 mg·kg−1) differed (P < 0.05), indicating that repeated administration of high dose KML29 leads to tolerance. Acute and chronic gabapentin did not differ, whereas the acute and chronic combination of KML29:gabapentin differed (P < 0.05), indicating partial tolerance developed in the low combination group.
To assess whether there was a change in efficacy of the mixtures to produce anti‐allodynia that went along with the potency shifts allodynia, the maximum effective doses of each drug alone and each of the three mixtures was compared by between‐subjects ANOVA. There was no significant effect of treatment condition on the maximal effect on either mechanical allodynia (P = 0.45) or cold allodynia (P = 0.20).
Chronic KML29:gabapentin reduces CB1 receptor density but not function in whole brain homogenates
The CB1/CB2 agonist CP‐55,940 produced concentration‐dependent and complete displacement of radiolabeled [3H]‐CP‐55,940 (i.e. homologous competition) from CB1 receptors expressed in brain homogenates and was best fit by a one‐site model. In mice injected daily with vehicle, [3H]‐CP‐55,940 bound to CB1 receptors with an affinity (KD) of 1.69 nM and a receptor density (BMAX) of 5.294 pmol·mg−1 protein. Repeated gabapentin administration failed to alter the BMAX of CB1 receptors, while repeated treatment with the MAGL inhibitor KML29 significantly decreased the density of CB1 receptors in brain homogenates by 32% [F(3, 27) = 5.69, P < 0.05; Figure 4; Table 2a]. Finally, in mice receiving KML29:gabapentin, the BMAX of CB1 receptors was reduced to levels comparable to that produced by KML29 alone (29%) when compared to vehicle‐treated controls. Chronic drug treatment produced slight but significant effects on the affinity (KD) of CP‐55,940 for CB1 receptors [F(3, 27) = 16.85, P < 0.05; Figure 4; Table 2a]. For example, KML29 treatment alone and in combination with gabapentin significantly reduced CP‐55,940 affinity from 1.69 nM in control animals, to 2.82 and 2.85 nM in KML29 and KML29:gabapentin‐treated mice respectively.
Figure 4.
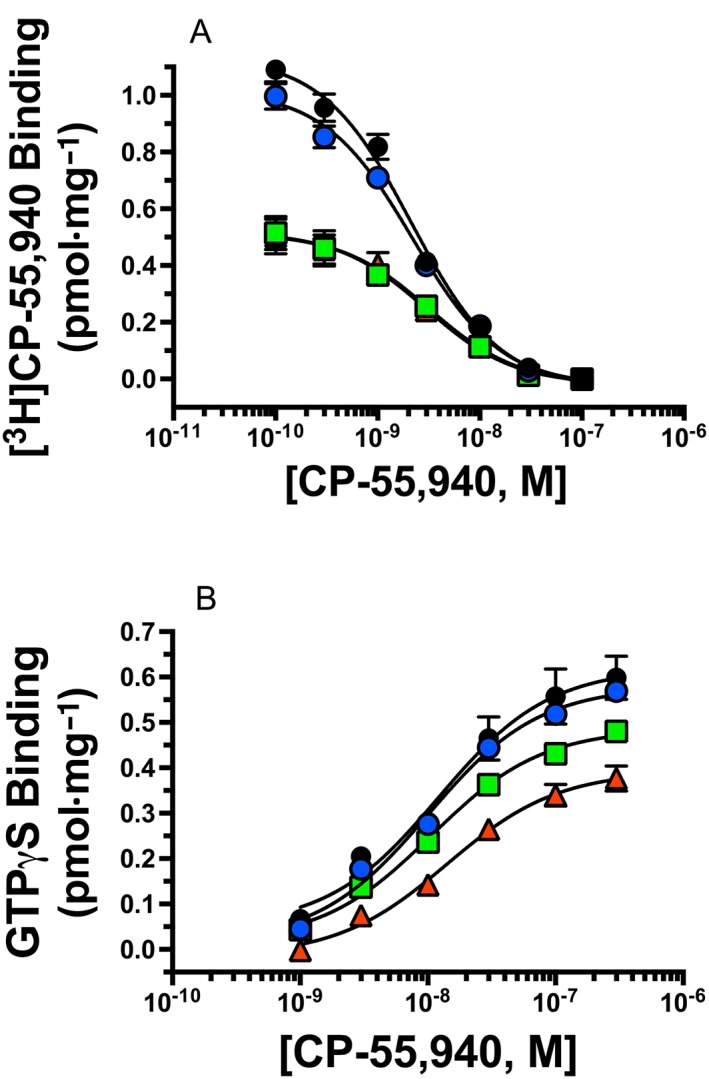
Homologous [3H]‐CP‐55,940 receptor binding and CP‐55,940 stimulated G‐protein activation in mouse whole brain homogenates. (A) Specific binding was determined as described in the Methods by incubating 0.5 nM [3H]‐CP‐55950 with increasing concentrations (10‐10to 10−7M) of CP‐55,940 and 50 μg of whole mouse brain membranes prepared from mice chronically treated with vehicle (black circles), gabapentin (blue circles), KML29 (red triangles) or a combination of gabapentin and KML29 (green squares). Receptor affinity (KD in nM) and density (BMAX in pmol·mg−1 protein) was determined by nonlinear regression analysis of specific [3H]‐CP‐55950 binding, and values are presented in Table 1. (B) Whole brain homogenates (10 μg) prepared from mice chronically treated as labelled were incubated in the presence of 0.1 nM [35S]‐GTPγS with increasing concentrations (10−9 to 3 × 10−7) of the full CB1 receptor agonist CP‐55,940 as described in the Methods. The potency (EC50 in nM) and efficacy (EMAX in pmol·mg−1 protein) of G‐protein activation was determined by nonlinear regression analysis, and values are presented in Table 2a. (n = 7–8).
Table 2a.
Homologous [3H]‐CP‐55,940 receptor binding in mouse whole brain homogenates
| Group | [3H]‐CP‐55,940 binding | |||
|---|---|---|---|---|
| KD (nM) | pKD (−Log[KD]) | BMAX (pmol·mg−1) | n | |
| Vehicle | 1.69 | 8.88 ± 0.04a,b | 5.29 ± 0.42a | 8 |
| GBP | 1.75 | 8.76 ± 0.03a,b | 4.81 ± 0.23a,b | 7 |
| KML29:GBP | 2.85 | 8.55 ± 0.03a,b | 3.76 ± 0.37a,b | 8 |
| KML29 | 2.82 | 8.56 ± 0.03a,b | 3.60 ± 0.31b | 8 |
pKD and BMAX values not sharing a letter are significantly different from values within the same column; That is, differences were observed with BMAX, but no differences were observed with pKD. P < 0.05, one‐way ANOVA, Tukey's post hoc test.
Table 2b.
CP‐55,940 stimulated [35S]‐GTPγS binding in mouse whole brain homogenates
| Treatment | [35S]‐GTPγS binding | |||
|---|---|---|---|---|
| Group | EC50 (nM) | pEC50 (−Log[EC50]) | EMAX (pmol·mg−1) | n |
| Vehicle | 9.6 | 8.03 ± 0.04a,b | 0.61 ± 0.06a | 8 |
| GBP | 9.7 | 8.03 ± 0.05a,b | 0.58 ± 0.04a,b | 7 |
| KML29:GBP | 10.4 | 8.01 ± 0.06a,b | 0.49 ± 0.05a,b | 8 |
| KML29 | 17.6 | 7.77 ± 0.05a,b | 0.41 ± 0.03b | 8 |
pEC50 and EMAX values not sharing a letter are significantly different from values within the same column; That is, differences were observed with EMAX, but no differences were observed with pEC50. P < 0.05, one‐way ANOVA, Tukey's post hoc test.
The CB1 cannabinoid receptor is a GPCR that produces intracellular effects via interaction with the Gi/Go‐subtype of G‐proteins (Dalton et al., 2009). Upon binding to CB1 receptors, agonists produce activation of G‐proteins that can be quantified in membrane preparations by measuring increases in agonist‐induced binding of [35S]‐GTPγS, a nonhydrolyzable GTP analogue (Harrison and Traynor, 2003). Therefore, to measure the function of CB1 receptors expressed in brain membranes of treated mice, the ability of increasing concentrations of CP‐55,940 to increase [35S]‐GTPγS binding was examined. In vehicle‐treated mice, CP‐55,940 produced a concentration‐dependent increase in [35S]‐GTPγS binding in brain membranes with a potency (EC50) of 9.6 nM and efficacy (EMAX) of 0.610 pmol·mg−1 protein. Although repeated treatment with gabapentin alone did not change the efficacy of CP‐55,940‐mediated G‐protein activation, daily injections of KML29 significantly decreased the EMAX of CP‐55,940 in mouse brain homogenates by 34% [F(3, 27) = 3.83, P < 0.05; Figure 4B]. Importantly, unlike that observed for effects on CB1 receptor density, the combination of gabapentin and KML29 produced a slight (19%), but non‐significant, reduction in the efficacy of CP‐55,940 to activate G‐proteins in brain homogenates.
Similar to effects on CP‐55,940 affinity (KD) repeated KML29 also had a slight but significant [F(3, 27) = 6.25, P < 0.05; Table 3] effect on the potency (EC50) of CP‐55,940 to activate G‐proteins. Specifically, KML29 treatment alone significantly reduced the potency of CP‐55,940 from 9.6 nM in controls to 17.6 nM in treated mice.
Table 3.
Eicosanoids in lumbar spinal cords
| Change with GBP relative to vehicle | Change with KML29 relative to vehicle | Change with combo relative to vehicle | Change with KML29 relative to GBP | Change with combo relative to GBP | Change with combo relative to KML | |
|---|---|---|---|---|---|---|
| N‐acyl ethanolamine | ||||||
| N‐palmitoyl ethanolamine | ↑↑ | |||||
| N‐stearoyl ethanolamine | ||||||
| N‐oleoyl ethanolamine | ||||||
| N‐linoleoyl ethanolamine | ↑↑↑ | |||||
| N‐arachidonoyl ethanolamine | ↑↑ | ↑ | ||||
| N‐docosahexaenoyl ethanolamine | ↑ | |||||
| N‐acyl glycine | ||||||
| N‐palmitoyl glycine | ||||||
| N‐stearoyl glycine | ||||||
| N‐oleoyl glycine | ||||||
| N‐arachidonoyl glycine | ↑↑↑↑ | |||||
| N‐acyl taurine | ||||||
| N‐arachidonoyl taurine | ↑ | |||||
| Free Fatty Acids | ||||||
| Oleic acid | ||||||
| Linoleic acid | ↑↑ | ↑↑ | ↑ | |||
| Arachidonic acid | ||||||
| 2‐acyl‐sn‐glycerol | ||||||
| 2‐palmitoyl‐sn‐glycerol | ↑↑ | |||||
| 2‐oleoyl‐sn‐glycerol | ↑↑ | ↑ | ||||
| 2‐linoleoyl‐sn‐glycerol | ↑↑↑ | ↑↑↑ | ↑↑ | ↑↑↑ | ||
| 2‐arachidonoyl‐sn‐glycerol | ↑↑↑↑ | ↑↑↑↑ | ↑↑↑↑ | ↑↑↑↑ | ↑ | |
| PGs | ||||||
| PGE2 | ↑ | ↓ | ↓↓ | ↓ | ↑ |
Summary of lumbar spinal cord levels of eicosanoids from mice treated repeatedly with KML29 (KML; 40 mg·kg−1), gabapentin (50 mg·kg−1) or KML29:gabapentin (combo; 13.33:4 mg·kg−1). Tissue was collected from mice subjected to CCI 2 h after drug administration. Arrows indicate fold increase or decrease ↑↑↑↑ (≥10), ↑↑↑↑ (3–9.99), ↑↑↑ (2–2.99), ↑↑ (1.5–1.99), ↑ (1–1.49), ↓ (1–1.49), ↓↓ (1.5–1.99).
The increase in [35S]‐GTPγS binding produced by CP‐55,940 in whole mouse brain homogenates was due to activation of CB1 receptors, because G‐protein activation is blocked by co‐incubation with the CB1 antagonist rimonabant (data not shown).
Chronic KML29:gabapentin increases 2‐AG, anandamide and NAEs in the spinal cord
Nineteen of the 26 lipids screened were present in all samples. The remaining seven were detectable in only some of the samples; therefore, those were not statistically analysed here. A series of comparisons of each of the 19 lipids in each of the four treatment groups was performed so that each of the three drug treatments was compared directly to the vehicle. We then compared each of the individual drug treatments to the combination treatments, and then, we compared the individual drug treatments. A full list of all lipids measured, the mean values for each and statistical comparisons for all interactions are available in supplemental data (Supporting Information Table S1). A composite of these series of comparisons is provided in Table 3.
Compared to vehicle controls, 40 mg·kg−1 of KML29 for 7 days caused a predictable increase in 2‐AG [F(3,27) = 70.58; P < 0.05] and 2‐linoleoyl glycerol (2‐LG) [F(3,27) = 8.39; P < 0.05]. In comparison to KML29 treatment, 50 mg·kg−1 gabapentin for 7 days showed an identical profile to high dose KML29, except gabapentin decreased PGE2 [F(3,27) = 8.19; P < 0.05;Figure 5D].
Figure 5.
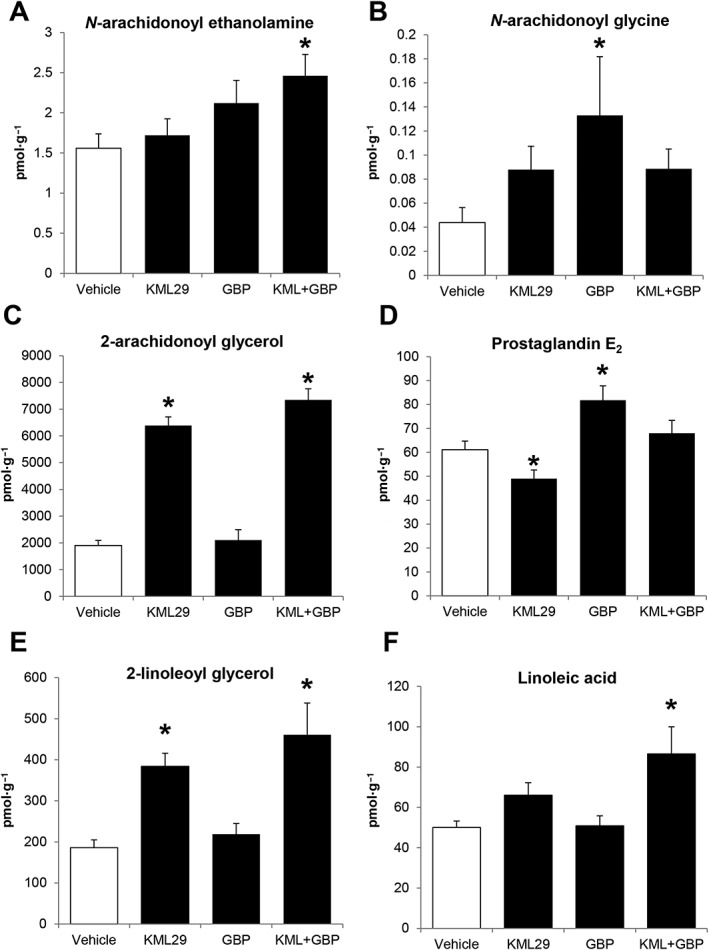
Lumbar spinal cord levels of eicosanoids expressed as mean (±SEM) in pmol·g−1. Tissue was collected from mice subjected to CCI and administered KML29 (40 mg·kg−1), gabapentin (50 mg·kg−1) or KML29:gabapentin (combo; 13.33:4 mg·kg−1) daily for 7 days. Samples were collected 2 h after the final drug administration. *P < 0.05 versus vehicle (n = 7–8).
The greatest changes in lipid levels occurred after the combination dose of KML29:gabapentin (13.33:4 mg·kg−1) was administered for 7 days. Unlike the treatment of KML29 or gabapentin alone, the combination therapy significantly increased levels of anandamide [F(3,27) = 3.07; P < 0.05; Figure 5A] and its endogenous structural analogues, the N‐acylethanolamines (NAEs) and N‐arachidonoyl taurine. Specifically, the individual NAEs increased by the combination therapy were N‐palmitoyl ethanolamine (PEA), N‐linoleoyl ethanolamine and N‐docosahexaenoyl ethanolamine (Table 3). Linoleic acid was also significantly increased [F(3,27) = 4.41; P < 0.05; Figure 5F]; however, no change was observed in levels of arachidonic acid. Levels of N‐arachidonoyl glycine (NAGly), which were significantly increased with gabapentin alone, were not significantly different from vehicle with the combination therapy. Conversely, levels of 2‐AG (Figure 5C) and 2‐LG (Figure 5E) that were significantly increased with KML29 treatment remained elevated. Interestingly, levels of 2‐oleoyl glycerol and 2‐palmitoyl glycerol that were unchanged with KML29 treatment alone were also significantly increased with KML29:gabapentin. Levels of PGE2, which were increased with gabapentin, were unchanged with the combination therapy.
Discussion
The main goal of this study was to evaluate the interaction between MAGL inhibition and gabapentin in a model of neuropathic pain in mice. The selective MAGL inhibitor, KML29, dose‐dependently attenuated mechanical and cold allodynia, as previously reported (Crowe et al., 2015). Gabapentin also dose‐dependently reduced mechanical and cold allodynia. Fixed‐dose proportions of KML29 and gabapentin additively reduced mechanical allodynia and synergistically attenuated cold allodynia. The reductions in allodynia were achieved by combining subthreshold doses of both drugs. Cannabinoid receptor involvement in the observed anti‐allodynia was also probed. The CB1 antagonist rimonabant, but not the CB2 antagonist SR144528, partially attenuated the anti‐allodynic effect of KML29:gabapentin in mechanical allodynia, indicating that the anti‐allodynic effects were at least partly mediated by CB1 receptors. However, neither rimonabant nor SR144528 affected cold allodynia, indicating a non‐cannabinoid mechanism, and possibly also a stronger contribution of gabapentin in reducing cold allodynia.
Using lower doses of candidate analgesics reduces drug exposure and may subsequently reduce the risk of drug tolerance. For example, chronic administration of high‐dose, but not low‐dose, MAGL inhibitors causes functional antagonism of CB1 receptors. Specifically, the MAGL inhibitor, JZL184 (≥16 mg·kg−1), causes down‐regulation and desensitization of CB1 receptors (Schlosburg et al., 2010; Kinsey et al., 2013), thereby reversing 2‐AG‐mediated antinociception. The tolerance to JZL184 is spared at lower doses (Kinsey et al., 2013). Similarly, the anti‐oedematous and anti‐allodynic effects of KML29 following carrageenan injection were reversed after repeated administration (Ignatowska‐Jankowska et al., 2014), indicating that the chronic blockade of MAGL with high‐dose KML29 produces tolerance. Although acute administration of a high‐dose MAGL inhibitor is beneficial for acute preclinical models of pain, chronic pain conditions, such as neuropathic pain, require long‐term treatments.
As expected, in the present study, prolonged MAGL inhibition by high‐dose KML29 produced analgesic tolerance in mechanical and cold allodynia, which was confirmed by a reduction in CB1 function (desensitization) and density (down‐regulation) in whole brain homogenates. In contrast to KML29, gabapentin produced anti‐allodynia that persisted following chronic administration in both tests. The combination treatment of low‐dose KML29:gabapentin did not show tolerance in mechanical allodynia, but tolerance may have developed in the cold allodynia test. Given the marginal effect, CB1 binding and GTPγS were assayed in whole brain homogenates. The combination reduced CB1 density but did not alter CB1 function. This reduced CB1 density may account for the incomplete anti‐allodynic tolerance of the drug combination evident in cold allodynia not observed in mechanical allodynia.
To further elucidate the mechanisms through which the KML29:gabapentin combination synergistically interact to attenuate allodynia, we quantified spinal cord lipid levels following repeated administration of each drug alone and in combination. High dose KML29 increased 2‐AG by threefold as compared to vehicle, while gabapentin increased NAGly and PGE2. Surprisingly, the combination of KML29:gabapentin not only increased 2‐AG but also increased PEA, N‐acyl glycines, 2‐acyl glycerols and N‐arachidonoyl taurine, which may indicate that dual administration of KML29 and gabapentin influences additional pathways other than those pathways activated by the two drugs alone. For example, KML29:gabapentin drive increases in all 2‐acyl glycerols tested. Levels of PGE2 are at baseline with combination treatment but were affected in opposite ways with individual treatment, suggesting that the suppression of PGE2 may be one of the ways the combination therapy is functioning. NAEs and N‐arachidonyl taurine are potent transient receptor potential cation channel subfamily V (TRPV) receptor ligands; however, the increases shown with the combination therapy was a low magnitude, which likely means that their effects on TRPVs would be minimal.
Mechanistically, we suggest that it is the dramatic and sustained increases in 2‐acyl glycerols, which appear to be involved in stabilizing PGE2 levels that is part of the lipid signalling driving the decrease in pain‐related behaviours observed with the combination therapy. Lipid‐based signalling molecules were quantified in the lumbar enlargement of the spinal cord. CCI surgery induces cFos expression in the lumbar spinal cord and activating CB receptors decreases cFos in the lumbar spinal cord (Rodella et al., 2005). Similarly, it is plausible that gabapentin‐induced reductions in pain pathway signalling indirectly alter endocannabinoid levels in discrete regions that were not captured in the present study, which examined homogenates of the lumber spinal cord. In rats, CCI surgery site specifically increases anandamide and 2‐AG in the lumbar spinal cord, as compared with sham operated rats (Petrosino et al., 2007). Because pain perception is mediated by afferent signalling in the dorsal spinal cord, quantifying endocannabinoid levels in the dorsal, lumbar spinal cord is a priority for future studies.
The efficacy of low dose gabapentin can be potentiated when combined with other non‐cannabinoid drugs. For example, patients with diabetic neuropathy or postherpetic neuralgia reported lower pain scores after receiving gabapentin and morphine than when administered morphine or gabapentin alone (Gilron et al., 2005). However, combining gabapentin and morphine was also associated with a higher incidence of constipation and dry mouth, compared to gabapentin or morphine alone respectively (Gilron et al., 2005). Similarly, patients reported an increase in pain relief after receiving a combined treatment of gabapentin and oxycodone but experienced an increase in opioid‐associated side effects (Hanna et al., 2008). In other words, the interaction of the combined drugs non‐selectively enhanced both analgesia and negative side effects of either drug. To evaluate potential changes in cannabimimetic side effects in the current study, KML29, gabapentin, KML29:gabapentin and vehicle were assessed for classic cannabinoid effects in the tetrad battery. None of the treatment groups elicited catalepsy, locomotor activity, tail immersion or body temperature differences, indicating that none of the treatments produced overt cannabimimetic effects. Moreover, the drug mixture results in the tetrad support that the synergistic antinociceptive effects were not due to a generalized suppression of all behaviour. However, an important limitation of the present study is that the treatment side effects were evaluated in non‐operated mice, and thus, it is possible that mice subjected to CCI may respond differently.
Patients with neuropathic pain tend to be over‐represented in pain clinics due to higher reported pain intensity, longer duration of pain and less effective pain relief, as compared with other forms of chronic pain (Torrance et al., 2006; 2007), underscoring the need to develop new, effective therapeutic treatments. Examining the effectiveness and safety of a MAGL inhibitor in clinical trials is an important step towards the development of new analgesics that target MAGL.
Clinically, synthetic cannabinoids are prescribed to cancer patients to stop emesis (Sticht et al., 2015); however, less is known about the efficacy of cannabinoids to control pain associated with chemotherapy‐induced neuropathy in patients. While there are relatively few clinical studies evaluating cannabinoids for chemotherapy‐induced neuropathy in humans, one study evaluated Sativex (an inhaled combination of Δ9‐THC and cannabidiol) in combination with patients' existing opioid treatment to increase the efficacy of reducing cancer pain (Johnson et al., 2010). Sativex, in combination with the existing opioid regimen, reduced pain severity, as compared to placebo, although it did not decrease the dose of opioids taken (Johnson et al., 2010). Murine models of chemotherapy‐induced neuropathic pain indicate that cannabinoids attenuate mechanical and cold allodynia (Hohmann, 2005; Guindon et al., 2013; Deng et al., 2015a,b) and prevent neuropathy when given in conjunction with chemotherapy (Ward et al., 2011; 2014; Rahn et al., 2014). Taken together, the results from clinical and preclinical research indicate that drugs targeting the endocannabinoid system alone and in conjunction with other non‐cannabinoid targets alleviate multiple types of neuropathic pain.
In summary, coadministration of KML29 and gabapentin additively reduced mechanical allodynia and synergistically reduced cold allodynia. Repeated administration of low dose KML29:gabapentin did not undergo tolerance in mechanical allodynia and did not alter CB1 receptor function in the brain. The synergistic interaction of the drugs was also evident in lipidomic analyses of the spinal cord, which indicate that the combination treatment activates pathways in addition to those altered by either drug alone. The combination of these drugs may be beneficial for increasing analgesia, while administered at relatively low doses. These data provide support to the strategy of targeting the endocannabinoid system in conjunction with non‐cannabinoid systems, to develop new candidate analgesics with limited negative side effects.
Author contributions
M.S.C., P.L.P., H.B.B., M.L.B. and S.G.K. participated in research design. M.S.C., C.D.W. and E.L. conducted the experiments. M.S.C., C.D.W., E.L., P.L.P., M.L.B., H.B.B. and S.G.K. performed the data analyses. M.S.C., E.L., P.L.P., H.B.B., M.L.B. and S.G.K. wrote or contributed to the writing of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Lumbar spinal cord levels of eicosanoids from mice treated repeatedly with KML29 (KML; 40 mg·kg−1), gabapentin (GBP; 50 mg·kg−1), or KML29: GBP (combo; 13.33:4 mg·kg−1). Tissue was collected from mice subjected to chronic constriction injury 2 h after drug administration Spinal cord levels are expressed as mean (SEM) in pmol·g−1. Tissue was collected from mice with CCI 2 h after drug administration. *P < 0.05 versus vehicle, Dunnett's post hoc test.
Acknowledgements
The authors thank Rachael Taylor, Sara Nass and Kristen Trexler for technical assistance.
This research was supported by the National Institutes of Health (AR066806, DA00668, DA037287, DA038714, DA039143, EY024625 and GM081741).
Crowe, M. S. , Wilson, C. D. , Leishman, E. , Prather, P. L. , Bradshaw, H. B. , Banks, M. L. , and Kinsey, S. G. (2017) The monoacylglycerol lipase inhibitor KML29 with gabapentin synergistically produces analgesia in mice. British Journal of Pharmacology, 174: 4523–4539. doi: 10.1111/bph.14055.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attal N, Cruccu G, Haanpaa M, Hansson P, Jensen TS, Nurmikko T et al (2006). EFNS guidelines on pharmacological treatment of neuropathic pain. Eur J Neurol 13: 1153–1169. [DOI] [PubMed] [Google Scholar]
- Beal B, Moeller‐Bertram T, Schilling JM, Wallace MS (2012). Gabapentin for once‐daily treatment of post‐herpetic neuralgia: a review. Clin Interv Aging 7: 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker L, Kinsey SG, Abdullah RA, Blankman JL, Long JZ, Ezzili C et al (2012). The fatty acid amide hydrolase (FAAH) inhibitor PF‐3845 acts in the nervous system to reverse LPS‐induced tactile allodynia in mice. Br J Pharmacol 165: 2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brents LK, Reichard EE, Zimmerman SM, Moran JH, Fantegrossi WE, Prather PL (2011). Phase I hydroxylated metabolites of the K2 synthetic cannabinoid JWH‐018 retain in vitro and in vivo cannabinoid 1 receptor affinity and activity. PLoS One 6: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brents LK, Gallus‐Zawada A, Radominska‐Pandya A, Vasiljevik T, Prisinzano TE, Fantegrossi WE et al (2012). Monohydroxylated metabolites of the K2 synthetic cannabinoid JWH‐073 retain intermediate to high cannabinoid 1 receptor (CB1R) affinity and exhibit neutral antagonist to partial agonist activity. Biochem Pharmacol 83: 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JW, Niphakis MJ, Lum KM, Cognetta AB, Wang C, Matthews ML et al. (2012). Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem Biol 19: 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaparro LE, Wiffen PJ, Moore RA, Gilron I (1996). Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst Rev 7 CD008943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Pogrel JW, Yaksh TL (1994). Role of voltage‐dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther 269: 1117–1123. [PubMed] [Google Scholar]
- Choi Y, Yoon YW, Na HS, Kim SH, Chung JM (1994). Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain 59: 369–376. [DOI] [PubMed] [Google Scholar]
- Crowe MS, Leishman E, Banks ML, Gujjar R, Mahadevan A, Bradshaw HB et al (2015). Combined inhibition of monoacylglycerol lipase and cyclooxygenases synergistically reduces neuropathic pain in mice. Br J Pharmacol 172: 1700–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Bass CE, Horn CV, Howletter AC (2009). Signal transduction via cannabinoid receptors. CNS Neurol Disord Drug Targets 8: 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87: 149–158. [DOI] [PubMed] [Google Scholar]
- Deng L, Cornett BL, Mackie K, Hohmann AG (2015a). CB1 knockout mice unveil sustained CB2‐mediated antiallodynic effects of the mixed CB1/CB2 agonist CP55,940 in a mouse model of paclitaxel‐induced neuropathic pain. Mol. Pharmacol. Mol Pharmacol 88: 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K, Hohmann AG (2015b). Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1‐dependent withdrawal. Biol Psychiatry 77: 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambelunghe C, Mariucci G, Tantucci M, Ambrosini MV (2005). Gas chromatography-tandem mass spectrometry analysis of gabapentin in serum. Biomed Chromatogr 19: 63–67. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Wise LE, Chen Y, Gujjar R, Mahadevan A, Cravatt BF et al (2013). The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci 92: 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL (2005). Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med 352: 1324–1334. [DOI] [PubMed] [Google Scholar]
- Gottrup H, Juhl G, Kristensen AD, Lai R, Chizh BA, Brown J et al (2004). Chronic oral gabapentin reduces elements of central sensitization in human experimental hyperalgesia. Anesthesiology 101: 1400–1408. [DOI] [PubMed] [Google Scholar]
- Grim TW, Ghosh S, Hsu K‐L, Cravatt BF, Kinsey SG, Lichtman AH (2014). Combined inhibition of FAAH and COX produces enhanced anti‐allodynic effects in mouse neuropathic and inflammatory pain models. Pharmacol Biochem Behav 124: 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Guijarro A, Piomelli D, Hohmann AG (2011). Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol 163: 1464–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Lai Y, Takacs SM, Bradshaw HB, Hohmann AG (2013). Alterations in endocannabinoid tone following chemotherapy‐induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty‐acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following c. Pharmacol Res 67: 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunduz O, Karadag HC, Ulugol A (2011). Synergistic anti‐allodynic effects of nociceptin/orphanin FQ and cannabinoid systems in neuropathic mice. Pharmacol Biochem Behav 99: 540–544. [DOI] [PubMed] [Google Scholar]
- Hanna M, O'Brien C, Wilson MC (2008). Prolonged‐release oxycodone enhances the effects of existing gabapentin therapy in painful diabetic neuropathy patients. Eur J Pain 12: 804–813. [DOI] [PubMed] [Google Scholar]
- Hao JX, Xu XJ, Urban L, Wiesenfeld‐Hallin Z (2000). Repeated administration of systemic gabapentin alleviates allodynia‐like behaviors in spinally injured rats. Neurosci Lett 280: 211–214. [DOI] [PubMed] [Google Scholar]
- Harrison C, Traynor JR (2003). The [35S]GTPγS binding assay: approaches and applications in pharmacology. Life Sci 74: 489–508. [DOI] [PubMed] [Google Scholar]
- Hohmann AG (2005). A cannabinoid pharmacotherapy for chemotherapy‐evoked painful peripheral neuropathy. Pain 118: 3–5. [DOI] [PubMed] [Google Scholar]
- Huggins JP, Smart TS, Langman S, Taylor L, Young T (2012). An efficient randomised, placebo‐controlled clinical trial with the irreversible fatty acid amide hydrolase‐1 inhibitor PF‐04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of th. Pain 153: 1837–1846. [DOI] [PubMed] [Google Scholar]
- Ignatowska‐Jankowska BM, Ghosh S, Crowe MS, Kinsey SG, Niphakis MJ, Abdullah RA et al (2014). In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br J Pharmacol 171: 1392–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska‐Jankowska BM, Baillie GL, Kinsey S, Crowe MS, Ghosh S, Allen Owens R et al (2015). A cannabinoid CB1 receptor positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology : 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JR, Burnell‐Nugent M, Lossignol D, Ganae‐Motan ED, Potts R, Fallon MT (2010). Multicenter, double‐blind, randomized, placebo‐controlled, parallel‐group study of the efficacy, safety, and tolerability of THC:CBD extract and THC extract in patients with intractable cancer‐related pain. J Pain Symptom Manage 39: 167–179. [DOI] [PubMed] [Google Scholar]
- Kazantzis NP, Casey SL, Seow PW, Mitchell VA, Vaughan CW (2016). Opioid and cannabinoid synergy in a mouse neuropathic pain model. Br J Pharmacol 173: 2521–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL et al (2009). Blockade of endocannabinoid‐degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther 330: 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, Cravatt BF, Lichtman AH (2010). Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti‐allodynic effects in mice through distinct cannabinoid receptor mechanisms. J Pain 11: 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Mahadevan A, Zhao B, Sun H, Naidu PS, Razdan RK et al (2011). The CB2 cannabinoid receptor‐selective agonist O‐3223 reduces pain and inflammation without apparent cannabinoid behavioral effects. Neuropharmacology 60: 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Wise LE, Ramesh D, Abdullah RA, Selley DE, Cravatt BF et al (2013). Repeated low‐dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1‐mediated antinociceptive and gastroprotective effects. J Pharmacol Exp Ther 345: 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TW (2005). Cannabinoid‐based drugs as anti‐inflammatory therapeutics. Nat Rev Immunol 5: 400–411. [DOI] [PubMed] [Google Scholar]
- Kukkar A, Bali A, Singh N, Jaggi AS (2013). Implications and mechanism of action of gabapentin in neuropathic pain. Arch Pharm Res 36: 237–251. [DOI] [PubMed] [Google Scholar]
- Leishman E, Cornett B, Spork K, Straiker A, MacKie K, Bradshaw HB (2016a). Broad impact of deleting endogenous cannabinoid hydrolyzing enzymes and the CB1 cannabinoid receptor on the endogenous cannabinoid‐related lipidome in eight regions of the mouse brain. Pharmacol Res 110: 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leishman E, Mackie K, Luquet S, Bradshaw HB (2016b). Lipidomics profile of a NAPE‐PLD KO mouse provides evidence of a broader role of this enzyme in lipid metabolism in the brain. Biochim Biophys Acta ‐ Mol Cell Biol Lipids 1861: 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Shelton CC, Advani T, Cravatt BF (2004). Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor‐mediated phenotypic hypoalgesia. Pain 109: 319–327. [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE et al (2009a). Selective blockade of 2‐arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X et al (2009b). Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci 106: 20270–20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK et al (1991). Behavioral, biochemical, and molecular modeling evalutions of cannabinoid analogs. Pharmacol Biochem Behav 40: 471–478. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RA, Wiffen PJ, Derry S, Toelle T, Rice ASC (2014). Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev 4: CD007938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu PS, Booker L, Cravatt BF, Lichtman AH (2009). Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther 329: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C, Navarro A, Saldana M, Masramon X, Perez M, Rejas J (2013). Clinical and resource utilization patterns in patients with refractory neuropathic pain prescribed pregabalin for the first time in routine medical practice in primary care settings in Spain. Pain Med 14: 1954–1963. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Palazzo E, de Novellis V, Bisogno T, Rossi F, Maione S et al (2007). Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology 52: 415–422. [DOI] [PubMed] [Google Scholar]
- Pinto CE, Moura E, Serrão MP, Martins MJ, Vieira‐Coelho MA (2010). Effect of (−)‐Delta(9)‐tetrahydrocannabinoid on the hepatic redox state of mice. Braz J Med Biol Res 43: 325–329. [DOI] [PubMed] [Google Scholar]
- Raffa RB (2001). Pharmacology of oral combination analgesics: rational therapy for pain. J Clin Pharm Ther 26: 257–264. [DOI] [PubMed] [Google Scholar]
- Rahn EJ, Hohmann AG (2009). Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics 6: 713–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn EJ, Deng L, Thakur GA, Vemuri K, Zvonok AM, Lai YY et al (2014). Prophylactic cannabinoid administration blocks the development of paclitaxel‐induced neuropathic nociception during analgesic treatment and following cessation of drug delivery. Mol Pain 10: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodella LF, Borsani E, Rezzani R, Ricci F, Buffoli B, Bianchi R (2005). AM404, an inhibitor of anandamide reuptake decreases Fos‐immunoreactivity in the spinal cord of neuropathic rats after non‐noxious stimulation. Eur J Pharmacol 508: 139–146. [DOI] [PubMed] [Google Scholar]
- Russo RC, Loverme J, Rana GL, Compton TR, Parrott J, Duranti A et al (2007). The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′‐carbamoylbiphenyl‐3‐yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther 322: 236–242. [DOI] [PubMed] [Google Scholar]
- Schlosburg JE, Kinsey SG, Lichtman AH (2009). Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J 11: 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG et al (2010). Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13: 1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Kinsey SG, Ignatowska‐Jankowska B, Ramesh D, Abdullah RA, Tao Q et al (2014). Prolonged monoacylglycerol lipase blockade causes equivalent cannabinoid receptor type 1 receptor‐mediated adaptations in fatty acid amide hydrolase wild‐type and knockout mice. J Pharmacol Exp Ther 350: 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sills GJ (2006). The mechanisms of action of gabapentin and pregabalin. Curr Opin Pharmacol 6: 108–113. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl SM, Porreca F, Taylor CP, Cheung R, Thorpe AJ, Clair A (2013). The diverse therapeutic actions of pregabalin: is a single mechanism responsible for several pharmacological activities? Trends Pharmacol Sci 34: 332–339. [DOI] [PubMed] [Google Scholar]
- Sticht MA, Rock EM, Limebeer CL, Parker LA (2015). Endocannabinoid mechanisms influencing nausea. Int Rev Neurobiol 125: 127–162. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ (2006). An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther 319: 1–7. [DOI] [PubMed] [Google Scholar]
- Torrance N, Smith BH, Bennett MI, Lee AJ (2006). The epidemiology of chronic pain of predominantly neuropathic origin. Results from a general population survey. J Pain 7: 281–289. [DOI] [PubMed] [Google Scholar]
- Torrance N, Smith BH, Watson MC, Bennett MI (2007). Medication and treatment use in primary care patients with chronic pain of predominantly neuropathic origin. Fam Pr 24: 481–485. [DOI] [PubMed] [Google Scholar]
- Tuchman M, Barrett JA, Donevan S, Hedberg TG, Taylor CP (2010). Central sensitization and Caνα2δ Ligands ligands in chronic pain syndromes: pathologic processes and pharmacologic effect. J Pain 11: 1241–1249. [DOI] [PubMed] [Google Scholar]
- Ward SJ, Ramirez MD, Neelakantan H, Walker A (2011). Cannabidiol prevents the development of cold and mechanical allodynia in paclitaxel‐treated female C57Bl6 mice. Anesth Analg 113: 947–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SJ, McAllister SD, Kawamura R, Murase R, Neelakantan H, Walker EA (2014). Cannabidiol inhibits paclitaxel‐induced neuropathic pain through 5‐HT 1A receptors without diminishing nervous system function or chemotherapy efficacy. Br J Pharmacol 171: 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Lumbar spinal cord levels of eicosanoids from mice treated repeatedly with KML29 (KML; 40 mg·kg−1), gabapentin (GBP; 50 mg·kg−1), or KML29: GBP (combo; 13.33:4 mg·kg−1). Tissue was collected from mice subjected to chronic constriction injury 2 h after drug administration Spinal cord levels are expressed as mean (SEM) in pmol·g−1. Tissue was collected from mice with CCI 2 h after drug administration. *P < 0.05 versus vehicle, Dunnett's post hoc test.