

Abstract
Background
Methylenetetrahydrofolate reductase (MTHFR) plays a crucial role in the hyperhomocysteinemia, which is a risk factor related to the occurrence of congenital heart defect (CHD). However, the association between MTHFR polymorphism and CHD has been inconclusive.
Methods
We conducted an updated meta-analysis to provide comprehensive evidence on the role of MTHFR A1298C polymorphism in CHD. Databases were searched and a total of 16 studies containing 2207 cases and 2364 controls were included.
Results
We detected that a significant association was found in the recessive model (CC vs. AA + AC: OR = 1.38, 95% CI: 1.10–1.73) for the overall population. Subgroup analysis showed that associations were found in patients without Down Syndrome in genetic models for CC vs. AA (OR = 1.47, 95% CI: 1.01–2.14), CC vs. AC (OR = 1.29, 95% CI: 1.00–1.66) and recessive model (OR = 1.44, 95% CI: 1.14–1.82). We conducted a meta-regression analysis, Galbraith plots and a sensitivity analysis to assess the sources of heterogeneity.
Conclusions
In summary, our present meta-analysis supports the MTHFR 1298C allele as a risk factor for CHD. However, further studies should be conducted to investigate the correlation of plasma homocysteine levels, enzyme activity, and periconceptional folic acid supplementation with the risk of CHD.
Electronic supplementary material
The online version of this article (10.1186/s13052-017-0425-1) contains supplementary material, which is available to authorized users.
Keywords: Methylenetetrahydrofolate reductase, Polymorphism, Congenital heart defects, Meta-analysis, Risk factor
Background
Congenital heart disease (CHD) is the most common defect in newborns, affecting approximately 8 to 10 per 1000 live births and causing 5%–10% of spontaneously aborted pregnancies [1, 2]. In addition, CHD is the leading cause of infant death all over the world, with mortality of 24.1% [3]. It has been proven that CHD is influenced by both genetic and environmental factors [4, 5]. Several genomewide association studies (GWASs) were conducted to identify the genetic factors in the development of CHD in the past decades [6, 7]. However, the etiology of CHD is still not fully known.
The methylenetetrahydrofolate reductase (MTHFR) gene, as one of the most crucial enzymes for the metabolism of folate/homocysteine, is located on chromosome 1p36.3. Specifically, this enzyme catalyzes the conversion of 5,10-methylenetetrahydrofolate into 5-methyltetrahydrofolate, which participates in the remethylation of homocysteine to methionine [8]. The lack of folic acid is known to result in hyperhomocysteinemia, which has been described as a possible risk factor for the occurrence of CHD [5, 7, 9, 10]. The MTHFR gene has 2 common mutations, C677T and A1298C, which result in the conversion of alanine to valine and glutamate to alanine, respectively. Moreover, these two mutations have been proved to decrease MTHFR enzyme activity, increase the level of homocysteine and eventually decrease the plasma concentration of folic acid.
Since Wenstrom first confirmed that MTHFR C677T polymorphism increased plasma homocysteine levels and the occurrence of CHD, several studies have been conducted to test this viewpoint. Several meta-analyses were also performed and suggested that MTHFR C677T polymorphism was associated with a susceptibility to CHD. However, few studies were carried out to investigate the association between MTHFR A1298C polymorphism and the risk of CHD. Xuan et al. conducted a correlation meta-analysis and found that the 1298C allele is a risk factor in the Caucasian population [11]. However, the included articles were published before Jun 2014. Therefore, we conducted an updated meta-analysis of all the available published data to integrate the results from case-control studies to provide comprehensive evidence on the role of MTHFR A1298C polymorphism in CHD.
Methods
Identification of relevant studies
We conducted an electronic search for relevant articles published before May 2016 in PubMed and Web of Science databases with the combination of the following terms: “methylenetetrahydrofolate reductase OR MTHFR”, “polymorphism OR snp OR variant” and “CHD OR congenital heart defect OR Malformation of heart OR Heart Abnormality OR Birth Defect OR Deformity OR Congenital Abnormality”. To expand the coverage of our searches, we further carried out searches in the Chinese National Knowledge Infrastructure (CNKI) and Wanfang databases with the translation of all English search terms. We also scanned for more qualifying studies from the reference lists of the retrieved articles.
Eligibility of relevant studies
We included case-control studies with human subjects that investigated the association between MTHFR A1298C polymorphism and CHD risk in both the English and the Chinese language. All phenotypes of CHDs, such as atrial septal defect, ventricular septal defect, patent formen ovale, patent ductus arteriosus, trilogy of fallot, coaratation of the aorta, and pulmonary valve stenosis, were included in this meta-analysis. Reviews, animal studies, simple commentaries, case reports and unpublished reports were excluded. Moreover, the studies that did not offer original data of allele frequencies in the initial publication were excluded after unsuccessful attempts to obtain this information by correspondence with the authors. Additionally, we usually retained the study with the most extensive data for the meta-analysis to avoid overlap in the information.
Data extraction
All data was extracted independently by two authors, and any disagreement was adjudicated by the corresponding author. The following information was extracted from each study: first author, year of publication, country of origin, ethnicity, type of CHD, and number of cases and controls, besides counts of alleles in case and control groups, and Hardy-Weinberg equilibrium were calculated (Table 1).
Table 1.
The main characteristics of all included studies for MTHFR A1298C polymorphism
| Study | Year | Country | CASE | CONTROL | HWE | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| AA | AC | CC | AA | AC | CC | |||||
| Storti | 2003 | Italy (Europe) | 45 | 47 | 11 | 101 | 86 | 13 | 0.347 | VSD, TOF, DORV, PA, TGA, AC |
| Galdieri | 2007 | Brasil (America) | 35 | 21 | 1 | 19 | 16 | 3 | 0.885 | CHD |
| van Driel | 2008 | Netherland (Europe) | 112 | 90 | 27 | 97 | 129 | 25 | 0.057 | TOF, TGA, ASD, VSD, CoA, AS, PS, HLHS |
| Locke | 2010 | USA (America) | 42 | 39 | 6 | 30 | 49 | 9 | 0.090 | ASD, VSD, AVSD |
| Obermann-Borst | 2010 | Netherland (Europe) | 69 | 57 | 13 | 75 | 90 | 18 | 0.227 | TOF, TGA, ASD, VSD, CoA, AS, PS, HLHS |
| Xu | 2010 | China (Asia) | 316 | 168 | 18 | 326 | 185 | 16 | 0.091 | Cyanotic Cardiac Disease, ASD, VSD, PDA, Left-sided Obstruction Defects |
| Božovic | 2011 | Europe | 30 | 22 | 2 | 25 | 30 | 3 | 0.113 | ASD, VSD, AVSD, PFO, TOF, PDA, Persistent truncus arteriosus |
| Christensen | 2013 | Canada (America) | 78 | 67 | 12 | 38 | 26 | 5 | 0.849 | VSD, TOF, AS, TGA, AVSD, DORV, PS, CoA, Truncus Arteriosus |
| Sahiner | 2013 | Turkey (Europe) | 45 | 68 | 24 | 31 | 54 | 8 | 0.022 | Obstruction in LV Output, Left-to-right Shunt, Conotruncal Anomalies,Complex Anomalies |
| Wang | 2013 | China (Asia) | 115 | 40 | 5 | 133 | 47 | 8 | 0.155 | CHD |
| Zidan | 2013 | Egypt (Africa) | 16 | 27 | 37 | 30 | 26 | 24 | 0.002 | ASD, VSD, PDA, Combined lesion, PS, TOF, HLHS |
| Chao | 2014 | China (Asia) | 13 | 2 | 2 | 15 | 19 | 0 | 0.024 | PDA |
| Huang | 2014 | China (Asia) | 111 | 56 | 3 | 146 | 54 | 6 | 0.712 | TOF |
| Sayin Kocakap | 2014 | Turkey (Europe) | 20 | 36 | 13 | 51 | 37 | 11 | 0.288 | PS, ASD, VSD, AVSD, TAPVR, TA, PA |
| Li | 2015 | China Asia) | 114 | 36 | 0 | 131 | 19 | 0 | 0.408 | VSD, ASD, PDA, TOF, CoA, AS, PS, TGA, DORV, Persistent truncus arteriosus |
| Koshy | 2015 | India (Asia) | 27 | 32 | 37 | 58 | 20 | 22 | 0 | TOF, DORV, PA-VS, TA, IAA |
AC Aortic Coarctation, AS Aortic Stenosis, ASD Atrial septal defect, AVSD Atrioventricular septal defect, CHD congenital heart disease, CoA Coarctation of the aorta, DORV Double outlet right ventricle, HLHS Hypoplastic left heart syndrome, IAA Interrupted aortic arch, PA Pulmonary atresia, PDA Patent ductus arteriosus, PS Pulmonary stenosis, TA Tricuspid atresia, TAPVR Total anomalous pulmonary venous return, TGA Transposition of great arteries, TOF Tetralogy of Fallot, VSD Ventricular septal defect
Statistical analysis
STATA (version 12.0; StataCorp, College Station, Texas, USA) was used for meta-analysis. All genotype models for the MTHFR A1298C polymorphisms were evaluated. The association between the A1298C polymorphism and CHD was compared using the odds ratio (OR) corresponding to a 95% confidence interval (95% CI). The pooled ORs were performed for C vs. A, CC vs. AA, AC vs. AA, CC vs. AC, dominant model (CC + AC vs. AA) and recessive model (CC vs. AA + AC). We used Q-test and the I 2 test to quantify the proportion of the total variation due to heterogeneity. I 2 ranges from 0 to 100%. A value of 0% means no observed heterogeneity, and larger values reflect increasing heterogeneity, with 25% regarded as low, 50% as moderate, and 75% as high heterogeneity. The pooled odds ratio (OR) was estimated with models based on fixed-effects or random-effects assumptions. If the effects were assumed to be heterogeneity (P < 0.1, I2 > 50%), the random-effects model was then used. Otherwise, a fixed-effect model was selected. And we used the Hardy–Weinberg equilibrium (HWE) to check the distributions of genotypes in the controls. Additionally, the stability of the results was assessed by sensitivity analysis and the influence of the individual study on the pooled ORs was reflected by deleting the single study involved in the meta-analysis each time.
The publication bias was estimated by Begg and Egger’s test (the significance was set at P < 0.05). Moreover, we used Egger’s plot of the MTHFR A1298C polymorphism to search for evidence of publication bias. The asymmetric funnel plots caused publication bias and the symmetric one did not.
Subgroup analyses were carried out by region (Asia, Europe, America or Africa), sample size (case number > 100 vs. <100), Down syndrome (without vs. with), HWE (consistent vs. inconsistent).
Results
Characteristics of eligible studies
The search strategy identified 3907 potentially relevant studies. Based on the inclusion criteria, a total of 16 relevant case-control [12–27] studies concerning MTHFR A1298C polymorphism and CHD, involving 2207 cases and 2364 controls, were included in this meta-analysis. The flow chart of the selected study is summarized in Fig. 1. Of these 16 articles, 6 were conducted in Asia [12, 15–17, 19, 20, 24, 25], 6 in Europe [21–23, 27], 3 in America [13, 14, 18] and 1 in Africa [12]. The distribution of the genotypes in the control groups was consistent with HWE except for 4 studies [12, 16, 20, 26]. The patients in 2 cases included studies accompanied by Down syndrome (DS) [27] [18]. The main characteristics of the included studies are presented in Table 1.
Fig. 1.
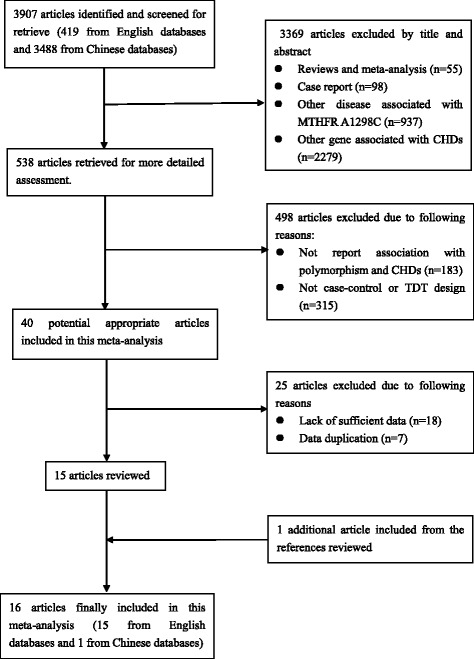
Flow chart of article screening and selection process
Results of the meta-analysis
We examined all genetic models of MTHFR A1298C polymorphism and the risk of CHD. Significant association was found in the recessive model (CC vs. AA + AC: OR = 1.38, 95% CI: 1.10–1.73; P heterogeneity = 0.289) when all eligible studies were pooled in the fixed-effect model (Fig. 2). In the subgroup analysis of region, obvious associations were found in Europe when relevant studies were pooled with the fixed-effect model for CC vs. AC (OR = 1.48, 95% CI: 1.05–2.09; P heterogeneity = 0.846) (Fig. 3) and recessive model (OR = 1.40, 95% CI: 1.01–1.94; P heterogeneity = 0.594) (Fig. 2). Moreover, remarkable associations were also found when the patients without DS were pooled with random- or fixed-effect models for CC vs. AA (OR = 1.47, 95% CI: 1.01–2.14; P heterogeneity = 0.021) (Fig. 4), CC vs. AC (OR = 1.29, 95% CI: 1.00–1.66; P heterogeneity = 0.461) (Fig. 5) and recessive model (OR = 1.44, 95% CI: 1.14–1.82; P heterogeneity = 0.308) (Fig. 6). The results of the subgroup analysis of the associations between MTHFR A1298C polymorphism and CHD are shown in Table 2.
Fig. 2.
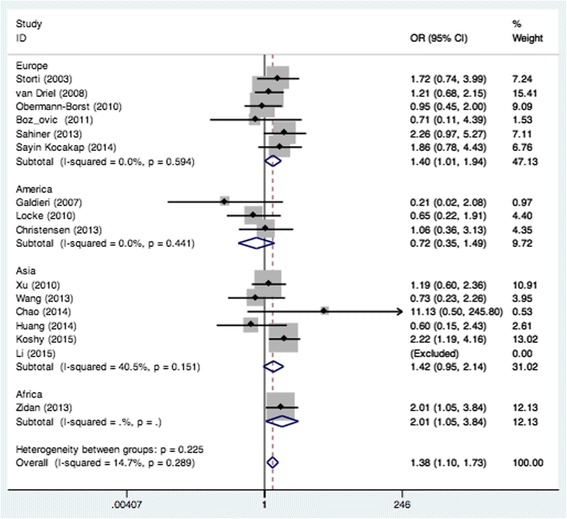
Pooled OR (recessive model) for the association between the MTHFR A1298C polymorphism and CHD in the overall and subgroups population
Fig. 3.
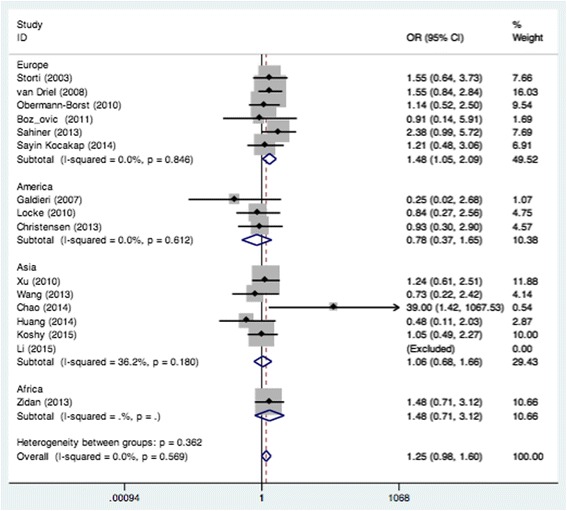
Pooled OR (CC vs. AC) for the association between the MTHFR A1298C polymorphism and CHD in the European population
Fig. 4.
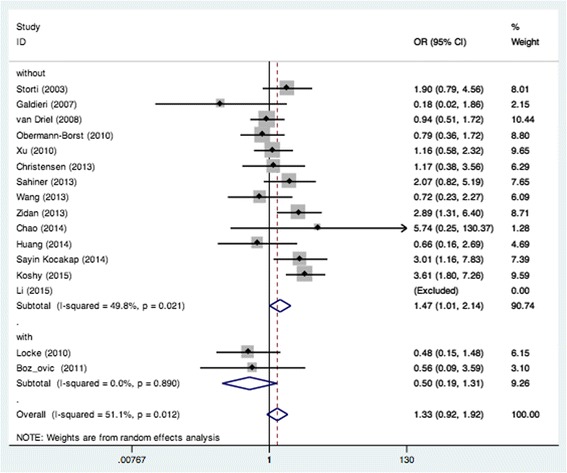
Pooled OR (CC vs. AA) for the association between the MTHFR A1298C polymorphism and CHD in the non-Down Syndrome population
Fig. 5.
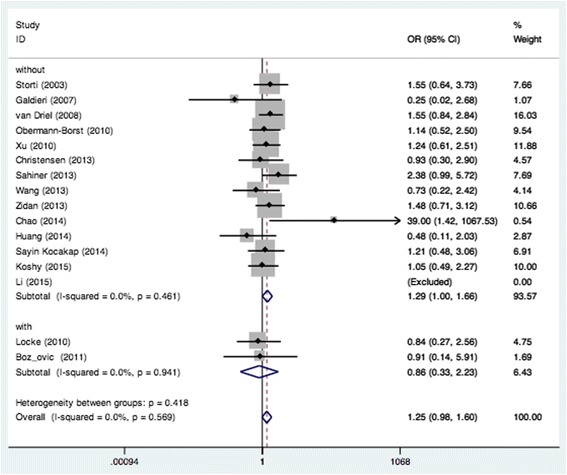
Pooled OR (CC vs. AC) for the association between the MTHFR A1298C polymorphism and CHD in the non-Down Syndrome population
Fig. 6.
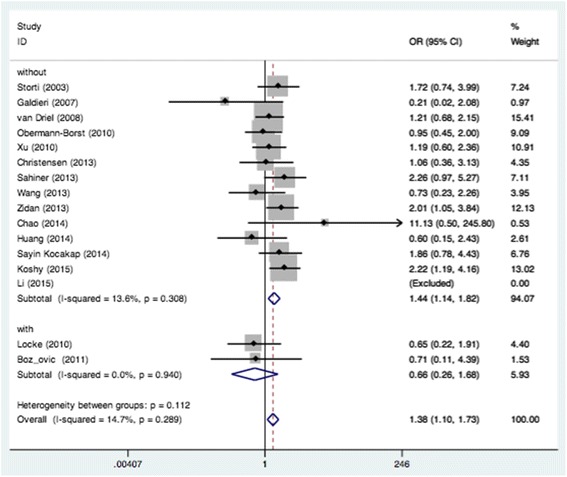
Pooled OR (recessive model) for the association between the MTHFR A1298C polymorphism and CHD in the non-Down Syndrome population
Table 2.
Subgroup analysis of the association between MTHFR A1298C polymorphism and CHD
| Subgroup analysis | Genetic model | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C vs. A | CC vs. AA | CC vs. AC | AC vs. AA | Dominant model | Recessive model | |||||||||||||
| No. | OR | 95% CI | No. | OR | 95% CI | No. | OR | 95% CI | No. | OR | 95% CI | No. | OR | 95% CI | No. | OR | 95% CI | |
| Summary | 16 | 1.13 | 0.92–1.37 | 15 | 1.33 | 0.92–1.92 | 15 | 1.25 | 0.98–1.60 | 16 | 1.05 | 0.81–1.36 | 16 | 1.10 | 0.84–1.43 | 15 | 1.38 | 1.10–1.73 |
| HWE | ||||||||||||||||||
| Consistent | 12 | 1.02 | 0.85–1.21 | 11 | 1.02 | 0.73–1.42 | 11 | 1.14 | 0.85–1.52 | 12 | 1.00 | 0.78–1.28 | 12 | 1.00 | 0.79–1.28 | 11 | 1.10 | 0.83–1.44 |
| Inconsistent | 4 | 1.55 | 0.94–2.57 | 4 | 2.97 | 1.89–4.66 | 4 | 1.59 | 1.01–2.50 | 4 | 1.11 | 0.40–3.06 | 4 | 1.37 | 0.56–3.36 | 4 | 2.21 | 1.49–3.27 |
| Down’s syndrome | ||||||||||||||||||
| Without | 14 | 1.20 | 0.98–1.47 | 13 | 1.47 | 1.01–2.14 | 13 | 1.29 | 1.00–1.66 | 14 | 1.14 | 0.86–1.50 | 14 | 1.19 | 0.90–1.57 | 13 | 1.44 | 1.14–1.82 |
| With | 2 | 0.69 | 0.48–0.98 | 2 | 0.50 | 0.19–1.31 | 2 | 0.86 | 0.33–2.23 | 2 | 0.59 | 0.36–0.95 | 2 | 0.57 | 0.36–0.92 | 2 | 0.66 | 0.26–1.68 |
| Region | ||||||||||||||||||
| Europe | 6 | 1.06 | 0.81–1.40 | 6 | 1.36 | 0.85–2.17 | 6 | 1.48 | 1.05–2.09 | 6 | 0.91 | 0.62–1.35 | 6 | 0.98 | 0.66–1.46 | 6 | 1.40 | 1.01–1.94 |
| Asia | 6 | 1.27 | 0.87–1.85 | 5 | 1.44 | 0.67–3.13 | 5 | 1.06 | 0.68–1.66 | 6 | 1.27 | 0.79–2.04 | 6 | 1.28 | 0.80–2.05 | 5 | 1.42 | 0.95–2.14 |
| America | 3 | 0.81 | 0.55–1.21 | 3 | 0.62 | 0.26–1.50 | 3 | 0.78 | 0.37–1.65 | 3 | 0.82 | 0.49–1.36 | 3 | 0.78 | 0.45–1.34 | 3 | 0.72 | 0.35–1.49 |
| Sample size | ||||||||||||||||||
| Large (>100) | 9 | 1.05 | 0.90–1.22 | 8 | 1.10 | 0.82–1.49 | 8 | 1.30 | 0.96–1.77 | 9 | 1.01 | 0.80–1.28 | 9 | 1.03 | 0.83–1.28 | 8 | 1.21 | 0.91–1.62 |
| Small (<100) | 7 | 1.13 | 0.69–1.87 | 7 | 1.60 | 0.74–3.45 | 7 | 1.17 | 0.78–1.76 | 7 | 1.02 | 0.51–2.05 | 7 | 1.09 | 0.54–2.21 | 7 | 1.68 | 1.17–2.41 |
Sensitivity analysis
Sensitivity analysis was carried out to detect the influence of the individual study on the pooled ORs by omitting a single study involved in the meta-analysis each time. The results showed that the corresponding pooled ORs were not substantially altered, except for one study for CC vs. AC [15], indicating the relatively stability of the results. The main results of the sensitivity analysis are shown in Table 3.
Table 3.
Sensitivity analysis of the association between MTHFR A1298C and CHD by omitting a single study each time
| Study omitted | Genetic model | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C vs. A | CC vs. AA | CC vs. AC | AC vs. AA | Dominant model | Recessive model | |||||||
| OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | |
| Storti | 1.11 | 0.90–1.38 | 1.28 | 0.86–1.91 | 1.23 | 0.96–1.59 | 1.04 | 0.79–1.37 | 1.08 | 0.82–1.43 | 1.35 | 1.07–1.71 |
| Galdieri | 1.16 | 0.95–1.41 | 1.39 | 0.97–2.00 | 1.28 | 1.00–1.63 | 1.07 | 0.82–1.41 | 1.13 | 0.86–1.48 | 1.40 | 1.12–1.76 |
| van Driel | 1.16 | 0.94–1.42 | 1.37 | 0.92–2.05 | 1.21 | 0.92–1.57 | 1.11 | 0.85–1.44 | 1.15 | 0.87–1.50 | 1.41 | 1.10–1.80 |
| Locke | 1.17 | 0.95–1.43 | 1.43 | 0.99–2.06 | 1.28 | 1.00–1.64 | 1.10 | 0.84–1.43 | 1.15 | 0.88–1.50 | 1.43 | 1.13–1.80 |
| Obermann-Borst | 1.16 | 0.94–1.42 | 1.39 | 0.94–2.06 | 1.27 | 0.98–1.64 | 1.10 | 0.83–1.43 | 1.14 | 0.86–1.50 | 1.43 | 1.13–1.81 |
| Xu | 1.14 | 0.91–1.42 | 1.33 | 0.88–2.01 | 1.26 | 0.97–1.63 | 1.06 | 0.79–1.43 | 1.11 | 0.82–1.50 | 1.40 | 1.10–1.78 |
| Božovic | 1.15 | 0.94–1.41 | 1.36 | 0.93–1.99 | 1.26 | 0.99–1.61 | 1.09 | 0.83–1.42 | 1.13 | 0.87–1.49 | 1.39 | 1.11–1.75 |
| Christensen | 1.12 | 0.91–1.39 | 1.33 | 0.90–1.98 | 1.27 | 0.99–1.63 | 1.04 | 0.79–1.37 | 1.09 | 0.82–1.44 | 1.39 | 1.11–1.76 |
| Sahiner | 1.12 | 0.91–1.38 | 1.27 | 0.86–1.89 | 1.19 | 0.92–1.53 | 1.07 | 0.81–1.41 | 1.10 | 0.83–1.46 | 1.33 | 1.05–1.68 |
| Wang | 1.14 | 0.93–1.41 | 1.38 | 0.94–2.03 | 1.28 | 1.00–1.65 | 1.06 | 0.80–1.40 | 1.11 | 0.84–1.47 | 1.41 | 1.12–1.78 |
| Zidan | 1.08 | 0.89–1.32 | 1.23 | 0.84–1.81 | 1.23 | 0.95–1.59 | 1.02 | 0.78–1.33 | 1.05 | 0.81–1.37 | 1.31 | 1.03–1.66 |
| Chao | 1.15 | 0.94–1.40 | 1.30 | 0.89–1.89 | 1.23 | 0.97–1.57 | 1.10 | 0.86–1.41 | 1.14 | 0.88–1.48 | 1.36 | 1.09–1.71 |
| Huang | 1.12 | 0.91–1.39 | 1.37 | 0.94–2.01 | 1.29 | 1.01–1.65 | 1.03 | 0.78–1.36 | 1.08 | 0.82–1.43 | 1.41 | 1.12–1.77 |
| Sayin Kocakap | 1.09 | 0.89–1.33 | 1.24 | 0.85–1.82 | 1.26 | 0.98–1.62 | 1.00 | 0.78–1.29 | 1.04 | 0.80–1.35 | 1.35 | 1.07–1.70 |
| Li | 1.09 | 0.89–1.33 | 1.33 | 0.92–1.92 | 1.25 | 0.98–1.60 | 1.00 | 0.78–1.30 | 1.05 | 0.81–1.37 | 1.38 | 1.10–1.73 |
| Koshy | 1.06 | 0.89–1.26 | 1.21 | 0.86–1.71 | 1.28 | 0.99–1.65 | 0.99 | 0.78–1.25 | 1.02 | 0.80–1.29 | 1.28 | 1.01–1.63 |
| Combined | 1.13 | 0.92–1.37 | 1.33 | 0.92–1.92 | 1.25 | 0.98–1.60 | 1.05 | 0.81–1.36 | 1.10 | 0.84–1.43 | 1.38 | 1.10–1.73 |
Heterogeneity analysis
Significant heterogeneity was detected in models of C vs. A (Q = 55.60; P = 0.000; I 2 = 73.0%), CC vs. AA (Q = 28.62; P = 0.012; I 2 = 51.1%), AC vs. AA (Q = 51.05; P = 0.000; I 2 = 70.6%) and CC + AC vs. AA (Q = 58.86; P = 0.000; I 2 = 74.5%), while no heterogeneity in models of CC vs. AC (Q = 12.047; P = 0.569; I 2 = 0.0%) and CC vs. AA + AC (Q = 16.42; P = 0.289; I 2 = 14.7%). Subsequently, a subgroup analysis was performed while obvious heterogeneity still existed. Therefore, we carried out a meta-regression with a Knapp-Hartung modification to identify the sources of heterogeneity and we found that inconsistencies with HWE in the control group may contribute to the heterogeneity in CC vs. AA (P = 0.002) and CC vs. AA + AC (P = 0.014). The main results of the heterogeneity analysis are shown in Table 4.
Table 4.
Heterogeneity analysis of the association between MTHFR A1298C polymorphism and CHD
| Subgroup analysis | Genetic model | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C vs. A | CC vs. AA | CC vs. AC | AC vs. AA | Dominant model | Recessive model | |||||||||||||
| Heterogeneity | Meta-regression | Heterogeneity | Meta-regression | Heterogeneity | Meta-regression | Heterogeneity | Meta-regression | Heterogeneity | Meta-regression | Heterogeneity | Meta-regression | |||||||
| I2 (%) | P | P | I2 (%) | P | P | I2 (%) | P | P | I2 (%) | P | P | I2 (%) | P | P | I2 (%) | P | P | |
| Summary | 73.0 | 0.000 | 51.1 | 0.012 | 0.0 | 0.569 | 70.6 | 0.000 | 74.5 | 0.000 | 14.7 | 0.289 | ||||||
| HWE | 0.076 | 0.002 | 0.248 | 0.558 | 0.308 | 0.014 | ||||||||||||
| Consistent | 58.4 | 0.006 | 20.3 | 0.251 | 0.0 | 0.859 | 63.7 | 0.001 | 65.3 | 0.001 | 0.0 | 0.709 | ||||||
| Inconsistent | 75.5 | 0.007 | 0.0 | 0.784 | 46.0 | 0.135 | 83.1 | 0.000 | 83.6 | 0.000 | 0.0 | 0.769 | ||||||
| Down’s syndrome | 0.111 | 0.106 | 0.433 | 0.186 | 0.143 | 0.149 | ||||||||||||
| Without | 73.2 | 0.000 | 49.8 | 0.021 | 0.0 | 0.461 | 71.7 | 0.000 | 75.1 | 0.000 | 13.6 | 0.308 | ||||||
| With | 0.0 | 0.908 | 0.0 | 0.890 | 0.0 | 0.941 | 0.0 | 0.886 | 0.0 | 0.856 | 0.0 | 0.940 | ||||||
| Region | 0.218 | 0.466 | 0.492 | 0.224 | 0.230 | 0.534 | ||||||||||||
| Europe | 68.0 | 0.008 | 38.9 | 0.147 | 0.0 | 0.846 | 68.5 | 0.007 | 72.0 | 0.003 | 0.0 | 0.594 | ||||||
| Asia | 79.2 | 0.000 | 60.3 | 0.039 | 36.2 | 0.180 | 78.9 | 0.000 | 80.9 | 0.000 | 40.5 | 0.151 | ||||||
| America | 44.7 | 0.164 | 20.4 | 0.285 | 0.0 | 0.612 | 40.5 | 0.186 | 50.2 | 0.134 | 0.0 | 0.441 | ||||||
| Sample size | 0.723 | 0.180 | 0.679 | 0.874 | 0.818 | 0.214 | ||||||||||||
| Large (>100) | 38.7 | 0.110 | 0.0 | 0.619 | 0.0 | 0.603 | 57.3 | 0.016 | 53.1 | 0.029 | 0.0 | 0.664 | ||||||
| Small (<100) | 83.8 | 0.000 | 64.0 | 0.011 | 12.0 | 0.338 | 80.7 | 0.000 | 84.5 | 0.000 | 37.1 | 0.145 | ||||||
Furthermore, we created Galbraith plots to graphically assess the sources of heterogeneity in models of C vs. A, CC vs. AA, AC vs. AA and CC + AC vs. AA (Additional file 1: Figure S1). A total of 5, 1, 5, and 6 studies were identified as the main sources of heterogeneity, respectively. After the outlier studies were omitted, the heterogeneity was effectively removed in models of C vs. A (Q = 11.78; P = 0.300; I 2 = 15.1%), CC vs. AA (Q = 20.56; P = 0.082; I 2 = 36.8%), AC vs. AA (Q = 13.57; P = 0.194; I 2 = 26.3%) and CC + AC vs. AA (Q = 11.71; P = 0.230; I 2 = 23.1%). Meanwhile, the corresponding ORs did not change substantially (C vs. A: OR = 0.97, 95% CI: 0.87–1.08; CC vs. AA: OR = 1.23, 95% CI: 0.95–1.59; AC vs. AA: OR = 0.96, 95% CI: 0.83–1.11; CC + AC vs. AA: OR = 0.95, 95% CI: 0.82–1.09).
Publication bias
Publication bias was assessed by Begg and Egger’s test (Additional file 2: Table S1). No publication biases for MTHFR A1298C polymorphism were detected in all genetic models. Egger’s funnel plots are shown in Additional file 3: Figure S2.
Discussion
According to statistics, nearly 18 million newborns come into the world each year and in China, about 0.9 million are born with a birth defect. Among them, up to 200,000 newborns are born with CHD, which has been the major birth defect for decades on the mainland of China. This is a serious problem, as the natural mortality rate of infants is 30 to 40% for one year without treatment. However, the etiology of CHD has not yet been fully understood and no effective interventions can be delivered. In most cases, surgery is the only way to save life, with this treatment costing tens of billions of dollars. Therefore, the prevention of CHD has great significance for public health.
As we known, MTHFR is a crucial regulatory enzyme in the metabolic pathway of folate/homocysteine and lack of it may cause hyperhomocysteinemia, which is one of the proven risk factors related to the occurrence of CHDs [28, 29]. MTHFR A1298C polymorphism results in the conversion of glutamate to alanine, which leads to a high level of homocysteine and low plasma concentration of folic acid. What is more, our previous meta-analysis demonstrated a positive association between maternal folate supplementation and a decreased risk of CHD [30]. From that date forward, many studies have explored the association between MTHFR A1298C polymorphism and the risk of CHD, yet the results are still controversial.
Recently, a meta-analysis for association between A1298C polymorphism and CHD was conducted and it was found that CC vs. AC (OR = 1.354, 95% CI: 1.022–1.793) and the recessive model (OR = 1.322, 95% CI: 1.015–1.732) increased the risk of CHD in the overall pediatric population. However, due to the lack of the analysis of the source of the heterogeneity and small sample size (1834 cases and 1744 controls), we believe that the results of the meta-analysis might not be stable. Thus, we performed an updated meta-analysis to integrate the same kinds of studies to increase the sample size and statistical power (2207 cases and 2364 controls), and then obtain a more authentic result.
Our results indicate that the CC homozygote increased susceptibility to CHD by 38% compared with AA + AC (95% CI: 1.10–1.73) in the overall population. Patients with DS are always accompanied by CHD and might be interfering with the results. Therefore, we excluded relevant studies and the results showed that the CC homozygote had a 47%, 29% and 44% elevated risk of CHD, when compared with AA (95% CI: 1.01–2.14), AC (95% CI: 1.00–1.66), or AA + AC (95% CI: 1.14–1.82) in children without DS, respectively. Meanwhile, compared with AC or AA + AC, CC homozygote also increased by 48% (95% CI: 1.05–2.09) and 40% (95% CI: 1.01–1.94) the risk of CHD to children in Europe, respectively. We conducted heterogeneity analyses, such as subgroup analysis, meta-regression, sensitivity analysis and Galbraith plots, to detect the source of heterogeneity, and found that the corresponding ORs did not change substantially after excluding the relevant studies. Therefore, our results are relatively reliable and stable; namely, CC homozygote is a risk factor for CHD, especially in children without DS or children in Europe.
However, there were still some limitations in our meta-analysis. First, all the data from studies were collected only in Chinese and English, which means that relevant studies performed in other languages, may be missed. Additionally, we extracted all our raw data from case-control studies, which were prone to information biases. Although no publication bias was found, heterogeneity existed, which may confuse the overall results. Therefore, we conducted meta-regression, Galbraith plots and sensitivity analysis to explore the sources of heterogeneity and proved that our results are stable. Second, almost all studies did not definitively classify the CHDs and used different types of heart defects in their papers. Thus, we have not included enough studies to perform a subgroup analysis with different CHD subtypes, which may have various etiologies. Third, the subjects of all the studies were from different countries and races, with different living environments, physical conditions and diet. Moreover, folate intake was the most crucial factor. Studies have demonstrated that periconceptional use of multivitamins containing folic acid can reduce the incidence of CHDs [31, 32]. Additionally, our previous meta-analysis showed that moderate supplementation with folic acid is associated with a significantly decreased risk of CHDs [30]. Thus, periconceptional folic acid supplementation and food fortification with folic acid should be taken in consideration. Finally, the number of case-controls is still not enough to draw a definite conclusion.
Conclusion
In conclusion, the results of this meta-analysis demonstrate that MTHFR A1298C polymorphism is significantly associated with CHD susceptibility. However, further larger sample studies are warranted to enable definitive conclusions, especially in plasma homocysteine levels, enzyme activity, and periconceptional folic acid supplementation. What is more, gene-gene and gene-environment interactions should be taken into account to further investigate the association between the MTHFR A1298C polymorphisms and CHD risk.
Additional files
Galbraith plots for models of C vs. A, CC vs. AA, AC vs. AA and CC + AC vs. AA. (TIFF 1442 kb)
Publication bias for MTHFR A1298C polymorphism. (DOCX 15 kb)
Egger's funnel plots in all genetic models for MTHFR A1298C polymorphism. (TIFF 1828 kb)
Acknowledgements
Not applicable.
Funding
The work was fully supported by grants from the National Natural Science Foundation of China (No. 81370277 & 81,370,279) and Jiangsu Provincial Special Program of Medical Science (BL2013003).
Availability of data and materials
All data generated or analyzed during this study are included in PubMed and Web of Science databases.
Authors’ contributions
XMM and DY conceived and designed the experiments. DY, ZLZ and XMM performed the experiments. DY, ZLZ and ZYW analyzed the data. XDZ contributed analysis tools. DY, ZLZ and ZYW wrote the paper. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13052-017-0425-1) contains supplementary material, which is available to authorized users.
Contributor Information
Di Yu, Email: 391122912@qq.com.
Zhulun Zhuang, Email: zhulunzhuang@126.com.
Zhongyuan Wen, Email: 353228540@qq.com.
Xiaodong Zang, Email: 963023105@qq.com.
Xuming Mo, Email: yd@njmu.edu.cn.
References
- 1.Hoffman JI. Incidence of congenital heart disease: I. Postnatal incidence. Pediatr Cardiol. 1995;16:103–113. doi: 10.1007/BF00801907. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman JI. Incidence of congenital heart disease: ii. Prenatal incidence. Pediatr Cardiol. 1995;16:155–165. doi: 10.1007/BF00801907. [DOI] [PubMed] [Google Scholar]
- 3.Gilboa SM, Salemi JL, Nembhard WN, Fixler DE, Correa A. Mortality resulting from congenital heart disease among children and adults in the united states, 1999 to 2006. Circulation. 2010;122:2254–2263. doi: 10.1161/CIRCULATIONAHA.110.947002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burn J, Brennan P, Little J, Holloway S, Coffey R, Somerville J, Dennis NR, Allan L, Arnold R, Deanfield JE, Godman M, Houston A, Keeton B, Oakley C, Scott O, Silove E, Wilkinson J, Pembrey M, Hunter AS. Recurrence risks in offspring of adults with major heart defects: results from first cohort of british collaborative study. Lancet. 1998;351:311–316. doi: 10.1016/S0140-6736(97)06486-6. [DOI] [PubMed] [Google Scholar]
- 5.Fredriksen A, Meyer K, Ueland PM, Vollset SE, Grotmol T, Schneede J. Large-scale population-based metabolic phenotyping of thirteen genetic polymorphisms related to one-carbon metabolism. Hum Mutat. 2007;28:856–865. doi: 10.1002/humu.20522. [DOI] [PubMed] [Google Scholar]
- 6.Cordell HJ, Bentham J, Topf A, Zelenika D, Heath S, Mamasoula C, Cosgrove C, Blue G, Granados-Riveron J, Setchfield K, Thornborough C, Breckpot J, Soemedi R, Martin R, Rahman TJ, Hall D, van Engelen K, Moorman AF, Zwinderman AH, Barnett P, Koopmann TT, Adriaens ME, Varro A, George AL, Jr, Dos Remedios C, Bishopric NH, Bezzina CR, O'Sullivan J, Gewillig M, Bu’Lock FA, Winlaw D, Bhattacharya S, Devriendt K, Brook JD, Mulder BJ, Mital S, Postma AV, Lathrop GM, Farrall M, Goodship JA, Keavney BD, et al. Nat Genet. 2013;45:822–824. doi: 10.1038/ng.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Z, Shi Y, Mo X, Xu J, Zhao B, Lin Y, Yang S, Xu Z, Dai J, Pan S, Da M, Wang X, Qian B, Wen Y, Wen J, Xing J, Guo X, Xia Y, Ma H, Jin G, Yu S, Liu J, Zhou Z, Wang X, Chen Y, Sha J, Shen HA. Genome-wide association study identifies two risk loci for congenital heart malformations in han chinese populations. Nat Genet. 2013;45:818–821. doi: 10.1038/ng.2636. [DOI] [PubMed] [Google Scholar]
- 8.Goyette P, Pai A, Milos R, Frosst P, Tran P, Chen Z, Chan M, Rozen R. Gene structure of human and mouse methylenetetrahydrofolate reductase (mthfr) Mamm Genome. 1998;9:652–656. doi: 10.1007/s003359900838. [DOI] [PubMed] [Google Scholar]
- 9.Hobbs CA, James SJ, Parsian A, Krakowiak PA, Jernigan S, Greenhaw JJ, Lu Y, Cleves MA. Congenital heart defects and genetic variants in the methylenetetrahydroflate reductase gene. J Med Genet. 2006;43:162–166. doi: 10.1136/jmg.2005.032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huhta JC, Hernandez-Robles JA. Homocysteine, folate, and congenital heart defects. Fetal Pediatr Pathol. 2005;24:71–79. doi: 10.1080/15227950591008240. [DOI] [PubMed] [Google Scholar]
- 11.Xuan C, Li H, Zhao JX, Wang HW, Wang Y, Ning CP, Liu Z, Zhang BB, He GW, Lun LM. Association between mthfr polymorphisms and congenital heart disease: a meta-analysis based on 9,329 cases and 15,076 controls. Sci Rep. 2014;4:7311. doi: 10.1038/srep07311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao CS, Wei J, Huang HW, Yang SC. Correlation between methyltetrahydrofolate reductase (mthfr) polymorphisms and isolated patent ductus arteriosus in taiwan. Heart Lung Circ. 2014;23:655–660. doi: 10.1016/j.hlc.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 13.Christensen KE, Zada YF, Rohlicek CV, Andelfinger GU, Michaud JL, Bigras JL, Richter A, Dube MP, Rozen R. Risk of congenital heart defects is influenced by genetic variation in folate metabolism. Cardiol Young. 2013;23:89–98. doi: 10.1017/S1047951112000431. [DOI] [PubMed] [Google Scholar]
- 14.Galdieri LC, Arrieta SR, Silva CM, Pedra CA, D'Almeida V. Homocysteine concentrations and molecular analysis in patients with congenital heart defects. Arch Med Res. 2007;38:212–218. doi: 10.1016/j.arcmed.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 15.Huang J, Mei J, Jiang L, Jiang Z, Liu H, Ding F. Mthfr rs1801133 c>t polymorphism is associated with an increased risk of tetralogy of fallot. Biomed Rep. 2014;2:172–176. doi: 10.3892/br.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koshy T, Venkatesan V, Perumal V, Hegde S, Paul SF. The a1298c methylenetetrahydrofolate reductase gene variant as a susceptibility gene for non-syndromic conotruncal heart defects in an indian population. Pediatr Cardiol. 2015;36:1470–1475. doi: 10.1007/s00246-015-1188-3. [DOI] [PubMed] [Google Scholar]
- 17.Li D, Yu K, Ma Y, Liu Y, Ji L. Correlationship between congenital heart disease and polymorphism of mthfr gene. Wei Sheng Yan Jiu. 2015;44:933–938. [PubMed] [Google Scholar]
- 18.Locke AE, Dooley KJ, Tinker SW, Cheong SY, Feingold E, Allen EG, Freeman SB, Torfs CP, Cua CL, Epstein MP, MC W, Lin X, Capone G, Sherman SL, Bean LJ. Variation in folate pathway genes contributes to risk of congenital heart defects among individuals with down syndrome. Genet Epidemiol. 2010;34:613–623. doi: 10.1002/gepi.20518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obermann-Borst SA, van Driel LM, Helbing WA, de Jonge R, Wildhagen MF, Steegers EA, Steegers-Theunissen RP. Congenital heart defects and biomarkers of methylation in children: a case-control study. Eur J Clin Investig. 2011;41:143–150. doi: 10.1111/j.1365-2362.2010.02388.x. [DOI] [PubMed] [Google Scholar]
- 20.Sahiner UM, Alanay Y, Alehan D, Tuncbilek E, Alikasifoglu M. Methylene tetrahydrofolate reductase polymorphisms and homocysteine level in heart defects. Pediatr Int. 2014;56:167–172. doi: 10.1111/ped.12222. [DOI] [PubMed] [Google Scholar]
- 21.Sayin Kocakap BD, Sanli C, Cabuk F, Koc M, Kutsal A. Association of mthfr a1298c polymorphism with conotruncal heart disease. Cardiol Young. 2015;25:1326–1331. doi: 10.1017/S1047951114002467. [DOI] [PubMed] [Google Scholar]
- 22.Storti S, Vittorini S, Iascone MR, Sacchelli M, Collavoli A, Ripoli A, Cocchi G, Biagini A, Clerico A. Association between 5,10-methylenetetrahydrofolate reductase c677t and a1298c polymorphisms and conotruncal heart defects. Clin Chem Lab Med. 2003;41:276–280. doi: 10.1515/CCLM.2003.043. [DOI] [PubMed] [Google Scholar]
- 23.van Driel LM, Verkleij-Hagoort AC, de Jonge R, Uitterlinden AG, Steegers EA, van Duijn CM, Steegers-Theunissen RP. Two mthfr polymorphisms, maternal b-vitamin intake, and chds. Birth Defects Res A Clin Mol Teratol. 2008;82:474–481. doi: 10.1002/bdra.20463. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Liu M, Yan W, Mao J, Jiang D, Li H, Chen Y. Association of snps in genes involved in folate metabolism with the risk of congenital heart disease. J Matern Fetal Neonatal Med. 2013;26:1768–1777. doi: 10.3109/14767058.2013.799648. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Xu X, Xue L, Liu X, Gu H, Cao H, Qiu W, Hu Z, Shen H, Chen Y. Mthfr c.1793g>a polymorphism is associated with congenital cardiac disease in a chinese population. Cardiol Young. 2010;20:318–326. doi: 10.1017/S1047951110000247. [DOI] [PubMed] [Google Scholar]
- 26.Zidan HE, Rezk NA, Mohammed D. Mthfr c677t and a1298c gene polymorphisms and their relation to homocysteine level in egyptian children with congenital heart diseases. Gene. 2013;529:119–124. doi: 10.1016/j.gene.2013.07.053. [DOI] [PubMed] [Google Scholar]
- 27.Bozovic IB, Vranekovic J, Cizmarevic NS, Mahulja-Stamenkovic V, Prpic I, Brajenovic-Milic B. Mthfr c677t and a1298c polymorphisms as a risk factor for congenital heart defects in down syndrome. Pediatr Int. 2011;53:546–550. doi: 10.1111/j.1442-200X.2010.03310.x. [DOI] [PubMed] [Google Scholar]
- 28.Gueant-Rodriguez RM, Gueant JL, Debard R, Thirion S, Hong LX, Bronowicki JP, Namour F, Chabi NW, Sanni A, Anello G, Bosco P, Romano C, Amouzou E, Arrieta HR, Sanchez BE, Romano A, Herbeth B, Guilland JC, Mutchinick OM. Prevalence of methylenetetrahydrofolate reductase 677t and 1298c alleles and folate status: a comparative study in mexican, west african, and european populations. Am J Clin Nutr. 2006;83:701–707. doi: 10.1093/ajcn.83.3.701. [DOI] [PubMed] [Google Scholar]
- 29.Kapusta L, Haagmans ML, Steegers EA, Cuypers MH, Blom HJ, Eskes TK. Congenital heart defects and maternal derangement of homocysteine metabolism. J Pediatr. 1999;135:773–774. doi: 10.1016/S0022-3476(99)70102-2. [DOI] [PubMed] [Google Scholar]
- 30.Feng Y, Wang S, Chen R, Tong X, Wu Z, Mo X. Maternal folic acid supplementation and the risk of congenital heart defects in offspring: a meta-analysis of epidemiological observational studies. Sci Rep. 2015;5:8506. doi: 10.1038/srep08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smedts HP, Rakhshandehroo M, Verkleij-Hagoort AC, de Vries JH, Ottenkamp J, Steegers EA, Steegers-Theunissen RP. Maternal intake of fat, riboflavin and nicotinamide and the risk of having offspring with congenital heart defects. Eur J Nutr. 2008;47:357–365. doi: 10.1007/s00394-008-0735-6. [DOI] [PubMed] [Google Scholar]
- 32.Verkleij-Hagoort AC, de Vries JH, Ursem NT, de Jonge R, Hop WC, Steegers-Theunissen RP. Dietary intake of b-vitamins in mothers born a child with a congenital heart defect. Eur J Nutr. 2006;45:478–486. doi: 10.1007/s00394-006-0622-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Galbraith plots for models of C vs. A, CC vs. AA, AC vs. AA and CC + AC vs. AA. (TIFF 1442 kb)
Publication bias for MTHFR A1298C polymorphism. (DOCX 15 kb)
Egger's funnel plots in all genetic models for MTHFR A1298C polymorphism. (TIFF 1828 kb)
Data Availability Statement
All data generated or analyzed during this study are included in PubMed and Web of Science databases.