

Abstract
The collecting duct in the mammalian kidney consists of principal cells (PCs) and intercalated cells (ICs), which regulate electrolyte/fluid and acid/base balance, respectively. The epigenetic regulators of PC and IC differentiation remain obscure. We previously used Aqp2 and V-ATPase B1B2 to label PCs and ICs, respectively. We found that mice with histone H3 K79 methyltransferase Dot1l disrupted in Aqp2-expressing cells (Dot1lAC) vs. Dot1lf/f possessed ~20% more ICs coupled with a similar decrease in PCs. Here, we performed multiple double immunofluorescence staining using various PC and IC markers and confirmed that this finding. Both α-IC and β-IC populations were significantly expanded in Dot1lAC vs. Dot1lf/f. These changes are associated with significantly upregulated V-ATPase B1 and B2, but not Aqp2, AE1, and Pendrin. Chromatin immunoprecipitation assay unveiled a significant reduction of Dot1l and H3K79 di-methylation bound at the Atp6v1b1 5′ flanking region. Overexpression of Dot1a significantly downregulated a stably-transfected luciferase reporter driven by the Atp6v1b1 promoter in IMCD3 cells. This downregulation was impaired, but not completely abolished when a methyltransferase-dead mutant was overexpressed. Taken together, our data suggest that Dot1l is a new epigenetic regulator of PC and IC differentiation and Atp6v1b1 is a new transcriptional target of Dot1l.
Keywords: V-ATPase, histone methyltransferase Dot1l, conditional knockout, principal cells, intercalated cells
INTRODUCTION
A critical role of the renal collecting duct (CD) in the mammalian kidney is the regulation of electrolyte, fluid and acid-base homeostasis. CD consists of principal cells (PCs) and intercalated cells (ICs) [1], which are structurally and functionally distinct. PCs reabsorb Na+ and water. Na+ enters into PCs from the urine through the apical epithelial Na+ channel (ENaC) and exits into blood through the basolateral Na+-K+-ATPase. A similar process occurs for water reabsorption mediated by the coordinated actions of water channels Aqp2 at the apical side and Aqp3 and Aqp4 at the basolateral side. These Na+ and water channels thus serves as the molecular markers of PCs. ICs are responsible for the acid-base balance and express carbonic anhydrase type II (CAII) and the proton-pumping V-ATPase. The latter contains 13 or more subunits such as A, B1 and B2. CAII and V-ATPase subunits are also used to identify ICs in the kidney. ICs are further divided into α-ICs and β-ICs. α-ICs are involved in acid secretion and marked by expression of AE1, the kidney variant of the band 3 Cl−/HCO3-exchanger. β-ICs secrete bicarbonate and are characterized by expression of the sodium-independent chloride/iodide transporter Pendrin [2].
The relative abundance of the three cell types in kidney can be affected by metabolism and the genes involved in the differentiation. For example, metabolic acidosis reduced β-ICs and increased α-ICs without changing the total IC population [3]. ICs as evaluated by detectable AE1 and H+-ATPase expression are severely depleted and replaced by PCs in CAII knockout mice. Hensin/DMBT1 encodes an extracellular matrix protein. Mice deficient in Hensin/DMBT1 contained no typical α-ICs [4]. Disruption of the forkhead transcription factor Foxi1 led to generation of a single cell type (Aqp2+ CAII+) that have no detectable expression of other IC markers including V-ATPase B1, Pendrin, and AE1 [5].
Disruptor of telomeric silencing (Dot1) was originally identified in yeast through a screen aimed to identify genes affecting telomeric silencing [6]. Biochemical analyses led to identification of Dot1 and its mammalian homologs (Dot1l) as the genes encoding a family of methyltransferases specific for histone H3 K79 [7–9]. Dot1l is involved in a variety of biological processes including development, erythropoiesis, differentiation, proliferation, and leukemogenesis [10–12], most likely by regulating transcription of target genes. General knockout of Dot1l results in embryonic lethality in mice [13].
Mouse Dot1l generates 5 alternative splicing variants that we named as Dot1a–e [9]. Among these variants, Dot1a and Dot1b are the only ones whose open reading frames have been characterized. Dot1a has been shown to promote H3 K79 methylation and represses the transcription of the epithelial Na+ channel αENaC and several other aldosterone-regulated genes in 293T cells, mouse cortical collecting duct M1 cells, mouse inner medulla collecting duct IMCD3 cells, and mouse kidney [14–16]. Consistently, Northern blotting and immunofluorescence staining analyses revealed strong expression of Dot1l mRNAs and H3 di-methyl K79 (m2K79) in mouse kidney [9, 16]. Most recently, using Aqp2Cre PAC transgene driver, we generated Dot1lAC mice that have disrupted Dot1l function in Aqp2-lineage cells [17]. Deletion of Dot1l in these cells was validated by complete loss of mono-, di-, and tri-methylated H3 K79 without affecting methylation in all other residues examined. Since Dot1l is the sole enzyme mediating H3 K79 methylation in mice, we used loss of di-methylated H3 K79 (H3m2K79) as a marker to trace Aqp2 lineage. We found that both PCs and most of ICs are derivatives of a common ancestral population that expresses Aqp2. Moreover, Dot1lAC vs. Dot1lf/f mice have ~20% less of PCs and ~16% more ICs, as evaluated by double immunofluorescence staining using two specific antibodies. One of them recognizes Aqp2 and the other labels both of V-ATPase subunits B1 and B2 [17]. Consistently, Dot1lAC vs. Dot1lf/f mice exhibited relatively hypoosmotic polyuria on a normal chow diet or after 24-h water deprivation [17, 18].
In this report, we further demonstrated that Dot1lAC vs. Dot1lf/f mice had decreased PCs and increased ICs including both α-ICs and β-ICs, reduced V-ATPase B1 mRNA levels, and diminished Dot1l and H3 K79 di-methylation bound at the Atp6v1b1 5′ flanking region. We also show that Dot1a inhibited Atp6v1b1 promoter activity, which is largely dependent on its methyltransferase activity, in IMCD3 cells. This study highlights the function of Dot1l in regulating PC and IC differentiation and addition of Atp6v1b1 as a new member in the growing list of Dot1l target for transcriptional regulation.
MATERIALS AND METHODS
Reagents
The primary antibodies used are chicken anti-Aqp2 LC54 (a gift from James B. Wade, University of Maryland School, Baltimore, MD), chicken anti-V-ATPase B1 [19], rabbit anti-AE1 (Alpha Diagnostic, 396768A3), anti-H3 di-methyl K79 (abcom), and five antibodies from Santa Cruz: goat anti-Aqp2 (sc-9882), goat anti-Aqp3 (sc-9885), goat anti-Pendrin (sc-16894), and rabbit anti-Aqp2 (sc-28629). The secondary antibodies are Dylight 594-AffiniPure goat anti-chicken IgG (Jackson ImmunoResearch LABORATORIE) and from Invitrogen Alexa Fluor 488-conjugated goat anti-mouse IgG (774904), Alexa Fluor 488-conjugated donkey anti-goat IgG (51475A), Alexa Fluor 594-conjugated donkey anti-mouse IgG (796011), and Alexa Fluor 594-conjugated donkey anti-rabbit IgG (A10042). Constructs expressing WT or methyltransferase-dead mutant Dot1a were described [9]. The Atp6v1b1 5′-flanking region (0.84 kb; −842 to −1, relative to the transcription start site) was amplified by PCR with the genomic DNA isolated from a Dot1lf/f mouse as the template. The fragment was inserted into pGL3Zeocin at the MluI-XhoI sites to generate pGL3Zeocin-0.84mVB1 for stable transfection.
Mouse models
Dot1lf/f, Aqp2Cre and Dot1lAC mice have been previously described [10, 20]. Littermates from the cross between Dot1lf/f and Dot1lAC mice were used for all of the current studies. Following weaning, mice were maintained with a standard pellet chow and used at the age of 2–5 months for immunofluorescence, real-time RT-qPCR, and chromatin immunoprecipitation (ChIP) analyses.
All animal experiments were approved by the University of Texas Health Science Center at Houston Animal Welfare Committee and carried out according to NIH Guides for the Care and Use of Laboratory Animals.
Immunofluorescence studies
Immunofluorescence (IF) staining was conducted and analyzed as we reported before [21, 22], with following modifications. IF images were examined and scored for the staining of each primary antibody with the aid of Adobe Photoshop CS4. To gauge the relative percentages of the 4 cell types in each double IF, we examined 3–4 mice/genotype in the related double IF experiments. Five to ten fields were examined from the whole kidney in each mouse. About 900 cells from >100 CNT/CD tubules in the cortex, outer medulla and inner medulla were counted and categorized on the basis of the staining of each primary antibody used. CNT/CD structures were defined by possessing at least one PC or IC. Any structures lacking a PC or IC were excluded from cell counting. In the IF experiments using anti-Aqp2 or anti-Aqp3 antibodies, CNT/CD segments were recognized by the existence of at least one Aqp2+ or Aqp3+ cells, respectively. It is possible that the populations of CNT/CD structures defined by the presence of at least one Aqp2+ or Aqp3+ cells are not necessarily identical, leading to fluctuations in the relative percentages of PCs and ICs in different IF experiments of the same genotype.
Genotyping, luciferase assay, real-time RT-qPCR, and ChIP assay
These assays were performed according to our published protocols [14, 16, 23, 24]. For luciferase assay, pGL3Zeocin-0.84mVB1 was transfected into IMCD3 cells. Cells were selected with Zeocin (800 μg/ml) for 3 weeks, with medium changes every 2–3 days. Survival cells were pooled to establish a stable cell line for transient transfection of Dot1a constructs. All primer sequences are available upon request.
Statistical analysis
Quantitative data are shown as mean±SEM. Statistical significance was evaluated via Student t-test. P<0.05 was considered significant.
RESULTS
Double immunofluorescence analyses revealed significantly reduced PCs and increased ICs in Dot1lAC vs. Dot1lf/f mice. Previous double immunofluorescence (IF) analyses with a rabbit anti-V-ATPase subunits B1 and B2 and a goat anti-Aqp2 revealed that Dot1lAC vs. Dot1lf/f mice have increased ICs to the detriment of PCs. To further solidify this finding, we first conducted double IF by replacing the anti-V-ATPase subunits B1 and B2 with an antibody specific for V-ATPase B1 (B1) only. This is because B1 and B2 display differential expression patterns, with B1 expression being restricted to ICs and B2 ubiquitous [25, 26].
The staining of the two antibodies differentiated the cells in the CNT/CD into 4 types: Aqp2+B1−, Aqp2-B1+, Aqp2+B1+ and Aqp2-B1−, each of which existed in the two genotypes. Representative IF images from outer medulla are shown in Figure 1A. Compared with Dot1lf/f mice, Dot1lAC mice exhibited significant reductions of PCs (Aqp2+B1−) by ~13% to ~22% and significant increases of ICs (Aqp2-B1+) by ~12% to 23%, depending on the regions examined (Figure 1, B–D). Aqp2+B1+ cells were detected occasionally (<4%) throughout the kidney in each genotype. The proportion of Aqp2+B1+ cells reached the highest (14%) in the outer medulla and the lowest (4%) in the inner medulla of Dot1lAC (Figure 1, B–D). The presence of Aqp2-B1− and Aqp2+B1+ cells is consistent with our hypothesis that ICs are derived from Aqp2+ progenitor cells. These two types of cells may represent the intermediate stages of the differentiation [17].
Figure 1.

Double immunofluorescence staining analyses of Aqp2 and V-ATPase B1 showing less PCs and more ICs in Dot1lAC than in control mice. (A) Representative IF images showing staining of Aqp2 (red) and V-ATPase B1 (green) as PC and IC markers, respectively, in the outer medulla of adult Dot1lf/f and Dot1lAC mice. The insert shows the enlarged boxed areas. Scale bar: 100 μM and 25 μM for insert. (B–D). Bar graphs showing the relative percentages of the 4 groups based on the staining of the two markers in the CNT/CD cells in the cortex (B), outer medulla (C), and inner medulla (D) of Dot1lf/f and Dot1lAC mice. In all cases, *P < 0.05 vs. Dot1lf/f.
Dot1lAC vs. Dot1lf/f mice have significantly increased α-ICs. ICs can be further divided into αICs and β-ICs, which express AE1 and Pendrin, respectively. To determine if the expansion of IC pool is associated with an enlarged population of α-ICs in Dot1lAC vs. Dot1lf/f, we carried out two double IF experiments. AE1 was used as the α-IC marker, along with either Aqp2 or Aqp3 to label the PCs.
Figure 2A shows the Aqp2/AE1 co-stained outer medulla of the two genotypes. Double positive cells were clearly seen and highlighted in the inserts. Compared with Dot1lf/f mice, Dot1lAC mice significantly increased the α-ICs (Aqp2-AE1+) by 8%–13%, in the three regions examined (Figure 2B–D). As expected, the proportion of PCs represented by Aqp2+AE1− was significantly less in Dot1lAC than in Dot1lf/f, regardless of the region examined. Such reductions ranged from 16% in the cortex to 22% in the inner medulla. In the cortex and outer medulla, Aqp2+AE1+ cells were not significantly different between the two genotypes, accounting for 3% to 4%. However, the percentages of Aqp2+AE1+ in the inner medulla of the two genotypes were much higher, reaching 10% and 15%, respectively. Similarly, the relative Aqp2-AE1− cell proportion was also significantly expanded throughout the whole kidneys in Dot1lAC vs. Dot1lf/f mice. Since Aqp2-AE1− cells in the cortex are most likely to be β-ICs, these observations raised the possibility that Dot1l deletion also facilitates β-IC specification. Indeed, our subsequent analyses supported this idea (see below).
Figure 2.

Double immunofluorescence staining analyses of Aqp2 and AE1 showing less PCs and more α-ICs in Dot1lAC than in control mice. (A) Representative IF images showing staining of Aqp2 (red) and AE1 (green) as PC and α-IC markers, respectively, in the outer medulla of adult Dot1lf/f and Dot1lAC mice. The insert shows the enlarged boxed areas. Scale bar: 50 μM and 25 μM for insert. (B–D). Bar graphs showing the relative percentages of the 4 groups based on the staining of the two markers in the CNT/CD cells in the cortex (B), outer medulla (C), and inner medulla (D) of Dot1lf/f and Dot1lAC mice. In all cases, *P < 0.05 vs. Dot1lf/f. Arrowhead: Aqp2+ AE1+ cells.
Aqp3/AE1 double staining unearthed all 4 types of CNT/CD cells throughout the kidneys of both genotypes. Representative images of the outer medulla are shown in Figure 3A. Like all aforementioned double IF experiments, Aqp3/AE1 staining also revealed significant reductions of the PCs (Aqp3+AE1−) by 12%–20% and similar increases of α-ICs (Aqp3-AE1+) in each of the three regions examined in Dot1lAC vs. Dot1lf/f mice. Double negative cells were also substantially detected, particularly in the cortex of the both genotypes. In the cortex, the percentage of the double negative cells significantly risen by 8% (20% in Dot1lAC vs. 12% in Dot1lf/f). These results are in line with what we observed in the Aqp2/AE1 staining experiment. Taken together, our data consistently suggest that the relative percentages of α-ICs (Aqp2-AE1+ and Aqp3-AE1+) and the potential β-ICs (Aqp2-AE1− and Aqp3-AE1− in the cortex) were significantly elevated in the expense of PCs (Aqp2+AE1− and Aqp3+AE1−) when Dot1l function is disrupted. We conclude that Dot1l deletion is advantageous to differentiation of the α-ICs and possibly β-ICs as well.
Figure 3.
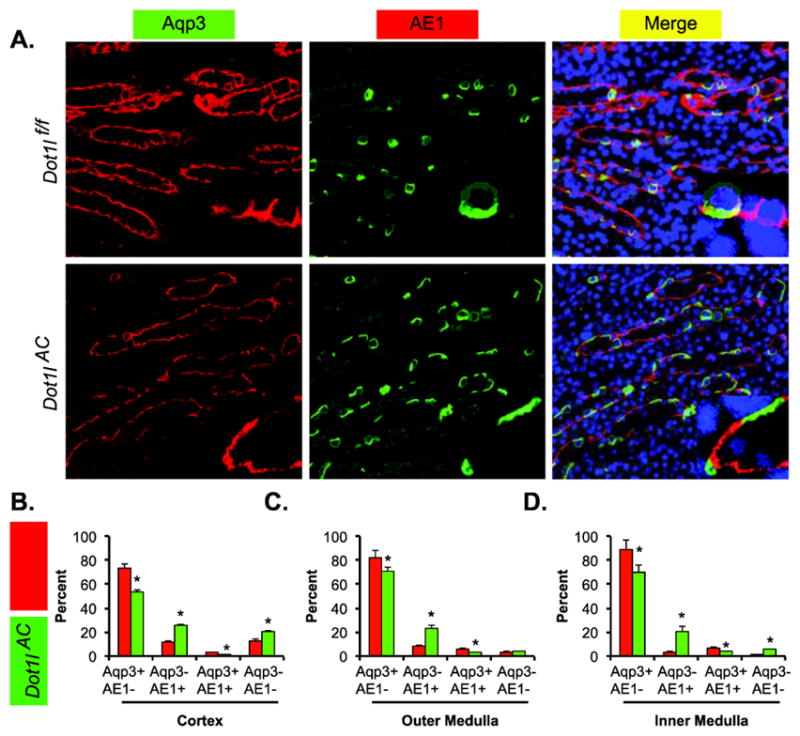
Double immunofluorescence staining analyses of Aqp3 and AE1 showing less PCs and more α-ICs in Dot1lAC than in control mice. (A) Representative IF images showing staining of Aqp3 (red) and AE1 (green) as PC and α-IC markers, respectively, in the outer medulla of adult Dot1lf/f and Dot1lAC mice. The insert shows the enlarged boxed areas. Scale bar: 100 μM and 25 μM for insert. (B–D). Bar graphs showing the relative percentages of the 4 groups based on the staining of the two markers in the CNT/CD cells in the cortex (B), outer medulla (C), and inner medulla (D) of Dot1lf/f and Dot1lAC mice. In all cases, *P < 0.05 vs. Dot1lf/f.
Dot1lAC vs. Dot1lf/f mice have significantly increased β-ICs. The impact of Dot1l inactivation on β-ICs was directly pursued through double IF using antibodies against Aqp2 and Pendrin. We focused on the cortex, the primary, if not the exclusive region where Pendrin+ β-ICs reside (Figure 4A). Clearly, the loss of Dot1l function facilitated the expansion of β-ICs (Aqp2− Pendrin+) population by 12%. This change was associated with a decrease in Aqp2+ Pendrin− by 19% and an increase in Aqp2− Pendrin− by 10% (Figure 4B). Since Aqp2− Pendrin− cells are presumably Aqp2− AE1+ α-ICs, the significantly increased Aqp2− Pendrin- cell percentage is consistent with the expanded α-ICs as shown in Figure 2B. β-ICs appeared very rarely in the outer and inner medulla in both groups. These observations suggest that the removal of Dot1l function can increase β-ICs in the cortex, but is not sufficient to do so in the medulla.
Figure 4.
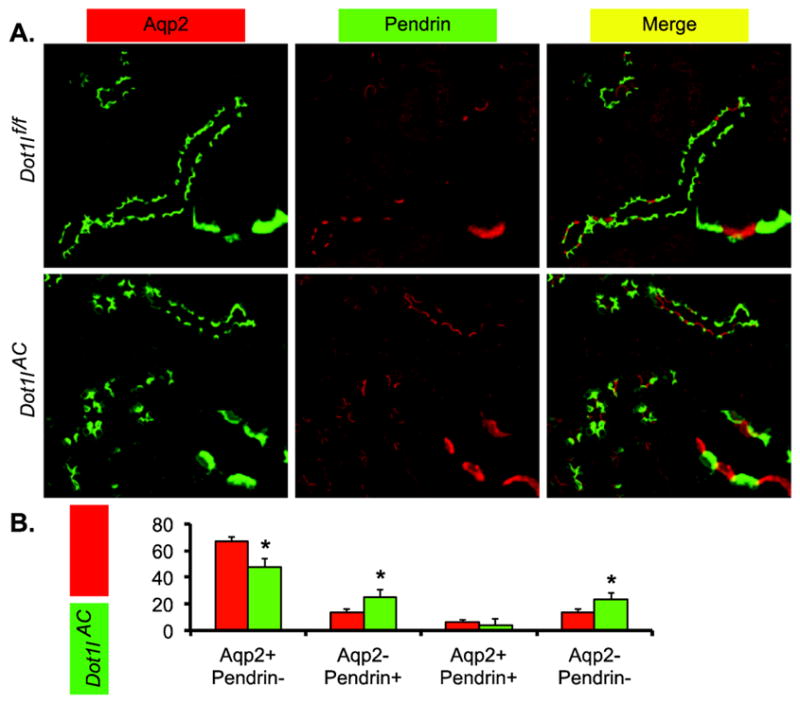
Double immunofluorescence staining analyses of Aqp2 and Pendrin showing less PCs and more β-ICs in Dot1lAC than in control mice. (A) Representative IF images showing staining of Aqp2 (green) and Pendrin (red) as PC and β-IC markers, respectively, in the cortex of adult Dot1lf/f and Dot1lAC mice. The insert shows the enlarged boxed areas. Scale bar: 100 μM and 25 μM for insert. (B–D). Bar graphs showing the relative percentages of the 4 groups based on the staining of the two markers in the CNT/CD cells in the cortex (B), outer medulla (C), and inner medulla (D) of Dot1lf/f and Dot1lAC mice. In all cases, *P < 0.05 vs. Dot1lf/f.
Dot1l downregulates expression of V-ATPase B1. To determine if the increased number of ICs is correlated with an overall increase in the mRNA expression of the IC markers, we conducted real-time RT-qPCR analysis of the total RNAs isolated from Dot1lf/f and Dot1lAC kidneys (n=6 mice/genotype). Dot1lAC vs. Dot1lf/f mice significantly increased mRNA levels of B1 by 48% and B2 by 55%, with slightly decreased Aqp2, mildly increased Pendrin, and no obvious changes in V-ATPase A and AE1 (Figure 5, A–F).
Figure 5.

V-ATPase B1 and B2 mRNA levels are significantly higher in Dot1lAC vs. Dot1lf/f mice. (A–F) Real-time RT-qPCR analyses of the total RNAs isolated from adult Dot1lf/f and Dot1lAC kidneys (n=6 mice/genotype) for expression of the genes as indicated, with β-actin as internal control and mRNA levels set to 1 in Dot1lf/f mice. In all cases, *P < 0.05 vs. Dot1lf/f.
To investigate if Dot1a represses Atp6v1b1, which encodes the V-ATPase B1, by promoting histone H3 di-methyl K79 (H3m2K79) at Atp6v1b1 5′ flanking region and the first part of gene body (exon 1), we performed chromatin immunoprecipitation (ChIP), complemented with real-time qPCR. Within the 5′ fragment spanning from −4470 to −1 (relative to the translation start site ATG), there is a 3.5-kb region full of repeat elements. The presence of the repeated elements excluded the possibility to design primers for PCR analysis. We divided the remaining sequences into subregions A–D (Figure 6A). ChIP signals of Dot1a and H3m2K79 were substantially detected in all subregions in Dot1lf/f mice and were significantly impaired in C and D, and mildly reduced in A and B in Dot1lAC vs. Dot1lf/f mice (Figure 6, B & C). Others and we have rigorously confirmed the specificities of Dot1a and H3m2K79 antibodies [16, 23, 24, 27]. Moreover, ChIP with normal rabbit IgG yielded barely detectable background signal (data not shown). Although our data are consistent with the notion that Dot1l plays a role in the initiation of transcription, the involvement of Dot1l in elongation, as suggested by genome-wide analyses (see discussion), cannot be ruled out. This is because we only examined the 5′ flanking region and the very first part of the gene body in our ChIP assay.
Figure 6.

Dot1l represses Atp6v1b1 at least in part by modulating targeted H3 K79 hypermethylation at the Atp6v1b1 promoter. (A) Diagram of the 5′ flanking region of Atp6v1b1 that encodes V-ATPase B1. (B & C) ChIP demonstrating impaired binding of Dot1l and H3m2K79 in the Atp6v1b1 5′ regulatory region. Chromatin from adult Dot1lf/f and Dot1lAC mice (n=6 mice/group) was immunoprecipitated by the rabbit antibodies specific for Dot1l (B) and H3m2K79 (C), followed by real-time qPCR with primers amplifying subregions A–D as shown in A. Relative ChIP efficiency was defined as the immunoprecipitated amount of materials present as compared to that of the initial input sample. *: P<0.05 vs. Dot1lf/f within the same subregion. (D) Luciferase assay showing that Dot1a represses a luciferase construct driven by a 0.84kb-promoter of Atp6v1b1 in IMCD3 cells. IMCD3 cells stably carrying the luciferase construct were transfected with an empty vector, or a construct expressing WT or methyltransferase-dead mutant Dot1a (MT) and analyzed for luciferase activity (n=3). *: P<0.05 vs. Vec.
To directly test if Dot1a restrains Atp6v1b1 promoter activity, we conducted luciferase assay. A luciferase reporter driven by an 840-bp promoter of Atp6v1b1 was constructed and stably transfected into IMCD3 cells. The resulting IMCD3 cells were then transiently transfected with an empty vector as control, or constructs expressing WT or mutant Dot1a. As shown in Figure 6D, overexpression of WT Dot1a significantly decreased the activity of the luciferase reporter to 45% of vector control. Such effect was largely compromised in the cells transfected with the methyltransferase-dead Dot1a mutant. Collectively, our data suggest that Dot1a resides at the Atp6v1b1 promoter and represses V-ATPase B1 mRNA expression, partially rather than completely in a methyltransferase-dependent manner.
DISCUSSION
In this study, we 1) performed a set of double IF using multiple PC and IC markers in various combinations and obtained consistent results supporting that inactivation of Dot1l leads to reduced PCs and increased ICs; 2) revealed that the increase of ICs is associated with increased proportions of both α-ICs and β-ICs; 3) defined Atp6v1b1 as a novel transcriptional target of Dot1l; and 4) determined the association of Dot1a and H3m2K79 with the Atp6v1b1 5′ regulatory regions as the potential mode of Dot1l-mediated repression.
Using loss of histone H3 K79 di-methylation, which is solely catalyzed by Dot1l, as the tracing marker, we have reported that Aqp2+ progenitor cells give rise to ICs in Dot1lAC mice and deletion of Dot1l favors this process, leading to less PCs and more ICs [17]. In our previous report, changes in the relative abundance of PCs and ICs were assessed only through the use of Aqp2 as the PC marker and an antibody recognizing V-ATPase B1 and B2 to label ICs [17]. Here, we extend these findings by using Aqp3 as an additional PC marker and by using more specific antibodies recognizing V-ATPase B1, AE1 and Pendrin to stain different subtypes of IC in whole Dot1lAC vs. Dot1lf/f kidneys. Our current study not only confirms that deletion of Dot1l leads to diminished PC and expanded IC populations, but also reveals that the expanded IC pool is coupled with increases in both α-IC and β-IC populations. Our result is also consistent with a recent study showing that H3 di- and tri-methyl K79 are remarkably upregulated during terminal renal epithelial differentiation [28].
While the detailed molecular mechanism by which derivation of ICs from Aqp2+ progenitor cells occurs is unclear, our current study suggests that inactivation of Dot1l favors this process. The reinforced notion that Dot1l ablation facilitates the specification of IC in the expense of PC may account at least in part for the impaired urine concentration ability in Dot1lAC vs. Dot1lf/f mice as we have previously reported [17, 18]. However, the increased IC is not sufficient to significantly change the urine pH, blood pH, and blood [HCO3−] [17], suggesting that Dot1lAC vs. Dot1lf/f mice are most likely able to maintain the acid-base balance.
We have shown that Dot1l is inactivated not only in PCs, but also in the most of ICs expressing V-ATPase A, B1, B2, CAII, AE1, and Pendrin in Dot1lAC kidney [17]. As a result, H3m2K79 became undetectable by immunofluorescence staining in these PCs and ICs. The remaining ICs have intact Dot1l function and thus robust H3m2K79 [17]. Therefore, IC markers including V-ATPase B1 can be produced in ICs with either WT or ablated Dot1l. To reconcile these findings, we hypothesized that Aqp2+ progenitor cells can differentiate into “ locked” PCs, “ locked” ICs, and “ plastic” PCs [17]. The “locked” cells eventually evolve into the canonical PCs and ICs, respectively. Deletion of Dot1l imposes little or no impact on these two processes. However, Dot1l ablation may induce specification of the plastic PCs into ICs, possibly by relief of Dot1a-mediated repression of target genes that regulate cell differentiation, leading to an increase in IC population. Whether induction of Atp6v1b1 is the cause or result of IC derivation remains unknown.
It can be speculated that Dot1a-mediated repression of Atp6v1b1 occurs in the locked and plastic PCs, but not in the locked ICs. In this regard, Dot1a-mediated H3m2K79 may serve as a signal for repression in a gene- and lineage-specific manner as does H3m3K27 [29, 30]. Nevertheless, we cannot distinguish if the increased V-ATPase B1 mRNA level results from more IC, more V-ATPases per cell, or both due to technical challenges. Since ~20% increase in IC is associated with 48% increase in V-ATPase B1 mRNA level, it is more likely that Dot1lAC vs. Dot1lf/f mice have higher levels of V-ATPase mRNA per cell.
Our current study adds Atp6v1b1 to the growing list of Dot1a target genes including αENaC, endothelin-1, and Aqp5 [18, 24, 31]. Our previous studies have shown that Af9 recruits Dot1a to the specific subregions of αENaC promoter [32]. Dot1la modulates targeted histone H3 K79 hypermethylation associated with these subregions and inhibits αENaC transcription. This repression can be partially relieved in an aldosterone-dependent and -independent manner [14, 15, 23, 24, 33]. Aldosterone relieves this repression by downregulating Dot1a and Af9 expression and by stimulating Sgk1, which, in turn, phosphorylates Af9 at S435 to attenuate Dot1a-Af9 interaction [24]. MR antagonizes Dot1a-Af9 action by competing with Dot1a for binding Af9 [34]. Aldosterone-independent relief comes from the action of Af17. Af17 enhances αENaC expression, at least in part by competing with Af9 for Dot1a binding and fostering Dot1a nuclear export [14]. As anticipated, Dot1lAC vs. Dot1lf/f mice have a higher level of αENaC mRNA expression despite of fewer PC [32]. This result suggests that αENaC, like Atp6v1b1, may increase the transcription per cell when Dot1l is ablated.
Since aldosterone-MR signaling network is operative in both PCs and ICs [35], future direction toward the understanding the role of Dot1a-Af9-Af17 axis in regulating IC markers such as V-ATPase B1 may shed new light into PC and IC differentiation. Moreover, we have not investigated the effect of Dot1l inactivation on the total number of PC and IC. It remains unclear if Dot1l plays a role in the control of branching morphogenesis. With Aqp2 as the PC marker, we have repeatedly found that PC accounts for ~75% in the cortex. This percentage is higher than those reported previously. For example, IC has been reported to make up of 30–40% of the cortical and outer medullary collecting duct cells [36–38], implying only 60–70% of cells being positive for Aqp2. The differences in the antibodies used to label the PC and IC as well as the differences in the genetic background of the mouse models examined may contribute to this discrepancy.
It should be noted that there are conflicting reports regarding the role of Dot1l-mediated H3K79 methylation in transcriptional regulation (reviewed in [39]). For example, chromatin immunoprecipitation (ChIP) coupled with gene expression microarray (ChIP–chip) in 3T3 cells and CD4+ T cells revealed that all H3K79 methylation marks reside within the body of transcribed genes, and that the abundance of the marks correlates with the level of gene expression [40, 41]. However, similar ChIP–chip in S. cerevisiae demonstrated no correlation between the amount of H3m3K79 and transcriptional activity [42]. ChIP coupled with deep sequencing (ChIP-seq) in human CD4+ T cells established a slight correlation between H3m1K79 and gene activation, a more close association of H3m3K79 with gene repression, and no correlation of H3m2K79 with transcriptional status [43]. Unlike genome-wide analyses, our gene-specific ChIP studies were focused on the upstream regions extending into the first exons of the genes examined including αENaC [16, 24, 44], Edn1 [31], Aqp5 [18], and Atp6v1b1 (Figure 6). We consistently found that Dot1l and H3m2K79 were specifically located in the regions examined. In line of these observations, Dot1l represses each of these genes.
The function of Dot1l in elongation is also controversial. Various elongation complexes have been purified. Some of them lack Dot1l [45, 46]. Others contain Dot1l as a component [47–49]. While the inconsistency between different studies may result from the different cell model systems, approaches, and procedures used in these studies, it cannot be ruled out that Dot1l and Dot1l-mediated H3 K79 methylation may regulate transcription initiation and elongation in a cell- and gene-specific manner. Such regulation may be dependent or independent of the methyltransferase activity.
Highlights.
Dot1lAC vs. Dot1lf/f mice have significantly reduced PCs and increased ICs.
Both α- and β-ICs are significantly increased in Dot1lAC vs. Dot1lf/f mice.
Dot1l downregulates expression of V-ATPase B1.
Dot1l and H3m2K79 bind the promoter of Atp6v1b1.
Atp6v1b1 is a new Dot1a target gene for transcriptional regulation.
Acknowledgments
We thank Dr. Hua A. Jenny Lu and Dr. Dennis Brown for sharing the chicken anti-V-ATPase B1 antibody and Dr. James Wade for the chicken Aqp2 antibody.
GRANTS.
This work was supported by the following grants: National Institutes of Health Grants 2R01 DK080236 06A1 (to W.Z.Z.) and 1R21DK104073-01 (to W.Z.Z.), and The National Natural Science Foundation of China (NSFC) grant 81173401 (to Q.L.Z.).
Footnotes
DISCLOSURES.
The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bagnis C, Marshansky V, Breton S, Brown D. Remodeling the cellular profile of collecting ducts by chronic carbonic anhydrase inhibition. American journal of physiology Renal physiology. 2001;280:F437–448. doi: 10.1152/ajprenal.2001.280.3.F437. [DOI] [PubMed] [Google Scholar]
- 2.Wall SM. Recent advances in our understanding of intercalated cells. Curr Opin Nephrol Hypertens. 2005;14:480–484. doi: 10.1097/01.mnh.0000168390.04520.06. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz GJ, Barasch J, Al-Awqati Q. Plasticity of functional epithelial polarity. Nature. 1985;318:368–371. doi: 10.1038/318368a0. [DOI] [PubMed] [Google Scholar]
- 4.Gao X, Eladari D, Leviel F, Tew BY, Miro-Julia C, Cheema F, Miller L, Nelson R, Paunescu TG, McKee M, Brown D, Al-Awqati Q. Deletion of hensin/DMBT1 blocks conversion of {beta}- to {alpha}-intercalated cells and induces distal renal tubular acidosis. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1010364107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, Brown R, Persson AE, Bergstrom GG, Enerback S. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest. 2004;113:1560–1570. doi: 10.1172/JCI20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150:613–632. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12:1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 8.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Hayashizaki Y, Kone BC. Structure and regulation of the mDot1 gene, a mouse histone H3 methyltransferase. Biochem J. 2004;377:641–651. doi: 10.1042/BJ20030839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang MJ, Wu H, Achille NJ, Reisenauer MR, Chou CW, Zeleznik-Le NJ, Hemenway CS, Zhang W. Histone H3 Lysine 79 Methyltransferase Dot1 Is Required for Immortalization by MLL Oncogenes. Cancer Res. 2010;70:10234–10242. doi: 10.1158/0008-5472.CAN-10-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jo SY, Granowicz EM, Maillard I, Thomas D, Hess JL. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. 2011;117:4759–4768. doi: 10.1182/blood-2010-12-327668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 13.Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, Baltus GA, Kadam S, Zhai H, Valdez R, Gonzalo S, Zhang Y, Li E, Chen T. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4:1–11. doi: 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reisenauer MR, Anderson M, Huang L, Zhang Z, Zhou Q, Kone BC, Morris AP, Lesage GD, Dryer SE, Zhang W. AF17 competes with AF9 for binding to Dot1a to up-regulate transcription of epithelial Na+ channel alpha. J Biol Chem. 2009;284:35659–35669. doi: 10.1074/jbc.M109.038448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H, Chen L, Zhou Q, Zhang W. AF17 facilitates Dot1a nuclear export and upregulates ENaC-mediated Na+ transport in renal collecting duct cells. PLoS One. 2011;6:e27429. doi: 10.1371/journal.pone.0027429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Xia X, Jalal DI, Kuncewicz T, Xu W, Lesage GD, Kone BC. Aldosterone-sensitive repression of ENaCalpha transcription by a histone H3 lysine-79 methyltransferase. Am J Physiol Cell Physiol. 2006;290:C936–946. doi: 10.1152/ajpcell.00431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu H, Chen L, Zhou Q, Zhang X, Berger S, Bi J, Lewis DE, Xia Y, Zhang W. Aqp2-expressing cells give rise to renal intercalated cells. Journal of the American Society of Nephrology : JASN. 2013;24:243–252. doi: 10.1681/ASN.2012080866. (Editorial in the same issue of J Am Soc Nephrol pg 163–165, Selection by Faculty of 1000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H, Chen L, Zhang X, Zhou Q, Li JM, Berger S, Borok Z, Zhou B, Xiao Z, Yin H, Liu M, Wang Y, Jin J, Blackburn MR, Xia Y, Zhang W. Aqp5 is a new transcriptional target of dot1a and a regulator of aqp2. PLoS One. 2013;8:e53342. doi: 10.1371/journal.pone.0053342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paunescu TG, Ljubojevic M, Russo LM, Winter C, McLaughlin MM, Wagner CA, Breton S, Brown D. cAMP stimulates apical V-ATPase accumulation, microvillar elongation, and proton extrusion in kidney collecting duct A-intercalated cells. American journal of physiology. Renal physiology. 2010;298:F643–654. doi: 10.1152/ajprenal.00584.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ronzaud C, Loffing J, Bleich M, Gretz N, Grone HJ, Schutz G, Berger S. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. Journal of the American Society of Nephrology : JASN. 2007;18:1679–1687. doi: 10.1681/ASN.2006090975. [DOI] [PubMed] [Google Scholar]
- 21.Reisenauer MR, Wang SW, Xia Y, Zhang W. Dot1a contains three nuclear localization signals and regulates the epithelial Na+ channel (ENaC) at multiple levels. American journal of physiology. Renal physiology. 2010;299:F63–76. doi: 10.1152/ajprenal.00105.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W, Xia X, Zou L, Xu X, LeSage GD, Kone BC. In vivo expression profile of a H+-K+-ATPase {alpha}2-subunit promoter-reporter transgene. American journal of physiology. Renal physiology. 2004;286:F1171–F1177. doi: 10.1152/ajprenal.00043.2003. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Wu H, Pochynyuk OM, Reisenauer MR, Zhang Z, Huang L, Zaika OL, Mamenko M, Zhang W, Zhou Q, Liu M, Xia Y, Zhang W. Af17 deficiency increases sodium excretion and decreases blood pressure. Journal of the American Society of Nephrology : JASN. 2011;22:1076–1086. doi: 10.1681/ASN.2010121270. (Selected by Faculty of 1000. Invited author interview by Hemodialysis.com) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest. 2007;117:773–783. doi: 10.1172/JCI29850. (Comment in the same issue of J Clin Invest pg 592–595, Selection by Faculty of 1000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Biochemistry. 2010;49:4715–4723. doi: 10.1021/bi100397s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paunescu TG, Da Silva N, Marshansky V, McKee M, Breton S, Brown D. Expression of the 56-kDa B2 subunit isoform of the vacuolar H(+)-ATPase in proton-secreting cells of the kidney and epididymis. Am J Physiol Cell Physiol. 2004;287:C149–162. doi: 10.1152/ajpcell.00464.2003. [DOI] [PubMed] [Google Scholar]
- 27.Vernimmen D, Lynch MD, De Gobbi M, Garrick D, Sharpe JA, Sloane-Stanley JA, Smith AJ, Higgs DR. Polycomb eviction as a new distant enhancer function. Genes Dev. 2011;25:1583–1588. doi: 10.1101/gad.16985411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McLaughlin N, Wang F, Saifudeen Z, El-Dahr SS. In situ histone landscape of nephrogenesis. Epigenetics. 2014;9:222–235. doi: 10.4161/epi.26793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yagi R, Zhu J, Paul WE. An updated view on transcription factor GATA3-mediated regulation of Th1 and Th2 cell differentiation. Int Immunol. 2011;23:415–420. doi: 10.1093/intimm/dxr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O’Shea JJ, Zhao K. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Q, Liu K, Wu H, Chen L, Pouranan V, Yuan M, Xiao Z, Peng W, Xiang A, Tang R, Zhang W. Spironolactone rescues Dot1a-Af9-mediated repression of endothelin-1 and improves kidney injury in streptozotocin-induced diabetic rats. PLoS One. 2012;7:e47360. doi: 10.1371/journal.pone.0047360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, Yu Z, Wu H, Chen L, Kong Q, Kone BC. An Af9 cis-element directly targets Dot1a to mediate transcriptional repression of the alphaENaC gene. American journal of physiology. Renal physiology. 2013;304:F367–375. doi: 10.1152/ajprenal.00537.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen L, Zhang X, Zhang W. Regulation of alphaENaC Transcription. Vitam Horm. 2015;98:101–135. doi: 10.1016/bs.vh.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Zhou Q, Chen L, Berger S, Wu H, Xiao Z, Pearce D, Zhou X, Zhang W. Mineralocorticoid receptor antagonizes Dot1a-Af9 complex to increase alphaENaC transcription. American journal of physiology. Renal physiology. 2013;305:F1436–1444. doi: 10.1152/ajprenal.00202.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shibata S, Rinehart J, Zhang J, Moeckel G, Castaneda-Bueno M, Stiegler AL, Boggon TJ, Gamba G, Lifton RP. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab. 2013;18:660–671. doi: 10.1016/j.cmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown D, Hirsch S, Gluck S. An H+-ATPase in opposite plasma membrane domains in kidney epithelial cell subpopulations. Nature. 1988;331:622–624. doi: 10.1038/331622a0. [DOI] [PubMed] [Google Scholar]
- 37.Teng-umnuay P, Verlander JW, Yuan W, Tisher CC, Madsen KM. Identification of distinct subpopulations of intercalated cells in the mouse collecting duct. Journal of the American Society of Nephrology : JASN. 1996;7:260–274. doi: 10.1681/ASN.V72260. [DOI] [PubMed] [Google Scholar]
- 38.Zharkikh L, Zhu X, Stricklett PK, Kohan DE, Chipman G, Breton S, Brown D, Nelson RD. Renal principal cell-specific expression of green fluorescent protein in transgenic mice. American journal of physiology. Renal physiology. 2002;283:F1351–1364. doi: 10.1152/ajprenal.0224.2001. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, Blobel GA, Vakoc CR. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 43.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 44.Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 Complex Mediates Histone H3 Lys-79 Hypermethylation and Repression of ENaC{alpha} in an Aldosterone-sensitive Manner. J Biol Chem. 2006;281:18059–18068. doi: 10.1074/jbc.M601903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 2010;17:198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 48.Mueller D, Garcia-Cuellar MP, Bach C, Buhl S, Maethner E, Slany RK. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009;7:e1000249. doi: 10.1371/journal.pbio.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, Slany RK. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]