

Abstract
Sperm motility is driven by motile cytoskeletal elements in the tail, called axonemes. The structure of axonemes consists of 9 + 2 microtubules, molecular motors (dyneins), and their regulatory structures. Axonemes are well conserved in motile cilia and flagella through eukaryotic evolution. Deficiency in the axonemal structure causes defects in sperm motility, and often leads to male infertility. It has been known since the 1970s that, in some cases, male infertility is linked with other symptoms or diseases such as Kartagener syndrome. Given that these links are mostly caused by deficiencies in the common components of cilia and flagella, they are called “immotile cilia syndrome” or “primary ciliary dyskinesia,” or more recently, “ciliopathy,” which includes deficiencies in primary and sensory cilia. Here, we review the structure of the sperm flagellum and epithelial cilia in the human body, and discuss how male fertility is linked to ciliopathy.
Keywords: Cilia, Ciliopathy, Dynein, Flagella, Sperm, Infertility
Abbreviations
- AK
Adenylate kinase
- ARMC
Armadillo repeat containing
- BB
Basal body
- BBS
Bardet‐Biedl syndrome
- CEP
Centrosomal protein
- CCDC
Coiled‐coil domain‐containing
- CP
Central pair apparatus
- DC
Docking complex
- DNAAF
Dynein axonemal assembly factor
- DNAH
Dynein, axonemal, heavy chain
- DNAI
Dynein, axonemal, intermediate chain
- DNALI
Dynein, axonemal, light intermediate chain
- DYNC2
Dynein, cytoplasmic 2
- DYX1C1
Dyslexia susceptibility 1 candidate 1
- FAP
Flagella‐associated protein
- FBB
Flagellar basal body
- FoxJ1
Forkhead box J1
- HC
Heavy chain
- HEATR
HEAT‐repeat containing
- IC
Intermediate chain
- IAD
Inner arm dynein
- IDA
Inner dynein arm
- IFT
Intraflagellar transport
- IMT
Intramanchette transport
- Iqcg
IQ motif containing G
- LC
Light chain
- LRRC
Leucine‐rich repeat containing
- MFN
Mitofusin
- MIA
Modifier of inner arms
- MKS
McKusick‐Kaufman syndrome
- MNS1
Meiosis‐specific nuclear structural 1
- N‐DRC
Nexin‐dynein regulatory complex
- NPHP
Nephronophthisis
- OAD
Outer arm dynein
- ODA
Outer dynein arm
- ODF
Outer dense fiber
- OFD
Oral‐facial‐digital syndrome
- PACRG
Parkin co‐regulated gene
- PCD
Primary ciliary dyskinesia
- Pcdp1
Primary ciliary dyskinesia protein 1
- PCP
Planar cell polarity
- PF
Paralyzed flagella
- PIH
Protein interacting with HSP90
- PKD
Polycystic kidney disease
- RFX
Regulatory factor X
- RPGR
Retinitis pigmentosa GTPase regulator
- RS
Radial spoke
- SPAG
Sperm‐associated antigen
- Spef2
Sperm flagellar protein 2
- TXNDC
Thioredoxin domain containing
- TZ
Transition zone
- VDAC3
Voltage‐dependent anion channel 3
- XLRP
X‐linked retinitis pigmentosa
- ZMYND
Zinc‐finger, MYND‐type containing protein
Introduction
Male infertility is caused by several factors ranging from psychological or behavioral problems to abnormal sperm function. Sperm dysfunction is the most obvious cause of male infertility that can be detected morphologically and physiologically. Sperm dysfunction is observed in several compartments of spermatozoa. In particular, defects in flagella directly affect sperm motility, and often lead to failure of fertilization. During the course of diagnosis of male patients with Kartagener syndrome, an important correlation between sperm dysfunction, bronchitis, and situs inversus was described [1, 2, 3]; the patients lacked dynein arms, which serve as molecular motors for motility of cilia and flagella. Because sperm flagella and motile cilia in the human body serve as centers of signal transduction, and have common internal structures for motility called axonemes, defects in the axonemal structure result not only in sperm dysfunction, but also in abnormalities of multiple ciliated cells and organs. Most such defects are caused by genetic mutations of ciliary proteins. Further investigations led to the proposal of the name “immotile‐cilia syndrome” or “primary ciliary dyskinesia” (PCD) [4]. More recently, the name “ciliopathy” has been used for the general symptoms caused by disorders of motile, primary and sensory cilia [4, 5]. In this review, we describe the structure and function of the axoneme, and illustrate how abnormalities of the axonemal structure affect the motility of cilia and flagella.
Structure and function of sperm flagella
Spermatozoa are male gametes that are structurally and functionally highly differentiated. Human spermatozoa are composed of a head (acrosome and nucleus) and a tail (a flagellum, containing a midpiece, principal piece, and end piece) (Fig. 1a) [6, 7, 8, 9]. The motile machinery of the flagellum, the axoneme, extends from the basal body (distal centriole) via the transition zone (TZ) and has a 9 + 2 microtubule structure. Mitochondria helically wrap around both the axoneme and outer dense fibers (ODFs) in the midpiece of the flagellum. In the principal piece, the axonemes are surrounded by ODF and fibrous sheath (FS) (Fig. 1b).
Figure 1.
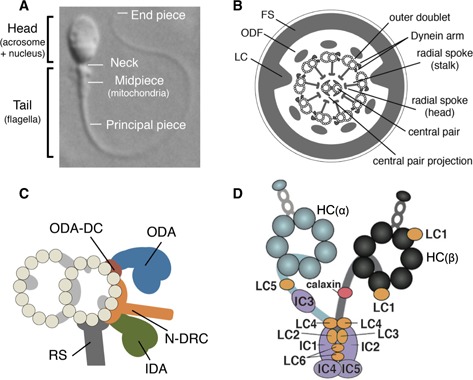
Structure of sperm flagellum. a Differential interference contrast microscopic image of human sperm. The tail is divided into the midpiece, principal piece, and end piece. b Internal structure of the flagellar principal piece (cross‐section). The central 9 + 2 structure, the axoneme, is the motile machinery of flagella. In mammalian sperm, it is surrounded by accessory structures, outer dense fiber (ODF) and the fibrous sheath (FS). A projection called the longitudinal column (LC) extends from the FS to two outer doublet microtubules (doublet numbers 3 and 7). c Detailed substructures of the axonemes. Each doublet microtubule is decorated by an outer dynein arm (ODA), inner dynein arm (IDA), nexin‐dynein regulatory complex (N‐DRC), and radial spoke (RS). The ODA docking complex (DC) is anchored at an interval of 24 nm on the microtubule. Each doublet microtubule is decorated by some structures inside the microtubule or between protofilaments. d Molecular structure of the ODA from Ciona sperm flagella. The ODA is composed of two HCs, five ICs, and six LCs. The HCs are named as α and β, which are orthologs of β‐ and γ‐type HC of Chlamydomonas ODA, respectively [8, 28, 37]. Calaxin is a Ca2+ sensor of opisthkont ODA that binds to Ciona β HC in the presence of Ca2+
Structure of the axoneme
The axonemes of motile cilia and flagella are supramolecular machinery composed of cytoskeletons and molecular motors [8, 10, 11]. The molecular motors for sperm motility are microtubule‐dependent ATPases called dyneins. Dyneins are classified into cytoplasmic and axonemal dyneins, both of which are composed of motor subunits (heavy chains, HCs) and associated subunits (intermediate chains, ICs and light chains, LCs), but are phylogenetically distinct from each other. Axonemal dyneins are observed as “arms,” which are attached to each of nine doublet microtubules (Fig. 1b, c). The sliding of doublet microtubules by axonemal dyneins is the basis of flagellar motility [12]. Outer arm dyneins (OADs) are involved in the increase of flagellar beat frequency and in the propagation of asymmetric flagellar waveforms. Inner arm dyneins (IADs) are responsible for flagellar bend formation and propagation [13]. The activities of axonemal dyneins are regulated by radial spokes (RSs) and by the central pair apparatus (CP) (central pair microtubules and the associated bridge and projections) [14, 15]. The green alga Chlamydomonas has been used to generate many useful mutants for studying cilia and flagella, including mutants for the outer and inner dynein arms (oda and ida) [13, 16] and other substructures (pf, paralyzed flagella) [17]. In animals, sperm from marine invertebrates have contributed to our understanding of the molecular architecture of flagella and the mechanism of their motility [18, 19, 20, 21, 22, 23, 24].
The molecular composition of axonemal dyneins has been widely examined in the green alga Chlamydomonas [25]. Outer arm dyneins (OADs) from Chlamydomonas flagella are composed of three heavy chains (HCs), two intermediate chains (ICs), and ten light chains (LCs). Work using sperm of tunicates (a subphylum of marine invertebrates) and sea urchins revealed similar molecular composition of the IADs but not the ODAs [26, 27, 28, 29]. The OAD from animal sperm has two HCs, two WD‐repeat ICs, a unique IC with thioredoxin and nucleoside diphosphate kinase domains [30, 31, 32], and regulatory LCs such as Tctex‐2 LC [33, 34] and a Ca2+‐binding protein named calaxin [35, 36, 37] (Fig. 1d). The precise subunits of the outer arm have been described in sea urchins and tunicates, but not in human sperm. The ODAs are arranged at 24‐nm intervals on each doublet microtubule, mediated by the molecular “ruler” called outer arm dynein docking complex (ODA‐DC) [38, 39]. In Chlamydomonas flagella, the ODA‐DC is composed of three subunits, DC1 (a coiled‐coil protein), DC2 (coiled‐coil protein), and DC3 (an EF‐hand protein). However, orthologs of DC1 and DC3 are not found in animal sperm flagella [28, 40].
Species of the IAD are more heterogeneous than the OAD in Chlamydomonas; one is two‐headed (two HCs) with three ICs and five LCs, and another six are single‐headed (single HC) with other subunits, such as actin, centrin, and a protein called p28 [41]. Only a few studies have been undertaken on the subunit structure of IAD in sperm [42, 43, 44]. From the base of IADs, two structures, dynein regulatory complex (DRC) and the nexin link, are observed. Both are now called “N‐DRC” and regarded as an integrated substructure. N‐DRC is bound to the base of the RS1, RS2 (Fig. 1c) [45, 46, 47, 48, 49]. Inside the cylinder of nine doublet microtubules, two types of structures are present in most motile axonemes: one is RS that extend from the A‐tubule of doublet microtubules, and the other is the CP composed of two singlet microtubules (C1 and C2), connected to each other by central bridges and decorated by central pair projections [50].
Each doublet microtubule has several structures between A‐ and B‐tubules and inside the tubule (Fig. 1c, light gray) [47, 51, 52]. The structures between A‐ and B‐tubules are called outer and inner junctions, both of which connect A‐ and B‐tubules and are thought to play roles in the initiation of doublet formation. The structures inside the A‐ and B‐tubules are composed of microtubule inner proteins (MIPs) [51] and the “beak” structure [52]. These structures are bound to the inside wall of microtubules with longitudinal periodicity of 16 and/or 48 nm and are thought to be involved in the assembly and stability of doublet microtubules [51].
The flagellum of mammalian sperm has accessory structures: ODF and FS [6]. These structures mechanically reinforce the axonemes, which is necessary for spermatozoa to move in highly viscous conditions. Multiple glycolytic enzymes are bound to the fibrous sheath and play important roles in the regulation of sperm motility [53, 54].
Structures of the flagellar base
The basal body (BB) is composed of nine triplet microtubules and is differentiated from the mother centriole (distal centriole) during the cell cycle. When cilia are formed, the mother centriole becomes anchored to the plasma membrane where the cilium starts to extend. Then the mother centriole comes to serve as the BB. Axonemes are nucleated from basal bodies via the TZ. In motile cilia, there are several appendages on the BB and the TZ [55, 56] (Fig. 2a). A striated (or ciliary) rootlet extends from the proximal site of the BB to the cytoplasm and is found in many but not all types of cilia. The basal foot, originated from the subdistal appendage in the mother centriole, directs the orientation of the beating plane [55, 57]. At the distal end of the BB, a structure called the transition fiber (TF), originated from the distal appendage of the mother centriole, links the basal body to the ciliary membranes. The TF serves as a diffusion barrier for ciliary proteins. In the transition region, the Y‐link extends from the microtubule to the plasma membrane (Fig. 2a).
Figure 2.
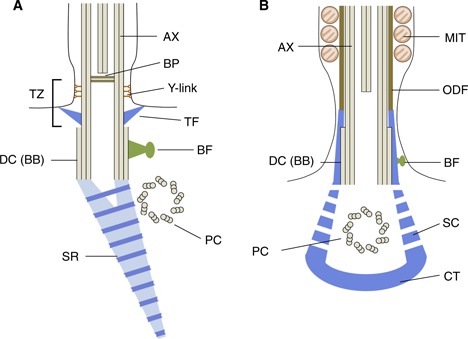
Structures at the base of epithelial cilia (a) and sperm flagella (b). AX axoneme, BP basal plate, TZ transition zone, TF transition fiber, BF basal foot, DC distal (mother) centriole, BB basal body, PC proximal (daughter) centriole, SR striated rootlet, MIT mitochondrion, ODF outer dense fiber, SC striated column, CT capitulum
Vesicles containing ciliary components are sorted by the Golgi network and transported to the base of the cilium (Fig. 3). A complex of Bardet–Biedl syndrome (BBS) proteins, called the BBSome (~450 kDa in molecular mass), is located at the base of a cilium and is involved in the tethering and transporting of membrane cargos to the cilium in a G‐protein (Rab, Ran, and Arf/Arl)‐dependent pathway [58, 59]. After passing through the TZ, ciliary components are then delivered by directional movement of intraflagellar transport (IFT) particles. IFT is a bidirectional movement driven by kinesin‐2 (anterograde) and cytoplasmic dynein 2 (DYNC2) (retrograde) to transport axonemal and membrane components for construction of cilia and flagella (Fig. 3). Two IFT particles, IFT‐B and IFT‐A complexes, are specifically involved in anterograde and retrograde transport, respectively [60, 61]. The detailed functions of BBSome, IFT‐A or IFT‐B in the transport of ciliary components, or their interactions, are not completely understood (Fig. 3).
Figure 3.
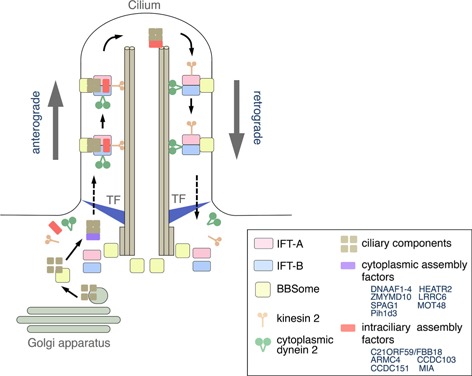
Simplified diagram of the assembly and transport of axonemal components. Ciliary proteins are first transported to the base of cilium by G‐protein‐ and BBSome‐dependent pathway. Substructural components, such as axonemal dyneins, are preassembled in the cytoplasm with the aid of assembly factors (purple square). They are transported by IFT particles (IFT‐A + IFT‐B) after passing through the transition fiber (TF). IFT is composed of anterograde and retrograde transports, driven by kinesin 2 and cytoplasmic dynein 2, respectively. Other assembly factors (red square) are involved in the intraciliary assembly of axonemal dyneins
The mechanism of formation of sperm flagella is considered to be similar to that of ciliogenesis, although some reports have demonstrated that sperm flagella are formed by a mechanism different from that of sensory cilia in Drosophila [62, 63]. Both proximal and distal centrioles are present in elongating spermatids, but the distal centriole and its associated structures are degenerated in mature sperm in mouse [64]. Human sperm have intact proximal centrioles, but the distal centrioles are largely degenerated [65]. The neck region of mature spermatozoa, called the connecting piece, is composed of two parts: the capitulum and striated collar (Fig. 2b). The latter is considered to be homologous to the striated rootlets of cilia and connected to the nucleus through the structure called the implantation fossa [6, 66]. The presence of a basal foot was described at least in spermatids, but its presence in mature spermatozoa is not clear.
Distribution of motile cilia in the human body
The name “ciliated epithelial cells” was originally used for cells bearing motile cilia for generation of water flow. In humans, most such cells are multiciliated (more than one cilium per cell), as seen in nasal and trachea epithelia (airway cilia), the lachrymal sac or Moll's gland, brain ventricles (ependymal cilia), male efferent ducts, and female oviducts (Fallopian tubes) (Fig. 4). The structures of the axonemes in motile multicilia have a 9 + 2 microtubule lattice with dynein arms, apparently the same as those in sperm flagella, although there are several reports showing minor differences in their components (e.g., [67]).
Figure 4.
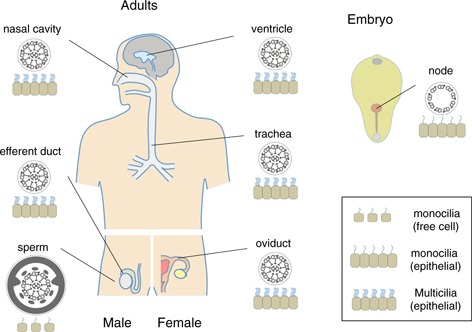
Distribution of motile cilia in the human body. Tissues or organs with motile cilia are shown with their types (monocilia or multicilia) and internal structures
Like airway cilia, some other epithelial tissues in humans have been reported to bear cilia. The epithelium of the middle ear and its connecting Eustachian tube bear multicilia. They have motile 9 + 2 structures and function in mucociliary clearance. A deficiency in motility results in less elimination of bacteria, leading to otitis media [3, 68]. Inner hair cell possesses stereociliary bundles, which are composed of a single kinocilium (monocilium) and 50–200 stereocilia (microvilli) [69]. The kinocilium has a 9 + 0 (lacking CP) or 9 + 2 structure, and seems not to have dynein arms [70]. However, it was reported that porcine kinocilium showed a 9 + 2 structure with ODAs [71]. The axonemal structure of human kinocilium is controversial.
It was thought that the cilia of the follicular epithelium of the human thyroid function in the mixing of the follicular colloid [72]. However, monocilia with a 9 + 0 structure observed in the follicular epithelium of the human thyroid lack dynein arms and appear immotile, although a weak uncoordinated movement was observed [73]. In a follicular carcinoma of the thyroid, a 9 + 2 structure of axonemes was observed, but dynein arms were still absent [73].
Efferent ducts are connected to the epididymis, where a series of events for sperm maturation occur. The epithelial cells of the epididymis bear immotile stereocilia [74]. Transport of sperm through the epididymis is considered to be driven both by hydrostatic pressure and by contraction of smooth muscle. The epithelia of vas deferens also bear immotile stereocilia (long microvilli), but no motile cilia [75, 76]. Apparent motile cilia with a 9 + 2 structure and dynein arms have been observed on the endometrium of the uterine lumen, but often with atypical axonemal structures. The population of ciliated cells varies due to turnover during the menstrual cycle [77, 78]. Movement of cilia is not the only driving force for sperm transport along the endometrium [79]. Other ciliated epithelia may be found in adults, children, growing embryos, and fetuses, but are not completely understood.
Most cells in human tissue possess nonmotile monocilia, called primary cilia, which serve as signaling centers for perception of extracellular signals for regulating cell proliferation and differentiation. Primary cilia possess 9 + 0 axonemes without dynein arms, and function as the antennae of cells, receiving several extracellular signals and transducing them into cells through, for example, pathways such as the Hedgehog and Wnt/planar cell polarity (PCP) pathways [80]. For example, loss of ciliary formation results in a disorder of cellular orientation, causing polycystic kidney disease due to nonpolarized cellular alignment. In addition, cilia on sensory organs play critical roles in perception of several stimuli, such as light, mechano‐ and chemostimuli [80].
Embryos and larvae in many aquatic invertebrates are ciliated for free‐swimming. In mammals, embryos develop without motile cilia and implant into the uterine wall. The only motile cilia seen in their early development are those on the node (Fig. 4, right). Nodal cilia have a 9 + 0 structure with dynein arms [81, 82]. Close observation of the axonemes strongly suggests that nodal cilia possess only outer arm dynein [41]. Nodal cilia are seen at the posterior side of the nodal cells, according to the planar cell polarity. They are tilted posteriorly and show rotational movement. The flow caused by nodal cilia determines left–right asymmetry [83, 84, 85]. Two types of cells are found in the node: pit cells and crown cells. The majority of crown cells bear immotile monocilia, which are proposed to have a sensory function. The presence of two types of cilia serves as a basis for the “two‐cilia model” for left–right asymmetry [86, 87]. In this model, a leftward flow is caused by cilia on the pit cells and is sensed by immotile cilia on the crown cells. Recent studies have shown that primary cilia are generated in early mouse embryos on nearly all cells in epiblasts (E5.5 and E6.0 embryos), definitive endoderm and node (E8.0), and limb mesenchymes and neural progenitors (E10.5). Trophoblast and extraembryonic endoderm stem cells have no cilia [88]. Cilia become differentiated in developing adult organs, and some of them are considered to play important roles not only in organ function but also in organ differentiation, such as heart development in mouse [89].
Ciliopathy due to deficiencies in transport and assembly of axonemal components
The formation of cilia and flagella initiates from the expression of master genes for ciliary and flagellar formation, such as FoxJ1 and the RFX family of transcription factors (for review, see [90]). Expression of these master genes is followed by maturing of the centriole into the BB, formation of the TZ, anchoring of the basal body to the plasma membrane, and transport of ciliary or flagellar proteins by the BBSome and IFT [91].
Ciliopathy is caused by recessive mutations in the genes necessary for formation of cilia and flagella. These encode (1) proteins for the gene expression and transport of ciliary and flagellar components, such as those of the BBSome, TZ, and IFT particles; (2) factors for cytoplasmic assembly of axonemal components; and (3) the elements of axonemal structures, such as the subunits of ODAa and IDAs, RSs, and the CP. Defects in these components result in ciliary dysfunction in a wide range of organs, represented by symptoms such as PCD, Kartagener syndrome, BBS, MKS, Meckel–Gruber syndrome, hydrocephalus, PKD, polycystic liver disease, nephronophthisis, Joubert syndrome, Alström syndrome, Jeune asphyxiating thoracic dystrophy, and laterality defects [92]. These diseases are often demonstrated experimentally using model organisms, such as knockout mice and knockdown embryos of zebrafish.
Two complexes for transport of ciliary proteins
Most of the mutations in the genes for ciliogenesis, i.e., those of the BBSome, TZ, and IFT particles, result in deficiency in ciliary and flagellar formation, causing ciliopathy. The BBSome and IFT are the transport systems of ciliary proteins at the base of the cilium and inside the cilium, respectively. Most mouse mutations in BBS genes are lethal, and the rest of the mutants show obesity, retinal degeneration, cystic kidneys or polydactyly [92, 93, 94]. Mutation in BBS2 or BBS4 causes male infertility due to the failure of sperm flagellar formation [95].
Molecular compositions of IFT particles have been studied extensively in Chlamydomonas [61]. Deficiency in IFT results in the failure of ciliary formation. The phenotypes of mouse mutants lacking IFT components are similar to those lacking the BBSome components. Mutation of an IFT‐B component, IFT172, results in the formation of cystic kidney in zebrafish [96]. Embryos in an IFT‐172‐knockout mouse show defects in cilia formation, resulting in the failure of brain development [97]. Mice with deletions of the gene encoding another IFT‐B component, IFT88/Polaris, develop PKD, hepatic fibrosis, defects in photoreceptor assembly and maintenance, and situs inversus [98, 99]. Mutants lacking some IFT‐A components have flagella with bulged membrane at the tips in Chlamydomonas [100, 101]. Mutations of IFT‐A components in mouse show a phenotype similar to bulged cilia [102]. They connect to abnormalities seen in human diseases, such as short‐rib polydactyly syndromes for WDR35 mutation, and Mainzer–Saldino syndrome (MSS) for IFT140 mutation [103, 104].
The transition zone and associated structures
Many proteins related to human ciliopathy are localized to the TZ, a barrier compartment where ciliary proteins are relayed from intracellular to intraciliary transports. Septins are polymerizing GTP‐binding proteins with cytoskeletal properties. They play roles in binding and organizing the membrane‐cytoskeleton network [105, 106]. The localization of septin 2 at the ciliary base indicates that the septin ring plays a role in the ciliary diffusion barrier to separate the ciliary and cytoplasmic compartments [107]. In mammalian spermatozoa, septin 4 and 12 are reported to be essential for spermatogenesis. In particular, septin 4 is a structural component of the sperm annulus, and septin 4‐null mice show abnormal morphology around the sperm neck region [108, 109]. Male patients with mutations in septin 12 genes show oligoasthenozoospermia or asthenoteratozoospermia. The sperm have a defective annulus with a bent tail [110].
CEP290, also known as NPHP6, is localized to the Y‐link in the transition zone and is involved in the assembly of these structures in Chlamydomonas [111]. Its mutation causes MKS in humans [112]. There are two other structures in the TZ, namely the basal foot (subdistal appendage) and TF (distal appendage). These are reported to contain ninein/ODF2 and ODF2/CEP164/CEP123, respectively [113, 114, 115, 116]. One of the basal foot proteins, ODF2, is necessary for both basal foot formation and orientation of the ciliary beat [117]. ODF2‐knockout mice show embryonic lethality [118], in agreement with the observation that ODF2 is a basic component of the mother centriole. ODF2 was originally found as a component of ODF of sperm tail [119]. Analysis of heterozygote mice indicates that ODF2 is essential for ODF formation and its defects result in production of spermatozoa with a bent tail in which one or more doublet microtubules are missing [120]. Several other proteins, such as those in the Tectonic module (TCTN1, TCTN2, and TCTN3) and MKS proteins, are localized to the transition zone and seem to help in ciliary formation and maintenance [91].
Other structures associated with basal bodies are not well understood. Major components of these structures so far identified include SF‐assemblin, a principal component of striated microtubule‐associated fiber at the basal apparatus of Chlamydomonas [121], and rootletin, a coiled‐coil protein component of ciliary rootlet [122]. Knockout mice lacking rootletin or its associated small GTPase, RAB28, show retinal disorders, indicating that the ciliary rootlet is especially involved in the ciliary transport in photoreceptors [123, 124].
Cytoplasmic assembly of axonemal components
Substructures in the axoneme, such as ODA/IDA and RSs, are composed of multiple subunits. They need factors for proper assembly and anchoring. Many axonemal proteins are transported to cilia or flagella after preassembly in the cytoplasm. Multiple factors for cytoplasmic assembly of axonemal components have been identified, especially those for OAD and IAD. Ktu (from the name of a gene kintoun)/PF13 was the first factor identified for the assembly of dynein arms and is now called DNAAF2 [125]. Both ODA and IDA are partially or completely absent in patients lacking DNAAF2. Respiratory cilia and sperm flagella are immotile in such patients. Some of the patients also show situs inversus. Ktu mutation causes the absence of DNAH5 and DNAI2, especially in the distal region of cilia. DNALI1 (Chlamydomonas p28) is also missing, resulting in the partial loss of IDA.
Mutations in DNAAF1/LRRC50/ODA7 cause deficiency in ciliary motility similar to those for DNAAF2 [126, 127]. Axonemes in patients with DNAAF1 mutations show significant loss of both ODAs and IDAs, and the extent is more severe than for DNAAF2 because the mutation in LRRC50/DNAAF1 results in the loss of both DNAH5 and DNAH9 and the IC, DNAI2, affecting the assembly of both proximal (type 1) and distal (type 2) ODAs. Loss of DNAAF3/PF22 results in ciliary defects similar to those for both DNAAF1 and DNAAF2 [128]. All of these assembly factors function together in the assembly of ODAs, but are considered to have distinct roles in the process. In conjunction with studies in Chlamydomonas, DNAAF1/ODA7, DNAAF2/PF13, and DNAAF3/PF22 have been shown to function as a chaperone complex for proper folding of the dynein‐HCs [128]. DYX1C1 (also named DNAAF4) is also an assembly factor for dynein. DYX1C1/DNAAF4 interacts with DNAAF2 but not with DNAAF1 or DNAAF3 [129].
Factors for cytoplasmic dynein assembly other than DNAAF1–4 have been reported. HEATR2 (recently called DNAAF5) interacts with DNAI2, but not with other DNAAFs or HSPs. It seems to play a role different from those of DNAAF1–4 at an early stage of dynein preassembly in the cytoplasm [130]. LRRC6 has structural and functional similarities to DNAAF1; its knockout mouse lacks both OAD and IAD [131]. LRRC6 interacts with another assembly factor, ZMYND10 [132, 133]. SPAG1 is also a cytoplasmic assembly factor for both ODA and IDA in respiratory cilia, similar to the function of DNAAF1, DNAAF3, LRRC6, and ZMYND10. SPAG1 mutant cilia lack the IDA component DNALI1 in the axoneme, suggesting further involvement of the assembly factors with the IAD [134]. In Chlamydomonas, two assembly factors for IADs are known. One is Chlamydomonas MOT48 (IDA10), which, like DNAAF2, contains a PIH domain, and is involved in the assembly of IAD species b, c, and d [135]. The other is the MIA complex [MIA1 (FAP100) plus MIA2 (FAP73)], which is located at the distal part of the IAD f/I1 and is involved in the assembly of this dynein [136].
Ciliopathy due to mutations in axonemal components
Mutations in axonemal components often result in the loss of substructure of the axoneme, leading to the dysfunction of motility in most cases. The spectrum of ciliopathy depends on how important the substructure is to the ciliary function of a tissue. For example, many patients or mutants lacking proteins of the RS/CP, such as HYDIN [137] and RSPH4 [138], suffer from defects in the motility of epithelial cilia but not from laterality defects because nodal cilia have 9 + 0 structures with no RS/CP (Fig. 4).
ODA and IDA
Approximately 70–80 % of PCD cases are caused by defects of ODA [139, 140]. OAD is involved in the elevation of ciliary beat frequency [19], force generation for penetration of sperm through the zona pellucida [141], metachronal synchrony of motile cilia [142], and propagation of asymmetric waveforms of sperm during chemotaxis [36]. The mutations in OAD reported so far include those in HCs (DNAH5, DNAH11), ICs (DNAI1, DNAI2, NME8), a LC (DNAL1), and a component of the DC (coiled‐coil domain containing protein 114, CCDC114; DC2 in Chlamydomonas) (Table 1). Compared with OAD, there are few mutants showing a lack of IAD components; these include DNAH7 (only in knockout mouse [143]) and DNALI1 [144]. Mouse mutants lacking Dnah7 show deficiency in ciliary and flagellar motility, but no apparent difference is found in the ultrastructure of OAD/IAD [143]. One interesting report is that mice lacking the gene for DNA polymerase lambda (Dpcd/poll) show situs inversus totalis and defects in motility of ependymal and respiratory cilia. The knockout male mice are infertile due to sperm immotility, but the female mice are fertile. Ultrastructures of cilia from λ−/− mice show a lack of IADs but not of OADs [145]. It is not known how mutation of λ−/− leads to the loss of IADs. The human ortholog (DPCD) was tested in 51 PCD patients (15 with IDA defects), and no mutation in the dpcd locus was discovered [146].
Table 1.
List of genes for human ciliopathy affecting specific axonemal structures and sperm motility
| Human gene | Protein | Localization | Chlamydomonas protein | Defect | References |
|---|---|---|---|---|---|
| DNAH5 | Dynein heavy chain | ODA | γ HC | ODA | [200] |
| DNAH11 | Dynein heavy chain (lrd dynein) | Putative ODA | Similar to β HC | n.d. | [149, 201] |
| DNAH7 | Dynein heavy chain | Putative IDA | DHC5 (β HC) | IDA, ODF a | [143, 202] |
| DNAI1 | Dynein intermediate chain | ODA | IC1/IC78 | ODA | [203] |
| DNAI2 | Dynein intermediate chain | ODA | IC2/IC69 | ODA | [204] |
| DNAL1 | Dynein light chain | ODA | LC1 | ODA a | [205] |
| TXNDC3/NME8 | Dynein intermediate chain | ODA | n.d. | ODA | [206] |
| TCTE3 | Dynein light chain | ODA | Tctex2 | ODA | [207] |
| DNALI1 | Dynein light chain | IDA | p28 | IDA | [144] |
| CCDC114 | Coiled‐coil protein | ODA | DCC2 | ODA | [153] |
| ARMC4 | Armadillo repeat containing protein 4 | ODA | n.d. | ODA | [160] |
| CCDC151 | Coiled‐coil domain containing 151 | ODA | ODA10 | ODA | [208] |
| CCDC103 | Coiled‐coil protein | ODA | CCDC103 | ODA | [161, 163] |
| RSPH1 | Radial spoke head protein | RS | RSP1 | RS, CP | [131] |
| RSPH9 | Radial spoke protein | RS | RSP9 | RS, CP | [138] |
| RSPH4A | Radial spoke protein | RS | RSP4 | RS, CP | [138] |
| HYDIN | Hydin (gene causing hydrocephalus) | CP (C2b) | Hydin | CP | [137, 209] |
| Spag6 | Sperm antigen 6 | CP | PF16 | CP a | [210] |
| Spag16L | Sperm antigen 16 | CP | PF20 | CP a | [175] |
| SPAG17 | Sperm antigen 17 | CP (C1) | PF6 | CP | [211] |
| Ak7 | Adenylate kinase 7 | CP | n.d. | CP, DMT | [212] |
| HSF‐1 | Heat shock transcription factor 1 | CP | n.d. | CP, DMT | [213] |
| Pacrg | Parkin co‐regulated gene product | DMT | PACRG | DMT | [172] |
| TECT2 | Tektin 2 | DMT | p58 b | IDA, DMT | [214] |
| HEATR2/DNAAF5 | HEAT‐repeat containing 2 | Cytoplasm | Cre03.g162400.t1.1 c | ODA | [130] |
| LRRC6 | Leucine‐rich repeat containing 6 | Cytoplasm | Cre17.g739850.t1.2 c | ODA, IDA | [131, 215] |
| DNAAF3 | Dynein axonemal assembly factor 3 | Cytoplasm | PF22 | ODA, IDA | [128] |
| ZMYND10 | Zinc finger, MYND‐type containing 10 | Cytoplasm | Cre08.g384628.t1.1 c | ODA, IDA | [216] |
| DNAAF4 | Dynein axonemal assembly factor 4 (DYX1C1) | Cytoplasm | Cre11.g467560.t1.1 c | ODA, IDA | [129] |
| SPAG1 | Sperm antigen 1 | Cytoplasm | Cre16.g682251.t1.1 c | ODA, IDA | [134] |
| C21ORF59 | Chromosome 21, ORF59 | Flagellar matrix | FBB18 | ODA, IDA | [163] |
| DNAAF1 | Dynein axonemal assembly factor 1 (LRRC50) | Cytoplasm | ODA7 | ODA, IDA | [126, 127] |
| DNAAF2 | Dynein axonemal assembly factor 2 (KTU) | Cytoplasm | PF13 | ODA, IDA | [125] |
| Pih1d3 | PIH1 domain‐containing protein | Cytoplasm | n.d. | ODA and IDA | [187] |
| CCDC39 | Coiled‐coil domain containing 39 | AX | FAP59 | IDA, DMT | [167] |
| CCDC40 | Coiled‐coil domain containing 40 | Cilia/cytoplasm | Cre05.g238270.t1.1 c | CP, IDA | [168] |
| CCDC164 | Coiled‐coil domain containing 164 | N‐DRC | DRC1 | N‐DRC | [169] |
| CCDC65 | Coiled‐coil domain containing 65 | N‐DRC | FAP250 | N‐DRC | [163] |
ODA outer dynein arm, IDA inner dynein arm, RS radial spoke, CP central apparatus, DMT doublet microtubule, N‐DRC nexin‐dynein regulatory complex, AX axoneme, ODF outer dense fiber, n.d. not defined
aObserved in mouse
bp58 is a homolog of tektin B1 (tektin 2) [217]
cID of the genes showing highest similarity in the Chlamydomonas reinhardtii genome (database v5.5)
Heterogeneity in the HCs of OAD
Several studies indicate that animal and Chlamydomonas flagella contain two and three HCs, respectively, in the OAD [11, 37]. From their molecular substructures, these are referred to as two‐headed or three‐headed dyneins. Unlike the heterogeneous IAD HCs, OAD had been thought to be present as a single form in an organism, lined along the doublet microtubule at intervals of 24 nm. However, recent studies suggest heterogeneities of OADs. Respiratory cilia in humans contain two types of OAD: type 1 OAD contains only DNAH5 (ortholog of Chlamydomonas ODAγ) and is present at the proximal region of cilia; type 2 OAD contains both DNAH5 and DNAH9 (ortholog of Chlamydomonas ODAβ) at the distal part of the cilia [147]. In sperm, however, DNAH9 is present along the entire length of the flagellum, whereas DNAH5 is only present in the proximal part of the flagellum. The motility defects in DNAH5‐mutated patients also differ between respiratory cilia and sperm flagella [147]. It is not clear whether type 1 OAD is present as a single‐headed dynein or a two‐headed dynein with another unknown HC.
Another interesting HC is DNAH11 (lrd). This HC was originally discussed in mouse, as it was first identified as the left–right dynein (product of lrd gene), which is mutated in inversus viscerum mouse. lrd shows high similarity to the β‐type HC of OAD and is specifically expressed in monocilia of the embryonic node [148]. DNAH11‐null male mice are fertile, and their tracheal cilia and sperm flagella show no apparent deficiency in ultrastructure and motility. Unlike in mice, human patients with mutations in the ortholog of DNAH11 show situs inversus and also immotile respiratory cilia [149, 150]. Knowledge of the precise composition of the OAD with respect to the HCs in nodal cilia would be critical to elucidate the mechanism of the unique phenotype of DNAH11 mutants.
Notes on the differences between sperm and Chlamydomonas
Chlamydomonas is an excellent model organism for studying the molecular composition and function of cilia and flagella. However, difference in the usage of axonemal proteins has been observed between Chlamydomonas and animal sperm [28, 37, 151]. The composition of the ODA‐DC is likely different between sperm and Chlamydomonas flagella [40]. DC2 is present in multiple forms in sperm of the ascidian Ciona intestinalis and humans. Studies on the molecular composition of the OAD in C. intestinalis indicate the presence of multiple forms of DC2 as IC4 and IC5 and a protein similar to DC2, Axp66.0 [40, 152]. Orthologs of DC1 and DC3 are not found in the Ciona or human genomes [40]. Male fertility was not greatly affected among human individuals carrying CCDC114 (DC2) mutations [153]. Although it has not been elucidated why, the redundancy of DC2 might compensate for the mutation of CCDC114 in sperm flagella. Similarly, knowledge has been accumulating showing that some axonemal components are specific to metazoans and do not exist in Chlamydomonas. For example, a calcium sensor for the OAD, calaxin, is specific to metazoan species [35, 37]. The calcium sensor for OAD in Chlamydomonas is dynein LC4, which is present in most bikont species [37]. Other examples of metazoan‐specific axonemal components include TXNDC3, an IC of the OAD, and RS proteins CMUB116, AK58, and NDK/DM44 [154].
Factors for intraciliary docking and regulation of OAD
Recent studies in Chlamydomonas suggest that ODA10 (CCDC151 in humans), ODA5 (homolog of a DC2‐like protein CCDC63), and ODA8 (LRRC56 in humans) form an accessory complex of the ODA‐DC [153, 155, 156]. The complex is associated with an adenylate kinase with sequence similarity to human cytosolic AK1 and AK5 [155]. ODA8 is localized to both the flagellar matrix and cytoplasm, similar to IFT components [157]. Thus, this accessory complex might be involved in IFT‐dependent assembly of the OAD before docking onto the doublet microtubules. Oda16/Wdr69 (WD‐repeat domain 65) is also an adaptor of the OAD for IFT [158]; Oda16 directly interacts with a component of IFT‐B, IFT46 [159], suggesting a role for IFT46 as a cargo‐specific adaptor between OAD and IFT‐B.
Other nonintegral components necessary for intraciliary docking of OADs have been reported. ARMC4 is a protein localized at the base of cilia and flagella. Patients with mutations in ARMC4 show reduced numbers of ODAs. ARMC4 interacts with DNAI2 and is considered to be involved in the specific targeting and anchoring of OAD [160]. CCDC103 binds tightly to microtubules and appears to have a role in attachment of OAD [161, 162]. C21orf59/FBB18 is a flagellar matrix protein and participates in dynein arm assembly. Loss of C21orf59/FBB18 in humans causes deficiency of both ODAs and IDAs [163].
Deficiency in other axonemal substructures
Because axonemes contain several nondynein structures for direct or indirect regulation of ciliary motors, mutations in the proteins of these structures may cause ciliopathy. RS and the CP are located in the center of 9 + 2 axonemes (Fig. 1b, c) and are involved in the activation or inactivation of dyneins on a specific doublet microtubule, resulting in the determination of the ciliary beating plane [14, 15, 164, 165]. Mutations in the genes for RS/CP leading to ciliopathy include RS proteins (RSPH1, RSPH4A, and RSPH9) and those in the CP (HYDIN and SPAG17) (Table 1). The DRC is located near RS2 and is connected to the nexin link (Fig. 1c) [46, 48, 166]. This “N‐DRC” (nexin‐DRC, Fig. 1c) is related to ciliopathy by defects in the regulation of ciliary motility. From studies in Chlamydomonas, this N‐DRC structure is thought potentially important in the regulation of IADs. Recent genetic and ultrastructural studies have identified the components of the N‐DRC, its associated proteins such as CCDC39, CCDC40, CCDC65, and DRC1 (CCDC164), and their connections with ciliopathy [163, 167, 168, 169].
In some cases, genetic diseases apparently unrelated to the function of axonemal dyneins cause abnormality in ciliary structure and motility. Mutations of RPGR cause XLRP. Several defects in axonemal structure and motility are observed in XLRP patients, including missing dynein arms and disorganization of ciliary orientation [132]. OFD1 is a coiled‐coil protein with a LisH domain, located at the base of the cilium [170]. Respiratory cilia with mutated OFD1 are motile with normal beat frequency, but show uncoordinated beating.
Mutations that affect sperm flagella but not cilia
Because the structure of axonemes and the mechanism of their formation are quite similar between somatic cilia and sperm flagella, many deficiencies lead to ciliopathy including male infertility. However, there are some cases of patients or animal mutants showing different phenotypes between somatic cilia and sperm flagella. This seems to be due to differences in the axonemal components [67], the presence of unique accessory structures in sperm, and degeneration of the basal body and its appendages during spermatogenesis.
Manchette‐dependent flagellar formation
Parkin and PACRG are genes located on mouse chromosome 17, and their deletions cause “quaking” mice, and male infertility [171, 172]. Studies in Chlamydomonas and Trypanosoma revealed that PACRG binds to EFHC1/RIB72 in the A‐tubule of a doublet microtubule [54, 173, 174]. In mice, PACRG is associated with MEIG1, which was identified as a binding partner of a CP protein SPAG16 (PF20) [175]. MEIG1‐knockout mice show male infertility, but no other phenotypes for ciliopathy are observed [176, 177, 178]. Further studies suggest that MEIG1/PACRG plays a critical role in the control of the structure called “manchette” [179], a microtubule‐based structure transiently formed during spermiogenesis. This structure is thought to play roles in the significant morphological changes of the nucleus and in flagellar formation. Manchette‐dependent protein transport, called IMT, is proposed to be essential in these changes [180].
Another example showing a possible role of manchette in the formation of sperm flagella is Iqcg [181]. Iqcg is highly expressed in ciliary tissues including trachea, oviduct, and testis, and its knockout male produces immotile sperm with highly disorganized axonemal structures [182]. However, the ciliary structures in the trachea and oviduct appear normal in the knockout mice. This raises the possibility that the deficiency in sperm flagella is caused by a spermatogenesis‐specific process for flagellar formation, such as IMT.
CP proteins Pcdp1 and Spef2
Testis‐ or sperm‐specific phenotypes are also seen in the knockout mice of another two CP proteins. One is Pcdp1, whose knockout results in hydrocephalus, male infertility, and slight defects in ciliary movement in trachea [183]. Studies in Chlamydomonas demonstrate that the Pcdp1 homolog, FAP221, is a CP protein that binds to calmodulin and is localized to the C1d projection [184]. Interestingly, sperm from Pcdp1‐null mice lack flagella, although respiratory epithelial cilia are apparently normal [183]. This suggests that Pcdp1 has a function specific to the formation of sperm flagella.
A similar phenotype with regard to ciliary structure is observed in the knockout mice of Spef2 [185]. Although the respiratory cilia in the knockout mice are normal in ultrastructure but have lower beat frequency, their sperm show shortened flagella with a disorganized axoneme and accessory structures. Spef2 is a homolog of Chlamydomonas CP protein Cpc1, which has an EF‐hand domain and a conserved adenylate kinase domain, and is suggested to function in ATP regeneration [186]. The fact that Spef2 interacts with an IFT‐B component, IFT20 in mouse testis [185], further suggests a specific function of CP proteins, like that of Pcdp1, in the formation of sperm flagella.
A candidate for a sperm‐specific assembly factor: Pih1d3
Two proteins for assembly of axonemal dynein, DNAAF2 and MOT48, are known to possess PIH domains. Another PIH protein, TWISTER, has a function in motile cilia in zebrafish [96]. A Twister‐like gene, Pih1d3, is specifically expressed in mouse testis. Knockout of this gene causes a defect in flagellar motility, although cilia of the respiratory tract and ependymal cells are normal in their motility and ultrastructure [187]. Most of the Pih1d3‐null mice lack both OADs and IADs in sperm flagella, and some show disorganized axonemes lacking a few doublet microtubules. Pih1d3 interacts with DNAI2, Hsp70, and Hsp90, but not with any of the DNAAFs, suggesting the involvement of this protein in a distinct process of dynein assembly in spermatogenesis. It is possible that Pih1d3 is involved in assembly coupled with a testis‐specific transport pathway, such as IMT. Alternatively, Pih1d3 may play a role in the assembly of axonemal dyneins mediated by a sperm‐specific component of the OAD and IAD [67].
The mitochondria sheath
The development of helical configuration of mitochondria is well coordinated with the elongation of the sperm tail [6, 188, 189, 190]. MFNs are the mediators for mitochondrial fusion. MFN2 is located on the mitochondrial outer membrane and participates in the fusion, maintenance, and operation of the mitochondrial network [191]. Loss of function of Fzo (fuzzy onions), the Drosophila homolog of MFN1 and MFN2, results in male sterility [192]. Recently, MFN2 was reported to form a complex with MNS1 in murine spermatogenic cells [193]. MFN2 is localized in the midpiece of spermatozoa, whereas MNS1 is present throughout the sperm tail. Knockout of MNS1 results in situs inversus, hydrocephalus, and partial loss of OADs in tracheal cilia, resulting in a motility defect. Interestingly, MNS1‐null sperm have abnormal and short flagella with disruption of outer doublet microtubules and the ODF [194]. These observations imply common mechanisms of axonemal formation between sperm and epithelia and an additional mitochondria/ODF‐dependent mechanism specific to axonemal formation in sperm flagella. It is hypothesized that a MNS1/MFN2 complex and ODF2 are involved in anchoring the mitochondria in the midpiece [193]. The involvement of mitochondria in the formation of axonemes is also suggested in mice lacking VDAC3, which show male infertility with structural loss of doublet microtubules from the sperm flagellar axonemes, but no structural abnormality in trachea cilia [195].
Conclusions
The connection between male infertility and ciliopathy was first disclosed by the observation of a common ultrastructural abnormality in sperm flagella and epithelial cilia in patients with Kartagener syndrome [1, 2, 196]. However, it also became evident that mutations in the proteins, such as Spef2 and Pih1d3, result in sperm dysfunction but not in ciliary defects of epithelia, suggesting a sperm‐specific mechanism leading to immotile flagella. Considering fertility, males are more affected by deficiencies of axonemal components than females, as seen in cases where females with Kartagener syndrome with dyneinless cilia were fertile [197, 198]. This further indicates a unique functional aspect of sperm flagella. In addition, some patients with typical clinical symptoms of Kartagener syndrome show normal sperm motility with normal axonemal ultrastructure [5, 199], suggesting some axonemal proteins whose deletion cannot be detected by electron microscopy but critically affects the flagellar motility.
Knockout mice are powerful models for studying ciliopathy. However, in most cases, loss of axonemal dyneins results in embryonic lethality. This is not compatible with the symptoms of human ciliopathy, in which patients are alive and show fertility, making it possible to carry out family studies. Further experiments are necessary with mouse models, including analyses with extensive N‐ethyl‐N‐nitrosourea mutants or dominant‐negative mutants, and the engineering of human point mutations in mice [94]. Studies using Chlamydomonas and marine invertebrates also shed light on the molecular structure and mechanism of cilia and flagella, and on the genetic diagnosis of ciliopathy.
Acknowledgments
This work was supported in part by Grants‐in‐aid #15H01201 for Scientific Research on Innovative Areas and #22370023 for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT). Studies with marine invertebrates cited in this paper were supported by the members of the Onagawa Field Research Center, Tohoku University; International Coastal Research Center, AORI, The University of Tokyo; and staff of the National Bioresource Project.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Human/animal studies
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1. Camner P, Mossberg B, Afzelius BA. Evidence of congenitally nonfunctioning cilia in the tracheobronchial tract in two subjects. Am Rev Respir Dis, 1975, 112, 807–809 [DOI] [PubMed] [Google Scholar]
- 2. Afzelius BA. A human syndrome caused by immotile cilia. Science, 1976, 193, 317–319 10.1126/science.1084576 [DOI] [PubMed] [Google Scholar]
- 3. Afzelius BA. “Immotile‐cilia” syndrome and ciliary abnormalities induced by infection and injury. Am Rev Respir Dis, 1981, 124, 107–109 [DOI] [PubMed] [Google Scholar]
- 4. Afzelius BA. Situs inversus and ciliary abnormalities. What is the connection?. Int J Dev Biol, 1995, 39, 839–844 [PubMed] [Google Scholar]
- 5. Afzelius BA. Cilia‐related diseases. J Pathol, 2004, 204, 470–477 10.1002/path.1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fawcett DW. The mammalian spermatozoon. Dev Biol, 1975, 44, 394–436 10.1016/0012‐1606(75)90411‐X [DOI] [PubMed] [Google Scholar]
- 7. Inaba K. Molecular architecture of the sperm flagella: molecules for motility and signaling. Zoolog Sci, 2003, 20, 1043–1056 10.2108/zsj.20.1043 [DOI] [PubMed] [Google Scholar]
- 8. Inaba K. Sperm flagella: comparative and phylogenetic perspectives of protein components. Mol Hum Reprod, 2011, 17, 524–538 10.1093/molehr/gar034 [DOI] [PubMed] [Google Scholar]
- 9. Darszon A, Nishigaki T, Beltran C, Treviño CL. Calcium channels in the development, maturation, and function of spermatozoa. Physiol Rev, 2011, 91, 1305–1355 10.1152/physrev.00028.2010 [DOI] [PubMed] [Google Scholar]
- 10. Gibbons IR. Cilia and flagella of eukaryotes. J Cell Biol, 1981, 91, 107s–124s 10.1083/jcb.91.3.107s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mohri H, Inaba K, Ishijima S, Baba SA. Tubulin–dynein system in flagellar and ciliary movement. Proc Jpn Acad Ser B Phys Biol Sci, 2012, 88, 397–415349108210.2183/pjab.88.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Summers KE, Gibbons IR. Adenosine triphosphate‐induced sliding of tubules in trypsin‐treated flagella of sea‐urchin sperm. Proc Natl Acad Sci USA, 1971, 68, 3092–309638959710.1073/pnas.68.12.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamiya R. Functional diversity of axonemal dyneins as studied in Chlamydomonas mutants. Int Rev Cytol, 2002, 219, 115–155 10.1016/S0074‐7696(02)19012‐7 [DOI] [PubMed] [Google Scholar]
- 14. Smith EF, Sale WS. Regulation of dynein‐driven microtubule sliding by the radial spokes in flagella. Science, 1992, 257, 1557–1559 10.1126/science.1387971 [DOI] [PubMed] [Google Scholar]
- 15. Smith EF, Yang P. The radial spokes and central apparatus: mechano‐chemical transducers that regulate flagellar motility. Cell Motil Cytoskeleton, 2004, 57, 8–17195094210.1002/cm.10155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamiya R. Mutations at twelve independent loci result in absence of outer dynein arms in Chlamydomonas reinhardtii . J Cell Biol, 1988, 107, 2253–2258 10.1083/jcb.107.6.2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang B, Piperno G, Luck DJ. Paralyzed flagella mutants of Chlamydomonas reinhardtii. Defective for axonemal doublet microtubule arms. J Biol Chem, 1979, 254, 3091–3099 [PubMed] [Google Scholar]
- 18. Gibbons BH, Gibbons IR. Flagellar movement and adenosine triphosphatase activity in sea urchin sperm extracted with triton X‐100. J Cell Biol, 1972, 54, 75–97210886510.1083/jcb.54.1.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gibbons BH, Gibbons IR. The effect of partial extraction of dynein arms on the movement of reactivated sea‐urchin sperm. J Cell Sci, 1973, 13, 337–357 [DOI] [PubMed] [Google Scholar]
- 20. Shingyoji C, Murakami A, Takahashi K. Local reactivation of Triton‐extracted flagella by iontophoretic application of ATP. Nature, 1977, 265, 269–270 10.1038/265269a0 [DOI] [PubMed] [Google Scholar]
- 21. Brokaw CJ. Direct measurements of sliding between outer doublet microtubules in swimming sperm flagella. Science, 1989, 243, 1593–1596 10.1126/science.2928796 [DOI] [PubMed] [Google Scholar]
- 22. Okuno M, Brokaw CJ. Inhibition of movement of trition‐demembranated sea‐urchin sperm flagella by Mg2+, ATP4−, ADP and P1 . J Cell Sci, 1979, 38, 105–123 [DOI] [PubMed] [Google Scholar]
- 23. Tang WJ, Bell CW, Sale WS, Gibbons IR. Structure of the dynein‐1 outer arm in sea urchin sperm flagella. I. Analysis by separation of subunits. J Biol Chem, 1982, 257, 508–515 [PubMed] [Google Scholar]
- 24. Ishijima S. Mechanical constraint converts planar waves into helices on tunicate and sea urchin sperm flagella. Cell Struct Funct, 2012, 37, 13–19 10.1247/csf.11019 [DOI] [PubMed] [Google Scholar]
- 25. King SM. The dynein microtubule motor. Biochim Biophys Acta, 2000, 1496, 60–75 10.1016/S0167‐4889(00)00009‐4 [DOI] [PubMed] [Google Scholar]
- 26. Gibbons IR, Gibbons BH, Mocz G, Asai DJ. Multiple nucleotide‐binding sites in the sequence of dynein beta heavy chain. Nature, 1991, 352, 640–643 10.1038/352640a0 [DOI] [PubMed] [Google Scholar]
- 27. Ogawa K. Four ATP‐binding sites in the midregion of the β heavy chain of dynein. Nature, 1991, 352, 643–645 10.1038/352643a0 [DOI] [PubMed] [Google Scholar]
- 28. Inaba K. Molecular basis of sperm flagellar axonemes: structural and evolutionary aspects. Ann N Y Acad Sci, 2007, 1101, 506–526 10.1196/annals.1389.017 [DOI] [PubMed] [Google Scholar]
- 29. Inaba K, Mizuno K, Shiba K Sawada H. Inoue N, Iwano M. Structure, function, and phylogenetic consideration of calaxin. Sexual reproduction in animals and plants, 2014. Springer: Berlin; 49–51 [Google Scholar]
- 30. Ogawa K, Takai H, Ogiwara A, Yokota E, Shimizu T, Inaba K et al. Is outer arm dynein intermediate chain 1 multifunctional?. Mol Biol Cell, 1996, 7, 1895–190727603810.1091/mbc.7.12.1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Padma P, Hozumi A, Ogawa K, Inaba K. Molecular cloning and characterization of a thioredoxin/nucleoside diphosphate kinase related dynein intermediate chain from the ascidian, Ciona intestinalis . Gene, 2001, 275, 177–183 10.1016/S0378‐1119(01)00661‐8 [DOI] [PubMed] [Google Scholar]
- 32. Sadek CM, Damdimopoulos AE, Pelto‐Huikko M, Gustafsson JA, Spyrou G, Miranda‐Vizuete A. Sptrx‐2, a fusion protein composed of one thioredoxin and three tandemly repeated NDP‐kinase domains is expressed in human testis germ cells. Genes Cells, 2001, 6, 1077–1090 10.1046/j.1365‐2443.2001.00484.x [DOI] [PubMed] [Google Scholar]
- 33. Inaba K, Morisawa S, Morisawa M. Proteasomes regulate the motility of salmonid fish sperm through modulation of cAMP‐dependent phosphorylation of an outer arm dynein light chain. J Cell Sci, 1998, 111, 1105–1115 [DOI] [PubMed] [Google Scholar]
- 34. Inaba K, Kagami O, Ogawa K. Tctex2‐related outer arm dynein light chain is phosphorylated at activation of sperm motility. Biochem Biophys Res Commun, 1999, 256, 177–183 10.1006/bbrc.1999.0309 [DOI] [PubMed] [Google Scholar]
- 35. Mizuno K, Padma P, Konno A, Satouh Y, Ogawa K, Inaba K. A novel neuronal calcium sensor family protein, calaxin, is a potential Ca2+‐dependent regulator for the outer arm dynein of metazoan cilia and flagella. Biol Cell, 2009, 101, 91–103 10.1042/BC20080032 [DOI] [PubMed] [Google Scholar]
- 36. Mizuno K, Shiba K, Okai M, Takahashi Y, Shitaka Y, Oiwa K et al. Calaxin drives sperm chemotaxis by Ca2+‐mediated direct modulation of a dynein motor. Proc Natl Acad Sci USA, 2012, 109, 20497–20502352857810.1073/pnas.1217018109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inaba K. Calcium sensors of ciliary outer arm dynein: functions and phylogenetic considerations for eukaryotic evolution. Cilia, 2015, 4, 6441524110.1186/s13630‐015‐0015‐z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takada S, Wilkerson CG, Wakabayashi K, Kamiya R, Witman GB. The outer dynein arm‐docking complex: composition and characterization of a subunit (oda1) necessary for outer arm assembly. Mol Biol Cell, 2002, 13, 1015–10299961610.1091/mbc.01‐04‐0201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Owa M, Furuta A, Usukura J, Arisaka F, King SM, Witman GB, Kamiya R, Wakabayashi K. Cooperative binding of the outer arm‐docking complex underlies the regular arrangement of outer arm dynein in the axoneme. Proc Natl Acad Sci USA, 2012, 111, 9461–9466 10.1073/pnas.1403101111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hozumi A, Satouh Y, Makino Y, Toda T, Ide H, Ogawa K et al. Molecular characterization of Ciona sperm outer arm dynein reveals multiple components related to outer arm docking complex protein 2. Cell Motil Cytoskeleton, 2006, 63, 591–603 10.1002/cm.20146 [DOI] [PubMed] [Google Scholar]
- 41. Kamiya R, Yagi T. Functional diversity of axonemal dyneins as assessed by in vitro and in vivo motility assays of Chlamydomonas mutants. Zoolog Sci, 2014, 31, 633–644 10.2108/zs140066 [DOI] [PubMed] [Google Scholar]
- 42. Wada S, Okuno M, Mohri H. Inner arm dynein ATPase fraction of sea urchin sperm flagella causes active sliding of axonemal outer doublet microtubule. Biochem Biophys Res Commun, 1991, 175, 173–178 10.1016/S0006‐291X(05)81216‐1 [DOI] [PubMed] [Google Scholar]
- 43. Yokota E, Mabuchi I. Isolation and characterization of a novel dynein that contains C and A heavy chains from sea urchin sperm flagellar axonemes. J Cell Sci, 1994, 107, 345–351 [DOI] [PubMed] [Google Scholar]
- 44. Hozumi A, Padma P, Toda T, Ide H, Inaba K. Molecular characterization of axonemal proteins and signaling molecules responsible for chemoattractant‐induced sperm activation in Ciona intestinalis . Cell Motil Cytoskeleton, 2008, 65, 249–267 10.1002/cm.20258 [DOI] [PubMed] [Google Scholar]
- 45. Huang B, Ramanis Z, Luck DJ. Suppressor mutations in Chlamydomonas reveal a regulatory mechanism for flagellar function. Cell, 1982, 28, 115–124 10.1016/0092‐8674(82)90381‐6 [DOI] [PubMed] [Google Scholar]
- 46. Gardner LC, O'Toole E, Perrone CA, Giddings T, Porter ME. Components of a “dynein regulatory complex” are located at the junction between the radial spokes and the dynein arms in Chlamydomonas flagella. J Cell Biol, 1994, 127, 1311–1325 10.1083/jcb.127.5.1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Downing KH, Sui H. Structural insights into microtubule doublet interactions in axonemes. Curr Opin Struct Biol, 2007, 17, 253–259 10.1016/j.sbi.2007.03.013 [DOI] [PubMed] [Google Scholar]
- 48. Heuser T, Raytchev M, Krell J, Porter ME, Nicastro D. The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J Cell Biol, 2009, 187, 921–933280632010.1083/jcb.200908067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bui KH, Sakakibara H, Movassagh T, Oiwa K, Ishikawa T. Asymmetry of inner dynein arms and inter‐doublet links in Chlamydomonas flagella. J Cell Biol, 2009, 186, 437–446272840610.1083/jcb.200903082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mitchell DR, Sale WS. Characterization of a Chlamydomonas insertional mutant that disrupts flagellar central pair microtubule‐associated structures. J Cell Biol, 1999, 144, 293–304213289610.1083/jcb.144.2.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nicastro D, Fu X, Heuser T, Tso A, Porter ME, Linck RW. Cryo‐electron tomography reveals conserved features of doublet microtubules in flagella. Proc Natl Acad Sci USA, 2011, 108, E845–E853319835410.1073/pnas.1106178108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hoops HJ, Witman GB. Outer doublet heterogeneity reveals structural polarity related to beat direction in Chlamydomonas flagella. J Cell Biol, 1983, 97, 902–908 10.1083/jcb.97.3.902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mukai C, Okuno M. Glycolysis plays a major role for adenosine triphosphate supplementation in mouse sperm flagellar movement. Biol Reprod, 2004, 71, 540–547 10.1095/biolreprod.103.026054 [DOI] [PubMed] [Google Scholar]
- 54. Miki K, Qu W, Goulding EH, Willis WD, Bunch DO, Strader LF et al. Glyceraldehyde 3‐phosphate dehydrogenase‐S, a sperm‐specific glycolytic enzyme, is required for sperm motility and male fertility. Proc Natl Acad Sci USA, 2004, 101, 16501–1650653454210.1073/pnas.0407708101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anderson RG. The three‐dimensional structure of the basal body from the rhesus monkey oviduct. J Cell Biol, 1972, 54, 246–265210888310.1083/jcb.54.2.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bornens M. Organelle positioning and cell polarity. Nat Rev Mol Cell Biol, 2008, 9, 874–886 10.1038/nrm2524 [DOI] [PubMed] [Google Scholar]
- 57. Boisvieux‐Ulrich E, Sandoz D. Determination of ciliary polarity precedes differentiation in the epithelial cells of quail oviduct. Biol Cell, 1991, 72, 3–14 10.1016/0248‐4900(91)90072‐U [DOI] [PubMed] [Google Scholar]
- 58. Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell, 2007, 129, 1201–1213 10.1016/j.cell.2007.03.053 [DOI] [PubMed] [Google Scholar]
- 59. Donaldson JG, Jackson CL. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat Rev Mol Cell Biol, 2011, 12, 362–375324555010.1038/nrm3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kozminski KG, Beech PL, Rosenbaum JL. The Chlamydomonas kinesin‐like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol, 1995, 131, 1517–1527 10.1083/jcb.131.6.1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol, 2002, 3, 813–825 10.1038/nrm952 [DOI] [PubMed] [Google Scholar]
- 62. Sarpal R, Todi SV, Sivan‐Loukianova E, Shirolikar S, Subramanian N, Raff EC et al. Drosophila KAP interacts with the kinesin II motor subunit KLP64D to assemble chordotonal sensory cilia, but not sperm tails. Curr Biol, 2003, 13, 1687–1696 10.1016/j.cub.2003.09.025 [DOI] [PubMed] [Google Scholar]
- 63. Han YG, Kwok BH, Kernan MJ. Intraflagellar transport is required in Drosophila to differentiate sensory cilia but not sperm. Curr Biol, 2003, 13, 1679–1686 10.1016/j.cub.2003.08.034 [DOI] [PubMed] [Google Scholar]
- 64. Manandhar G, Sutovsky P, Joshi HC, Stearns T, Schatten G. Centrosome reduction during mouse spermiogenesis. Dev Biol, 1998, 203, 424–434 10.1006/dbio.1998.8947 [DOI] [PubMed] [Google Scholar]
- 65. Manandhar G, Simerly C, Schatten G. Highly degenerated distal centrioles in rhesus and human spermatozoa. Hum Reprod, 2000, 15, 256–263 10.1093/humrep/15.2.256 [DOI] [PubMed] [Google Scholar]
- 66. Fawcett DW, Phillips DM. The fine structure and development of the neck region of the mammalian spermatozoon. Anat Rec, 1969, 165, 153–164 10.1002/ar.1091650204 [DOI] [PubMed] [Google Scholar]
- 67. Konno A, Shiba K, Cai C, Inaba K. Branchial cilia and sperm flagella recruit distinct axonemal components. PLoS One, 2015, 10, e0126005442745610.1371/journal.pone.0126005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li X, Xu L, Li J, Li B, Bai X, Strauss JF 3rd et al. Otitis media in sperm‐associated antigen 6 (Spag6)‐deficient mice. PLoS One, 2014, 9, e112879423107310.1371/journal.pone.0112879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dabdoub A, Kelley MW. Planar cell polarity and a potential role for a Wnt morphogen gradient in stereociliary bundle orientation in the mammalian inner ear. J Neurobiol, 2005, 64, 446–457 10.1002/neu.20171 [DOI] [PubMed] [Google Scholar]
- 70. Sobkowicz HM, Slapnick SM, August BK. The kinocilium of auditory hair cells and evidence for its morphogenetic role during the regeneration of stereocilia and cuticular plates. J Neurocytol, 1995, 24, 633–653 10.1007/BF01179815 [DOI] [PubMed] [Google Scholar]
- 71. Kikuchi T, Takasaka T, Tonosaki A, Watanabe H. Fine structure of guinea pig vestibular kinocilium. Acta Otolaryngol, 1989, 108, 26–30 10.3109/00016488909107388 [DOI] [PubMed] [Google Scholar]
- 72. Sobrinho‐Simões M, Johannessen JV. Scanning electron microscopy of the normal human thyroid. J Submicrosc Cytol, 1981, 13, 209–222 [PubMed] [Google Scholar]
- 73. Martin A, Hedinger C, Häberlin‐Jakob M, Walt H. Structure and motility of primary cilia in the follicular epithelium of the human thyroid. Virchows Arch B Cell Pathol Incl Mol Pathol, 1988, 55, 159–166 [DOI] [PubMed] [Google Scholar]
- 74. Robaire B, Hinton BT, Orgebin‐Crist MC Neill JD. The epididymis. Knobil and Neill's physiology of reproduction, 2006. 3 Elsevier: Amsterdam; 1071–1148 [Google Scholar]
- 75. Kormanko M, Reijonen K. Microvascular structure of human epididymis. Am J Anat, 1976, 145, 23–32 10.1002/aja.1001450103 [DOI] [PubMed] [Google Scholar]
- 76. Paniagua R, Regadera J, Nistal M, Abaurrea MA. Histological, histochemical and ultrastructural variations along the length of the human vas deferens before and after puberty. Acta Anat, 1982, 111, 190–203 10.1159/000145467 [DOI] [PubMed] [Google Scholar]
- 77. Hando T, Okada DM, Zamboni L. Atypical cilia in human endometrium. J Cell Biol, 1968, 39, 475–481210752410.1083/jcb.39.2.475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Denholm RB, More IA. Atypical cilia of the human endometrial epithelium. J Anat, 1980, 131, 309–3151233270 [PMC free article] [PubMed] [Google Scholar]
- 79. Eliyahu S, Shalev E. A fertile woman with Kartagener's syndrome and three consecutive pregnancies. Hum Reprod, 1996, 11, 683 10.1093/HUMREP/11.3.683 [DOI] [PubMed] [Google Scholar]
- 80. Singla V, Reiter JF. The primary cilium as the cell's antenna: signaling at a sensory organelle. Science, 2006, 313, 629–633 10.1126/science.1124534 [DOI] [PubMed] [Google Scholar]
- 81. Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y et al. Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell, 1998, 95, 829–837 10.1016/S0092‐8674(00)81705‐5 [DOI] [PubMed] [Google Scholar]
- 82. Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left–right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A−/− mice analysis. J Cell Biol, 1999, 145, 825–836213317710.1083/jcb.145.4.825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Marshall WF, Nonaka S. Cilia: tuning into the cell's antenna. Curr Biol, 2006, 16, R604–R614 10.1016/j.cub.2006.07.012 [DOI] [PubMed] [Google Scholar]
- 84. Hirokawa N, Tanaka Y, Okada Y, Takeda S. Nodal flow and the generation of left–right asymmetry. Cell, 2006, 125, 33–45 10.1016/j.cell.2006.03.002 [DOI] [PubMed] [Google Scholar]
- 85. Shinohara K, Kawasumi A, Takamatsu A, Yoshiba S, Botilde Y, Motoyama N et al. Two rotating cilia in the node cavity are sufficient to break left–right symmetry in the mouse embryo. Nat Commun, 2012, 3, 622 10.1038/ncomms1624 [DOI] [PubMed] [Google Scholar]
- 86. McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left–right asymmetry in the mouse. Cell, 2003, 114, 61–73 10.1016/S0092‐8674(03)00511‐7 [DOI] [PubMed] [Google Scholar]
- 87. Babu D, Roy S. Left–right asymmetry: cilia stir up new surprises in the node. Open Biol, 2013, 3, 130052386686810.1098/rsob.130052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bangs FK, Schrode N, Hadjantonakis AK, Anderson KV. Lineage specificity of primary cilia in the mouse embryo. Nat Cell Biol, 2015, 17, 113–122440623910.1038/ncb3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Slough J, Cooney L, Brueckner M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev Dyn, 2008, 237, 2304–2314291924010.1002/dvdy.21669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Thomas J, Morlé L, Soulavie F, Laurençon A, Sagnol S, Durand B. Transcriptional control of genes involved in ciliogenesis: a first step in making cilia. Biol Cell, 2010, 102, 499–513 10.1042/BC20100035 [DOI] [PubMed] [Google Scholar]
- 91. Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep, 2012, 13, 608–618338878410.1038/embor.2012.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tobin JL, Beales PL. The nonmotile ciliopathies. Genet Med., 2009, 11, 386–402 10.1097/GIM.0b013e3181a02882 [DOI] [PubMed] [Google Scholar]
- 93. Zaghloul NA, Katsanis N. Mechanistic insights into Bardet–Biedl syndrome, a model ciliopathy. J Clin Invest, 2009, 119, 428–437264868510.1172/JCI37041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Norris DP, Grimes DT. Mouse models of ciliopathies: the state of the art. Dis Model Mech, 2012, 5, 299–312333982410.1242/dmm.009340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B et al. Bardet–Biedl syndrome type 4 (BBS4)‐null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci USA, 2004, 101, 8664–866942325210.1073/pnas.0402354101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development, 2004, 131, 4085–4093 10.1242/dev.01240 [DOI] [PubMed] [Google Scholar]
- 97. Gorivodsky M, Mukhopadhyay M, Wilsch‐Braeuninger M, Phillips M, Teufel A, Kim C et al. Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev Biol, 2009, 325, 24–32261385810.1016/j.ydbio.2008.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left–right axis determination. Development, 2000, 127, 2347–2355 [DOI] [PubMed] [Google Scholar]
- 99. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol, 2000, 151, 709–718218558010.1083/jcb.151.3.709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Iomini C, Babaev‐Khaimov V, Sassaroli M, Piperno G. Protein particles in Chlamydomonas flagella undergo a transport cycle consisting of four phases. J Cell Biol, 2001, 153, 13–24218552210.1083/jcb.153.1.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Iomini C, Li L, Esparza JM, Dutcher SK. Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii . Genetics, 2009, 183, 885–896277898410.1534/genetics.109.101915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Liem KF Jr, Ashe A, He M, Satir P, Moran J, Beier D et al. The IFT‐A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J Cell Biol, 2012, 197, 789–800337340010.1083/jcb.201110049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MA et al. Human and mouse mutations in WDR35 cause short‐rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet, 2011, 88, 508–515307192210.1016/j.ajhg.2011.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F et al. Mainzer–Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet, 2012, 90, 864–870337654810.1016/j.ajhg.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Byers B, Goetsch L. A highly ordered ring of membrane‐associated filaments in budding yeast. J Cell Biol, 1976, 69, 717–721 10.1083/jcb.69.3.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bezanilla M, Gladfelter AS, Kovar DR, Lee WL. Cytoskeletal dynamics: a view from the membrane. J Cell Biol, 2015, 209, 329–337442779310.1083/jcb.201502062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science, 2010, 329, 436–439309279010.1126/science.1191054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ihara M, Kinoshita A, Yamada S, Tanaka H, Tanigaki A, Kitano A et al. Cortical organization by the septin cytoskeleton is essential for structural and mechanical integrity of mammalian spermatozoa. Dev Cell, 2005, 8, 343–352 10.1016/j.devcel.2004.12.005 [DOI] [PubMed] [Google Scholar]
- 109. Kissel H, Georgescu MM, Larisch S, Manova K, Hunnicutt GR, Steller H. The Sept4 septin locus is required for sperm terminal differentiation in mice. Dev Cell, 2005, 8, 353–364 10.1016/j.devcel.2005.01.021 [DOI] [PubMed] [Google Scholar]
- 110. Kuo YC, Lin YH, Chen HI, Wang YY, Chiou YW, Lin HH et al. SEPT12 mutations cause male infertility with defective sperm annulus. Hum Mutat, 2012, 33, 710–719 10.1002/humu.22028 [DOI] [PubMed] [Google Scholar]
- 111. Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol, 2010, 190, 927–940293556110.1083/jcb.201006105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet, 2007, 81, 170–179195092910.1086/519494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ishikawa H, Kubo A, Tsukita S, Tsukita S. Odf2‐deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nat Cell Biol, 2005, 7, 517–524 10.1038/ncb1251 [DOI] [PubMed] [Google Scholar]
- 114. Delgehyr N, Sillibourne J, Bornens M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci, 2005, 118, 1565–1575 10.1242/jcs.02302 [DOI] [PubMed] [Google Scholar]
- 115. Singla V, Romaguera‐Ros M, Garcia‐Verdugo JM, Reiter JF. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell, 2010, 18, 410–424284106410.1016/j.devcel.2009.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sillibourne JE, Specht CG, Izeddin I, Hurbain I, Tran P, Triller A et al. Assessing the localization of centrosomal proteins by PALM/STORM nanoscopy. Cytoskeleton, 2011, 68, 619–627 10.1002/cm.20536 [DOI] [PubMed] [Google Scholar]
- 117. Kunimoto K, Yamazaki Y, Nishida E, Shinohara K, Ishikawa H, Hasegawa T et al. Coordinated ciliary beating requires Odf2‐mediated polarization of basal bodies via basal feet. Cell, 2012, 148, 189–200 10.1016/j.cell.2011.10.052 [DOI] [PubMed] [Google Scholar]
- 118. Salmon NA, Reijo Pera RA, Xu EY. A gene trap knockout of the abundant sperm tail protein, outer dense fiber 2, results in preimplantation lethality. Genesis, 2006, 44, 515–522303865610.1002/dvg.20241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Shao X, Tarnasky HA, Schalles U, Oko R, van der Hoorn FA. Interactional cloning of the 84‐kDa major outer dense fiber protein Odf84. Leucine zippers mediate associations of Odf84 and Odf27. J Biol Chem, 1997, 272, 6105–6113 10.1074/jbc.272.10.6105 [DOI] [PubMed] [Google Scholar]
- 120. Tarnasky H, Cheng M, Ou Y, Thundathil JC, Oko R, Hoorn FA. Gene trap mutation of murine Outer dense fiber protein‐2 gene can result in sperm tail abnormalities in mice with high percentage chimaerism. BMC Dev Biol, 2010, 10, 67289478010.1186/1471‐213X‐10‐67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lechtreck KF, Melkonian M. Striated microtubule‐associated fibers: identification of assemblin, a novel 34‐kD protein that forms paracrystals of 2‐nm filaments in vitro. J Cell Biol, 1991, 115, 705–716 10.1083/jcb.115.3.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Yang J, Liu X, Yue G, Adamian M, Bulgakov O, Li T. Rootletin, a novel coiled‐coil protein, is a structural component of the ciliary rootlet. J Cell Biol, 2002, 159, 431–440217307010.1083/jcb.200207153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gilliam JC, Chang JT, Sandoval IM, Zhang Y, Li T, Pittler SJ, Chiu W et al. Three‐dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell, 2012, 151, 1029–1041358233710.1016/j.cell.2012.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Roosing S, Rohrschneider K, Beryozkin A, Sharon D, Weisschuh N, Staller J et al. Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal‐recessive cone‐rod dystrophy. Am J Hum Genet, 2013, 93, 110–117371076110.1016/j.ajhg.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Omran H, Kobayashi D, Olbrich H, Tsukahara T, Loges NT, Hagiwara H et al. Ktu/PF13 is required for cytoplasmic pre‐assembly of axonemal dyneins. Nature, 2008, 456, 611–616327974610.1038/nature07471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Duquesnoy P, Escudier E, Vincensini L, Freshour J, Bridoux AM, Coste A et al. Loss‐of‐function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am J Hum Genet, 2009, 85, 890–896279056910.1016/j.ajhg.2009.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Loges NT, Olbrich H, Becker‐Heck A, Häffner K, Heer A, Reinhard C et al. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am J Hum Genet, 2009, 85, 883–889279580110.1016/j.ajhg.2009.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mitchison HM, Schmidts M, Loges NT, Freshour J, Dritsoula A, Hirst RA et al. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat Genet, 2012, 44, 381–389331561010.1038/ng.1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tarkar A, Loges NT, Slagle CE, Francis R, Dougherty GW, Tamayo JV et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat Genet, 2013, 45, 995–1003400044410.1038/ng.2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Horani A, Druley TE, Zariwala MA, Patel AC, Levinson BT, Arendonk LG et al. Whole‐exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am J Hum Genet, 2012, 91, 685–693348450510.1016/j.ajhg.2012.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kott E, Legendre M, Copin B, Papon JF, Dastot‐Le Moal F, Montantin G et al. Loss‐of‐function mutations in RSPH1 cause primary ciliary dyskinesia with central‐complex and radial‐spoke defects. Am J Hum Genet, 2013, 93, 561–570376992410.1016/j.ajhg.2013.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Moore A, Escudier E, Roger G, Tamalet A, Pelosse B, Marlin S et al. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J Med Genet, 2006, 43, 326–333256322510.1136/jmg.2005.034868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zariwala MA, Gee HY, Kurkowiak M, Al‐Mutairi DA, Leigh MW, Hurd TW et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am J Hum Genet, 2013, 93, 336–345373882710.1016/j.ajhg.2013.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Knowles MR, Ostrowski LE, Loges NT, Hurd T, Leigh MW, Huang L et al. Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms. Am J Hum Genet, 2013, 93, 711–720379125210.1016/j.ajhg.2013.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yamamoto R, Hirono M, Kamiya R. Discrete PIH proteins function in the cytoplasmic preassembly of different subsets of axonemal dyneins. J Cell Biol, 2010, 190, 65–71291166810.1083/jcb.201002081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yamamoto R, Song K, Yanagisawa HA, Fox L, Yagi T, Wirschell M et al. The MIA complex is a conserved and novel dynein regulator essential for normal ciliary motility. J Cell Biol, 2013, 201, 263–278362851510.1083/jcb.201211048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Olbrich H, Schmidts M, Werner C, Onoufriadis A, Loges NT, Raidt J et al. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left–right body asymmetry. Am J Hum Genet, 2012, 91, 672–684348465210.1016/j.ajhg.2012.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Castleman VH, Romio L, Chodhari R, Hirst RA, Castro SC, Parker KA et al. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central‐microtubular‐pair abnormalities. Am J Hum Genet, 2009, 84, 197–209266803110.1016/j.ajhg.2009.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Papon JF, Coste A, Roudot‐Thoraval F, Boucherat M, Roger G, Tamalet A et al. A 20‐year experience of electron microscopy in the diagnosis of primary ciliary dyskinesia. Eur Respir J, 2010, 35, 1057–1063 10.1183/09031936.00046209 [DOI] [PubMed] [Google Scholar]
- 140. Shoemark A, Dixon M, Corrin B, Dewar A. Twenty‐year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia. J Clin Pathol, 2012, 65, 267–271 10.1136/jclinpath‐2011‐200415 [DOI] [PubMed] [Google Scholar]
- 141. Wolf JP, Feneux D, Escalier D, Rodrigues D, Frydman R, Jouannet P. Pregnancy after subzonal insemination with spermatozoa lacking outer dynein arms. J Reprod Fertil, 1993, 97, 487–492 10.1530/jrf.0.0970487 [DOI] [PubMed] [Google Scholar]
- 142. Rompolas P, Patel‐King RS, King SM. An outer arm dynein conformational switch is required for metachronal synchrony of motile cilia in planaria. Mol Biol Cell, 2010, 21, 3669–3679296568410.1091/mbc.E10‐04‐0373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Neesen J, Kirschner R, Ochs M, Schmiedl A, Habermann B, Mueller C et al. Disruption of an inner arm dynein heavy chain gene results in asthenozoospermia and reduced ciliary beat frequency. Hum Mol Genet, 2001, 10, 1117–1128 10.1093/hmg/10.11.1117 [DOI] [PubMed] [Google Scholar]
- 144. Shoemark A, Ives A, Becker‐Heck A, Burgoyne T, Dixon M, Bilton D et al. Inner dynein arm defects in primary ciliary dyskinesia. J Genet Syndr Gene Ther, 2013, 4, 7 10.4172/2157‐7412.1000163 [Google Scholar]
- 145. Kobayashi Y, Watanabe M, Okada Y, Sawa H, Takai H, Nakanishi M et al. Hydrocephalus, situs inversus, chronic sinusitis, and male infertility in DNA polymerase lambda‐deficient mice: possible implication for the pathogenesis of immotile cilia syndrome. Mol Cell Biol, 2002, 22, 2769–277613374010.1128/MCB.22.8.2769‐2776.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zariwala M, O'Neal WK, Noone PG, Leigh MW, Knowles MR, Ostrowski LE. Investigation of the possible role of a novel gene, DPCD, in primary ciliary dyskinesia. Am J Respir Cell Mol Biol, 2004, 30, 428–434 10.1165/rcmb.2003‐0338RC [DOI] [PubMed] [Google Scholar]
- 147. Fliegauf M, Olbrich H, Horvath J, Wildhaber JH, Xariwala MA, Kennedy M et al. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am J Respir Crit Care Med, 2005, 171, 1343–1349271847810.1164/rccm.200411‐1583OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Supp DM, Witte DP, Potter SS, Brueckner M. Mutation of an axonemal dynein affects left–right asymmetry in inversus viscerum mice. Nature, 1997, 389, 963–966180058810.1038/40140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Bartoloni L, Blouin JL, Pan Y, Gehrig C, Maiti AK, Scamuffa N et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci USA, 2002, 99, 10282–1028612490510.1073/pnas.152337699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Schwabe GC, Hoffmann K, Loges NT, Birker D, Rossier C, Santi MM et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat, 2008, 29, 289–298 10.1002/humu.20656 [DOI] [PubMed] [Google Scholar]
- 151. Inaba K Hirose K, Amos LA. Regulatory subunits of axonemal dynein. Handbook of dynein, 2011. Singapore: Pan Stanford; 304–324 [Google Scholar]
- 152. Ushimaru Y, Konno A, Kaizu M, Ogawa K, Satoh N, Inaba K. Association of a 66 kDa homolog of Chlamydomonas DC2, a subunit of the outer arm docking complex, with outer arm dynein of sperm flagella in the ascidian Ciona intestinalis . Zoolog Sci, 2006, 23, 679–687 10.2108/zsj.23.679 [DOI] [PubMed] [Google Scholar]
- 153. Onoufriadis A, Paff T, Antony D, Shoemark A, Micha D, Kuyt B et al. Splice‐site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am J Hum Genet, 2013, 92, 88–98354245510.1016/j.ajhg.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Satouh Y, Inaba K. Proteomic characterization of sperm radial spokes identifies a novel spoke protein with an ubiquitin domain. FEBS Lett, 2009, 583, 2201–2207 10.1016/j.febslet.2009.06.016 [DOI] [PubMed] [Google Scholar]
- 155. Wirschell M, Pazour G, Yoda A, Hirono M, Kamiya R, Witman GB. Oda5p, a novel axonemal protein required for assembly of the outer dynein arm and an associated adenylate kinase. Mol Biol Cell, 2004, 15, 2729–274142009710.1091/mbc.E03‐11‐0820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Dean AB, Mitchell DR. Chlamydomonas ODA10 is a conserved axonemal protein that plays a unique role in outer dynein arm assembly. Mol Biol Cell, 2013, 24, 3689–3696384299510.1091/mbc.E13‐06‐0310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Desai PB, Freshour JR, Mitchell DR. Chlamydomonas axonemal dynein assembly locus ODA8 encodes a conserved flagellar protein needed for cytoplasmic maturation of outer dynein arm complexes. Cytoskeleton, 2015, 72, 16–28436136710.1002/cm.21206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ahmed NT, Mitchell DR. ODA16p, a Chlamydomonas flagellar protein needed for dynein assembly. Mol Biol Cell, 2005, 16, 5004–5012123709910.1091/mbc.E05‐07‐0627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ahmed NT, Gao C, Lucker BF, Cole DG, Mitchell DR. ODA16 aids axonemal outer row dynein assembly through an interaction with the intraflagellar transport machinery. J Cell Biol, 2008, 183, 313–322256802610.1083/jcb.200802025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Hjeij R, Lindstrand A, Francis R, Zariwala MA, Liu X, Li Y et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am J Hum Genet, 2013, 93, 357–367373882810.1016/j.ajhg.2013.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Panizzi JR, Becker‐Heck A, Castleman VH, Al‐Mutairi DA, Liu Y, Loges NT et al. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat Genet, 2012, 44, 714–719337165210.1038/ng.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. King SM, Patel‐King RS. The oligomeric outer dynein arm assembly factor CCDC103 is tightly integrated within the ciliary axoneme and exhibits periodic binding to microtubules. J Biol Chem, 2015, 290, 7388–7401436724910.1074/jbc.M114.616425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Austin‐Tse C, Halbritter J, Zariwala MA, Gilberti RM, Gee HY, Hellman N et al. Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am J Hum Genet, 2013, 93, 672–686379126410.1016/j.ajhg.2013.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Sale WS. The axonemal axis and Ca2+‐induced asymmetry of active microtubule sliding in sea urchin sperm tails. J Cell Biol, 1986, 102, 2042–2052 10.1083/jcb.102.6.2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Nakano I, Kobayashi T, Yoshimura M, Shingyoji C. Central‐pair‐linked regulation of microtubule sliding by calcium in flagellar axonemes. J Cell Sci, 2003, 116, 1627–1636 10.1242/jcs.00336 [DOI] [PubMed] [Google Scholar]
- 166. Piperno G, Mead K, Shestak W. The inner dynein arms I2 interact with a “dynein regulatory complex” in Chlamydomonas flagella. J Cell Biol, 1992, 118, 1455–1463 10.1083/jcb.118.6.1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Merveille AC, Davis EE, Becker‐Heck A, Legendre M, Amirav I, Bataille G et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat Genet, 2011, 43, 72–78350978610.1038/ng.726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Becker‐Heck A, Zohn IE, Okabe N, Pollock A, Lenhart KB, Sullivan‐Brown J et al. The coiled‐coil domain containing protein CCDC40 is essential for motile cilia function and left–right axis formation. Nat Genet, 2011, 43, 79–84313218310.1038/ng.727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Wirschell M, Olbrich H, Werner C, Tritschler D, Bower R, Sale WS et al. The nexin‐dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat Genet, 2013, 45, 262–268 10.1038/ng.2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M et al. A novel X‐linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral‐facial‐digital type I syndrome. Hum Genet, 2006, 120, 171–178 10.1007/s00439‐006‐0210‐5 [DOI] [PubMed] [Google Scholar]
- 171. Bennett WI, Gall AM, Southard JL, Sidman RL. Abnormal spermiogenesis in quaking, a myelin‐deficient mutant mouse. Biol Reprod, 1971, 5, 30–58 [DOI] [PubMed] [Google Scholar]
- 172. Lorenzetti D, Bishop CE, Justice MJ. Deletion of the Parkin coregulated gene causes male sterility in the quakingviable mouse mutant. Proc Natl Acad Sci USA, 2004, 101, 8402–840742040610.1073/pnas.0401832101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Dawe HR, Farr H, Portman N, Shaw MK, Gull K. The Parkin co‐regulated gene product, PACRG, is an evolutionarily conserved axonemal protein that functions in outer‐doublet microtubule morphogenesis. J Cell Sci, 2005, 118, 5421–5430 10.1242/jcs.02659 [DOI] [PubMed] [Google Scholar]
- 174. Ikeda K, Ikeda T, Morikawa K, Kamiya R. Axonemal localization of Chlamydomonas PACRG, a homologue of the human Parkin‐coregulated gene product. Cell Motil Cytoskeleton, 2007, 64, 814–821 10.1002/cm.20225 [DOI] [PubMed] [Google Scholar]
- 175. Zhang Z, Kostetskii I, Tang W, Haig‐Ladewig L, Sapiro R, Wei Z et al. Deficiency of SPAG16L causes male infertility associated with impaired sperm motility. Biol Reprod, 2006, 74, 751–759 10.1095/biolreprod.105.049254 [DOI] [PubMed] [Google Scholar]
- 176. Zhang Z, Shen X, Gude DR, Wilkinson BM, Justice MJ, Flickinger CJ et al. MEIG1 is essential for spermiogenesis in mice. Proc Natl Acad Sci USA, 2009, 106, 17055–17060274612410.1073/pnas.0906414106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Salzberg Y, Eldar T, Karminsky OD, Itach SB, Pietrokovski S, Don J. Meig1 deficiency causes a severe defect in mouse spermatogenesis. Dev Biol, 2010, 338, 158–167 10.1016/j.ydbio.2009.11.028 [DOI] [PubMed] [Google Scholar]
- 178. Teves ME, Jha KN, Song J, Nagarkatti‐Gude DR, Herr JC, Foster JA et al. Germ cell‐specific disruption of the Meig1 gene causes impaired spermiogenesis in mice. Andrology, 2013, 1, 37–46412026010.1111/j.2047‐2927.2012.00001.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Li W, Tang W, Teves ME, Zhang Z, Zhang L, Li H et al. A MEIG1/PACRG complex in the manchette is essential for building the sperm flagella. Development, 2015, 142, 921–930435297810.1242/dev.119834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Kierszenbaum AL. Spermatid manchette: plugging proteins to zero into the sperm tail. Mol Reprod Dev, 2001, 59, 347–349 10.1002/mrd.1040 [DOI] [PubMed] [Google Scholar]
- 181. Pan Q, Zhu YJ, Gu BW, Cai X, Bai XT, Yun HY et al. A new fusion gene NUP98‐IQCG identified in an acute T‐lymphoid/myeloid leukemia with a t(3;11)(q29q13;p15)del(3)(q29) translocation. Oncogene, 2008, 27, 3414–3423 10.1038/sj.onc.1210999 [DOI] [PubMed] [Google Scholar]
- 182. Li RK, Tan JL, Chen LT, Feng JS, Liang WX, Guo XJ et al. Iqcg is essential for sperm flagellum formation in mice. PLoS One, 2014, 9, e98053402979110.1371/journal.pone.0098053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Lee L, Campagna DR, Pinkus JL, Mulhern H, Wyatt TA, Sisson JH et al. Primary ciliary dyskinesia in mice lacking the novel ciliary protein Pcdp1. Mol Cell Biol, 2008, 28, 949–957222340510.1128/MCB.00354‐07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. DiPetrillo CG, Smith EF. Pcdp1 is a central apparatus protein that binds Ca2+‐calmodulin and regulates ciliary motility. J Cell Biol, 2010, 189, 601–612286729510.1083/jcb.200912009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Sironen A, Hansen J, Thomsen B, Andersson M, Vilkki J, Toppari J, Kotaja N. Expression of SPEF2 during mouse spermatogenesis and identification of IFT20 as an interacting protein. Biol Reprod, 2010, 82, 580–590 10.1095/biolreprod.108.074971 [DOI] [PubMed] [Google Scholar]
- 186. Zhang H, Mitchell DR. Cpc1, a Chlamydomonas central pair protein with an adenylate kinase domain. J Cell Sci, 2004, 117, 4179–4188152502110.1242/jcs.01297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Dong F, Shinohara K, Botilde Y, Nabeshima R, Asai Y, Fukumoto A et al. Pih1d3 is required for cytoplasmic preassembly of axonemal dynein in mouse sperm. J Cell Biol, 2014, 204, 203–213389717710.1083/jcb.201304076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Tokuyasu KT. Dynamics of spermiogenesis in Drosophila melanogaster: 3. Relation between axoneme and mitochondrial derivatives. Exp Cell Res, 1974, 84, 239–250 10.1016/0014‐4827(74)90402‐9 [DOI] [PubMed] [Google Scholar]
- 189. Otani H, Tanaka O, Kasai K, Yoshioka T. Development of mitochondrial helical sheath in the middle piece of the mouse spermatid tail: regular dispositions and synchronized changes. Anat Rec, 1988, 222, 26–33 10.1002/ar.1092220106 [DOI] [PubMed] [Google Scholar]
- 190. Ho HC, Wey S. Three dimensional rendering of the mitochondrial sheath morphogenesis during mouse spermiogenesis. Microsc Res Tech, 2007, 70, 719–723 10.1002/jemt.20457 [DOI] [PubMed] [Google Scholar]
- 191. Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J et al. Mitofusin‐2 determines mitochondrial network architecture and mitochondrial metabolism. J Biol Chem, 2003, 278, 17190–17197 10.1074/jbc.M212754200 [DOI] [PubMed] [Google Scholar]
- 192. Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell, 1997, 90, 121–129 10.1016/S0092‐8674(00)80319‐0 [DOI] [PubMed] [Google Scholar]
- 193. Vadnais ML, Lin AM, Gerton GL. Mitochondrial fusion protein MFN2 interacts with the mitostatin‐related protein MNS1 required for mouse sperm flagellar structure and function. Cilia, 2014, 3, 5403805910.1186/2046‐2530‐3‐5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zhou J, Yang F, Leu NA, Wang PJ. MNS1 is essential for spermiogenesis and motile ciliary functions in mice. PLoS Genet, 2012, 8, e1002516329153410.1371/journal.pgen.1002516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Sampson MJ, Ross L, Decker WK, Craigen WJ. A novel isoform of the mitochondrial outer membrane protein VDAC3 via alternative splicing of a 3‐base exon. Functional characteristics and subcellular localization. J Biol Chem, 1998, 273, 30482–30486 10.1074/jbc.273.46.30482 [DOI] [PubMed] [Google Scholar]
- 196. Pedersen H, Mygind N. Absence of axonemal arms in nasal mucosa cilia in Kartagener's syndrome. Nature, 1976, 262, 494–495 10.1038/262494a0 [DOI] [PubMed] [Google Scholar]
- 197. Bleau G, Richer C‐L, Bousquet D. Absence of dynein arms in cilia of endocervical cells in a fertile woman. Fertil Steril, 1978, 30, 362–363 [DOI] [PubMed] [Google Scholar]
- 198. Jean Y, Langlais J, Roberts KD, Chapdelaine A, Bleau G. Fertility of a woman with nonfunctional ciliated cells in the Fallopian tubes. Fertil Steril, 1979, 31, 349–350 [DOI] [PubMed] [Google Scholar]
- 199. Escudier E, Escalier D, Homasson J‐P, Pinchon MC, Bernaudin JF. Unexpectedly normal cilia and normal spermatozoa in an infertile man with Kartagener's syndrome. Eur J Respir Dis, 1987, 70, 180–183 [PubMed] [Google Scholar]
- 200. Olbrich H, Haffner K, Kispert A, Volkel A, Volz A, Sasmaz G et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left–right asymmetry. Nat Genet, 2002, 30, 143–144 10.1038/ng817 [DOI] [PubMed] [Google Scholar]
- 201. Supp DM, Witte DP, Potter SS, Brueckner M. Mutation of an axonemal dynein affects left–right asymmetry in inversus viscerum mice. Nature, 1997, 389, 963–966180058810.1038/40140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Zhang YJ, O'Neal WK, Randell SH, Blackburn K, Moyer MB, Boucher RC et al. Identification of dynein heavy chain 7 as an inner arm component of human cilia that is synthesized but not assembled in a case of primary ciliary dyskinesia. J Biol Chem, 2002, 277, 17906–17915 10.1074/jbc.M200348200 [DOI] [PubMed] [Google Scholar]
- 203.Pennarun G, Escudier E, Chapelin C, Bridoux AM, Cacheux V, Roger G, et al. Loss‐of‐function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am J Hum Genet. 1999; 65:1508–19. [DOI] [PMC free article] [PubMed]
- 204. Loges NT, Olbrich H, Fenske L, Mussaffi H, Horvath J, Fliegauf M et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet, 2008, 83, 547–558266802810.1016/j.ajhg.2008.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Mazor M, Alkrinawi S, Chalifa‐Caspi V, Manor E, Sheffield VC, Aviram M et al. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am J Hum Genet, 2011, 88, 599–607314673110.1016/j.ajhg.2011.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Duriez B, Duquesnoy P, Escudier E, Bridoux AM, Escalier D, Rayet I et al. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc Natl Acad Sci USA, 2007, 104, 3336–3341180556010.1073/pnas.0611405104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Rashid S, Grzmil P, Drenckhahn JD, Meinhardt A, Adham I, Engel W et al. Disruption of the murine dynein light chain gene Tcte3–3 results in asthenozoospermia. Reproduction, 2010, 139, 99–111 10.1530/REP‐09‐0243 [DOI] [PubMed] [Google Scholar]
- 208. Hjeij R, Onoufriadis A, Watson CM, Slagle CE, Klena NT, Dougherty GW et al. CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am J Hum Genet, 2014, 95, 257–274415714610.1016/j.ajhg.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Lechtreck KF, Witman GB. Chlamydomonas reinhardtii hydin is a central pair protein required for flagellar motility. J Cell Biol, 2007, 176, 473–482206398210.1083/jcb.200611115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Sapiro R, Kostetskii I, Olds‐Clarke P, Gerton GL, Radice GL, Strauss IJ. Male infertility, impaired sperm motility, and hydrocephalus in mice deficient in sperm‐associated antigen 6. Mol Cell Biol, 2002, 22, 6298–630513401010.1128/MCB.22.17.6298‐6305.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Teves ME, Zhang Z, Costanzo RM, Henderson SC, Corwin FD, Zweit J et al. Sperm‐associated antigen‐17 gene is essential for motile cilia function and neonatal survival. Am J Respir Cell Mol Biol, 2013, 48, 765–772372787710.1165/rcmb.2012‐0362OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Fernandez‐Gonzalez A, Kourembanas S, Wyatt TA, Mitsialis SA. Mutation of murine adenylate kinase 7 underlies a primary ciliary dyskinesia phenotype. Am J Respir Cell Mol Biol, 2009, 40, 305–313264552810.1165/rcmb.2008‐0102OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Takaki E, Fujimoto M, Nakahari T, Yonemura S, Miyata Y, Hayashida N et al. Heat shock transcription factor 1 is required for maintenance of ciliary beating in mice. J Biol Chem, 2007, 282, 37285–37292 10.1074/jbc.M704562200 [DOI] [PubMed] [Google Scholar]
- 214. Tanaka H, Iguchi N, Toyama Y, Kitamura K, Takahashi T, Kaseda K et al. Mice deficient in the axonemal protein Tektin‐t exhibit male infertility and immotile‐cilium syndrome due to impaired inner arm dynein function. Mol Cell Biol, 2004, 24, 7958–796451505410.1128/MCB.24.18.7958‐7964.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Horani A, Ferkol TW, Shoseyov D, Wasserman MG, Oren YS, Kerem B et al. LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects. PLoS One, 2013, 8, e59436360230210.1371/journal.pone.0059436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Moore DJ, Onoufriadis A, Shoemark A, Simpson MA, zur Lage PI, de Castro SC et al. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am J Hum Genet, 2013, 93, 346–356373883510.1016/j.ajhg.2013.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Yanagisawa H, Kamiya R. A Tektin homologue is decreased in Chlamydomonas mutants lacking an axonemal inner‐arm dynein. Mol Biol Cell., 2004, 15, 2105–211540400810.1091/mbc.E03‐11‐0854 [DOI] [PMC free article] [PubMed] [Google Scholar]