

Abstract
17β‐Estradiol (E2), as the main circulating estrogen hormone, regulates many tissue and organ functions in physiology. The effects of E2 on cells are mediated by the transcription factors and estrogen receptor (ER)α and ERβ that are encoded by distinct genes. Localized at the peri‐membrane, mitochondria, and the nucleus of cells that are dependent on estrogen target tissues, the ERs share similar, as well as distinct, regulatory potentials. Different intracellular localizations of the ERs result in dynamically integrated and finely tuned E2 signaling cascades that orchestrate cellular growth, differentiation, and death. The deregulation of E2–ER signaling plays a critical role in the initiation and progression of target tissue malignancies. A better understanding of the complex regulatory mechanisms that underlie ER actions in response to E2 therefore holds a critical trajectory for the development of novel prognostic and therapeutic approaches with substantial impacts on the systemic management of target tissue diseases.
Keywords: estrogen, estrogen receptor, molecular mechanism, signaling, structure
1. Introduction
Nuclear hormone receptors (NHRs) are members of a large nuclear receptor family that acts as transcription factors. These are distributed throughout the body and play diverse roles in cellular processes.1, 2 Nuclear hormone receptors include the androgen receptor, glucocorticoid receptor (GR), progesterone receptor, mineralcorticoid receptor, estrogen receptor (ER)α, and ERβ.1, 2 The activity of NHRs is modulated by steroid hormones that are derived from cholesterol. Due to their hydrophobic nature, steroid hormones diffuse across the plasma membrane, enabling systemic extracellular signals to regulate tissue‐specific intracellular events.1, 2
Estrogens are one class of steroid hormones that includes estrone, estradiol (E2), and estriol.3, 4 17β‐Estradiol, the most potent estrogen hormone in the circulation, is involved in a wide variety of vital physiological functions that range from the development and maintenance of reproductive organs to the regulation of cardiovascular, musculoskeletal, immune, and central nervous system homeostasis.3, 4 Estradiol also contributes to the initiation and development of target tissue malignancies.3, 4
The effects of E2 are mediated by ERα (NR3A1) and ERβ (NR3A2). The dissection of the ER‐mediated E2 signaling in estrogen target tissues largely stems from knock‐out (KO) animal models.5, 6, 7 Species‐specific differences in tissue distribution withstanding, it appears that ERα predominates, whereas ERβ plays a minor role, in the uterus, mammary glands, pituitary gland, skeletal muscle, adipose tissue, and bone. Estrogen receptor β, in contrast, is found to be critical in mediating E2 signaling in the ovary, prostate, lung, cardiovascular and central nervous systems. Even within a single tissue, the expression pattern of each subtype is cell type‐specific. In the ovary, for example, ERβ is expressed in the granulosa cells but ERα is more abundant in the theca cells.5, 6, 7 Reflecting the different ER‐subtype distribution patterns, ERα‐KO and ERβ‐KO mice show different phenotypes. The ERα‐KO female mice are, for example, infertile with a hypotrophic uterus, as well as with anovulatory and hemorrhagic ovaries.5, 6, 7 In contrast, the ERβ‐KO female mice are subfertile and display reduced ovulation, probably as a result of a retardation in granulosa cell differentiation.5, 6, 7
Although significant progress has been made towards understanding the mechanism of ERβ signaling since its discovery in 1996,8, 9 many aspects of ERβ's actions and its role in the physiology and pathophysiology of E2 signaling remain unknown.5, 10, 11 This is due to, as indicated by one study,10 at least in part, because of the lack of established experimental cell models that synthesize ERβ endogenously and of receptor‐specific antibodies. Nevertheless, accumulating evidence from in vitro, in cellula, and in vivo systems has broadened the understanding of both ER subtype actions in E2 signaling. This communication aims to summarize a current state of understanding of E2–ER signaling by pointing out the similarities, as well as the differences, between the receptor subtypes.
2. Estrogen Receptor Structure
Estrogen receptors, as other members of the NHR family, are modular proteins in that distinct structural region of the receptors that display unique functional features.12, 13 Both ERα and ERβ are encoded by two distinct genes and are expressed in the same and different tissues at varying levels. The human ERα gene (ESR1) is a large genomic segment that spans ~300 kb and is located at q24‐q27 of chromosome 6.14, 15, 16 ESR1 includes eight exons that encode the full‐length 66 kDa protein that is composed of 595 amino‐acids.14, 15, 16 Similarly, the ERβ gene (ESR2), mapping to q22‐24 of chromosome 14, is a large genome segment and spans 254 kb with eight encoding exons.17 It consists of 530 amino acids, with a molecular mass (MM) of 60 kDa.18
Estrogen receptors share structural characteristics that are responsible for similar functional features. Distinct amino‐acid compositions at various structural regions also render the receptors with subtype‐specific properties in conveying E2 signaling. Estrogen receptors, as other members of the NHRs, are subdivided into six functionally distinct domains.10, 11, 12, 13 The structurally distinct amino‐terminal A/B domains share a 17% amino‐acid identity between the ERs. The near‐identical central C region (97%) is the DNA‐binding domain (DBD). The flexible hinge, or D, domain (36%) contains a nuclear localization signal (NLS) and links the C domain to the multifunctional carboxyl‐terminal (E) domain. Also called the “ligand‐binding domain” (LBD), E shows 56% amino‐acid homology between the ERs. The LBD is a globular region that harbors a hormone‐binding site, a dimerization interface (homo‐ and hetero‐dimerization), and a ligand‐dependent co‐regulator interaction function (activation function, AF‐2). Sharing an 18% amino‐acid identity, the F domain is located at the extreme carboxyl‐terminus of the receptors (Fig. 1).
Figure 1.
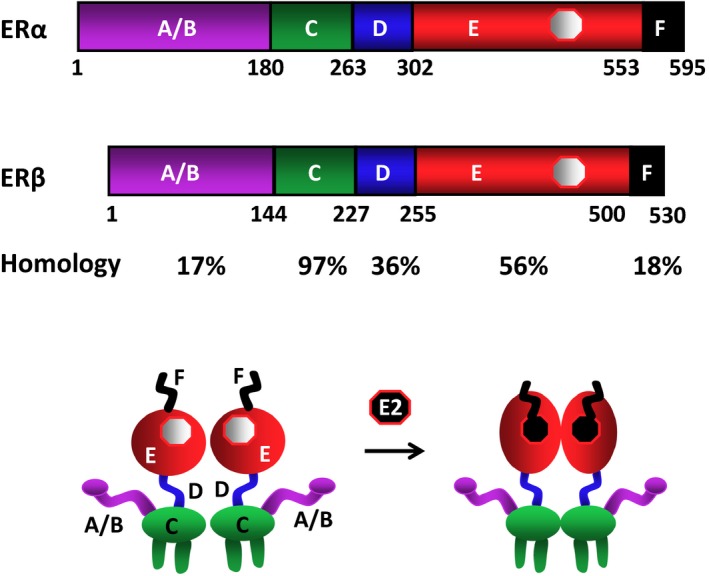
Schematics of the estrogen receptor (ER)α and ERβ structural regions. Estrogen receptor α is composed of 595 amino acids, while ERβ contains 530 amino acids. The structurally distinct amino terminal A/B domains share a 17% amino‐acid identity between the ERs. The near‐identical central C region (97%) is the DNA‐binding domain. The flexible hinge, or D, domain (36%) contains a nuclear localization signal and links the C domain to the multifunctional carboxyl terminal (E) domain, which shows 56% amino‐acid homology between the ERs. The carboxyl‐terminal F domain shares an 18% amino‐acid identity. The ERs are dimers with or without the endogenous ligand, 17β‐estradiol, the binding of which induces conformational changes in the receptors. The figure is modified from Muyan, et al168
In both the ERα and ERβ genes, exon 1 encodes the A/B region. The C region, the DBD, is encoded by exons 2 and 3, with an intron located between the two fingers. Exon 4 encodes a part of the C region, all of the D region, and part of the E region. The hormone‐binding domain (E/F) is encoded by exons 4–8 (also see Fig. 2).14, 17
Figure 2.
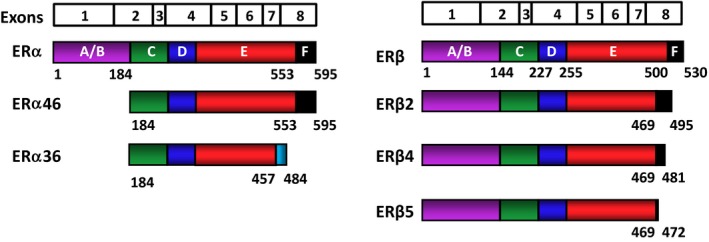
Schematic representation of the estrogen receptor (ER) isoforms. The ERs are encoded by eight exons. The exon boundaries (lines) correspond to the regions of the ERs that are depicted with colored and labeled (A‐F) structural domains. Estrogen receptor α is 595 amino acids long, whereas ERβ is composed of 530 amino acids. Estrogen receptor α46, which is generated by an alternative splicing event, lacks the amino‐terminal A/B region and acts as a competitive inhibitor of ERα. Estrogen receptor α36 is generated from a promoter in the first intron of the ERα gene, together with alternative splicing events that result in a truncated protein with a unique 27 amino‐acid carboxyl‐terminus (light blue) that replaces the last 138 amino acids that are encoded by exons 7 and 8 of wild‐type (WT)‐ERα. Estrogen receptor α36 lacks both activation function (AF)1 and AF‐2. Palmitoylated ERα36 localizes to the plasma membrane and cytoplasm, plays a role in the membrane‐initiated 17β‐estradiol (E2) signaling and adversely affects WT‐ERα‐mediated events. The ERβ isoforms are formed from alternative splicing of exon 8, resulting in carboxyl‐terminally truncated ERβ2, ERβ4, and ERβ5 variants with varying molecular masses. These variants cannot bind ligand and lack AF‐2, but they could adversely affect E2 signaling by heterodimerizing with WT‐ERα or WT‐ERβ when co‐synthesized
The binding of E2 is the pivotal step in the cellular action of the ERs that are present as dimers at the peri‐membrane, mitochondria, and nucleus.19, 20, 21, 22, 23 Estradiol binding induces a major structural re‐organization of the LBD that converts the inactive ER to the functionally active form by generating surfaces for enhanced stability of the ER dimer24 and of the interacting co‐regulatory proteins.25
Due to the central importance of ERs in the physiology and pathophysiology of estrogen target tissues, a short review of the structural features of the receptors could provide the critical prelude for a better understanding of E2 signal transmission to cells that results in dramatic alterations in phenotypic features. The practical consequences of this understanding would be the development of new research modalities that uncover the mechanisms of E2–ER actions in order to design function‐specific steroidal drugs for therapeutic use.
2.1. Structure of the estrogen receptor–ligand‐binding domains
The LBDs of NHRs display a three‐layered antiparallel α‐helical fold.26, 27 This fold is universal within the receptor superfamily and is formed with 10‐12 helices, depending on the receptor species, with the same numbering scheme used for all NHRs.26, 27 The ERα‐LBD has 12 helices (Fig. 3). The antiparallel α‐helical fold, comprising a central core layer of three helices (H5/6, H9, and H10), is sandwiched between two additional layers of helices (H1‐4 and H7, H8, and H11). This helical arrangement creates a scaffold that maintains a ligand‐binding cavity. The remaining secondary structural elements, a small two‐stranded antiparallel β‐sheet and the dynamically mobile H12,28, 29 flank the main three‐layered motif.26, 27 The overall structure of the LBD of ERβ shows a close similarity to that of ERα.30 Both ERs also contain a relatively unstructured carboxyl‐terminal extension, or F domain. The secondary structure of the ERα‐F domain appears to contain an α‐helical region and an extended β‐strand separated by regions of random coil, with a short extended region near the extreme carboxyl‐terminus.31 On the other hand, the F domain of ERβ exhibits a random coil, with only a very short extended region near the extreme carboxyl‐terminus of the protein.31 Although the role of ERβ‐F is unclear, the F domain of ERα appears to modulate the transcriptional activity, co‐activator interactions, dimerization, and stability of the receptor.32, 33, 34
Figure 3.
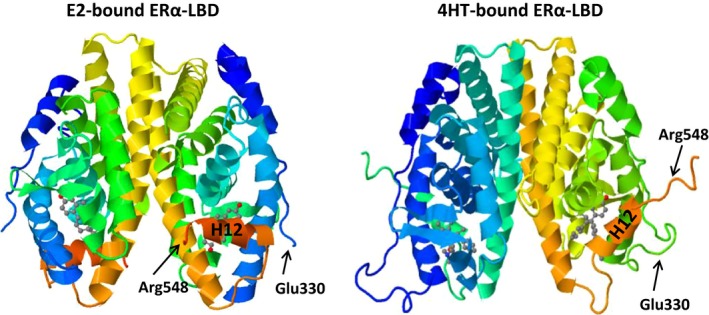
Tertiary structure of the estrogen receptor (ER)α‐ligand‐binding domain (LBD) dimer that is bound to 17β‐estradiol (E2) or 4‐hydroxytamoxifen (4HT). The binding of E2 induces a conformational change in the LBD that positions the dynamically mobile H12 over the ligand‐binding cavity. This positioning generates a surface for the interactions with co‐activators to establish a competent activation function 2 (Protein Data Bank [PDB] identification [ID]: 1ERE; Brzozowski, et al.26). The binding of 4‐HT, a selective estrogen receptor modulator, prevents H12 from docking in agonist conformation, effectively preventing co‐activator binding and transcription activation (PDB ID: 3ERT; Shiau, et al.38). H12 and the residues at the amino and carboxyl‐termini of the tertiary structures are indicated for comparison
Dimer formation is essential for ERα function, as mutations that interfere with dimerization render the receptor transcriptionally inactive.35 Although the DBD of each monomer also contributes to the dimerization of ERα, the predominant dimerization interface is formed by the H11 helices of each ERα‐LBD monomer.26, 27, 36 The LBDs interact via a stretch of conserved hydrophobic residue at their amino‐terminal ends, with additional dimer interactions provided by the residues of H8 and the loop between H9 and H10.26, 27 Ligand recognition is achieved through hydrogen bonds and the complementarity of the hydrophobic residues that line the cavity to the non‐polar nature of the ER ligands.26, 27, 37 It appears that E2 binding positions the dynamically mobile H12 over the cavity perpendicular to the dimerization interface and is packed against H3, H5/6, and H11, forming a lid on the binding cavity (Fig. 3).26, 27, 28, 29 This positioning of H12 is a prerequisite for transcriptional activation as it generates a competent ligand‐dependent activation function (AF)‐2 that is capable of interacting with the co‐activators.26, 27, 37 In this conformation, the E2‐bound LBD can accept a short helical segment, the LXXLL motif (where “L” is leucine and “X” is any residue) from a variety of co‐activator proteins, which is exemplified by the members of the p160 steroid receptor co‐activator (SRC) family, including SRC1‐3.25, 38, 39
Estrogen receptors also bind to various molecules with agonist, mixed agonist–antagonist, or full antagonist properties.40, 41 Mixed agonist–antagonists, also called “selective estrogen receptor modulators” (SERMs), display distinct pharmacological effects, depending on the estrogen target tissue. Tamoxifen, for example, has been used widely for clinical treatment of breast cancers as an antagonist, yet it acts as an agonist in most estrogen target tissues. Raloxifene, in contrast, has protective effects on bone and displays antiproliferative effects on breast cancer cells. Pure antagonists of estrogenic compounds, exemplified by fulvestrant, also referred to as the “selective estrogen receptor down‐regulators” (SERDs), act as complete antagonists. Although most of the key amino acids in the ligand‐binding cavity that are responsible for binding SERMs or SERDs are identical, a large side chain emanating from the core of the ligand prevents the H12 of ERα from docking in agonist conformation (Fig. 3).26, 38 This conformational shift in H12 leads to the occupation of the LXXLL‐binding cleft, thereby preventing co‐activator binding and transcription activation.26, 38 Independent of intracellular locations, the AF‐2 of the ER–LBD is indispensable in receptor actions. An AF‐2 mutant knock‐in (KI) mouse model bearing point mutations or deletions in the AF‐2 region to disrupt the AF‐2‐mediated transactivation ability of ERα is shown to display female and male phenotypes that are indistinguishable from those of the ERα‐KO mouse model.42, 43
In other NHRs, antagonist binding locates H12 to a position outside the AF‐2 region, leading to an interaction with the corepressor/nuclear receptor (CoRNR) consensus motif (LIL; where “L” is leucine, “I” is isoleucine, and “X” is any residue) of the corepressor proteins.44 Unlike most NHRs, however, the importance of NR corepressors in ER signaling remains unclear. Nevertheless, studies have indicated that both agonist‐ and antagonist‐bound ERs are able to recruit a variety of proteins that can repress receptor activity.45 A search for a mechanism identified a previously unrecognized internal CoRNR motif within H12.46 This motif is able to compete with corepressors to bind to the AF‐2 surface, thereby reducing or preventing the ability of ERα to directly interact with the corepressors. This suggests that corepressor proteins might not require CoRNR motifs for recruitment to the antagonist‐bound ERα.46 Furthermore, dynamic modeling of tamoxifen‐occupied ERα suggests that, in the presence of tamoxifen, the ERα‐LBD assumes flexible conformations that fluctuate between agonist and antagonist confirmation.47 These fluctuating conformations could underlie the mixed agonist–antagonist property of the compound.47 In addition to blocking ER‐cofactor interactions,48 fulvestrant (as an effective SERD) prevents the binding of ERα to DNA by altering the stability, turnover, and intra‐nuclear location of the receptor.49, 50, 51, 52, 53
Although the structural features of the LBDs of ERα and ERβ largely overlap, the ligand‐binding pocket of ERβ differs from that of ERα, with only two amino‐acid positions. This, together with distinct residues outside of the LBD, generates differences in the size of the pockets that allow the binding of a ligand to receptors in a subtype‐specific manner, exemplified by ERβ‐specific agonist diarylpropionitrile.54 Studies using subtype‐selective agonists and antagonists have been critical in determining the biological actions that are specific to ERα or ERβ, extending the findings from ER‐KO animal models.
2.2. Structure of the estrogen receptor‐DNA‐binding domain
The nuclear ERs interact with chromatin target sites through two distinct modes: estrogen response element (ERE)‐dependent and ERE‐independent pathways. The EREs are permutations of the 5′‐GGTCAnnnTGACC‐3′ DNA palindrome, wherein ‘n’ denotes a non‐specific three‐nucleotide spacer, located at various distances from the transcription start site and/or within a gene locus.20, 55 The regulation of gene expression by the binding of E2–ER to the EREs is referred to as the “ER‐dependent signaling pathway.”19, 56, 57, 58, 59, 60 On the other hand, the transcriptional modulation of target genes through the interactions of E2–ERα with transcription factors, exemplified by stimulatory protein (SP) 1 and activator protein (AP) 1, bound to their cognate regulatory elements on DNA, denotes the ERE‐independent signaling pathway.57, 58, 59, 60 The underlying mechanism of the ERE‐independent signaling pathway is unclear. However, the ER has been suggested to establish direct or indirect, via co‐regulatory proteins, interactions with transcription factors through regions that also encompass the DBD, while the integrated effects of the amino‐ and carboxyl‐termini are responsible for the regulation of transcription.19, 56, 57, 58, 59, 60
The DBDs mediate the ability of ERs to bind to EREs. The centrally positioned DBDs, which are highly conserved among NHRs,28 share the same three‐dimensional structure (Fig. 4). The DBD of ERα contains two zinc‐binding motifs and each motif contains an α‐helix that is nucleated at its amino‐terminus through binding a zinc ion.61 Two helices are oriented perpendicularly to each other and cross at their mid‐points.61 The DBD makes phosphate contacts on both sides of the major groove.61 Each DBD of the ERα dimer makes analogous contacts with one of the inverted motifs, resulting in a rotationally symmetrical structure.61 Two monomers of the DBD bind to adjacent major grooves from one side of the DNA double helix. Distinct residues in a region of the first zinc‐finger module of DBD, the P‐box, particularly Glu203, Gly204, and Ala207, determine the DNA‐binding specificity that is critical for sequence discrimination12, 62, 63 and binding to the ERE.64 The residues in the second zinc finger‐like module, the D‐box, are involved in the discrimination of half‐site spacing through a protein–protein interaction between two ER monomers.12, 62, 63
Figure 4.
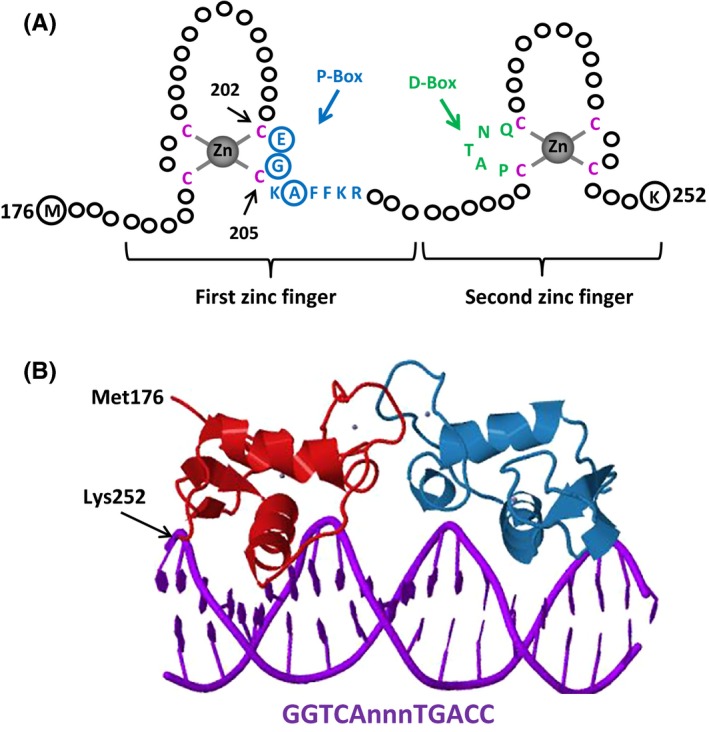
A, The DNA‐binding domain (DBD) of estrogen receptors (ERs). Schematized is the ERα–DBD. The DBD of ERα contains two zinc (Zn)‐binding motifs that are formed by a Zn ion (grey) that is coordinated by four cysteine residues (red). A region of the first Zn‐finger module, the P‐box, which contains amino acids (blue), particularly glutamic acid (E), glycine (G), and alanine (A) at positions 203, 204, and 207, respectively (circularized blue), determine the DNA‐binding specificity that is critical for sequence discrimination and binding to the estrogen response element. The residues (green) in the second Zn‐finger module, the D‐box, are involved in the discrimination of half‐site spacing. B, The tertiary structure of ERα–DBD (residues Met176–Lys252) as dimer‐bound to the consensus DNA sequence, GGTCAnnnTGACC (estrogen response element), with three non‐specific (n) intervening bases (Protein Data Bank identification: 1HCQ; Schwabe, et al61)
2.3. Structure of the estrogen receptor amino‐terminus
The highly divergent amino‐terminal domain of many members of the NHR family contains an AF‐1 region.65 Studies have indicated that the AF‐1 of ERα functions independently of the AF‐2‐containing carboxyl region in yeast and chicken cells in a ligand‐ and promoter‐dependent manner. However, AF‐1 is ineffective in altering transcription in mammalian cells when separated from the carboxyl‐terminus.66, 67, 68, 69 The function of the AF‐1 domain of ERα is therefore dependent on the structural integrity of the hormone‐binding domain, agonist nature of the ligand, and the cellular‐context.66, 67, 68, 69 Studies have further shown that the functional integration of both AF‐1 and AF‐2 is required for the full activity of the receptor.69, 70, 71, 72 These results have been confirmed by the findings derived from mouse KI models.42, 43
Despite the important functions of the amino‐terminus in ER activity, the biochemical and structural features of the underlying mechanism of AF‐1 action are incomplete. This is because the amino‐termini of NHRs, including ERα, are intrinsically disordered.65, 73, 74, 75 It has been proposed that this intrinsic disorder leads to the formation of a large collection of rapidly inter‐converting receptor conformations.65, 73, 74, 75 An intrinsic disorder allows the amino‐terminus to rapidly and reversibly adopt various configurations. These conformational changes are controlled by allosteric cooperativity between different domains and interactions with proteins and post‐translational modifications, particularly phosphorylation.76 For example, the TATA box‐binding protein was shown to interact with and induce a more ordered structure in the amino‐terminus of ERα.75 Similarly, the phosphorylation of serine 118 in the amino‐terminus of ERα that was bound to E2 or SERM (tamoxifen) by growth factor signaling resulted in the recruitment of the peptidyl prolyl cis/trans isomerase, Pin1, that isomerizes the serine 118–proline 119 bond from a cis to a trans isomer. This isomerization appears to lead to a local conformational change that promotes the ligand‐independent and agonist‐ or SERM‐inducible activity of ERα.76 These protein interaction‐mediated conformational changes are critical for stable interactions with other co‐regulatory proteins in order to establish an effective transcription.65, 73, 74
In contrast to ERα, the amino‐terminus of ERβ impairs the receptor ERE interactions,77 does not contain an AF‐1,19, 72, 78, 79, 80 and does not interact with the carboxyl‐terminus.72
3. 17β‐Estradiol–Estrogen Receptor Signaling
A plethora of factors, including the amount and type of ERs and/or complementary proteins that are required for receptor actions in a particular cell, is ultimately responsible for the manifestation of E2‐mediated cellular changes. However, the presence of ERs in various locations in cells implies that the exertion of E2 effects on cellular phenotypes involves dynamically integrated and finely tuned ER‐mediated signaling cascades. For example, the so‐called “extra‐nuclear” or “membrane‐initiated’’ E2 signaling not only mediates the second‐to‐minute (or rapid) transcription‐independent effects of ERs but also post‐translationally modulates the actions of nuclear ERs, transcription factors, co‐regulatory proteins, and chromatin complexes. It is therefore imperative that E2 signaling from intracellular locations is viewed as integrated, rather than discrete, alternative events (Fig. 5). Nevertheless, a dissected review of the relative contribution of these compartmentalized ER locations to E2 signaling is a necessary prelude in order to provide a current short story of the mechanism of ER actions at various levels, for which there exist excellent reviews.10, 40, 81, 82, 83, 84, 85, 86, 87, 88, 89
Figure 5.
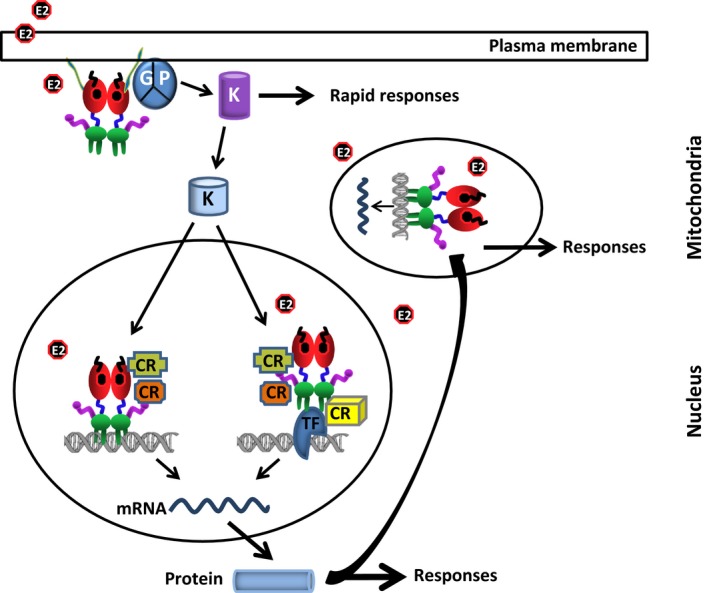
Integrated model of 17β‐estradiol (E2)–estrogen receptor (ER)‐mediated signaling. In the membrane‐initiated signaling, the E2‐bound and palmitoylated (green) ER interacts with a G protein (GP) that results in the activation of kinases, which in turn phosphorylate substrates, including membrane‐based ion channels and secondary messenger systems, leading to rapid cellular responses. The activated kinases also phosphorylate the protein components of the nuclear E2 signaling, including ERs, co‐regulatory proteins, transcription factors (TFs), and chromatin proteins, that result in alterations in responsive gene expression. In the mitochondria, E2‐ER alters the mitochondrial functions by mediating gene expression through a direct interaction with the mtDNA, as well as increasing manganese superoxide dismutase. The mitochondrial functions also are modulated by the nuclear E2‐ERs through the expression of genes, whose protein products are involved in mitochondrial functions. In the nuclear signaling, the ER mediates E2 action with two distinct modes: estrogen response element (ERE)‐dependent and ERE‐independent pathways. The ERE‐dependent signaling route involves the interactions of E2–ER with EREs on DNA and the subsequent regulation of gene expression. The ERE‐independent signaling pathway entails the modulation of responsive gene expression by a direct or indirect, through co‐regulatory proteins (CRs), interaction of E2–ER with transcription factors that are bound to their cognate responsive elements on DNA
3.1. Estrogen receptor‐mediated membrane signaling
The exposure of target tissue cells, including pituitary, uterus, ovary, vascular epithelium, bone, and breast, to E2 can rapidly induce ion fluxes and the activation of many protein kinases across the plasma membrane, independently of protein synthesis. These observations have led to the recognition of a membrane‐associated ER signaling pathway. The role of various ER isoforms and the G protein‐coupled estrogen receptor (or GPR30, which is a member of the G‐protein‐coupled receptor 1 family and localizes to the membrane endoplasmic reticulum) in rapid E2 signaling notwithstanding,90, 91, 92 membrane ERs have been established to be the same protein products of the genes that encode nuclear ERs.21, 82, 93 The palmitoylation of the Cys447 residue of ERα‐LBD and the Cys399 residue of ERβ‐LBD, with the aid of heat shock protein 27, appears to result in the interaction of ERs with the caveolin‐1 protein that serves as the transporter of ERs to caveolae rafts within the cell membrane.21, 94, 95, 96, 97, 98 The palmitoylated ERs are translocated to the membrane as monomers and the dimerization of the membrane ERs occurs within seconds of E2 exposure, which results in the activation of Gα and Gβγ proteins in a cell‐type dependent manner.99, 100 This leads to rapid E2 signaling.94, 95, 96, 97, 98 The E2‐dependent depalmitoylation of, at least, ERα decreases receptor‐caveolin‐1 association. This allows ER redistribution and its association with adaptors and/or signaling proteins, including the proline‐, glutamic acid‐, and leucine‐rich protein 1, modulator of non‐genomic activity of ERs (PELP1/MNAR), tyrosine kinase src, and tyrosine kinase receptors.94, 100, 101, 102 This, in turn, contributes to the activation of the ERK/MAPK and PI3K/AKT signaling cascades, impacting cellular proliferation, migration, and many other processes.100, 101, 102
Despite the well‐established protective role of E2 in the cardiovascular system, the mechanism by which E2 mediates its effect has been largely unclear. Recent studies have used a novel selective ER modulator, termed the “estrogen dendrimer conjugate,” or EDC,103 in a mouse model. The findings revealed that the membrane‐initiated ER signaling regulates processes that could be central to cardiovascular health and disease.99 The EDC, which possesses a minimal capacity to induce genomic activities because of its size and charge, stimulates endothelial cells, but not other cell types, and their proliferation and migration by inducing ERα‐G protein interaction.99 This interaction leads to the activation of endothelial nitric oxide synthase and nitric oxide production.99
The development of mouse models that synthesize the ligand‐binding domain (E) of ERα in order to target the domain exclusively to the cell membrane104 or the mutation of the Cys451 residue to Ala105, 106 in order to prevent palmitoylation, and hence the trafficking of the receptor to the membrane, are supportive of E2‐mediated membrane signaling. Disjointing the nuclear ER‐mediated transcriptional events, these in vivo models display infertility, abnormal ovaries, abnormal pituitary hormone regulation, stunted mammary gland ductal development, and altered vascular events.105, 106 Importantly, the cells that were isolated from the affected organs or tissues of these mice showed profoundly affected membrane signaling in response to E2.105, 106
3.2. Estrogen receptor‐mediated mitochondrial events
The mitochondria are essential for adenosine triphosphate (ATP) production, heme biosynthesis, β‐oxidation, the metabolism of certain amino acids, and steroid synthesis.84 The mitochondria also are involved in the control and mediation of apoptosis that is induced by several stimuli, including those that increase reactive oxygen species (ROS).84
Accumulating evidence suggests that the mitochondria are important targets of E2 actions, which inhibit the early stages of apoptosis.81, 107 Both ERα and ERβ are shown to localize to the mitochondria in various tissue and cell types that include the uterus, ovary,108 cardiomyocytes,22 breast adenocarcinoma‐derived MCF‐7 cells, and endothelial cells23 in a cell‐ and ER subtype‐dependent manner. It appears, for example, that although both ERs are localized primarily to the nucleus, ERβ is highly enriched in the mitochondria of MCF‐7 cells, whereas ERα resides in the mitochondria of endothelial cells at higher amounts, compared to ERβ, as both ERs are also present in the nucleus.23 Derived from the same genes encoding nuclear ERs,23 the presence of ERs in the mitochondria in the cells of various tissues suggests that mitochondrial ERs could directly mediate the effects of E2 within the mitochondria. However, the mechanism by which ERs are translocated into the mitochondria is unclear. The nuclear‐encoded mitochondrial proteins contain signal sequences that target them to the mitochondria through chaperone proteins.109, 110 The translocation of some mitochondrial proteins also occurs co‐translationally, such that mitochondrial proteins that are synthesized on cytosolic ribosomes are imported to the organelle.109, 110 Although ERs lack a sequence that targets them to the mitochondria, co‐translational translocation of ERs is a plausible mechanism for mitochondrial residency of the receptors.
Mitochondrial DNA (mtDNA) is a 16.5 kb circular genome that encodes 13 mRNAs, two rRNAs, and 22 tRNAs.111, 112 Thirteen of the 80 proteins of the electron transport chain (ETC) complexes I, II, III, IV, and V are encoded by mtDNA.111, 112 The remaining subunits of the ETC, as well as other proteins that are involved in mtDNA metabolism and function, are nuclear‐encoded.111, 112 The mtDNA transcription is initiated at two promoters (PL and PH) that are located in the D‐loop regulatory region through binding of the mitochondrial RNA polymerase and the mitochondrial transcription factors (TFAMs) (Transcription Factor A, Mitochondrial DNA Maintenance Factor), TFB1M and TFB2M (transcription factors b1 and b2, mitochondrial).111, 112 The TFAMs, TFB1M, and TFB2M are nuclear‐encoded genes whose transcription is regulated by Nuclear Respiratory Factor (NRF)‐1.111, 112
17β‐Estradiol is shown to augment the mitochondrial DNA‐encoded mRNAs, including the mitochondrial ATP synthase subunit E, COVII, and a number of other genes.111 These, together with the observations that ERα and ERβ bind to ERE‐like sequences that are present in the D‐loop of mouse and human mtDNA,113 suggest that the effects of E2 in the mitochondria are mediated through ER‐regulated transcriptions. Moreover, it has been shown that E2‐ERα, but not E2‐ERβ, induces NRF‐1 expression through a direct interaction with the DNA in the nucleus, resulting in an increased transcription of TFAM, TFB1, and TFB2, as well as the MRC genes in the cell models of breast and lung carcinomas.114 Based on these observations, it was suggested that, in addition to the transcriptional regulation of some of the mitochondrial genes through nuclear E2‐ER signaling, the protein products of NRF‐1‐regulated genes enter into the mitochondria in order to increase the expression of the mtDNA‐encoded genes, mitochondrial biogenesis, and oxidative phosphorylation.81, 114 This leads to increased ATP and ROS production. It should be noted that the superoxide of ROS that is generated by the mitochondrial respiratory chain is normally detoxified by mitochondrial antioxidant systems, including manganese superoxide dismutase (MnSOD). As E2‐ER also induces MnSOD expression and activity,23 the increased superoxide generation by E2 signaling can be detoxified by the increased MnSOD activity, thereby preventing apoptosis.
Moreover, recently it was reported that the accumulation of proteins in the inter‐membrane space (IMS) of the mitochondria in a breast adenocarcinoma cell model activates a distinct unfolded protein response.115, 116 On IMS stress, overproduction of ROS and phosphorylation of AKT kinase activates the nuclear ERα through the phosphorylation of Ser167.115, 116 This activated ERα is suggested to further augment the transcription of NRF‐1, as well as the expression of IMS protease HtrA serine peptidase 2 (HTRA2) in order to overcome mitochondrial dysfunction and to maintain cellular integrity.115, 116
3.3. Estrogen receptor‐mediated nuclear signaling
The integration of ER signaling that is generated from various cellular locations appears to be critical in the regulation of cellular proliferation, differentiation, motility, and death, dependent on the estrogen target tissue. However, the nuclear ERs are clearly the dominant players in the manifestation of cellular responses to E2. The NLS that is located in the D region is required for the translocation of the ER to the nucleus. Although the mechanism by which the ER is translocated to the nucleus remains unclear, the import of nuclear hormone receptors to the nucleus is controlled by a multimeric chaperone machinery.117 The interaction of NLS with importins and microtubule‐associated molecular motor proteins appears to mediate NHR transport to the nucleus.117 It also was reported that ERα contains a leucine‐rich nuclear export sequence (NES) in the LBD.118, 119 The NES, through binding to an exportin, was suggested to modulate the nucleocytoplasmic shuttling of ERα.118, 119
3.3.1. Estrogen response element‐dependent signaling pathway
The nuclear unliganded ERs are highly mobile molecules that are dynamically partitioned between target sites on chromatin and nuclear matrix.51, 53 As mentioned, ERs mediate E2 action in the nucleus with two distinct modes: ERE‐dependent and ERE‐independent signaling pathways. In the ERE‐dependent signaling route, ERs interact with a 5′‐GGTCAnnnTGACC‐3′ DNA palindrome sequence, the consensus ERE. Estrogen‐responsive genes, however, contain single or multiple copies of EREs that deviate from the consensus by one or more nucleotides.20, 48, 120 Although these EREs confer estrogen responsiveness that is mediated by the ER, they are less potent regulators of transcription than the consensus ERE.20, 48, 120 This is related to the ERE‐induced conformational change in the DBD of ERα.48, 121, 122 A single nucleotide change in the consensus ERE, for example, requires the formation of new interconnected hydrogen bonds between the response element and the DBD of ERα, thereby altering the conformation of the region.123 The unliganded ERs associate with the EREs.77, 124, 125 Kinetic studies using a well‐characterized estrogen‐responsive gene, Trefoil Factor 1 (TFF1, or pS2) promoter, as a model indicate that the engagement of the unliganded ERα with ERE occurs cyclically, with short periods requiring both activating and repressing epigenetic processes.126, 127, 128 The unliganded ERα through its amino‐ and carboxyl‐termini interacts, albeit inefficiently,48 with highly mobile heterogeneous co‐regulator complexes.51 These complexes include protein and chromatin modifiers that contain histone acetyl transferase (HAT), histone methyl transferase (HMT), and/or ATP‐dependent remodeling activities.122, 123, 124, 125 However, in the absence of a ligand, RNA polymerase II is not recruited to the promoter and, consequently, the transcription cannot begin. Further protein alterations, encompassing the ubiquitination of ERα and associated co‐regulators, disassemble the transcription complex,122, 123, 124, 125 followed by promoter remodeling through the association of modifiers with basal transcription factors. This oscillating promoter restructuring is suggested to provide a mechanism that enables a rapid adaptation of transcription to the E2 signal.122, 123, 124, 125 Kinetic studies further indicate that the interaction of ERα on binding to E2 initiates a series of interdependent events that result in an extended periodicity of cyclic engagement.122, 123, 124, 125 An interconnected ensemble of multisubunit transcription factor complexes governs transcriptional activation. The ERE binding of the E2‐ERα complex is followed immediately by the recruitment of the Switch–Sucrose Non‐fermentable chromatin remodeling complex that locks the nucleosomes into a stable orientation. This is followed by the recruitment of HMT and HATs to modify histones. The E2‐ERα recruits members of the p160 co‐activator family that includes SRC‐1, transcription intermediary factor‐2, and amplified in breast cancer‐1. The AF‐2 domain of ERα interacts with an amphipathic α‐helix that contains the sequence LXXLL, in the so‐called “nuclear receptor interacting domains” (NRIDs) of a co‐factor. These NRIDs serve as signal input domains by anchoring the members of the p160 family co‐activators to the promoter and connecting these proteins with the upstream end of the signaling pathway. The p160 family co‐activators are also HATs that acetylate the chromosomal histone proteins. This results in destabilization of the histone‐DNA contacts and chromatin decompaction in order to allow the positional phasing of the nucleosomes. These ERα‐associated co‐activators subsequently serve as a platform for the recruitment of p300, a co‐integrator with HAT activity. The recruitment of p300 coincides with an increased level of histone acetylation and with the recruitment of the initiation‐competent (unphosphorylated) form of RNA polymerase II and subsequent transcription initiation. Also, p300 appears to participate in the initiation of transcription.122, 123, 124, 125 This recruitment of p300 is thought to catalyze chromatin modifications that prime the promoter for multiple rounds of transcription.122, 123, 124, 125 The subsequent phosphorylation of the RNA polymerase II by a component of the basal transcription complex converts the polymerase to an elongation‐competent form.122, 123, 124, 125 Following these events, the dissociation of p300 from, and the subsequent binding of, cAMP response element‐binding (CREB) protein and p300/CBP‐associated factor (pCAF) to the complex take place. The CREB protein alone or together with pCAF further modifies chromatin through histone acetylation and/or methylation. These extensive alterations in the chromatin architecture provide the necessary scaffold for the ER complexes to enhance transcription through multiple rounds of transcription re‐initiation.126, 127, 128, 129 Studies also suggest that DNA methylation, particularly of CpG dinucleotides, occurs during the initial phase of every productive cycle and is associated with the recruitment of methyl CpG‐binding protein 2 and DNA (cytosine‐5‐)‐methyltransferase 1 to the promoter, which coincides with the recruitment of the remodeling complex, nucleosome remodeling deacetylase.130, 131, 132 Moreover, E2‐ERα‐mediated restructuring and transcriptional competence of the responsive gene promoter appear to require the generation of a DNA double‐stranded DNA break that is promoted by topoisomerase II.133 Thus, ERα‐mediated transcriptional events are tightly associated with induced local structural changes in chromatin. These changes encompass the positional phasing of nucleosomes and post‐translational modification of nucleosomes, the methylation status of CpG dinucleotides, and the formation of DNA breaks. The CREB protein also appears to be involved in the termination of transactivation by acetylating the acetyltransferases. The acetylation of the p160 proteins by the CREB protein leads to the disruption of the p160 co‐regulator–receptor complex.122, 123, 124, 125 This results in the termination of transcription and the remodeling of chromatin for recycling for transcription and/or proteasomal degradation.134
Although the events that are associated with the initiation and termination of ERE‐dependent genomic signaling could be similar between ERα and ERβ, the mode and extent of transcription that are mediated by the ERs through the ERE‐dependent signaling pathway differ significantly.135, 136 Comparative studies using heterologous reporter systems that emulate the ERE‐dependent signaling pathway and endogenous ERE‐driven gene responses72, 78, 137, 138 indicate that ERβ, in response to E2, displays considerably less potency than ERα in inducing transcription in the ERE‐dependent genomic signaling pathway. Estrogen receptor α–AF‐1, as discussed above, operates in cooperation with the carboxyl‐terminus in a cell and promoter context‐dependent manner.48, 70, 139, 140 It appears that the ability of the A/B domain to recruit72, 141, 142 and exchange143 co‐regulatory proteins is critical not only for AF‐1, but also for the functional integration of both AF‐1 and AF‐2 of ERα to mediate transcription at full capacity in response to E2 in a tissue‐specific manner.70, 71, 72, 142 Consistent with these studies, mouse KI models suggest that, although AF‐1 of ERα is dispensable for the vasculoprotective effects of E2, including the acceleration of the re‐endothelialization process and the prevention of atheroma, both AF‐1 and AF‐2 of ERα are necessary for uterine physiology.42, 43 In contrast to ERα, the amino‐terminus of human ERβ impairs the receptor‐ERE interactions,77 lacks an activation function,19, 72, 78, 79, 80 and is incapable of interacting with the carboxyl‐terminus.72 Therefore, this indicates that the distinct amino‐termini of ERs define the differences in the magnitude of transcriptional responses that are mediated through the ERE‐dependent E2‐ER signaling pathway. Nevertheless, the ability of ERβ to bind to an ERE with a lower affinity than ERα48, 144 and to interact with a different set of proteins48, 145 also contributes to distinct ER actions in the ERE‐dependent signaling pathway.
3.3.2. Estrogen response element‐independent signaling pathway
The ability of E2‐ER to mediate gene expression by functional interactions with, for example AP‐1 and Sp‐1, transcription factors bound to their cognate element on DNA, constitutes the ERE‐independent signaling pathway.19, 56, 57, 58, 59, 60 This pathway is dependent on the receptor subtype, nature of the ER ligand, and the cell context.19, 56, 57, 58, 59, 60
The AP‐1 transcription factor consists of members of the Jun, Fos, activating transcription factor, and musculoaponeurotic fibrosarcoma basic region leucine zipper motif protein families. The leucine zipper domain allows the dimerization of the Jun‐Jun and Jun‐Fos members to regulate gene expression. Once dimerized, their basic regions interact with the consensus TGAGTCA sequence, known as 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA), ‐response elements (TREs).146 Specificity protein 1, on the other hand, belongs to the Sp/KLF zinc‐finger transcription factor family that binds to the consensus (G/T)GGGCGG(G/A)(G/A)(C/T) sequence, referred to as the “GC box element.”146, 147 Both the AP‐1 and SP‐1 proteins play critical roles in cellular proliferation, differentiation, and death.146, 147
Studies have indicated that AP‐1 activity can be induced by E2 treatment and reduced by anti‐estrogens without increasing in c‐Fos and c‐Jun expression.148 Subsequent studies further showed that ERα60, 149 or ERβ60 does not bind directly to TREs, but the receptors are recruited by protein–protein interactions to c‐Jun through a region encompassing the ER‐DBD. It appears that ERα‐mediated transcription is dependent on the AF‐1 and AF‐2 functions of ERα as the receptor that lacks AF‐1 or AF‐2 fails to modulate the transcription from a TRE site.60, 150, 151 Although ERα and AP‐1 proteins use similar co‐regulators, as exemplified with the p160 proteins and CREB protein,60, 150, 151 in transcription at the ERE and TRE sites, the different combinatorial assembly of co‐regulatory proteins appears to be critical for ERα‐mediated signaling events through the TRE‐dependent pathway. Indeed, the observations that SERMs and SERDs can activate, rather than repress, the transcriptional responses that are mediated by ERα, but not ERβ, at a TRE site60, 150, 151 suggest that the altered pharmacology of ER ligands could be explained by differences in the amount and/or type of the co‐regulatory proteins, which show variations in cells from different tissues of origin.152
Moreover, ERα, but not ERβ, in response to E2 cross‐talks with the SP‐1 transcription factor to modulate the transcription of a variety of estrogen‐responsive genes.58, 153, 154 This interaction is mediated by the tethering of ERα to the GC box response element‐bound SP‐1 protein.58, 153, 154 Moreover, it appears that the amino‐terminal of region ERα is critical for responses from GC box element‐bearing promoters.58, 153, 154
In cellula and in vivo studies have attempted to understand the importance of the ERE‐independent pathway in E2‐ER signaling by dissecting nuclear ER signaling pathways. Studies, as discussed, indicated that Glu203, Gly204, and Ala207 residues, of the P‐box in the DNA‐binding helix of human ERα, determine the DNA‐binding specificity that is critical for sequence discrimination12, 62, 63 and binding to ERE.64 Changing Glu203 and Gly204 residues to Ala in the DNA‐binding helix of the human155 and the corresponding residues of the mouse149 ERα generates a mutant receptor that is capable of mediating E2 signaling only through the ERE‐independent pathway. Analogous mutations in the DBD of the human ERβ59, 156 also render the receptor functional only in the ERE‐independent signaling pathway. Studies with a mouse KI model of the P‐box in the DNA‐binding helix of mouse ERα (ERαAA) provide compelling support for the importance of the ERE‐independent pathway in the regulation of various tissue functions, albeit in a tissue‐specific manner.157, 158, 159 On the other hand, in an attempt to correlate the genomic responses from the ERE‐independent signaling pathway to alterations in cellular phenotypes, the authors found that changing Glu203Ala and Gly204Ala human ERα reduces, but does not prevent, the functional features of ERα in the ERE‐dependent signaling pathway.64 Moreover, Glu203Ala and Gly204Ala mutations could alter the response element specificity of ERα, as indicated by studies using ERαAA mouse uteri, which showed that the ERαAA mutant binds to hormone‐responsive motifs that are normally occupied by the progesterone receptor, leading to E2 regulation of uterine transcripts that are normally progesterone‐responsive.160
Previous studies indicated that a network of protein–DNA hydrogen bonds confers the binding specificity and stability of the human ERα to DNA.61, 161 For the consensus ERE, the network involves residues Glu203, Lys207, Lys210, and Arg211.161 Although the recognition of a non‐consensus ERE is achieved by a rearrangement of the side chains of various residues of ERα‐DBD, particularly Lys207 and Lys210, the interactions of Glu203 and Arg211 with DNA remain unaltered.161 Based on these observations, the replacement of positively charged Arg211, which is a conserved residue among NHRs, with the negatively charged Glu residue in the ERα203/204 mutant generated an ERE‐binding defective ERα mutant (or ERαEBD) that abolished the in vitro and in cellula ability of ERαEBD to interact with and to modulate transcription from an ERE while retaining the functionality at simulated ERE‐independent signaling pathways in various cell lines.64 Furthermore, the ERαEBD in response to E2 mediated a subset of estrogen‐responsive genes in a manner that was similar to E2‐ERα, but it was insufficient to alter the phenotypic features of the cell models, in contrast to E2‐ERα.64 Identical results were observed with an ERE‐binding mutant of ERβ.156 This suggests that the genomic responses from the ERE‐independent signaling pathway can be dissociated from the induction of phenotypic alterations. These findings also imply that the ERE‐dependent pathway is a required signaling route for E2‐ERs to induce cellular responses. This conclusion is supported by the observations that were derived from a mutant KI mouse (ERαEAAE) model bearing mutations at the DBD that synthesize an ERE‐binding defective ERα mutant that is incapable of modulating transcription from the ERE‐dependent signaling pathway but that is effectively regulating gene expression at the ERE‐independent signaling route.162 Displaying hypoplastic uteri, hemorrhagic ovaries, impaired mammary gland development, and liver function, the phenotypic features of the ERαEAAE mouse162 resembled the general loss‐of‐function phenotype of the ERα‐KO mouse models.
The critical importance of the ERE‐dependent signaling pathway in inducing cellular alterations is also supported by experimental studies that used oligonucleotide decoys, ER‐specific electrophilic agents, or designer transcription factors.163, 164, 165 Short sequences of DNA containing a response element for a transcription factor have been used as “decoys” to bind the cognate transcription factor in cellula or in vivo. The binding of a transcription factor to decoy DNA sequesters the transcription factor away from the endogenous binding sites. This renders the transcription factor ineffective to regulate target gene expression in a variety of systems. The use of a synthetic consensus ERE as the decoy in transfected ER‐positive breast cancer cell models was shown to prevent the growth of the cells in response to E2.163 Similarly, the prevention of an ER–ERE interaction by ER‐specific electrophilic agents that preferentially disrupted the zinc fingers of ERα effectively suppressed the E2‐mediated growth of ER‐positive breast cancer cell models in cellula and in vivo.164, 165 Moreover, the authors previously have shown that the intrinsic specificity of the DNA‐binding domain of ERα to interact with ERE sequences can be exploited in order to engineer a monomeric ERE‐binding module by co‐joining two DNA binding domains with the hinge domain.166 The integration of strong transcription activation domains from other transcription factors into the ERE‐binding module generated monomeric transcription factors, or monotransregulators, with constitutive activity at ERE‐driven gene promoters.77, 167, 168 These monotransregulators, but not the ERE‐binding defective counterparts, altered the cellular phenotypes by mimicking the effects of E2‐ERα on the gene transcriptions that required ERE interactions.
4. Increasing the Repertoire of Estrogen Receptor Actions
4.1. Estrogen receptorαβ heterodimer
Due to the shared and distinct regulatory potentials of ERα and ERβ, the repertoire of ER activity in response to E2 is expected to expand through the heterodimerization of ERs in cells that synthesize both subtypes. Early studies showed that ERα and ERβ, when co‐synthesized through transient transfection in mammalian cells, form the ERαβ heterodimer, the extent of which depends on the relative amount of each ER subtype.24, 137, 169, 170, 171 The ERαβ heterodimer interacts with DNA and modulates gene transcription in reporter systems, as well as in the chromatin context.24, 137, 169, 170, 171 As ER subtypes are not functionally equivalent, deciphering the role of the heterodimer, ERαβ, in E2 signaling is difficult because of the presence of ER homodimers. In order to address this issue, studies introduced DNA‐binding specificity‐altered ER mutants172 and single‐chain ER171 approaches. The ER mutants with an alteration in the DNA‐binding specificity were based on the observations that the ERE‐binding specificity of ERα (conversely, of GR) can be converted to glucocorticoid‐responsive element (GRE or conversely to ERE) by changing the Glu204, Gly204 and Ala207 residues of the ERα P‐box to those of the GR P‐box.62, 63 The co‐expression of a wild‐type (WT)‐ERβ, for example, with the GRE‐binding ERα (ERαGRE) allows for the measurement of the transcriptional properties of the ERβ‐ERαGRE heterodimer from a hybrid response element that is composed of an ERE and a GRE half‐site without interference from the ER homodimers. The single‐chain ER approach, in contrast, used a genetic fusion strategy to generate a homogeneous population of a homodimer or the heterodimer of ERs by the joining of ERα and/or ERβ cDNAs to produce single‐chain ER proteins in order to simulate an ER homodimer or the heterodimer protein.171 As ERα and ERβ are present on the same polypeptide chain, thereby circumventing the pivotal dimerization step in receptor action, the approach allows the generation of only the ERαβ single‐chain in heterodimer configuration without contaminating the ERα and ERβ homodimers. These studies have suggested that although ERα is the dominant partner in the ERαβ heterodimer, ERαβ also contributes new attributes to E2 signaling by combining distinct functional properties of both contributing partners.171, 172
Although studies have highlighted the overlapping and distinct functional features of ERs, addressing the roles of endogenous ER dimers in the physiology and pathophysiology of E2 signaling has been hampered by the absence of appropriate in cellula and in vivo models. Nevertheless, adenoviral infections, stable transfections, as well as engineered cell systems that allow the synthesis of one or both ER subtype(s), have expanded the previous findings to indicate a dynamic interplay among ER dimers. The findings indicate that ER dimers generate similar, as well as unique, genome‐wide expression profiles through mechanisms that involve shared chromatin‐binding sites and also alterations in their chromatin binding as a result of competition, restriction, and site shifting that are dependent on the nature of the ER ligands.173, 174, 175, 176, 177, 178
4.2. Estrogen receptor variants
Alterations in the expression of ER subtypes by epigenetic events, as well as by the generation of ER‐variant proteins through alternative splicing, are important elements that alter the dynamic regulation of tissue functions and also contribute to the initiation and/or the development of malignancies (Fig. 2).
Seven different promoters that are located upstream of the first coding exon are involved in the transcription of the ERα gene.14, 15, 16 The detection of distinct ERα transcripts in different tissues suggests that the composition of regulatory promoter elements is critical for tissue‐specific expression of the ERα gene. Although the different promoter usage gives rise to ERα transcript variants that differ in their 5′‐untranslated region, all the transcript variants encode the full‐length 66 kDa protein. Similarly, the promoter regions of the ERβ gene contain various regulatory elements179, 180, 181 that allow a versatile use of regulatory signals that are critical for tissue‐specific expression.15, 17 It appears that differential splicing of the 5′‐untranslated regions of the ERβ gene generates at least seven ERβ transcripts with various sizes of untranslated 5′ exonic sequences.17
The promoter regions of the ERs are also GC‐rich, implying a susceptibility to change in methylation status, an event that is associated with altered gene expression and an increased risk of disease.15, 17 Both the ERα and ERβ genes undergo changes in promoter methylation during development and under normal and pathological conditions. For example, methylation of the ERα gene promoter is reported to occur in vascular tissue and might play a role in atherogenesis and aging of the vascular system.182 Epigenetic dysregulation of ERβ gene expression is also suggested to contribute to the development of atherosclerosis and the aging of the vascular system, wherein ERβ plays a critical physiological role.183 Changes in the expression of ERα and ERβ also are reported to be associated with the progression of numerous types of cancerous tissues, including breast and lung. Up to one‐third of breast cancers that initially express ERα lose ERα expression during tumor progression as a result of methylation‐mediated ERα gene silencing.184 Similarly, studies suggest that a decrease in ERβ gene expression could be associated with breast tumorigenesis and that DNA methylation is an important mechanism for ERβ gene silencing in breast cancer.185 In prostate cancer, in contrast, the ERβ promoter is hyper‐methylated, resulting in decreased expression of the ERβ gene.181
Alternative splicing events and distinct translation initiation sites generate ERα and ERβ variants. These variant ERs are found to be present in both normal and neoplastic estrogen target tissues, adding further complexity to the biological responses to estrogens, as they can form homodimers or heterodimers with the WT‐ERs.88, 186, 187, 188 Despite the presence of a great variety of ERα mRNA splice forms in various estrogen target tissues, the function of ERα isoforms is derived primarily from experimental studies. For example, the removal of exon 4 results in an in‐frame deletion mutant ERα lacking the nuclear localization signal and part of the hormone‐binding domain, resulting in an ERα variant with a MM of 55 kDa. The resulting mutant lacks the DNA‐ and hormone‐binding abilities, transactivation function, as well as the ability to interfere with the activity of WT‐ERα.189 Whereas, the deletion of exon 5 results in the introduction of a new stop codon within the hormone‐binding domain, giving rise to an ERα variant protein with a MM of 52 kDa.190 This carboxyl‐terminally truncated ERα lacks the hormone‐binding function but retains the DNA‐binding function. The ERα exon 5‐deleted mutant is reported to show a constitutive activity in some breast cancer cell lines.190, 191, 192, 193 Likewise, the splice variant of exon 7 expresses an ERα in the normal breast tissue that lacks both transactivation and hormone‐binding functions. This variant binds to DNA and behaves like a dominant negative isoform for both ERα and ERβ and thus regulates estrogen responsiveness.190, 193
Evidence for the existence of endogenous ERα variant proteins, on the other hand, is limited to a few. For example, an ERα variant with a MM of 46 kDa (ERα46) is found to be present in human primary osteoblasts,194 an analog of which is also present in the bone of the original ERα‐KO mice model.194, 195 This isoform, expressed at a level similar to WT‐ERα, is generated by an alternative splicing of the ERα gene, which results in exon 1 being skipped, with a start codon in exon 2 being used to initiate translation of the protein. Consequently, the AF‐1 of this ERα isoform is absent. Functional analyses suggest that this amino‐terminally truncated ERα is able to heterodimerize with WT‐ERα and also with WT‐ERβ. ERα46 is a strong inhibitor of WT‐ERα when co‐synthesized and represses cellular proliferation in response to E2.194, 195 ERα36 is generated from a promoter in the first intron of the ERα gene and continues from exon 2 to exon 6 and skips exons 7 and 8.196, 197 This results in a unique carboxyl‐terminus of 27 amino acids that replaces the last 138 amino acids of full‐length ERα.196, 197 ERα36 is localized mainly in the cytoplasm and the plasma membrane. Palmitoylation of ERα36 could contribute to the membrane localization of the variant. It appears that ERα36 mediates membrane‐initiated E2 signaling and adversely affects the events that are mediated by both ERα and ERβ.196, 197
Multiple ERβ transcripts exist as a result of the alternative usage of untranslated exons in the 5′ of the gene, alternative splicing of the last coding exons, or deletion of one or more coding exons. For example, although very rare among the population,198 an ERβ testis cDNA that encodes an amino‐terminally extended ERβ isoform was reported.199 This variant ERβ results from the presence of an additional A–T base pair in the 5′‐untranslated region of the ERβ gene that generates an early ATG initiation codon that extends the amino‐terminus of ERβ by 18 additional amino acids; hence, it is referred to as “ERβ548.”199 Interestingly, ERβ548 displays a more robust activity than WT‐ERβ in inducing transcription in an ERE‐dependent reporter system. Moreover, tamoxifen and raloxifene appear to act as agonists for ERβ548, in contrast with their action as antagonists for WT‐ERβ.199 In addition, many ERβ variants that have resulted from alternative splicing events also have been reported in normal and pathological estrogen target tissues. Although the importance of these ERβ splice variants remains unclear, several major variants have been described to alter E2–ER signaling in experimental systems.200, 201, 202, 203 Of these, ERβ2, 4, and 5, which contain exons 1–7 of the human ERβ gene, followed by alternatively spliced exon 8, have been studied in detail. The studies indicate that although these carboxyl‐terminally truncated variants cannot bind ligand and lack co‐activator‐recruiting helix 12, they can heterodimerize with WT‐ERβ, as well as with WT‐ERα, and modulate estrogen‐mediated transcriptional activities of the receptors, raising the possibility that when co‐synthesized, ERβ isoforms could adversely alter ERα and ERβ signaling.200, 201, 202, 203
5. Epilogue
Despite a large number of experimental studies indicating that ERα and ERβ show similar, as well as distinct, regulatory potentials in cells of different estrogen target tissues, the physiological role of ERβ in E2‐mediated signaling remains elusive. However, one consensus is that rather the subtype, the relative level of synthesis of ERs and ER variants, particularly in cases wherein both subtypes are synthesized, can have profound effects on the dynamic and integrated network of cellular events in both the physiology and pathophysiology of target tissues. Although beyond the scope of this paper, and there are many excellent reviews,204, 205, 206, 207, 208, 209, 210 one important integrated network involves the cross‐talk of E2–ER with growth factor signaling pathways (GFSPs). These GFSPs modify, and are modified by, E2–ER signaling.201, 202 Adding further complexity to E2–ER signaling, are phosphorylation, glycosylation, ubiquitination, and acetylation events that not only modulate unliganded or liganded ER functions at every level but also alter the ligand pharmacology.204, 205, 206, 207, 208, 209, 210 The deregulation of growth factor signaling appears to play a vital role in ER‐driven neoplastic processes and also the development of endocrine resistance in the treatment of estrogen target tissue malignancies, exemplified by breast cancers.204, 205, 206, 207, 208, 209, 210 Consequently, a better understanding of the complex regulatory mechanisms that underlie ER actions holds considerable promise for the development of novel biomarkers and predictors, as well as therapeutic approaches that could have a substantial impact on the systemic management of estrogen target tissue malignancies.
Disclosure
Conflict of interest: The authors declare no conflict of interest. Human and animal studies: This article does not contain any study with humans or animals that was performed by any of the authors.
Acknowledgements
The work in the laboratory was supported by grants from the Scientific and Technological Research Council of Turkey (TUBITAK) (TUBITAK‐KBAG 212T031 and 114Z243), Ankara, Turkey, and by the Middle East Technical University (METU‐BAP‐08‐11‐2015‐019), Ankara, Turkey. Gamze Ayaz was supported by a graduate study fellowship from TUBITAK.
Yaşar, P. , Ayaz, G. , User, S. D. , Güpür, G. and Muyan, M. (2017), Molecular mechanism of estrogen–estrogen receptor signaling. Reproductive Medicine and Biology, 16: 4–20. doi: 10.1002/rmb2.12006.
References
- 1. Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ribeiro RC, Kushner PJ, Baxter JD. The nuclear hormone receptor gene superfamily. Annu Rev Med. 1995;46:443–453. [DOI] [PubMed] [Google Scholar]
- 3. Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and actions of estrogens. N Engl J Med. 2002;346:340–352. [DOI] [PubMed] [Google Scholar]
- 4. Nelson LR, Bulun SE. Estrogen production and action. J Am Acad Dermatol. 2001;45:S116–S124. [DOI] [PubMed] [Google Scholar]
- 5. Harris HA. Estrogen receptor‐beta: recent lessons from in vivo studies. Mol Endocrinol. 2007;21:1–13. [DOI] [PubMed] [Google Scholar]
- 6. Hamilton KJ, Arao Y, Korach KS. Estrogen hormone physiology: reproductive findings from estrogen receptor mutant mice. Reprod Biol. 2014;14:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hewitt SC, Winuthayanon W, Korach KS. What's new in estrogen receptor action in the female reproductive tract. J Mol Endocrinol. 2016;56:R55–R71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuiper GG, Enmark E, Pelto‐Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392:49–53. [DOI] [PubMed] [Google Scholar]
- 10. Deroo BJ, Buensuceso AV. Minireview: estrogen receptor‐beta: mechanistic insights from recent studies. Mol Endocrinol. 2010;24:1703–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haldosén L‐A, Zhao C, Dahlman‐Wright K. Estrogen receptor beta in breast cancer. Mol Cell Endocrinol. 2014;382:665–672. [DOI] [PubMed] [Google Scholar]
- 12. Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–951. [DOI] [PubMed] [Google Scholar]
- 13. Green S, Kumar V, Krust A, Walter P, Chambon P. Structural and functional domains of the human estrogen receptor. Cold Spring Harb Symp Quant Biol. 1986;51:751–758. [DOI] [PubMed] [Google Scholar]
- 14. Ponglikitmongkol M, Green S, Chambon P. Genomic organization of the human oestrogen receptor gene. EMBO J. 1988;7:3385–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sand P, Luckhaus C, Schlurmann K, Götz M, Deckert J. Untangling the human estrogen receptor gene structure. J Neural Transm. 2002;109:567–583. [DOI] [PubMed] [Google Scholar]
- 16. Kos M, Reid G, Denger S, Gannon F. Minireview: genomic organization of the human ERalpha gene promoter region. Mol Endocrinol. 2001;15:2057–2063. [DOI] [PubMed] [Google Scholar]
- 17. Enmark E, Pelto‐Huikko M, Grandien K, et al. Human estrogen receptor beta‐gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab. 1997;82:4258–4265. [DOI] [PubMed] [Google Scholar]
- 18. Ogawa S, Inoue S, Watanabe T, et al. The complete primary structure of human estrogen receptor beta (hER beta) and its heterodimerization with ER alpha in vivo and in vitro. Biochem Biophys Res Commun. 1998;243:122–126. [DOI] [PubMed] [Google Scholar]
- 19. Huang J, Li X, Hilf R, Bambara RA, Muyan M. Molecular basis of therapeutic strategies for breast cancer. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:379–396. [DOI] [PubMed] [Google Scholar]
- 20. Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER. Plasma membrane estrogen receptors exist and functions as dimers. Mol Endocrinol. 2004;18:2854–2865. [DOI] [PubMed] [Google Scholar]
- 22. Yang S‐H, Liu R, Perez EJ, et al. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci USA. 2004;101:4130–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pedram A, Razandi M, Wallace DC, Levin ER. Functional estrogen receptors in the mitochondria of breast cancer cells. Mol Biol Cell. 2006;17:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bai Y, Giguére V. Isoform‐selective interactions between estrogen receptors and steroid receptor coactivators promoted by estradiol and ErbB‐2 signaling in living cells. Mol Endocrinol. 2003;17:589–599. [DOI] [PubMed] [Google Scholar]
- 25. Mak HY, Hoare S, Henttu PM, Parker MG. Molecular determinants of the estrogen receptor–coactivator interface. Mol Cell Biol. 1999;19:3895–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brzozowski AM, Pike AC, Dauter Z, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. [DOI] [PubMed] [Google Scholar]
- 27. Pike ACW, Brzozowski AM, Hubbard RE. A structural biologist's view of the oestrogen receptor. J Steroid Biochem Mol Biol. 2000;74:261–268. [DOI] [PubMed] [Google Scholar]
- 28. Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rastinejad F, Huang P, Chandra V, Khorasanizadeh S. Understanding nuclear receptor form and function using structural biology. J Mol Endocrinol. 2013;51:T1–T21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pike AC, Brzozowski AM, Hubbard RE, et al. Structure of the ligand‐binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwartz JA, Zhong L, Deighton‐Collins S, Zhao C, Skafar DF. Mutations targeted to a predicted helix in the extreme carboxyl‐terminal region of the human estrogen receptor‐alpha alter its response to estradiol and 4‐hydroxytamoxifen. J Biol Chem. 2002;277:13202–13209. [DOI] [PubMed] [Google Scholar]
- 32. Montano MM, Müller V, Trobaugh A, Katzenellenbogen BS. The carboxy‐terminal F domain of the human estrogen receptor: role in the transcriptional activity of the receptor and the effectiveness of antiestrogens as estrogen antagonists. Mol Endocrinol. 1995;9:814–825. [DOI] [PubMed] [Google Scholar]
- 33. Koide A, Zhao C, Naganuma M, et al. Identification of regions within the F domain of the human estrogen receptor alpha that are important for modulating transactivation and protein–protein interactions. Mol Endocrinol. 2007;21:829–842. [DOI] [PubMed] [Google Scholar]
- 34. Arao Y, Hamilton KJ, Coons LA, Korach KS. Estrogen receptor α L543A, L544A mutation changes antagonists to agonists which correlates with the ligand binding domain dimerization associated with DNA binding activity. J Biol Chem. 2013;288:21105–21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lees JA, Fawell SE, White R, Parker MG. A 22‐amino‐acid peptide restores DNA‐binding activity to dimerization‐defective mutants of the estrogen receptor. Mol Cell Biol. 1990;10:5529–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA. Estrogen receptor dimerization: ligand binding regulates dimer affinity and dimer dissociation rate. Mol Endocrinol. 2002;16:2706–2719. [DOI] [PubMed] [Google Scholar]
- 37. Vajdos FF, Hoth LR, Geoghegan KF, et al. The 2.0 A crystal structure of the ERalpha ligand‐binding domain complexed with lasofoxifene. Protein Sci. 2007;16:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. [DOI] [PubMed] [Google Scholar]
- 39. Tetel MJ. Nuclear receptor coactivators: essential players for steroid hormone action in the brain and in behaviour. J Neuroendocrinol. 2009;21:229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McDonnell DP, Wardell SE. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr Opin Pharmacol. 2010;10:620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jordan VC, O'Malley BW. Selective estrogen‐receptor modulators and antihormonal resistance in breast cancer. J Clin Oncol. 2007;25:5815–5824. [DOI] [PubMed] [Google Scholar]
- 42. Arao Y, Hamilton KJ, Ray MK, Scott G, Mishina Y, Korach KS. Estrogen receptor alpha AF‐2 mutation results in antagonist reversal and reveals tissue selective function of estrogen receptor modulators. Proc Natl Acad Sci USA. 2011;108:14986–14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Billon‐Galés A, Krust A, Fontaine C, et al. Activation function 2 (AF2) of estrogen receptor‐alpha is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci USA. 2011;108:13311–13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. [DOI] [PubMed] [Google Scholar]
- 45. Dobrzycka KM, Townson SM, Jiang S, Oesterreich S. Estrogen receptor corepressors – a role in human breast cancer? Endocr Relat Cancer. 2003;10:517–536. [DOI] [PubMed] [Google Scholar]
- 46. Heldring N, Pawson T, McDonnell D, Treuter E, Gustafsson JÅ, Pike ACW. Structural insights into corepressor recognition by antagonist‐bound estrogen receptors. J Biol Chem. 2007;282:10449–10455. [DOI] [PubMed] [Google Scholar]
- 47. Chakraborty S, Levenson AS, Biswas PK. Structural insights into resveratrol's antagonist and partial agonist actions on estrogen receptor alpha. BMC Struct Biol. 2013;13:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yi P, Driscoll MD, Huang J, et al. The effects of estrogen‐responsive element‐ and ligand‐induced structural changes on the recruitment of cofactors and transcriptional responses by ER alpha and ER beta. Mol Endocrinol. 2002;16:674–693. [DOI] [PubMed] [Google Scholar]
- 49. Dauvois S, Danielian PS, White R, Parker MG. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci USA. 1992;89:4037–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dauvois S, White R, Parker MG. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J Cell Sci. 1993;106:1377–1388. [DOI] [PubMed] [Google Scholar]
- 51. Stenoien DL, Patel K, Mancini MG, et al. FRAP reveals that mobility of oestrogen receptor‐alpha is ligand‐ and proteasome‐dependent. Nat Cell Biol. 2001;3:15–23. [DOI] [PubMed] [Google Scholar]
- 52. Long X, Nephew KP. Fulvestrant (ICI 182,780)‐dependent interacting proteins mediate immobilization and degradation of estrogen receptor‐alpha. J Biol Chem. 2006;281:9607–9615. [DOI] [PubMed] [Google Scholar]
- 53. Muyan M, Callahan LM, Huang Y, Lee AJ. The ligand‐mediated nuclear mobility and interaction with estrogen‐responsive elements of estrogen receptors are subtype specific. J Mol Endocrinol. 2012;49:249–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor‐β potency‐selective ligands: structure–activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–4251. [DOI] [PubMed] [Google Scholar]
- 55. Lin C‐Y, Vega VB, Thomsen JS, et al. Whole‐genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. [DOI] [PubMed] [Google Scholar]
- 57. Kushner P, Agard D, Greene G, et al. Estrogen receptor pathways to AP‐1. J Steroid Biochem Mol Biol. 2000;74:311–317. [DOI] [PubMed] [Google Scholar]
- 58. Safe S. Transcriptional activation of genes by 17 beta‐estradiol through estrogen receptorSp1 interactions. Vitam Horm. 2001;62:231–252. [DOI] [PubMed] [Google Scholar]
- 59. Björnström L, Sjöberg M. Mutations in the estrogen receptor DNA‐binding domain discriminate between the classical mechanism of action and cross‐talk with Stat5b and activating protein 1 (AP‐1). J Biol Chem. 2002;277:48479–48483. [DOI] [PubMed] [Google Scholar]
- 60. Cheung E, Acevedo ML, Cole PA, Kraus WL. Altered pharmacology and distinct coactivator usage for estrogen receptor‐dependent transcription through activating protein‐1. Proc Natl Acad Sci USA. 2005;102:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schwabe JWR, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA‐binding domain bound to DNA: how receptors discriminate between their response elements. Cell. 1993;75:567–578. [DOI] [PubMed] [Google Scholar]
- 62. Green S, Kumar V, Theulaz I, Wahli W, Chambon P. The N‐terminal DNA‐binding “zinc finger” of the oestrogen and glucocorticoid receptors determines target gene specificity. EMBO J. 1988;7:3037–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mader S, Kumar V, de Verneuil H, Chambon P. Three amino acids of the oestrogen receptor are essential to its ability to distinguish an oestrogen from a glucocorticoid‐responsive element. Nature. 1989;338:271–274. [DOI] [PubMed] [Google Scholar]
- 64. Nott SL, Huang Y, Li X, et al. Genomic responses from the estrogen‐responsive element‐dependent signaling pathway mediated by estrogen receptor alpha are required to elicit cellular alterations. J Biol Chem. 2009;284:15277–15288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kumar R, Thompson EB. Transactivation functions of the N‐terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol Endocrinol. 2003;17:1–10. [DOI] [PubMed] [Google Scholar]
- 66. Tora L, White J, Brou C, et al. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. 1989;59:477–487. [DOI] [PubMed] [Google Scholar]
- 67. Metzger D, Ali S, Bornert JM, Chambon P. Characterization of the amino‐terminal transcriptional activation function of the human estrogen receptor in animal and yeast cells. J Biol Chem. 1995;270:9535–9542. [DOI] [PubMed] [Google Scholar]
- 68. Bocquel MT, Kumar V, Stricker C, Chambon P, Gronemeyer H. The contribution of the N‐ and C‐terminal regions of steroid receptors to activation of transcription is both receptor and cell‐specific. Nucleic Acids Res. 1989;17:2581–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tasset D, Tora L, Fromental C, Scheer E, Chambon P. Distinct classes of transcriptional activating domains function by different mechanisms. Cell. 1990;62:1177–1187. [DOI] [PubMed] [Google Scholar]
- 70. Kraus WL, McInerney EM, Katzenellenbogen BS. Ligand‐dependent, transcriptionally productive association of the amino‐ and carboxyl‐terminal regions of a steroid hormone nuclear receptor. Proc Natl Acad Sci USA. 1995;92:12314–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Benecke A, Chambon P, Gronemeyer H. Synergy between estrogen receptor alpha activation functions AF1 and AF2 mediated by transcription intermediary factor TIF2. EMBO Rep. 2000;1:151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yi P, Bhagat S, Hilf R, Bambara RA, Muyan M. Differences in the abilities of estrogen receptors to integrate activation functions are critical for subtype‐specific transcriptional responses. Mol Endocrinol. 2002;16:1810–1827. [DOI] [PubMed] [Google Scholar]
- 73. Kumar R, Litwack G. Structural and functional relationships of the steroid hormone receptors’ N‐terminal transactivation domain. Steroids. 2009;74:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kumar R, Zakharov MN, Khan SH, et al. The dynamic structure of the estrogen receptor. J Amino Acids. 2011;2011:812540, doi: 10.4061/2011/812540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wärnmark A, Wikström A, Wright APH, Gustafsson JÅ, Härd T. The N‐terminal regions of estrogen receptor α and β are unstructured in vitro and show different tbp binding properties. J Biol Chem. 2001;276:45939–45944. [DOI] [PubMed] [Google Scholar]
- 76. Rajbhandari P, Finn G, Solodin NM, et al. Regulation of estrogen receptor N‐terminus conformation and function by peptidyl prolyl isomerase Pin1. Mol Cell Biol. 2012;32:445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Huang J, Li X, Maguire CA, Hilf R, Bambara RA, Muyan M. Binding of estrogen receptor beta to estrogen response element in situ is independent of estradiol and impaired by its amino terminus. Mol Endocrinol. 2005;19:2696–2712. [DOI] [PubMed] [Google Scholar]
- 78. Cowley SM, Parker MG. A comparison of transcriptional activation by ER alpha and ER beta. J Steroid Biochem Mol Biol. 1999;69:165–175. [DOI] [PubMed] [Google Scholar]
- 79. Delaunay F, Pettersson K, Tujague M, Gustafsson JA. Functional differences between the amino‐terminal domains of estrogen receptors alpha and beta. Mol Pharmacol. 2000;58:584–590. [DOI] [PubMed] [Google Scholar]
- 80. McInerney EM, Weis KE, Sun J, Mosselman S, Katzenellenbogen BS. Transcription activation by the human estrogen receptor subtype beta (ER beta) studied with ER beta and ER alpha receptor chimeras. Endocrinology. 1998;139:4513–4522. [DOI] [PubMed] [Google Scholar]
- 81. Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105:1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Levin ER. Plasma membrane estrogen receptors. Trends Endocrinol Metab. 2009;20:477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Levin ER. Extranuclear estrogen receptor's roles in physiology: lessons from mouse models. Am J Physiol Endocrinol Metab. 2014;307:E133–E140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen JQ, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta. 2009;1793:1540–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yager JD, Chen JQ. Mitochondrial estrogen receptors – new insights into specific functions. Trends Endocrinol Metab. 2007;18:89–91. [DOI] [PubMed] [Google Scholar]
- 86. Zhao C, Dahlman‐Wright K, Gustafsson JÅ. Estrogen signaling via estrogen receptor β. J Biol Chem. 2010;285:39575–39579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Thomas C, Gustafsson J.‐Å. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11:597–608. [DOI] [PubMed] [Google Scholar]
- 88. Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. [DOI] [PubMed] [Google Scholar]
- 89. Acconcia F, Marino M. Synergism between genomic and non genomic estrogen action mechanisms. IUBMB Life. 2003;55:145–150. [DOI] [PubMed] [Google Scholar]
- 90. Wang ZY, Yin L. Estrogen receptor alpha‐36 (ER‐α36): a new player in human breast cancer. Mol Cell Endocrinol. 2015;418:193–206. [DOI] [PubMed] [Google Scholar]
- 91. Prossnitz ER, Barton M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol. 2014;389:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Prossnitz ER, Hathaway HJ. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol. 2015;153:114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol. 1999;13:307–319. [DOI] [PubMed] [Google Scholar]
- 94. Marino M, Ascenzi P, Acconcia F. S‐palmitoylation modulates estrogen receptor α localization and functions. Steroids. 2006;71:298–303. [DOI] [PubMed] [Google Scholar]
- 95. Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. S‐palmitoylation modulates human estrogen receptor‐α functions. Biochem Biophys Res Commun. 2004;316:878–883. [DOI] [PubMed] [Google Scholar]
- 96. Galluzzo P, Caiazza F, Moreno S, Marino M. Role of ERbeta palmitoylation in the inhibition of human colon cancer cell proliferation. Endocr Relat Cancer. 2007;14:153–167. [DOI] [PubMed] [Google Scholar]
- 97. Marino M, Ascenzi P. Membrane association of estrogen receptor α and β influences 17β‐estradiol‐mediated cancer cell proliferation. Steroids. 2008;73:853–858. [DOI] [PubMed] [Google Scholar]
- 98. Pedram A, Razandi M, Sainson RCA, Kim JK, Hughes CC, Levin ER. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007;282:22278–22288. [DOI] [PubMed] [Google Scholar]
- 99. Kumar P, Wu Q, Chambliss KL, et al. Direct interactions with G α i and G βγ mediate nongenomic signaling by estrogen receptor α. Mol Endocrinol. 2007;21:1370–1380. [DOI] [PubMed] [Google Scholar]
- 100. Galluzzo P, Ascenzi P, Bulzomi P, Marino M. The nutritional flavanone naringenin triggers antiestrogenic effects by regulating estrogen receptor α‐palmitoylation. Endocrinology. 2008;149:2567–2575. [DOI] [PubMed] [Google Scholar]
- 101. Razandi M, Pedram A, Park ST, Levin ER. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. 2003;278:2701–2712. [DOI] [PubMed] [Google Scholar]
- 102. Sanchez AM, Flamini MI, Baldacci C, Goglia L, Genazzani AR, Simoncini T. Estrogen receptor‐alpha promotes breast cancer cell motility and invasion via focal adhesion kinase and N‐WASP. Mol Endocrinol. 2010;24:2114–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Harrington WR, Kim SH, Funk CC, et al. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006;20:491–502. [DOI] [PubMed] [Google Scholar]
- 104. Pedram A, Razandi M, Kim JK, et al. Developmental phenotype of a membrane only estrogen receptor α (MOER) mouse. J Biol Chem. 2009;284:3488–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Adlanmerini M, Solinhac R, Abot A, et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue‐specific roles for membrane versus nuclear actions. Proc Natl Acad Sci USA. 2014;111:E283–E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pedram A, Razandi M, Lewis M, Hammes S, Levin ER. Membrane‐localized estrogen receptor alpha is required for normal organ development and function. Dev Cell. 2014;29:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Simpkins JW, Yang SH, Sarkar SN, Pearce V. Estrogen actions on mitochondria – Physiological and pathological implications. Mol Cell Endocrinol. 2008;290:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Monje P, Boland R. Subcellular distribution of native estrogen receptor alpha and beta isoforms in rabbit uterus and ovary. J Cell Biochem. 2001;82:467–479. [DOI] [PubMed] [Google Scholar]
- 109. Lee J, Sharma S, Kim J, Ferrante RJ, Ryu H. Mitochondrial nuclear receptors and transcription factors: who's minding the cell? J Neurosci Res. 2008;86:961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen J‐Q, Yager JD, Russo J. Regulation of mitochondrial respiratory chain structure and function by estrogens/estrogen receptors and potential physiological/pathophysiological implications. Biochim Biophys Acta. 2005;1746:1–17. [DOI] [PubMed] [Google Scholar]
- 112. Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. [DOI] [PubMed] [Google Scholar]
- 113. Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF‐7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem. 2004;93:358–373. [DOI] [PubMed] [Google Scholar]
- 114. Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM. Estradiol stimulates transcription of nuclear respiratory factor‐1 and increases mitochondrial biogenesis. Mol Endocrinol. 2008;22:609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Papa L, Germain D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci. 2011;124:1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Germain D. Estrogen carcinogenesis in breast cancer. Endocrinol Metab Clin North Am. 2011;40:473–484. [DOI] [PubMed] [Google Scholar]
- 117. Echeverria PC, Picard D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim Biophys Acta. 2010;1803:641649. [DOI] [PubMed] [Google Scholar]
- 118. Lombardi M, Castoria G, Migliaccio A, et al. Hormone‐dependent nuclear export of estradiol receptor and DNA synthesis in breast cancer cells. J Cell Biol. 2008;182:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Giraldi T, Giovannelli P, Di Donato M, Castoria G, Migliaccio A, Auricchio F. Steroid signaling activation and intracellular localization of sex steroid receptors. J Cell Commun Signal. 2010;4:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Driscoll MD, Sathya G, Muyan M, Klinge CM, Hilf R, Bambara RA. Sequence requirements for estrogen receptor binding to estrogen response elements. J Biol Chem. 1998;273:29321–29330. [DOI] [PubMed] [Google Scholar]
- 121. Loven MA, Wood JR, Nardulli AM. Interaction of estrogen receptors alpha and beta with estrogen response elements. Mol Cell Endocrinol. 2001;181:151–163. [DOI] [PubMed] [Google Scholar]
- 122. Hall JM, McDonnell DP, Korach KS. Allosteric regulation of estrogen receptor structure, function, and coactivator recruitment by different estrogen response elements. Mol Endocrinol. 2002;16:469–486. [DOI] [PubMed] [Google Scholar]
- 123. Freedman LP, Luisi BF. On the mechanism of DNA binding by nuclear hormone receptors: a structural and functional perspective. J Cell Biochem. 1993;51:140–150. [DOI] [PubMed] [Google Scholar]
- 124. Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone‐induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. [DOI] [PubMed] [Google Scholar]
- 125. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor‐regulated transcription. Cell. 2000;103:843–852. [DOI] [PubMed] [Google Scholar]
- 126. Métivier R, Penot G, Hübner MR, et al. Estrogen receptor‐α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. [DOI] [PubMed] [Google Scholar]
- 127. Métivier R, Penot G, Carmouche RP, et al. Transcriptional complexes engaged by apo‐estrogen receptor‐alpha isoforms have divergent outcomes. EMBO J. 2004;23:3653–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Reid G, Denger S, Kos M, Gannon F. Human estrogen receptor‐alpha: regulation by synthesis, modification and degradation. Cell Mol Life Sci. 2002;59:821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kraus WL, Kadonaga JT. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev. 1998;12:331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kangaspeska S, Stride B, Métivier R, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. [DOI] [PubMed] [Google Scholar]
- 131. Reid G, Gallais R, Métivier R. Marking time: the dynamic role of chromatin and covalent modification in transcription. Int J Biochem Cell Biol. 2009;41:155–163. [DOI] [PubMed] [Google Scholar]
- 132. Métivier R, Gallais R, Tiffoche C, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. [DOI] [PubMed] [Google Scholar]
- 133. Ju B‐G, Lunyak VV, Perissi V, et al. A topoisomerase IIβ–mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. [DOI] [PubMed] [Google Scholar]
- 134. Lonard DM, Nawaz Z, Smith CL, O'Malley BW. The 26S proteasome is required for estrogen receptor‐alpha and coactivator turnover and for efficient estrogen receptor‐alpha transactivation. Mol Cell. 2000;5:939–948. [DOI] [PubMed] [Google Scholar]
- 135. Li X, Huang J, Fluharty BR, Huang Y, Nott SL, Muyan M. What are comparative studies telling us about the mechanism of ERbeta action in the ERE‐dependent E2 signaling pathway? J Steroid Biochem Mol Biol. 2008;109:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Huang J, Li X, Qiao T, Bambara RA, Hilf R, Muyan M. A tale of two estrogen receptors (ERs): how differential ER‐estrogen responsive element interactions contribute to subtype‐specific transcriptional responses. Nucl Recept Signal. 2006;4:e015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Hall JM, McDonnell DP. The estrogen receptor beta‐isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140:5566–5578. [DOI] [PubMed] [Google Scholar]
- 138. Huang YF, Li XD, Muyan M. Estrogen receptors similarly mediate the effects of 17 beta‐estradiol on cellular responses but differ in their potencies. Endocrine. 2011;39:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Tora L, Mullick A, Metzger D, Ponglikitmongkol M, Park I, Chambon P. The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J. 1989;8:1981–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tzukerman MT, Esty A, Santiso‐Mere D, et al. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol Endocrinol. 1994;8:21–30. [DOI] [PubMed] [Google Scholar]
- 141. Yi P, Wang Z, Feng Q, et al. Structure of a biologically active estrogen receptor–coactivator complex on DNA. Mol Cell. 2015;57:1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Webb P, Nguyen P, Shinsako J, et al. Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol Endocrinol. 1998;12:1605–1618. [DOI] [PubMed] [Google Scholar]
- 143. Zhu P, Baek SH, Bourk EM, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. [DOI] [PubMed] [Google Scholar]
- 144. Kulakosky PC, McCarty MA, Jernigan SC, Risinger KE, Klinge CM. Response element sequence modulates estrogen receptor alpha and beta affinity and activity. J Mol Endocrinol. 2002;29:137–152. [DOI] [PubMed] [Google Scholar]
- 145. Ivanova M, Abner S, Pierce W, Klinge C. Ligand‐dependent differences in estrogen receptor beta‐interacting proteins identified in lung adenocarcinoma cells corresponds to estrogenic responses. Proteome Sci. 2011;9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Karin M, Liu ZG, Zandi E. AP‐1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. [DOI] [PubMed] [Google Scholar]
- 147. Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer. 2005;41:2438–2448. [DOI] [PubMed] [Google Scholar]
- 148. Philips A, Chalbos D, Rochefort H. Estradiol increases and anti‐estrogens antagonize the growth factor‐induced activator protein‐1 activity in MCF7 breast cancer cells without affecting c‐fos and c‐jun synthesis. J Biol Chem. 1993;268:14103–14108. [PubMed] [Google Scholar]
- 149. Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL. Estrogen receptor binding to DNA Is not required for its activity through the nonclassical AP1 pathway. J Biol Chem. 2001;276:13615–13621. [DOI] [PubMed] [Google Scholar]
- 150. Teyssier C, Belguise K, Galtier F, Chalbos D. Characterization of the physical interaction between estrogen receptor alpha and JUN proteins. J Biol Chem. 2001;276:36361–36369. [DOI] [PubMed] [Google Scholar]
- 151. Webb P, Nguyen P, Valentine C, et al. The estrogen receptor enhances AP‐1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol. 1999;13:1672–1685. [DOI] [PubMed] [Google Scholar]
- 152. McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. [DOI] [PubMed] [Google Scholar]
- 153. Saville B, Wormke M, Wang F, et al. Ligand‐, cell‐, and estrogen receptor subtype (alpha/beta)‐dependent activation at GC‐rich (Sp1) promoter elements. J Biol Chem. 2000;275:5379–5387. [DOI] [PubMed] [Google Scholar]
- 154. Safe S, Kim K. Non‐classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein‐1 signaling pathways. J Mol Endocrinol. 2008;41:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. DeNardo DG, Cuba VL, Kim H, Wu K, Lee AV, Brown PH. Estrogen receptor DNA binding is not required for estrogen‐induced breast cell growth. Mol Cell Endocrinol. 2007;277:13–25. [DOI] [PubMed] [Google Scholar]
- 156. Li X, Nott SL, Huang Y, et al. Gene expression profiling reveals that the regulation of estrogen‐responsive element‐independent genes by 17 beta‐estradiol–estrogen receptor beta is uncoupled from the induction of phenotypic changes in cell models. J Mol Endocrinol. 2008;40:211–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Glidewell‐Kenney C, Hurley LA, Pfaff L, Weiss J, Levine JE, Jameson JL. Nonclassical estrogen receptor alpha signaling mediates negative feedback in the female mouse reproductive axis. Proc Natl Acad Sci USA. 2007;104:8173–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Christian CA, Glidewell‐Kenney C, Jameson JL, Moenter SM. Classical estrogen receptor α signaling mediates negative and positive feedback on gonadotropin‐releasing hormone neuron firing. Endocrinology. 2008;149:5328–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. McDevitt MA, Glidewell‐Kenney C, Jimenez MA, et al. New insights into the classical and non‐classical actions of estrogen: evidence from estrogen receptor knock‐out and knock‐in mice. Mol Cell Endocrinol. 2008;290:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Hewitt SC, Li L, Grimm SA, et al. Novel DNA motif binding activity observed in vivo with an estrogen receptor α mutant mouse. Mol Endocrinol. 2014;28:899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Schwabe JW, Chapman L, Rhodes D. The oestrogen receptor recognizes an imperfectly palindromic response element through an alternative side‐chain conformation. Structure. 1995;3:201–213. [DOI] [PubMed] [Google Scholar]
- 162. Ahlbory‐Dieker DL, Stride BD, Leder G, et al. DNA binding by estrogen receptor‐alpha is essential for the transcriptional response to estrogen in the liver and the uterus. Mol Endocrinol. 2009;23:1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Wang LH, Yang XY, Zhang X, Mihalic K, Xiao W, Farrar WL. The cis decoy against the estrogen response element suppresses breast cancer cells via target disrupting c‐fos not mitogen‐activated protein kinase activity. Cancer Res. 2003;63:2046–2051. [PubMed] [Google Scholar]
- 164. Wang LH, Yang XY, Zhang X, et al. Suppression of breast cancer by chemical modulation of vulnerable zinc fingers in estrogen receptor. Nat Med. 2004;10:40–47. [DOI] [PubMed] [Google Scholar]
- 165. Wang LH, Yang XY, Zhang X, et al. Disruption of estrogen receptor DNA‐binding domain and related intramolecular communication restores tamoxifen sensitivity in resistant breast cancer. Cancer Cell. 2006;10:487–499. [DOI] [PubMed] [Google Scholar]
- 166. Huang J, Li X, Yi P, Hilf R, Bambara RA, Muyan M. Targeting estrogen responsive elements (EREs): design of potent transactivators for ERE‐containing genes. Mol Cell Endocrinol. 2004;218:65–78. [DOI] [PubMed] [Google Scholar]
- 167. Nott SL, Huang Y, Kalkanoglu A, et al. Designer monotransregulators provide a basis for a transcriptional therapy for de novo endocrine‐resistant breast cancer. Mol Med. 2010;16:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Muyan M, Gupur G, Yasar P, et al. Modulation of estrogen response element‐driven gene expressions and cellular proliferation with polar directions by designer transcription regulators. PLoS ONE. 2015;10:e0136423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Cowley SM, Hoare S, Mosselman S, Parker MG. Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem. 1997;272:19858–19862. [DOI] [PubMed] [Google Scholar]
- 170. Pace P, Taylor J, Suntharalingam S, Coombes RC, Ali S. Human estrogen receptor beta binds DNA in a manner similar to and dimerizes with estrogen receptor alpha. J Biol Chem. 1997;272:25832–25838. [DOI] [PubMed] [Google Scholar]
- 171. Li X, Huang J, Yi P, Bambara RA, Hilf R, Muyan M. Single‐chain estrogen receptors (ERs) reveal that the ERalpha/beta heterodimer emulates functions of the ERalpha dimer in genomic estrogen signaling pathways. Mol Cell Biol. 2004;24:7681–7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Tremblay GB, Tremblay A, Labrie F, Giguère V. Dominant activity of activation function 1 (AF‐1) and differential stoichiometric requirements for AF‐1 and ‐2 in the estrogen receptor alpha–beta heterodimeric complex. Mol Cell Biol. 1999;19:1919–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Charn TH, Liu ET‐B, Chang EC, Lee YK, Katzenellenbogen JA, Katzenellenbogen BS. Genome‐wide dynamics of chromatin binding of estrogen receptors alpha and beta: mutual restriction and competitive site selection. Mol Endocrinol. 2010;24:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Liu Y, Gao H, Marstrand TT, et al. The genome landscape of ERalpha‐ and ERbeta‐binding DNA regions. Proc Natl Acad Sci USA. 2008;105:2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Monroe DG, Secreto FJ, Subramaniam M, Getz BJ, Khosla S, Spelsberg TC. Estrogen receptor alpha and beta heterodimers exert unique effects on estrogen‐ and tamoxifen‐dependent gene expression in human U2OS osteosarcoma cells. Mol Endocrinol. 2005;19:1555–1568. [DOI] [PubMed] [Google Scholar]
- 176. Chang EC, Charn TH, Park S‐H, et al. Estrogen receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol Endocrinol. 2008;22:1032–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–4842. [DOI] [PubMed] [Google Scholar]
- 178. Papoutsi Z, Zhao C, Putnik M, Gustafsson JA, Dahlman‐Wright K. Binding of estrogen receptor alpha/beta heterodimers to chromatin in MCF‐7 cells. J Mol Endocrinol. 2009;43:65–72. [DOI] [PubMed] [Google Scholar]
- 179. Li LC, Yeh CC, Nojima D, Dahiya R. Cloning and characterization of human estrogen receptor beta promoter. Biochem Biophys Res Commun. 2000;275:682–689. [DOI] [PubMed] [Google Scholar]
- 180. Zhang X, Leung Y‐K, Ho S‐M. AP‐2 regulates the transcription of estrogen receptor (ER)‐beta by acting through a methylation hotspot of the 0N promoter in prostate cancer cells. Oncogene. 2007;26:7346–7354. [DOI] [PubMed] [Google Scholar]
- 181. Zhu X, Leav I, Leung Y‐K, et al. Dynamic regulation of estrogen receptor‐beta expression by DNA methylation during prostate cancer development and metastasis. Am J Pathol. 2004;164:2003–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Post WS, Goldschmidt‐Clermont PJ, Wilhide CC, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. 1999;43:985–991. [DOI] [PubMed] [Google Scholar]
- 183. Kim J, Kim JY, Song KS, et al. Epigenetic changes in estrogen receptor beta gene in atherosclerotic cardiovascular tissues and in‐vitro vascular senescence. Biochim Biophys Acta. 2007;1772:72–80. [DOI] [PubMed] [Google Scholar]
- 184. Yang X, Yan L, Davidson NE. DNA methylation in breast cancer. Endocr Relat Cancer. 2001;8:115–127. [DOI] [PubMed] [Google Scholar]
- 185. Zhao C, Lam EW‐F, Sunters A, et al. Expression of estrogen receptor beta isoforms in normal breast epithelial cells and breast cancer: regulation by methylation. Oncogene. 2003;22:7600–7606. [DOI] [PubMed] [Google Scholar]
- 186. Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25:869–898. [DOI] [PubMed] [Google Scholar]
- 187. Sommer S, Fuqua SA. Estrogen receptor and breast cancer. Semin Cancer Biol. 2001;11:339–352. [DOI] [PubMed] [Google Scholar]
- 188. Leygue E, Murphy LC. A bi‐faceted role of estrogen receptor beta in breast cancer. Endocr Relat Cancer. 2013;20:R127–R139. [DOI] [PubMed] [Google Scholar]
- 189. Koehorst SGA, Cox JJ, Donker GH, et al. Functional analysis of an alternatively spliced estrogen receptor lacking exon 4 isolated from MCF‐7 breast cancer cells and meningioma tissue. Mol Cell Endocrinol. 1994;101:237–245. [DOI] [PubMed] [Google Scholar]
- 190. Fuqua SA, Fitzgerald SD, Allred DC, et al. Inhibition of estrogen receptor action by a naturally occurring variant in human breast tumors. Cancer Res. 1992;52:483–486. [PubMed] [Google Scholar]
- 191. Fuqua SA, Fitzgerald SD, Chamness GC, et al. Variant human breast tumor estrogen receptor with constitutive transcriptional activity. Cancer Res. 1991;51:105–109. [PubMed] [Google Scholar]
- 192. Omoto Y, Iwase H, Iwata H, et al. Expression of estrogen receptor alpha exon 5 and 7 deletion variant in human breast cancers. Breast Cancer. 2000;7:27–31. [DOI] [PubMed] [Google Scholar]
- 193. García Pedrero JM, Zuazua P, Martínez‐Campa C, Lazo PS, Ramos S. The naturally occurring variant of estrogen receptor (ER) ERDeltaE7 suppresses estrogen‐dependent transcriptional activation by both wild‐type ERalpha and ERbeta. Endocrinology. 2003;144:2967–2976. [DOI] [PubMed] [Google Scholar]
- 194. Denger S, Reid G, Kos M, et al. ERalpha gene expression in human primary osteoblasts: evidence for the expression of two receptor proteins. Mol Endocrinol. 2001;15:2064–2077. [DOI] [PubMed] [Google Scholar]
- 195. Kos M, Denger S, Reid G, Korach KS, Gannon F. Down but not out? A novel protein isoform of the estrogen receptor alpha is expressed in the estrogen receptor alpha knockout mouse. J Mol Endocrinol. 2002;29:281–286. [DOI] [PubMed] [Google Scholar]
- 196. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor‐alpha36, a novel variant of human estrogen receptor‐alpha66. Biochem Biophys Res Commun. 2005;336:1023–1027. [DOI] [PubMed] [Google Scholar]
- 197. Rao J, Jiang X, Wang Y, Chen B. Advances in the understanding of the structure and function of ER‐α36, a novel variant of human estrogen receptor‐alpha. J Steroid Biochem Mol Biol. 2011;127:231–237. [DOI] [PubMed] [Google Scholar]
- 198. Xu L, Pan‐Hammarstrom Q, Forsti A, et al. Human estrogen receptor beta 548 is not a common variant in three distinct populations. Endocrinology. 2003;144:3541–3546. [DOI] [PubMed] [Google Scholar]
- 199. Wilkinson HA, Dahllund J, Liu H, et al. Identification and characterization of a functionally distinct form of human estrogen receptor beta. Endocrinology. 2002;143:1558–1561. [DOI] [PubMed] [Google Scholar]
- 200. Ogawa S, Inoue S, Watanabe T, et al. Molecular cloning and characterization of human estrogen receptor betacx: a potential inhibitor of estrogen action in human. Nucleic Acids Res. 1998;26:3505–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Peng B, Lu B, Leygue E, Murphy LC. Putative functional characteristics of human estrogen receptor‐beta isoforms. J Mol Endocrinol. 2003;30:13–29. [DOI] [PubMed] [Google Scholar]
- 202. Leung Y‐K, Mak P, Hassan S, Ho S‐M. Estrogen receptor (ER)‐beta isoforms: a key to understanding ER‐beta signaling. Proc Natl Acad Sci. 2006;103:13162–13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Moore JT, McKee DD, Slentz‐Kesler K, et al. Cloning and characterization of human estrogen receptor beta isoforms. Biochem Biophys Res Commun. 1998;247:75–78. [DOI] [PubMed] [Google Scholar]
- 204. Giuliano M, Trivedi MV, Schiff R. Bidirectional crosstalk between the estrogen receptor and human epidermal growth factor receptor 2 signaling pathways in breast cancer: molecular basis and clinical implications. Breast Care. 2013;8:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Levin ER. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol. 2003;17:309–317. [DOI] [PubMed] [Google Scholar]
- 206. Treviño LS, Weigel NL. Phosphorylation: a fundamental regulator of steroid receptor action. Trends Endocrinol Metab. 2013;24:515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Anbalagan M, Huderson B, Murphy L, Rowan BG. Post‐translational modifications of nuclear receptors and human disease. Nucl Recept Signal. 2012;10:e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Le Romancer M, Poulard C, Cohen P, Sentis SP, Renoir JM, Corbo L. Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr Rev. 2011;32:597–622. [DOI] [PubMed] [Google Scholar]
- 209. Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone‐regulated cancer therapy. Nat Rev Cancer. 2014;14:26–38. [DOI] [PubMed] [Google Scholar]
- 210. Leung YK, Lee MT, Lam HM, Tarapore P, Ho SM. Estrogen receptor‐beta and breast cancer: translating biology into clinical practice. Steroids. 2012;77:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]