

Abstract
Background and Purpose
Elevated angiotensin II (Ang II) and sympathetic activity contributes to a high risk of ventricular arrhythmias in heart disease. The rapidly activating delayed rectifier K+ current (I Kr) carried by the hERG channels plays a critical role in cardiac repolarization, and decreased I Kr is involved in increased cardiac arrhythmogenicity. Stimulation of α1A‐adrenoreceptors or angiotensin II AT1 receptors is known to inhibit I Kr via PKC. Here, we have identified the PKC isoenzymes mediating the inhibition of I Kr by activation of these two different GPCRs.
Experimental Approach
The whole‐cell patch‐clamp technique was used to record I Kr in guinea pig cardiomyocytes and HEK293 cells co‐transfected with hERG and α1A‐adrenoreceptor or AT1 receptor genes.
Key Results
A broad spectrum PKC inhibitor Gö6983 (not inhibiting PKCε), a selective cPKC inhibitor Gö6976 and a PKCα‐specific inhibitor peptide, blocked the inhibition of I Kr by the α1A‐adrenoreceptor agonist A61603. However, these inhibitors did not affect the reduction of I Kr by activation of AT1 receptors, whereas the PKCε‐selective inhibitor peptide did block the effect. The effects of angiotensin II and the PKCε activator peptide were inhibited in mutant hERG channels in which 17 of the 18 PKC phosphorylation sites were deleted, whereas a deletion of the N‐terminus of the hERG channels selectively prevented the inhibition elicited by A61603 and the cPKC activator peptide.
Conclusions and Implications
Our results indicated that inhibition of I Kr by activation of α1A‐adrenoreceptors or AT1 receptors were mediated by PKCα and PKCε isoforms respectively, through different molecular mechanisms.
Abbreviations
- Ang II
angiotensin II
- IKr
rapidly activating delayed rectifier K+ current
Introduction
The human ether‐a‐go‐go‐related gene (hERG) encodes the pore‐forming subunit of the channel (Kv11.1) responsible for the rapidly activating delayed rectifier potassium current (I Kr) (Sanguinetti et al., 1995). Outward I Kr is an important determinant of cardiac action potential repolarization, and a reduction of the current has been implicated in inherited and acquired long QT syndrome (LQT), which is characterized by a prolongation in action potential duration and QT interval on the surface ECG, and an increased risk for ‘torsades de pointes’ ventricular arrhythmias and sudden cardiac death (Marban, 2002). Recent studies have revealed that hERG channels are modulated by GPCRs including the angiotensin II AT1 receptors and α‐adrenoceptors through the intracellular second messengers, such as cAMP, protein kinase A (PKA) (Cui et al., 2000) and PKC (Thomas et al., 2003; Cockerill et al., 2007). Our previous study has shown that stimulation of AT1 receptors produces an acute inhibitory effect on I Kr/hERG currents via a PKC pathway in guinea pig ventricular myocytes and HEK293 cells (Wang et al., 2008). Meanwhile, activation of α1A‐adrenoceptors also results in an instant inhibition of I Kr/hERG current through PKC signalling (Thomas et al., 2004; Urrutia et al., 2016). Accordingly, hERG channels represent a target for modulation by autonomic neurotransmitters and hormones, and thus, PKC may become a candidate mediator of the pathological cardiac electrophysiological remodelling. It is known that sympathetic overactivity and elevated levels of angiotensin II (Ang II) are major contributors to many cardiac diseases including arrhythmias (Chen et al., 2001). Ang II inhibitors (including angiotensin‐converting enzyme inhibitors or AT1 receptor antagonists) significantly decrease sudden cardiac death in patients with heart failure (Brooksby et al., 1999; Lindholm et al., 2003), and α1‐adrenoreceptor blockade has an anti‐arrhythmic effect in an animal model of LQT2 (Mow et al., 2015). Blockade of the reduction of I Kr/hERG current by Ang II inhibitors and α1A‐adrenoreceptor antagonists may contribute to their beneficial effects.
Both α1A‐adrenoreceptors and AT1 receptors produce effects through the Gq‐coupled PKC pathway. However, it has been noted that the modulation of the hERG channels by these two GPCRs shows a different mode of action. The activation of α1A‐adrenoreceptors shifts the voltage‐dependent activation curve to the right (Thomas et al., 2004; Urrutia et al., 2016), whereas AT1 receptor agonists do not (Wang et al., 2008). It is known that PKCs can be classified into three subgroups based on their structure and activation requirements. The conventional PKCs (cPKC including α, βI, βII and γ) require calcium, DAG and phosphatidylserine (PS) for activation. Novel PKCs (including δ, ε, π and θ) can be activated by DAG and PS but are not sensitive to calcium. Atypical PKCs (including ζ and ι/λ) are unresponsive to calcium or DAG but require PS for activation (Ron and Kazanietz, 1999). Many PKC isoenzymes have been found in the heart in different animal species (Takeishi et al., 1999; Zhang et al., 2013). There is accumulating evidence to indicate that PKC isoforms in the heart are differentially regulated by various stimuli under physiological and pathological conditions. The individual PKC isoforms have distinct roles on channel activity (Makary et al., 2011; Radresa et al., 2014). Thus, it is possible that activation of α1A‐adrenoreceptors or AT1 receptors may modulate the channel through distinct PKC isoforms, which produce different effects on voltage‐dependent activation of hERG channels.
To address the above hypothesis, the present study was designed to investigate the effects of the α1A‐adrenoreceptor selective agonist A61603 (Knepper et al., 1995) and Ang II on I Kr/hERG currents in native cardiomyocytes and a heterologous expression system, in order to identify the PKC isoforms involved and the molecular mechanism(s) involved in their specific regulatory actions on hERG channels, using the whole‐cell patch‐clamp technique and Western blots.
Methods
Animals
All animal care and experimental procedures were approved by the Animal Care and Ethical Committee of Hebei Medical University (Shijiazhuang, China). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).The experiments were performed in the Department of Pharmacology, Hebei Medical University. Before experiments, animals were housed under controlled conditions at 25°C in a 12 h light/dark cycle with ad libitum access to water and food.
Guinea pig ventricular cardiomyocyte isolation
Twelve male guinea pigs (weight 200–250 g, 6–8 weeks old) were supplied by the Experimental Animal Center of Hebei Medical University. Single ventricular myocytes were enzymically dissociated from the hearts of adult guinea pigs, as described previously (Wang et al., 2008). Animals were injected with heparin (1.0 U·kg−1) and were anaesthetized with pentobarbital sodium (30–35 mg·kg−1) by i.p. injection. Hearts were excised and retrogradely perfused with Ca2+‐free modified Tyrode solution composed of (in mM) NaCl, 140; KCl, 5.4; MgCl2, 1; HEPES, 10 and glucose, 10 (pH 7.4 with NaOH). After 5 min of perfusion, the solution was switched to one containing Type II collagenase (Worthington Biochemical Corporation, Lakewood, NJ, USA, 0.4 mg·mL−1), and hearts were removed after 10–15 min of perfusion from the perfusion apparatus. Left ventricular free wall was cut into small pieces in high K+ solution, which contained (in mM) KOH, 80; KCl, 40; KH2PO4, 25; MgSO4, 3; glutamic acid, 50; taurine, 20; EGTA, 0.5; HEPES, 10 and glucose, 10 (pH 7.3 with KOH). Single cardiomyocytes were harvested and used for electrophysiological recording within 6–8 h after isolation.
Transfection of hERG and GPCR cDNA
The hERG‐WT cDNA plasmid (kindly provided by Dr. Chiamvimonvat Nipavan from the University of California, Davis, CA, USA) was transfected into HEK293 cells by the lipofectamine method. A stably transfected hERG‐HEK cell line was built up by G418 treatment.HEK cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin solution and kept at 37°C in a 5% CO2 incubator. The hERG‐△PKC cDNA (in which 17 of the 18 serine or threonine residues of the PKC phosphorylation sites were mutated to alanine, kindly provided by Dr Dierk Thomas from Department of Cardiology, Medical University Hospital Heidelberg (Bergheimerstrasse, Germany), hERG‐NTK cDNA (a mutant whose 2–354 amino acids containing the PAS domain and T74 have been removed, kindly provided by Michael C Sanguinetti, University of Utah (Salt Lake City, UT, USA), GFP, α1A‐adrenoreceptor, and AT1 receptor cDNA (Invitrogen, Grand Island, NY, USA) were transiently transfected using Lipofectamine 2000 Reagent kit (Invitrogen). GFP‐positive cells were identified using the epifluorescence system and studied within 24–48 h of transfection.
Patch‐clamp recordings
The I Kr/hERG currents were recorded using the conventional whole‐cell patch‐clamp technique (Hamill et al., 1981). Borosilicate glass electrodes had tip resistances of 1–3 MΩ when filled with the pipette solution. Uncompensated capacitance currents in response to small hyperpolarizing voltage steps were recorded for offline integration, as a means of measuring cell capacitance. All experiments were performed at room temperature (22–25°C) using an Axon patch 700B amplifier and pClamp 10.2 software (Molecular Devices, USA). The electrical signals were sampled at 2.5–10 kHz and filtered at 1 kHz using a low‐pass filter and digitized with an A/D converter (Digidata 1322; Molecular Devices).
Western blot analysis
Isolated ventricular myocytes were used to perform immunoblots by using whole cell homogenates. The whole‐cell protein (50 μg) was denatured and fractionated on 8% SDS‐PAGE and then transferred electrophoretically to Immobilon‐P polyvinylidene fluoride membranes. Membranes were blocked with 3% BSA for 2 h and incubated with primary antibodies overnight at 4°C. After being washed and reblocked, membranes were incubated with goat anti‐rabbit (1:5000; Rockland Immunochemicals, Philadelphia, USA) secondary antibodies. Quantification of the signals was performed by Odyssey Infrared Imaging System (LICOR 9120; Li‐COR, Lincoln, NE, USA). The protein bands were normalized to the GAPDH band in each sample. The value was then averaged from all the different sets of experiments. The primary antibodies, anti‐p‐PKCα and anti‐p‐PKCε (Abcam, USA), were used, and anti‐GAPDH (Santa Cruz, USA) was used as internal loading control.
Solutions and chemicals
For I Kr recordings in ventricular myocytes, the external solution contained (in mM) NaCl, 132; KCl, 4; CaCl2, 1.8; MgCl2, 1.2; glucose, 5 and HEPES, 10 (pH 7.4 with NaOH). The pipette solution contained (in mM) CaCl2, 0.5; KCl, 140; Mg‐ATP, 4; MgCl2, 1; EGTA, 5 and HEPES 10 (pH 7.2 with KOH). The concentration of free Ca2+ in the pipette solution was calculated to be approximately 1.7 × 10−8 M. It should be noted that conventional PKC isoforms can be functionally activated by the presence of intracellular free Ca2+ concentration on the order of 10−8 M in guinea pig cardiac myocytes (Hool, 2000). Nimodipine (1 μM) was added to the external solution to block the L‐type Ca2+ current. Na+ and T‐type Ca2+ currents were inactivated by holding potential of −40 mV. The slowly activating delayed rectifier potassium current (I Ks) was blocked by the addition of 20 μM chromanol 293B. For hERG recording in HEK cells, the external solution contained (in mM) NaCl, 140; KCl, 5.4; MgCl2, 1; CaCl2, 2; glucose, 10 and HEPES, 10 (pH 7.4 with NaOH). The pipette solution was the same as that for recording I Kr in ventricular myocytes.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Blinding was not used since all the measurements were not subjective but strictly quantitative data. To avoid bias, the raw data were assessed independently by two co‐authors to ensure the correctness of the measurements. All data are shown as means ± SEM. Data were normalized to eliminate the differences in basal current amplitudes recorded from different cells, and in some cases, currents were normalized to the maximal tail current in a given cell to study the voltage‐dependent effects. The currents were analysed and fitted using Clampfit 10.2 (Molecular device, USA). The concentration–response curve was fitted with the logistic equation: y = A2 + (A1–A2)/[1 + (x/x0)nH], where y is the response, A1 and A2 are the maximum and minimum response, respectively, x is the drug concentration and nH is the Hill coefficient. The current activation curves were generated by plotting the normalized tail current amplitudes against the step potentials and were fitted with a Boltzmann function: y = A/{1 + exp[(Vh–Vm)/k]}. Here, A is the maximum current amplitude, Vh is the voltage for half‐maximal activation, Vm is the test potential and k is the slope factor. Group sizes are equal by design. However, the cell number varied in different treatment groups due to some failures in patch clamp recordings in the study. Graphical and statistical analysis was carried out using ORIGIN8.1 (OriginLab Corporation, USA) software. Group comparisons were performed with unpaired Student's t‐tests (for single two‐group comparisons) and ANOVA with Dunnett's post hoc tests (for multiple‐group comparisons). The differences were considered significant at P < 0.05.
Materials
Ang II (Sigma Aldrich, Saint Louis, MO, USA) was prepared as a 1 mM stock solution in water, and A61603 (Sigma) was prepared as a 10 mM stock solution in water. Chromanol 293B (Sigma) was prepared as a 50 mM stock solution in DMSO. Nimodipine (Sigma) was prepared as a 50 mM stock solution in DMSO. E‐4031 (Sigma) was prepared as a 1 mM stock in water. WB4101 (Sigma) was prepared as a 10 mM stock solution in water. All of the stock solutions were stored at −20°C. H‐89 and Bis‐1 were supplied by Sigma‐Aldrich. The highest final concentration of DMSO in external solution was ≤0.1%, a concentration that had no effect on I Kr. The PKCα inhibitor peptide (αC2‐4, SLNPEYRQT), PKCε inhibitor peptide (εV1‐2, EAVSLKPT), cPKC activator peptide (cPKC‐AP, SVEIWD), PKCε activator peptide (ε‐AP, HDAPIGYD) and scrambled peptides were synthesized by AngTai Biotechnology Company (HangZhou, China) and prepared as 100 μM stock solutions in water and stored at −20°C.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Effect of α1A‐adrenoreceptor stimulation on IKr current in guinea pig ventricular myocytes
The protocol for I Kr recording has been described previously (Wang et al., 2008). Briefly, ventricular myocytes were depolarized from a holding potential of −40 mV to various prepulse potentials of −40 to +60 mV for 2 s and repolarized to −40 mV to evoke outward tail currents in the presence of I Ks blocker, chromanol 293B (20 μM). The tail currents were abolished by a specific I Kr blocker, E‐4031 (2 μM) (Figure 1A). I Kr was recorded in the control conditions and after application of A61603 in the bath. The results showed that A61603 significantly reduced the tail currents during repolarization (Figure 1A). The time course of the inhibitory action of A61603 on I Kr was determined by measuring the amplitude of tail currents elicited on repolarization to a test potential of −40 mV after a 2 s prepulse potential of 40 mV every minute (Figure 1B). The reduction of I Kr tail current occurred rapidly and reached saturation about 10 min after the addition of A61603 (1 μM) to the bath. The inhibitory effect of A61603 on I Kr was not reversible after washout. The voltage dependence of the activation is shown in Figure 1C. At more positive potentials (from −10 to 60 mV), A61603 showed a significant inhibition of the tail currents. The percentage inhibition of I Kr current amplitude was calculated and plotted against concentrations of A61603 (Figure 1D). The mean data were well described by a logistic equation with an IC50 value of 185 nM.
Figure 1.
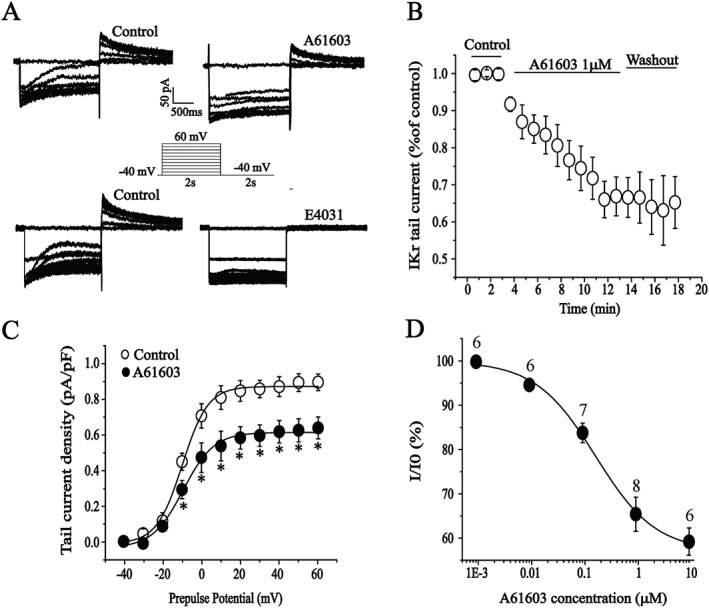
Effects of the α1A‐adrenoreceptor agonist A61603 on IKr in isolated guinea pig ventricular cardiomyocytes. (A) Representative current traces recorded under pulse protocol shown in inset before (left) and after application of A61603 (1 μM) or E‐4031 (2 μM) (right). (B) The time course of the I Kr current reduction by A61603. n = 8. (C) The effect of A61603 on I Kr was quantitatively evaluated by measuring the percentage of decrease in the tail currents (n = 8). *P < 0.05, significantly different from control. (D) The concentration–response curve for the effect of A61603; the IC50 = 185 nM. The number above the data points represents the cell numbers assayed at each concentration. All of the cardiomyocytes were taken from five hearts.
To confirm that the I Kr response to A61603 was not the result of a direct channel block, we tested the effect of A61603 on the I Kr tail current in the presence of the selective α1A‐adrenoreceptor antagonist WB4101 (O‐Uchi et al., 2008). As illustrated in Figure 2, WB4101 (200 nM) abolished the inhibitory effect of A61603, confirming the involvement of α1A‐adrenoreceptors in its action. Activation of α1A‐adrenoreceptors is classically coupled to proteins and signals through phospholipase C, Ca2+ and PKC pathway. To test whether α1A‐adrenoreceptor‐dependent depression of I Kr current was mediated by PKC, the non‐specific PKC inhibitor Bis‐1 (300 nM) or PKA inhibitor H‐89 (10 μM) was applied to the bath for 5 min and then co‐applied with A61603 (1 μM). The inhibitory effects of A61603 on I Kr were significantly attenuated by pretreatment of ventricular myocytes with Bis‐1, but not H‐89. The results suggest an important role for PKC in I Kr modulation by A61603. With H‐89, there was a trend towards a delay in the time course of the inhibitory action of A61603 and a reduction in the overall inhibition produced by this agent (Figure 2), but these effects did not reach the level of statistical significance. Previous studies have indicated that in addition to PKC, PKA is also involved in the regulation of hERG by α1A‐adrenoreceptor activation (Thomas et al., 2004; Wang et al., 2009). Therefore, while inhibition of hERG by activation of these receptors appears to be primarily mediated through PKC in ventricular myocytes, it remains possible that there is a small contribution of PKA to this effect.
Figure 2.
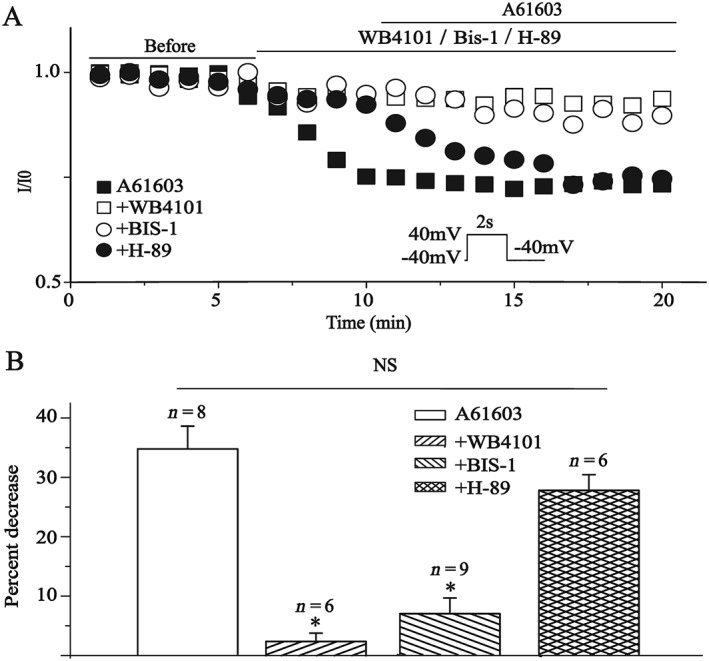
Effects of α1A‐adrenoreceptor antagonist WB4101, PKC inhibitor Bis‐1 and PKA inhibitor H‐89 on the action of A61603 in isolated guinea pig ventricular cardiomyocytes. (A) Time course of I Kr tail current amplitude measured at +40 mV prepulse after application of A61603 (1 μM) or co‐application with WB4101 (200 nM), Bis‐1 (300 nM) or H‐89 (10 μM). (B) Summary data for the percentage decrease in I Kr tail current amplitude. *P < 0.05, significantly different from A61603. All of the cardiomyocytes were extracted from five hearts.
PKC isoenzymes involved in the inhibitory effects of A61603 and Ang II on IKr
By using selective PKC inhibitors, we examined the PKC isoenzyme(s) mediating the effect of A61603. The results showed that the action of A61603 was significantly blocked when ventricular myocytes were pre‐incubated with Gö6976 (100 nM, selectively inhibiting PKCα, β and γ) or Gö6983 (100 nM, selectively inhibiting PKCα, β, γ, δ and ζ) (Figure 3A, B). The results implied that the cPKCs were involved. We further tested the effect of isoenzyme‐selective inhibitor peptides (Dorn et al., 1999; Hellberg et al., 2009), which were added into the pipette solution and allowed to diffuse into the cytosol for 10 min after a whole cell recording was established. As shown in Figure 3A, B, the PKCα inhibitor peptide αC2‐4 (200 nM) completely antagonized the I Kr inhibition of A61603. On the contrary, the PKCε inhibitor peptide (εV1‐2) had no effect. The results suggest that the PKCα isoform mediates the inhibitory effect of α1A‐adrenoreceptor activation on I Kr.
Figure 3.
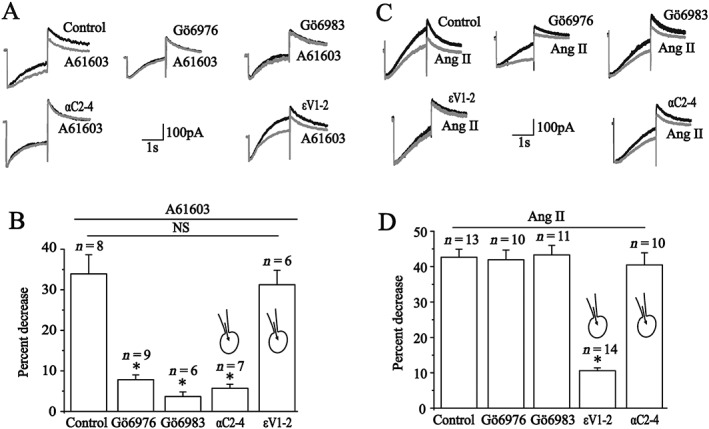
Effects of PKC inhibitors and isoenzyme‐specific inhibitor peptides on the action of A61603 and Ang II in isolated guinea pig ventricular cardiomyocytes. (A, C) Representative current traces of IKr recorded under a depolarizing prepulse +40 mV for 2 s and then repolarizing to −40 mV in the presence of Gö6976, Gö6983 or the PKCα and PKCε inhibitor peptides before (black) and after application of 1 μM A61603 (A, grey) or 100 nM Ang II (C, grey). (B) Summary data for the percentage decrease obtained from A. *P < 0.05, significantly different from control. (D) Summary data for the percentage decrease obtained from C. *P < 0.05, significantly different from control). All of cardiomyocytes were taken from five hearts.
Similar to α1A‐adrenoreceptors, the AT1 receptors are also a Gq‐coupled receptor, and we previously reported that Ang II inhibited I Kr in ventricular myocytes via a AT1 receptor‐PKC pathway (Wang et al., 2008). Here, we tested the effects of selective PKC inhibitors and found that neither Gö6976 (100 nM) nor Gö6983 (100 nM) significantly affected the inhibition of I Kr by Ang II (100 nM) (Figure 3C, D). The result indicated that the cPKCs were not implicated in this process. Furthermore, the inhibitory effect of Ang II on I Kr was significantly reduced when PKCε inhibitor peptide (εV1‐2) (200 nM) was pre‐added into the pipette solution, but the PKCα inhibitor peptide (αC2‐4, 200 nM) did not affect the inhibitory effect of Ang II on I Kr (Figure 3C, D). These findings pointed to an important role of PKCε in mediating the inhibitory effect of Ang II on I Kr.
These patch‐clamp recordings supported the notion that the PKCα and PKCε isoenzymes mediated the inhibition of I Kr by activation of α1A‐adrenoreceptors or AT1 receptors respectively. Subsequently, active forms of PKCα or PKCε protein levels were quantified by Western blotting. It is known that PKC activity is dependent on its phosphorylation status, and studies have shown that phosphorylation of the hydrophobic motif of PKC regulates the enzyme's stability, phosphatase sensitivity, subcellular localization and catalytic function (Bornancin and Parker, 1997). Accordingly, anti‐phospho‐PKC antibodies have been used to demonstrate PKC activation following cell stimulation (Walker and Plows, 2003). In this study, whole cell protein extracts were obtained from freshly isolated ventricular myocytes, and phospho‐PKC antibodies were used to detect the activated PKC isoenzymes. As shown in Figure 4, the abundance of p‐PKCα (phospho‐S657) was increased after ventricular myocytes were incubated with A61603 (1 μM) for 10 min, but there was no change in p‐PKCε levels. While Ang II (100 nM) significantly increased p‐PKCε (phospho‐S729) and p‐PKCα, there was a greater enhancement for p‐PKCε than for p‐PKCα. These molecular biological results provide evidence that stimulation of α1A‐adrenoreceptors selectively activated the PKCα isoenzyme, whereas activation of AT1 receptors activated PKCε more potently than the PKCα isoenzyme.
Figure 4.
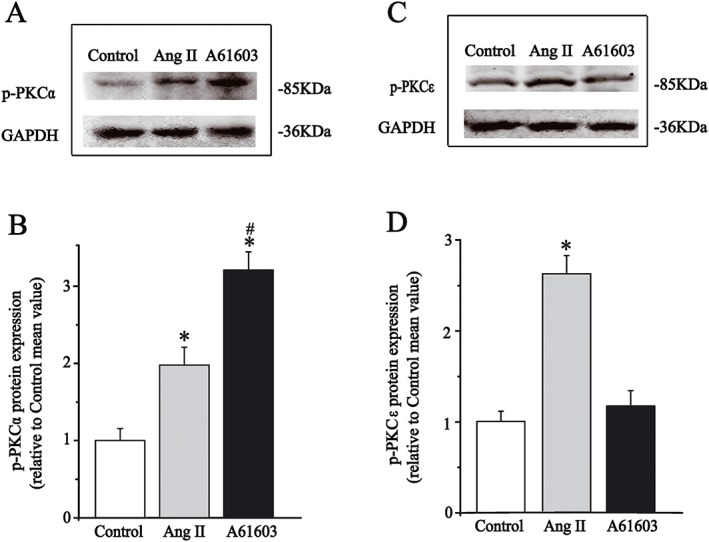
Effects of A61603 and Ang II on the expression of phosphorylated PKC isoforms in isolated guinea pig ventricular cardiomyocytes. (A, C) Representative immunoblots for phosphorylated PKCα (left) and PKCε (right) along with internal standard GAPDH after application of A61603 or Ang II for 10 min. (B, D) Summary data for expression levels of phosphorylated PKC isoforms that were presented as fold change compared to control group mean, and the quantification of the band intensities were normalized with GAPDH. All data were obtained from five guinea pig hearts, *P < 0.05, significantly different from control; # P < 0.05, significantly different from Ang II.
Effects of activating specific PKC isoenzyme on hERG current in a heterologous expression system
Up to now, little is known about the specific regulatory effect on the hERG channels by directly activating individual PKC isoenzymes (Radresa et al., 2014). By using PKC‐specific activator peptides, we observed the action of direct activation of cPKC by pseudo‐RACK1 (Ron et al., 1995) and PKCε by pseudo‐ε RACK (Dorn et al., 1999) on hERG channels in HEK293 cells. Peptides were added to the pipette solution, and the tail current amplitudes were measured at 10 min after obtaining stable recordings. Corresponding scrambled peptides were also used to test non‐specific action of the peptides on the hERG current. As shown in Figure 5A, the hERG current was elicited from a holding potential of −80 mV to prepulses from −60 to 50 mV for a 4 s duration and was followed by a test pulse to −50 mV to evoke large, slowly decaying outward tail currents. Compared to cells treated with respective scrambled peptide, the hERG currents were decreased in the presence of cPKC or PKCε activator peptides (200 nM). The current–voltage curves showed that both peptides inhibited the tail current with a significantly higher potency at more positive potentials (Figure 5B, D). Meanwhile, the half‐maximal activation voltage (V1/2) of the currents was shifted from −16.7 to −9.0 mV in the presence of the cPKC activator peptide (Figure 5C), but the PKCε activator peptide did not alter the V1/2 of the hERG channels (Figure 5E). There were no differences in hERG currents between the absence and presence of scrambled peptides (data not shown). These findings indicate that cPKC and PKCε activator peptides have different effects on the voltage‐dependent activation of the hERG channels, although they both reduce this current. To exclude the possibility that data taken from two groups of cells brought error because of variable current densities, we also studied the effect of these PKC activator peptides by recording the current from the same cell at different times after whole cell break‐in (Supporting Information Figure S1). A decline in the current densities was observed as the cPKC or PKCε activator peptides diffused into the cells and was absent in cells treated with the scrambled peptides. These results confirm that these activator peptides inhibited the hERG current.
Figure 5.
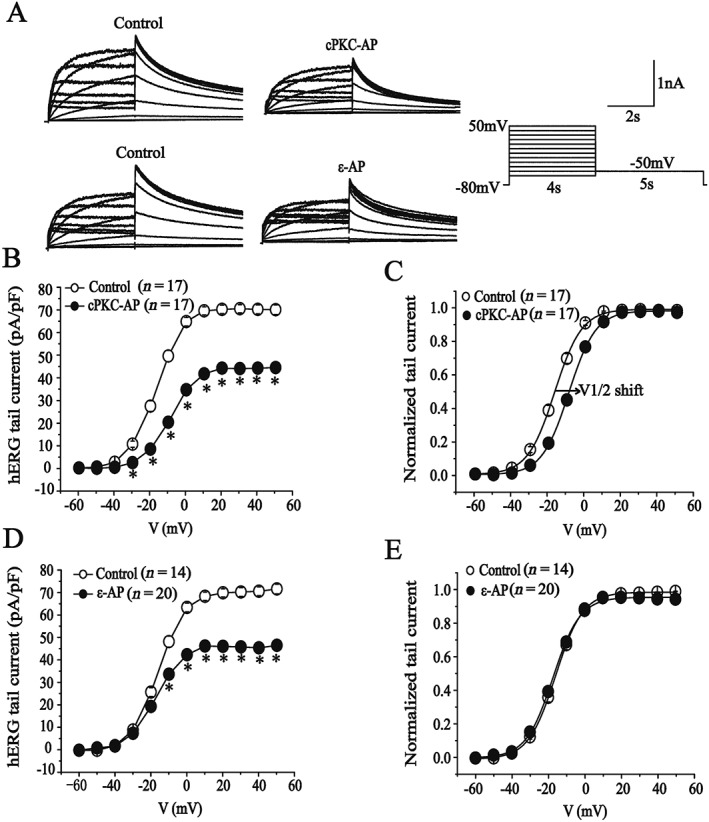
Effects of cPKC and PKCε‐specific activator peptides on hERG‐WT current. (A) Representative current traces showing the effects of cPKC (cPKC‐AP) and PKCε activator peptides (ε‐AP). (B, D) I–V relationships for tail currents in the presence of scrambled peptides (control) or cPKC activator peptide (200 nM). *P < 0.05, significantly different from control and corresponding voltage dependence of activation. (C, E) I–V relationships for tail currents in the control and the presence of the PKCε activator peptide (200 nM). *P < 0.05, significantly different from control) and corresponding voltage dependence of activation.
Then we investigated the effects of A61603 and Ang II on hERG currents in HEK cells co‐expressing hERG cDNA and α1A‐adrenoreceptor cDNA or AT1 receptor cDNA. Supporting Information Figure S2 shows that the effect of A61603 was similar to the effect of cPKC activator peptide. It inhibited the hERG current at voltage range from −30 to 50 mV and shifted V1/2 from −16.8 to −11.8 mV (P < 0.05). Similar responses to A61603 and the cPKC activator peptide suggest that the inhibition of hERG channels produced by α1A‐adrenoreceptor activation is mediated primarily through the PKCα isoform. In addition, Ang II exhibited a similar action to the PKCε activator peptide on the hERG current, that is, no effect on the voltage‐dependent activation of the channel, although the hERG current was decreased. These results indicate that PKCε mediates the inhibitory effects of Ang II on the hERG current.
Molecular mechanism underlying the modulation on hERG channels by PKCα and PKCε isoenzymes
A frequently observed mechanism of regulating ion channel function for PKC isoforms is direct phosphorylation of the ion channel (Dai et al., 2009; Ferreira et al., 2012). There are 18 PKC‐specific phosphorylation sites in the hERG channels protein (Thomas et al., 2003). First, we used a mutant channel hERG‐△PKC, in which 17 (except T74) of the 18 serine or threonine residues of the PKC phosphorylation sites were mutated to alanine (Thomas et al., 2003), to investigate how PKCα and PKCε isoenzymes modulated the hERG channels. The half‐maximal activation voltage in hERG‐△PKC was similar to that in hERG‐WT (data not shown).The cPKC activator peptide significantly depressed the hERG‐△PKC tail current in the potential range from −30 to +50 mV (Figure 6A, B). However, the PKCε activator peptide did not affect the currents (Figure 6C). The percentage inhibition of the current by cPKC and PKCε activator peptides were calculated at 0 mV voltage for hERG‐WT and hERG‐△PKC channels and are shown in Figure 6D. There was no difference for in the effects of the cPKC activator peptide between WT and mutant currents , whereas the inhibitory action of PKCε activator peptide on the hERG‐△PKC current was significantly less than on the WT current. Next, we observed the effects of the activator peptides in hERG‐NTK, a mutant whose 2‐354 amino acids containing the PAS domain and T74 have been removed (Thomas et al., 2003).The hERG‐NTK transiently expressed in HEK293 cells had functional currents with fast deactivation kinetics and similar activation half voltage compared to hERG‐WT (Cockerill et al., 2007).The cPKC activator peptide failed to inhibit hERG‐NTK tail current (Figure 6E, F). However, the PKCε activator peptide significantly depressed hERG‐NTK tail current at the potential range from −10 to +50 mV (Figure 6G). The percentage inhibition of the hERG‐WT and hERG‐NTK current by cPKC and PKCε activator peptides were calculated at 0 mV voltage and shown in Figure 6H. There was no significant difference for the PKCε activator peptide (41.3% for hERG‐NTK vs. 33.1% for hERG‐WT), whereas the inhibitory action of cPKC activator peptide on the current in hERG‐NTK channels almost disappeared. These results indicate that the cPKC and PKCε activator peptides regulate channel function through different mechanisms.
Figure 6.
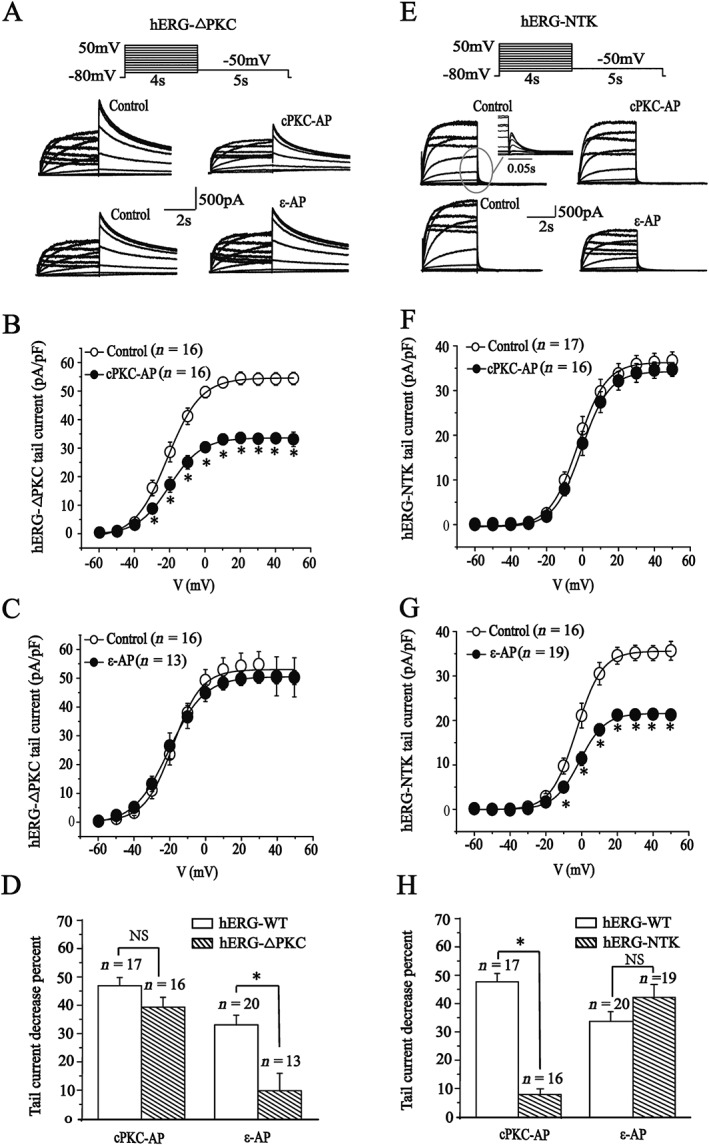
Effects of cPKC and PKCε activator peptides on mutant hERG‐ΔPKC and hERG‐NTK currents. (A, E) Representative current traces evoked using the voltage protocol shown in the presence of the scrambled peptides (control) or cPKC (cPKC‐AP) and PKCε activator peptides (ε‐AP). The inset shows an amplification of the tail currents. (B, F) I–V relationships for tail currents in the control and the presence of cPKC activator peptide (200 nM). (C, G) I–V relationships for tail currents in the control and the presence of PKCε activator peptide (200 nM). (D, H) Percentage decrease of tail currents measured at 0 mV depolarizing prepulse on the hERG‐WT and mutant currents. *P < 0.05, significantly different from control.
Then we further investigated the effects of activation of the two GPCRs, on hERG‐△PKC and hERG‐NTK channels. As shown in Supporting Information Figure S3, A61603 significantly reduced the current at most positive voltages and the percentage decrease of the current measured at potential of 0 mV showed no significant difference to that of the hERG‐WT. That is, the inhibitory effects of α1A‐adrenoreceptor activation was still preserved in the hERG‐△PKC channels. However, the inhibitory action of these receptors was significantly decreased in the hERG‐NTK channels. In contrast, Ang II had no significant effect on the hERG‐△PKC currents, but the inhibitory action of AT1 receptor activation was still preserved for the hERG‐NTK currents. The results demonstrated that the responses to agonists for the α1A‐adrenoreceptors and the AT1 receptors were similar to those for cPKC and PKCε activator peptides respectively. The findings provide further evidence that activation of α1A‐adrenoreceptors or AT1 receptors inhibits the hERG current through different PKC isoforms. It is known that both sympathetic overactivity and elevated Ang II are important contributors to ion channel dysfunction in many cardiac diseases. Based on our findings, we deduced that the inhibition effects on the hERG current by activation of α1A‐adrenoreceptors or AT1 receptors should be additive. We tested the effects of sequential addition of A61603 and Ang II, in different order, on I Kr current in guinea pig ventricular myocytes. The result revealed that no matter whether A61603 (1 μM) was applied first and then together with Ang II (100 nM) or vice versa, the two GPCR activators showed an additive inhibitory effect on I Kr currents (Figure 7).
Figure 7.
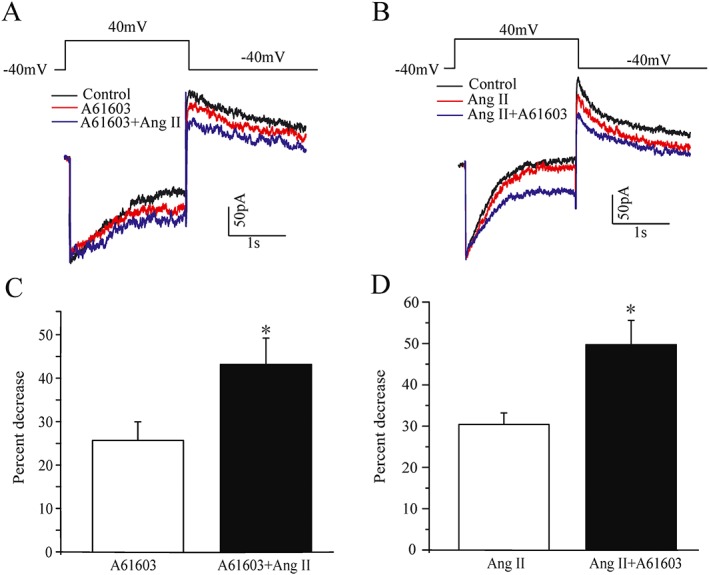
Effects of co‐application of A61603 and Ang II on IKr in guinea pig isolated ventricular cardiomyocytes. (A, B) Representative I Kr traces in the presence of A61603 (1 μM) and Ang II (100 nM) alone or in combination. (C, D) Summary of the results obtained from A (n = 6); *P < 0.05, significantly different from A61603: from B (n = 9; *P < 0.05, significantly different from Ang II).
Discussion
Previous researchers have shown that activation of both α1A‐adrenoreceptors or AT1 receptors inhibit I Kr/hERG current. This study has for the first time provided evidence showing that the two Gq‐coupled receptors modulate the hERG channels by a distinct PKC isoform and through different molecular mechanisms.
PKCα isoenzyme mediates the inhibitory effect of activating α1A‐adrenoreceptors on the IKr/hERG current
In animals and humans, α1A‐adrenoreceptors and α1B‐adrenoreceptors are the predominant α1‐adrenoreceptor subtypes in the myocardium. Stimulation of α1‐adrenoreceptors regulates cardiac function, mainly through α1A‐adrenoreceptors (O'Connell et al., 2014). Studies have demonstrated that the α1‐adrenoreceptor agonist phenylephrine inhibits I Kr current in ventricular cardiomyocytes (Wang et al., 2014). Meanwhile, stimulation of α1A‐adrenoreceptors significantly decreased the cloned hERG current (Thomas et al., 2004). Further, the PKC‐dependent signalling pathway is involved in the hERG current modulation by α1A‐adrenoreceptor activation. However, a specific PKC isoenzyme mediating the effect has not been identified.
Consistent with these results, we found that a selective α1A‐adrenoreceptor agonist, A61603, decreased the amplitude of I Kr in a concentration‐dependent manner through specific activation of α1A‐adrenoreceptors in guinea pig ventricular myocytes. The non‐specific PKC inhibitor Bis‐1 and the cPKC inhibitors Gö6983 or Gö6976 markedly attenuated the inhibitory action of A61603 on I Kr. In addition, the PKCα‐specific inhibitor peptide antagonized the inhibition, but the PKCε‐selective inhibitor peptide did not. The findings suggest that PKCα mediates the inhibition of I Kr by α1A‐adrenoreceptor activation.
To further define the effect of activating PKCα on hERG channels, we observed the effects of the cPKC‐specific activator peptide in a HEK cell expression system. The cPKC activator peptide significantly inhibited the hERG current, with a shift in voltage dependence of activation towards more positive potentials. The findings support that cPKC inhibits the current by regulating voltage‐dependence activation of the hERG channels. Moreover, we found that A61603 produced a similar effect to the cPKC‐specific activator peptide. The results are consistent with the previous reports that PKC activators or phenylephrine cause a depolarizing shift in the voltage‐dependent activation of the hERG channels (Thomas et al., 2003, 2004; Urrutia et al., 2016). Western blot analysis showed that the phospho‐PKCα, but not phospho‐PKCε, was increased in the ventricular myocytes after α1A‐adrenoreceptor stimulation. The result provided evidence for PKCα isoform activation selectively coupling to α1A‐adrenoreceptor stimulation at the molecular level. However, the isoform‐specific dynamic translocation of PKC in HEK cells by α1A‐adrenoreceptor stimulation was recently measured by confocal microscopy and was found to activate PKCα, β and ε (O‐Uchi et al., 2015). The inconsistency may result from the fact that there are differences between species and tissue distribution for individual PKC isozymes.
PKCε isoenzyme mainly mediates the inhibitory effect of activating AT1 receptors on the IKr/hERG current
Like the α1A‐adrenoreceptors, the AT1 receptors also belongs to the family of Gq‐protein coupled receptors. We have reported that Ang II inhibited I Kr in ventricular myocytes via a AT1 receptor‐PKC pathway (Wang et al., 2008). In this paper, our results demonstrated that Gö6976 and Gö6983 as well as PKCα inhibitor peptide did not affect the inhibitory effect of AT1 receptor activation on I Kr, whereas the PKCε inhibitor peptide significantly antagonized the inhibitory effect, pointing to an important role of PKCε for mediating the effect of Ang II. In addition, for cloned hERG potassium channels, activation of PKCε isoform by the PKCε‐selective activator peptide produced a significant inhibition on the hERG current, without affecting voltage‐dependent activation of the channel, implying a different regulation mechanism to that mediating the effects of PKCα activation. Ang II elicited a similar response to PKCε activator peptide, providing further evidence that AT1 receptor stimulation inhibited the hERG current mainly through the PKCε isoform. Moreover, Western blot analyses showed that Ang II significantly increased the level of phospho‐PKCε, indicating the activation of PKCε. However, it was noted that in addition to PKCε, Ang II also enhanced the abundance of phospho‐PKCα. This result appears to be inconsistent with the aforementioned electrophysiological data. Based on the fact that Ang II produced a greater increase in phospho‐PKCε than in phospho‐PKCα, it is possible that there may be a threshold concentration for the activated PKC isoform to modulate the channel and the amount of activated PKCα isoform may not have reached this threshold concentration. This possibility needs to be further tested. Taken together, our data indicate that AT1 receptors and α1A‐adrenoreceptors are functionally coupled to distinct PKC isoforms that modulate the hERG channels.
Regulation of the hERG potassium channels by PKCα and PKCε occurs through different molecular mechanism
Up to now, the cellular mechanisms of the PKC isoform‐mediated modulation on the hERG channels are still not completely understood. It has been suggested that this process did ont involve direct phosphorylation of the channel by PKC, because modulation of the hERG channels by phorbol 12‐myristate 13‐acetate, a non‐selective PKC activator, is still present in the hERG‐△PKC mutant channels where 17 of the 18 potential PKC‐phosphorylation sites have been removed (Thomas et al., 2003). However, deletion of the N‐terminus of the hERG channels (hERG‐NTK) that selectively removes the remaining potential PKC phosphorylation site (T74) abolishes the inhibition of the hERG current produced by the PKC activator 1‐oleoyl 2‐acetylglycerol (Cockerill et al., 2007). Thus, Cockerill et al. (2007) have suggested that hERG channels may be phosphorylated by PKC at T74. In this study, both cPKC activator peptide and A61603 showed comparable inhibition of WT and the hERG‐△PKC mutant channels. However, they failed to inhibit hERG current in the hERG‐NTK channels. This result is similar to a recent study by Urrutia et al., which has shown that the stimulation of α1‐adrenoreceptors by phenylephrine reduces the amplitude of hERG current through a positive shift in the activation half voltage, and that this effect is absent in cells expressing the hERG‐NTK mutant channels (Urrutia et al., 2016). These findings suggest that cPKC may modulate the hERG channels by phosphorylating T74. However, it is still possible that cPKC phosphorylates a different molecule which interacts with the PAS domain or elsewhere in the N‐terminus of the channel. In contrast, the responses to the PKCε activator peptide and Ang II were lost in the hERG‐△PKC channels but were preserved in cells expressing hERG‐NTK channels. Our data have provided evidence that PKCε activation depresses the hERG currents via a mechanism that does not involve T74 and is likely to be mediated through phosphorylating one or more of the other potential 17 phosphorylation sites. Based on these results, we have outlined the processes for the modulation of hERG channels by activation of α1A‐adrenoreceptors or AT1 receptors in Figure 8.
Figure 8.
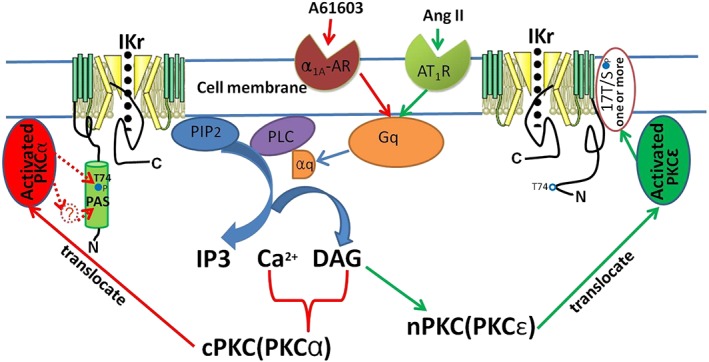
Diagram of possible mechanisms underlying the modulation of IKr induced by activation of α1A‐adrenoreceptors or AT1 receptors via PKC isoform‐specific signalling. The activation of α1A‐adrenoreceptors or AT1 receptors at the plasma membrane initiates downstream signalling through the Gq proteins, leading to activation of PLC, hydrolysis of phosphatidylinositol 4,5‐bisphosphate into IP3 and DAG (1,2‐diacylglycerol), and increases in IP3/calcium signalling, and then mainly activate PKCα and PKCε respectively. The activated PKCα depresses I Kr through phosphorylating T74 or a different molecule which interacts with the PAS domain or elsewhere in the N‐terminus of the channel, whereas PKCε acts through one or more of the other 17 potential phosphorylation sites.
Furthermore, our data showed that co‐activation of α1A‐adrenoreceptors or AT1 receptors produced additive inhibitory effects on I Kr because the two GPCRs modulated the channel through different signalling pathways. Thus, the inhibitory effects of adrenergic stimulation and Ang II on I Kr may jointly contribute to the pathological decrease of the current. The result suggests that a combination of Ang II inhibitors with α1A‐adrenoreceptor blockers may be more effective than either single drug against cardiac arrhythmias in pathophysiological conditions.
One clear limitation of this study is that it did not generate single PKC phosphorylation site mutations of hERG cDNA, so the sites of hERG channels phosphorylation were not precisely defined. Further studies using such mutations are required to further clarify the phosphorylation sites of the hERG channels, modulated by different PKC isoforms. Moreover, in this study, in addition to inhibitor and activator peptides, some pharmacological inhibitors and agonists were used to identify PKC isoforms. However, those inhibitors and agonists may have non‐specific or off‐target effects. Further investigations are needed of the mechanisms of hERG channels modulation, using more specific methods such as siRNA knockdown.
In addition to the acute modulations of the hERG channels, PKCs also mediate the effects of sustained α1A‐adrenoreceptor and AT1 receptor stimulation on hERG channels abundance, which may occur over hours to days. We have previously demonstrated that sustained stimulation of AT1 receptors by Ang II reduces the mature hERG channels protein via accelerating channel degradation involving the PKC pathway (Cai et al., 2014). In contrast, chronic stimulation of α1A‐adrenoreceptors with phenylephrine or direct activation of PKC increase hERG channels protein abundance and K+ current density by accelerating synthesis or translation of channel protein (Chen et al., 2010). Further investigation into the chronic effect of PKC isoforms on hERG protein abundance needs to be carried out.
In conclusion, our present study indicates that activation of α1A‐adrenoreceptors or AT1 receptors inhibited hERG current through different molecular mechanisms that involved PKCα and PKCε isoforms respectively. The findings have provided novel insights into mechanisms by which sympathetic over‐activity and elevated Ang II are involved in the occurrence of arrhythmias under cardiac pathophysiological conditions.
Author contributions
X.L., Y.W., H.Z. and L.S. performed the research. Y.X. designed the research study. X.L. and Y.W. analysed the data. X.L., Y.W. and Y.X. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effects of cPKC and PKCε‐specific activator peptides on the hERG current. (A) Representative current traces recorded under a single pulse protocol from a same cell at 2 (black line) and 10 min (red line) after whole cell break‐in. The cPKC (cPKC‐AP) and PKCε activator peptides (ε‐AP) or the scrambled peptides were added into the pipette solution. (B) Summary bar graph showing the percentage change in the tail currents in the presence of cPKC and PKCε activator peptides (*P < 0.05).
Figure S2 Effects of A61603 and Ang II on hERG‐WT current. (A and B) I‐V relationships for tail currents in the control and the presence of 1 μM A61603 and corresponding activation curves (n = 6, *P < 0.05 versus control). (C and D) I‐V relationships for tail currents in the control and the presence of 100 nM Ang II and corresponding activation curves (n = 5, *P < 0.05 versus control).
Figure S3 Effects of A61603 and Ang II on hERG‐ΔPKC and hERG‐NTK currents. (A and B) I‐V relationships for tail currents in the control and the presence of 1 μM A61603 for 10 min (*P < 0.05, versus control). (C) Summary bar graph showing the percentage decrease in the tail currents of hERG‐WT, hERG‐ΔPKC and hERG‐NTK in the present of 1 μM A61603 for 10 min and corresponding activation curves (*P < 0.05, versus hERG‐WT). (D and E) I‐V relationships for tail currents in the control and the presence of 100 nM Ang II for 10 min (*P < 0.05, versus control). (F) Summary bar graph showing the percentage decrease in the tail currents of hERG‐WT, hERG‐ΔPKC and hERG‐NTK in the present of 100 nM Ang II (*P < 0.05, versus hERG‐WT).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31271210 to Y.X.) and Program for University Innovation Team Leading Talent of Hebei Province (LJRC019 to Y.X.) and the National Natural Science Foundation of Hebei Province (C2013206032 to Y.X.)
Liu, X. , Wang, Y. , Zhang, H. , Shen, L. , and Xu, Y. (2017) Different protein kinase C isoenzymes mediate inhibition of cardiac rapidly activating delayed rectifier K+ current by different G‐protein coupled receptors. British Journal of Pharmacology, 174: 4464–4477. doi: 10.1111/bph.14049.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornancin F, Parker PJ (1997). Phosphorylation of protein kinase C‐alpha on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase‐resistant state. J Biol Chem 272: 3544–3549. [DOI] [PubMed] [Google Scholar]
- Brooksby P, Robinson PJ, Segal R, Klinger G, Pitt B, Cowley AJ (1999). Effects of losartan and captopril on QT dispersion in elderly patients with heart failure. ELITE study group. Lancet 354: 395–396. [DOI] [PubMed] [Google Scholar]
- Cai Y, Wang Y, Xu J, Zuo X, Xu Y (2014). Down‐regulation of ether‐a‐go‐go‐related gene potassium channel protein through sustained stimulation of AT1 receptor by angiotensin II. Biochem Biophys Res Commun 452: 852–857. [DOI] [PubMed] [Google Scholar]
- Chen J, Chen K, Sroubek J, Wu ZY, Thomas D, Bian JS et al (2010). Post‐transcriptional control of human ether‐a‐go‐go‐related gene potassium channel protein by alpha‐adrenergic receptor stimulation. Mol Pharmacol 78: 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PS, Chen LS, Cao JM, Sharifi B, Karagueuzian HS, Fishbein MC (2001). Sympathetic nerve sprouting, electrical remodeling and the mechanisms of sudden cardiac death. Cardiovasc Res 50: 409–416. [DOI] [PubMed] [Google Scholar]
- Cockerill SL, Tobin AB, Torrecilla I, Willars GB, Standen NB, Mitcheson JS (2007). Modulation of hERG potassium currents in HEK‐293 cells by protein kinase C. Evidence for direct phosphorylation of pore‐forming subunits. J Physiol 581: 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Melman Y, Palma E, Fishman GI, McDonald TV (2000). Cyclic AMP regulates the HERG K(+) channel by dual pathways. Curr Biol 10: 671–674. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Hall DD, Hell JW (2009). Supramolecular assemblies and localized regulation of voltage‐gated ion channels. Physiol Rev 89: 411–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ et al (1999). Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proc Natl Acad Sci U S A 96: 12798–12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira JC, Mochly‐Rosen D, Boutjdir M (2012). Regulation of cardiac excitability by protein kinase C isozymes. Front Biosci (Schol Ed) 4: 532–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981). Improved patch‐clamp techniques for high‐resolution current recording from cells and cell‐free membrane patches. Pflugers Arch 391: 85–100. [DOI] [PubMed] [Google Scholar]
- Hellberg C, Schmees C, Karlsson S, Ahgren A, Heldin CH (2009). Activation of protein kinase C alpha is necessary for sorting the PDGF beta‐receptor to Rab4a‐dependent recycling. Mol Biol Cell 20: 2856–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool LC (2000). Hypoxia increases the sensitivity of the L‐type Ca(2+) current to beta‐adrenergic receptor stimulation via a C2 region‐containing protein kinase C isoform. Circ Res 87: 1164–1171. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA (1995). A‐61603, a potent alpha 1‐adrenergic receptor agonist, selective for the alpha 1A receptor subtype. J Pharmacol Exp Ther 274: 97–103. [PubMed] [Google Scholar]
- Lindholm LH, Dahlof B, Edelman JM, Ibsen H, Borch‐Johnsen K, Olsen MH et al (2003). Effect of losartan on sudden cardiac death in people with diabetes: data from the LIFE study. Lancet 362: 619–620. [DOI] [PubMed] [Google Scholar]
- Makary S, Voigt N, Maguy A, Wakili R, Nishida K, Harada M et al (2011). Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine‐regulated potassium channels in atrial remodeling. Circ Res 109: 1031–1043. [DOI] [PubMed] [Google Scholar]
- Marban E (2002). Cardiac channelopathies. Nature 415: 213–218. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mow T, Frederiksen K, Thomsen MB (2015). Assessment of anti‐arrhythmic activity of antipsychotic drugs in an animal model: influence of non‐cardiac alpha(1)‐adrenergic receptors. Eur J Pharmacol 748: 10–17. [DOI] [PubMed] [Google Scholar]
- O'Connell TD, Jensen BC, Baker AJ, Simpson PC (2014). Cardiac alpha1‐adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev 66: 308–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O‐Uchi J, Sasaki H, Morimoto S, Kusakari Y, Shinji H, Obata T et al (2008). Interaction of alpha1‐adrenoceptor subtypes with different G proteins induces opposite effects on cardiac L‐type Ca2+ channel. Circ Res 102: 1378–1388. [DOI] [PubMed] [Google Scholar]
- O‐Uchi J, Sorenson J, Jhun BS, Mishra J, Hurst S, Williams K et al (2015). Isoform‐specific dynamic translocation of PKC by alpha1‐adrenoceptor stimulation in live cells. Biochem Biophys Res Commun 465: 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radresa O, Guia A, Baroudi G (2014). Roles of PKC isoforms in PMA‐induced modulation of the hERG channel (Kv11.1). J Biomol Screen 19: 890–899. [DOI] [PubMed] [Google Scholar]
- Ron D, Kazanietz MG (1999). New insights into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J 13: 1658–1676. [PubMed] [Google Scholar]
- Ron D, Luo J, Mochly‐Rosen D (1995). C2 region‐derived peptides inhibit translocation and function of beta protein kinase C in vivo. J Biol Chem 270: 24180–24187. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT (1995). A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81: 299–307. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeishi Y, Jalili T, Ball NA, Walsh RA (1999). Responses of cardiac protein kinase C isoforms to distinct pathological stimuli are differentially regulated. Circ Res 85: 264–271. [DOI] [PubMed] [Google Scholar]
- Thomas D, Wu K, Wimmer AB, Zitron E, Hammerling BC, Kathofer S et al (2004). Activation of cardiac human ether‐a‐go‐go related gene potassium currents is regulated by alpha(1A)‐adrenoceptors. J Mol Med (Berl) 82: 826–837. [DOI] [PubMed] [Google Scholar]
- Thomas D, Zhang W, Wu K, Wimmer AB, Gut B, Wendt‐Nordahl G et al (2003). Regulation of HERG potassium channel activation by protein kinase C independent of direct phosphorylation of the channel protein. Cardiovasc Res 59: 14–26. [DOI] [PubMed] [Google Scholar]
- Urrutia J, Alday A, Gallego M, Malagueta‐Vieira LL, Arechiga‐Figueroa IA, Casis O et al (2016). Mechanisms of IhERG/IKr modulation by alpha1‐adrenoceptors in HEK293 cells and cardiac myocytes. Cell Physiol Biochem 40: 1261–1273. [DOI] [PubMed] [Google Scholar]
- Walker AJ, Plows LD (2003). Bacterial lipopolysaccharide modulates protein kinase C signalling in Lymnaea stagnalis haemocytes. Biol Cell 95: 527–533. [DOI] [PubMed] [Google Scholar]
- Wang S, Min XY, Pang SS, Qian J, Xu D, Guo Y (2014). Adrenergic regulation of the rapid component of delayed rectifier K+ currents in guinea pig cardiomyocytes. J Thorac Dis 6: 1778–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xu DJ, Cai JB, Huang YZ, Zou JG, Cao KJ (2009). Rapid component I (Kr) of cardiac delayed rectifier potassium currents in guinea‐pig is inhibited by alpha(1)‐adrenoreceptor activation via protein kinase A and protein kinase C‐dependent pathways. Eur J Pharmacol 608: 1–6. [DOI] [PubMed] [Google Scholar]
- Wang YH, Shi CX, Dong F, Sheng JW, Xu YF (2008). Inhibition of the rapid component of the delayed rectifier potassium current in ventricular myocytes by angiotensin II via the AT1 receptor. Br J Pharmacol 154: 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Xiao Z, Yao J, Zhao G, Fa X, Niu J (2013). Participation of protein kinase C in the activation of Nrf2 signaling by ischemic preconditioning in the isolated rabbit heart. Mol Cell Biochem 372: 169–179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of cPKC and PKCε‐specific activator peptides on the hERG current. (A) Representative current traces recorded under a single pulse protocol from a same cell at 2 (black line) and 10 min (red line) after whole cell break‐in. The cPKC (cPKC‐AP) and PKCε activator peptides (ε‐AP) or the scrambled peptides were added into the pipette solution. (B) Summary bar graph showing the percentage change in the tail currents in the presence of cPKC and PKCε activator peptides (*P < 0.05).
Figure S2 Effects of A61603 and Ang II on hERG‐WT current. (A and B) I‐V relationships for tail currents in the control and the presence of 1 μM A61603 and corresponding activation curves (n = 6, *P < 0.05 versus control). (C and D) I‐V relationships for tail currents in the control and the presence of 100 nM Ang II and corresponding activation curves (n = 5, *P < 0.05 versus control).
Figure S3 Effects of A61603 and Ang II on hERG‐ΔPKC and hERG‐NTK currents. (A and B) I‐V relationships for tail currents in the control and the presence of 1 μM A61603 for 10 min (*P < 0.05, versus control). (C) Summary bar graph showing the percentage decrease in the tail currents of hERG‐WT, hERG‐ΔPKC and hERG‐NTK in the present of 1 μM A61603 for 10 min and corresponding activation curves (*P < 0.05, versus hERG‐WT). (D and E) I‐V relationships for tail currents in the control and the presence of 100 nM Ang II for 10 min (*P < 0.05, versus control). (F) Summary bar graph showing the percentage decrease in the tail currents of hERG‐WT, hERG‐ΔPKC and hERG‐NTK in the present of 100 nM Ang II (*P < 0.05, versus hERG‐WT).