


Abstract
During DNA replication, the single-stranded DNA binding protein (SSB) wraps single-stranded DNA (ssDNA) with high affinity to protect it from degradation and prevent secondary structure formation. Although SSB binds ssDNA tightly, it can be repositioned along ssDNA to follow the advancement of the replication fork. Using all-atom molecular dynamics simulations, we characterized the molecular mechanism of ssDNA association with SSB. Placed in solution, ssDNA–SSB assemblies were observed to change their structure spontaneously; such structural changes were suppressed in the crystallographic environment. Repeat simulations of the SSB–ssDNA complex under mechanical tension revealed a multitude of possible pathways for ssDNA to come off SSB punctuated by prolonged arrests at reproducible sites at the SSB surface. Ensemble simulations of spontaneous association of short ssDNA fragments with SSB detailed a three-dimensional map of local affinity to DNA; the equilibrium amount of ssDNA bound to SSB was found to depend on the electrolyte concentration but not on the presence of the acidic tips of the SSB tails. Spontaneous formation of ssDNA bulges and their diffusive motion along SSB surface was directly observed in multiple 10-µs-long simulations. Such reptation-like motion was confined by DNA binding to high-affinity spots, suggesting a two-step mechanism for SSB diffusion.
INTRODUCTION
During DNA replication and repair, each nucleotide of a parent DNA molecule is read and copied to make up the DNA molecule of the progeny. Errors in DNA metabolism can lead to mutations and are at the origin of many diseases, including cancer (1,2). To replicate DNA with high-fidelity, a number of proteins work concertedly in a loose complex known as the replisome (3,4). A helicase protein separates duplex DNA into two complementary single strands. A DNA polymerase protein traverses each strand and synthesizes the complementary daughter strand. Because the DNA synthesis occurs in the 5′-to-3′ direction, one of the two polymerase proteins needs to ‘backstitch’ every 100 to 1000 nucleotides (nts) on one of the strands (the lagging strand) in order to keep up with the motion of the helicase. As a result, a significant amount of single-stranded DNA (ssDNA) is present between the helicase and the DNA polymerase, which can pose a problem for high-fidelity DNA replication, as ssDNA is vulnerable to enzymatic and oxidative degradation (5,6). Furthermore, reanealing of the DNA strands emerging from the helicase or in the self-complementary regions of the lagging strand can stall the replication process or introduce deletion errors (5,6). To protect against these effects, all organisms have proteins that sequester ssDNA. In bacteria, these proteins are known as single-stranded DNA binding proteins (SSBs) (7,8).
Escherichia coli SSB is a homotetramer, Figure 1A. Each monomer features a structured DNA-binding domain (residues 1–112) and a long and disordered C-terminal tail (residues 116–177) containing a highly acidic tip. The amount of ssDNA bound to a single SSB protein was experimentally found to depend on the concentrations of ions and free SSB in the surrounding solution (8,9). At low concentration of monovalent salt (<10 mM), one SSB protein binds 35 nts to form the SSB35 complex, Figure 1B. At high salt concentrations (>200 mM), about 65 nts bind to SSB, forming the SSB65 complex, Figure 1C. Intermediate binding states (involving between 35 and 65 nts per SSB) have been reported in biochemical studies (10) and examined in detail through single molecule experiments (11). When SSB is abundant in the solution, many copies of the protein can bind to a long ssDNA molecule cooperatively, forming a fiber made up of the SSB35 complexes, even at high ion concentrations (12). Likewise, when the concentration of DNA nucleotides is greater than 70 per SSB tetramer, SSB65 will form even under somewhat low salt conditions (11). It remains unclear which binding mode is of greatest biological significance, and it has been hypothesized that the relevant binding mode may depend on the process (e.g. replication versus repair). The ability of SSB to cluster may be biologically significant because SSB, through its disordered C-terminal tails, recruits numerous DNA metabolism enzymes (13–17). Protein clustering can promote enzymatic activity, as shown theoretically for general processes (18) and experimentally in vivo for the model process of transcription (19). Hence, SSB might provide the cell with a passive mechanism for amplifying DNA repair or replication activity in response to excess ssDNA through the promotion of enzymatic clusters. Besides being an essential component of DNA metabolism, SSB molecules have recently been used in nanopore sensor applications to slow the translocation of DNA (20) and as a model system for targeted protein detection (21).
Figure 1.

Microscopic models of the ssDNA–SSB complex. (A) The structure of the SSB protein. The structure is shown using a cartoon representation for backbone atoms resolved in the crystal structure (PDB accession code: 1EYG) (22) enclosed by a semi-transparent surface indicating the protein volume inaccessible to solvent. The arginine residues of the DNA binding grooves between loops L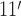 and L23 (DBGs) and the arginine-rich patches near the N-terminus loops are shown using orange and yellow van der Waals (vdW) spheres, respectively. The intrinsically disordered C-terminal domains, which were partly truncated and otherwise unresolved in the crystal structure, are drawn schematically. (B) A model of the SSB35 complex where 40 DNA nucleotides wrap once around SSB such that the DNA ends emerge on the opposite sides of the protein. (C) A model of the SSB65 complex where 70 DNA nucleotides double back around the SSB surface such that the two ends of ssDNA emerge near one another. In panels B and C, the DNA backbone and base atoms are respectively depicted using green and light-green vdW spheres, and the protein is depicted as a molecular surface colored by the charge of the nearest-to-surface atoms. The nucleotides are numbered in ascending order from the 5′ end to the 3′ end. The leftmost images show the complex from the same viewpoint as in panel A. The models of SSB35 and SSB65 were created by the crystallographers based on the atomic coordinates resolved in the x-ray structure (22).
and L23 (DBGs) and the arginine-rich patches near the N-terminus loops are shown using orange and yellow van der Waals (vdW) spheres, respectively. The intrinsically disordered C-terminal domains, which were partly truncated and otherwise unresolved in the crystal structure, are drawn schematically. (B) A model of the SSB35 complex where 40 DNA nucleotides wrap once around SSB such that the DNA ends emerge on the opposite sides of the protein. (C) A model of the SSB65 complex where 70 DNA nucleotides double back around the SSB surface such that the two ends of ssDNA emerge near one another. In panels B and C, the DNA backbone and base atoms are respectively depicted using green and light-green vdW spheres, and the protein is depicted as a molecular surface colored by the charge of the nearest-to-surface atoms. The nucleotides are numbered in ascending order from the 5′ end to the 3′ end. The leftmost images show the complex from the same viewpoint as in panel A. The models of SSB35 and SSB65 were created by the crystallographers based on the atomic coordinates resolved in the x-ray structure (22).
Raghunathan, Kozlov, Lohman and Waksman solved the structure of a crystal formed by two 35 nts ssDNA fragments and an SSB tetramer lacking the disordered C-terminal tails (PDB accession code: 1EYG) (22). The crystal structure reveals that Trp54 and Phe60—residues identified by mutagenesis studies as important for DNA binding (8,23,24)—line a DNA-bound groove in the structured domain of each monomer that we refer to as the ‘DNA binding groove between loops L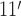 and L23’ (DBG) that is also flanked by a number of arginine residues, see Figure 1A. The fragments of ssDNA resolved in the structure did not represent the arrangement of ssDNA in either complex, SSB35 or SSB65. The crystallographic information was, however, sufficient to obtain a model for each complex by connecting and extending the resolved DNA fragments (22). These models are shown in Figure 1B and C.
and L23’ (DBG) that is also flanked by a number of arginine residues, see Figure 1A. The fragments of ssDNA resolved in the structure did not represent the arrangement of ssDNA in either complex, SSB35 or SSB65. The crystallographic information was, however, sufficient to obtain a model for each complex by connecting and extending the resolved DNA fragments (22). These models are shown in Figure 1B and C.
Experiments showed that SSB binds ssDNA with high affinity (estimated 1011 M−1 in 200 mM electrolyte) and dissociates at a very slow rate (0.0059 s−1) (25). During DNA repair, dissociation of SSB can be induced by the polymerization of a RecA filament (26), but no protein has been identified that actively removes SSB during DNA replication. The slow rate of dissociation is at apparent odds with the requirements of rapid replication (>500 bp per second in E. coli) (27), provoking the following questions: how does SSB move out of the way of other components of the replication machinery, and how is SSB removed when a polymerase back-stitches on the lagging strand in the replisome?
Single-molecule studies have demonstrated the ability of SSB to diffuse along short stretches of ssDNA (28,29). Three models for such diffusion have been considered: sliding, where all contacts between ssDNA and the protein shift in a concerted manner; sliding by reptation, where a bulge of ssDNA forms and propagates around the SSB surface, and rolling diffusion, where ssDNA at one end of the complex peels off to expose a patch of the SSB surface for subsequent binding of ssDNA from the other end of the strand (29–31). Single-molecule fluorescence experiments indicated that reptation was the dominant mode of diffusion (29). Diffusion along a short piece of ssDNA was found to occur through steps averaging 3 nts in length that occurred on average 60 times per second (28). While SSB has been studied extensively through experimental methods, only a few computational studies focusing on E. coli SSB have been published. In particular, a coarse-grained molecular dynamics (MD) model indicated that the positively charged SSB residues, not the aromatic residues, stabilize ssDNA in the geometry that was revealed by x-ray crystallographers (32). Another study simulated the translocation of SSB through a nanopore decorated with ssDNA on the nanosecond timescale (21). Finally, ligand docking algorithms were combined with 100 ns all-atom MD simulations to demonstrate the importance of arginine residues for ssDNA binding to a monomeric Lactococcus bacteriophage SSB, a protein having similar overall structure to monomeric E. coli SSB (33). In addition to simulation studies, surveys of DNA-binding proteins have attempted to distinguish features of single- and double-stranded DNA binding proteins (34).
Here, we report the results of all-atom MD simulations that examined the interaction of SSB with ssDNA. Through equilibrium simulations of SSB–ssDNA complexes in solution and crystallographic environments, we demonstrate how one can reconcile the seemingly opposing observations of SSB diffusion along ssDNA and the fact that ssDNA bound to SSB can be resolved by x-ray crystallography. Next, we elucidate the process of forced unwrapping of ssDNA from SSB, showing that ssDNA comes off SSB stochastically but in discrete steps. By simulating spontaneous association of ssDNA fragment to SSB, we determine how solution conditions affect the process and whether acidic tips of the disordered tails can compete with ssDNA for binding to SSB. Finally, we report 10 μs-long MD trajectories of SSB65 complexes under various solution conditions that detail spontaneous rearrangements of DNA conformations consistent with a reptation model of SSB diffusion along ssDNA.
MATERIALS AND METHODS
All MD simulations employed the TIP3P model of water (35), periodic boundary conditions and particle-mesh Ewald (PME) full electrostatics with a PME grid density of about 1 Å per grid point. The temperature was held constant at 291 K (except where specified) by applying Langevin forces (36) to all non-hydrogen atoms; the Langevin damping constant was set to 0.1 ps−1. For simulations performed in the NPT ensemble (constant number of atoms N, pressure P and temperature T), the pressure was maintained at 1 bar using the Nos -Hoover Langevin piston pressure control (37,38). Integration was performed using 2–2–6 fs multiple timestepping (39), and vdW and short-range electrostatic energies were calculated using a smooth (7–8 Å) cutoff (40). Except where specified, simulations were performed using the program NAMD (39), the CHARMM36 (41,42) parameters with the CMAP corrections (43), standard parameters for ions (44) and CUFIX corrections for non-bonded interactions between charged groups (45,46).
-Hoover Langevin piston pressure control (37,38). Integration was performed using 2–2–6 fs multiple timestepping (39), and vdW and short-range electrostatic energies were calculated using a smooth (7–8 Å) cutoff (40). Except where specified, simulations were performed using the program NAMD (39), the CHARMM36 (41,42) parameters with the CMAP corrections (43), standard parameters for ions (44) and CUFIX corrections for non-bonded interactions between charged groups (45,46).
Some SSB loops include residues that are not resolved in the crystallographic structure (22) (PDB accession code: 1EYG). During assembly of the all-atom systems, these residues were modeled using homology of different subunits of the homotetramer and, where needed, another E. coli SSB structure (47) (PDB accession code: 1QVC). The crystallographic model of SSB was obtained using a deletion mutant that was missing 42 residues of the 64-residue C-terminal tails (22). Most of the remaining residues of the tails were not resolved in the structure and hence were not included in the structural model unless explicitly specified. Water molecules were added using the solvate plugin of VMD (48). Then neutralizing counterions were added to each system. Finally, a number of ions were added to provide the desired concentration c using the expression Nion = 0.018 c Nwater, where Nion and Nwater are the numbers of ions per species and water molecules, respectively. Except where specified, ions were added using the autoionize plugin, which adds each ion by replacing a randomly selected water molecule. After system assembly, at least 1200 steps of minimization were performed via the conjugate gradient method to remove steric clashes. During the first nanoseconds of equilibration, the ions in the system redistribute so that the ion concentration of bulk-like solution increases. Accordingly, throughout the manuscript we report the effective bulk ion concentration as the concentration of counterions in the solution 25 Å away from the protein and DNA atoms.
ssDNA–SSB complexes in solution and crystal environments
An all-atom model of SSB65 in solution was assembled from a model of the complex based on the x-ray structure of SSB and ssDNA (PDB accession code: 1EYG) created by and obtained from the crystallographers (22). The model contained exactly 70 dC nts, and each SSB monomer was extended up to Arg115. The structure was then submerged in a pre-equilibrated volume of water 98 Å on each side. After adding neutralizing potassium counterions, chloride and potassium ions were added to a concentration of 120 mM. During subsequent equilibration, the ions quickly redistribute resulting in an increase of the concentration of bulk-like ions (those at least 25 Å away from the SSB particles) to ∼160 mM. Following minimization, the system was simulated in the NPT ensemble. After 4 ns of simulation with restraints on backbone atoms (kspring = 1390 pN/nm), the system was simulated without any restraints on the D.E. Shaw Research Anton supercomputer (49). The last part of the ‘Materials and Methods’ section provides detailed information about the simulation procedures used.
The complete crystallographic unit cell of SSB complexed with DNA contains four symmetry-related complexes, each formed by an SSB tetramer and a pair of dC35 fragments (22). An all-atom model of the complete crystallographic unit cell was assembled in several steps. DNA nucleotides that were not resolved in the original x-ray structure were modeled by homology. Four copies of the complex were transformed and combined in accord with the unit cell parameters of the structure. The Solvate program (50) was used to pack water molecules in energetically favorable conformations around the protein–DNA complex, and the cionize program was used to place neutralizing potassium counterions in energetically preferred locations. The box of water was trimmed to fit the unit cell; randomly selected water molecules were replaced by potassium or chloride ions to bring the concentration to 100 mM, which was estimated according to the Donnan equilibrium equation, 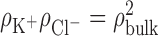 , where
, where 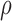 denotes concentration. The system was equilibrated in the NVT ensemble (constant number of atoms N, volume V and temperature T), having all crystallographically-resolved non-hydrogen atoms restrained about their crystal structure coordinates for the first 15 ns (kspring = 1390 pN/nm). During the next 25 ns of simulation, only crystallographically-resolved backbone and water oxygen atoms were restrained (kspring = 1390 pN/nm). Thereafter the system was used in an unrestrained equilibrium simulation lasting ∼500 ns.
denotes concentration. The system was equilibrated in the NVT ensemble (constant number of atoms N, volume V and temperature T), having all crystallographically-resolved non-hydrogen atoms restrained about their crystal structure coordinates for the first 15 ns (kspring = 1390 pN/nm). During the next 25 ns of simulation, only crystallographically-resolved backbone and water oxygen atoms were restrained (kspring = 1390 pN/nm). Thereafter the system was used in an unrestrained equilibrium simulation lasting ∼500 ns.
Forced dissociation of ssDNA from SSB
All-atom models of SSB35 and SSB65 in solution were prepared from models of the complexes as described in the previous section, except that no attempt was made to model missing C-terminal residues. The model of SSB35 contained exactly 40 dC nts.
After 25 ns of simulation with restraints on backbone atoms, the systems were used in equilibration simulations that were carried out in the absence of all restraints and lasted 810 and 690 ns for systems containing SSB35 and SSB65, respectively. The MD simulations of forced unbinding of ssDNA from SSB were performed using, as the starting condition for the simulations, the instantaneous conformations of the protein–DNA complex extracted at three different times (after about 100, 300 and 500 ns) from the equilibration trajectories of the SSB35 and SSB65 systems. Specifically, the snapshot of the equilibration system was rotated so that the vector connecting the center of mass (CoM) of the DNA ends laid along the 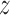 -axis of the system. This axis was chosen arbitrarily to be the pulling axis during subsequent simulations. The dimensions were obtained for the smallest cube containing all atoms within 6 Å of the SSB–ssDNA complex and with axes laying along the system axes. Molecules laying entirely outside that cube were removed from the system. The starting systems for the forced unbinding simulations were obtained by adding solvent and ions to fill a 90 × 90 × 130 Å3 volume with a 120 mM KCl electrolyte. After equilibration, the average bulk-like ion concentration away from SSB particles increased to ∼150 mM.
-axis of the system. This axis was chosen arbitrarily to be the pulling axis during subsequent simulations. The dimensions were obtained for the smallest cube containing all atoms within 6 Å of the SSB–ssDNA complex and with axes laying along the system axes. Molecules laying entirely outside that cube were removed from the system. The starting systems for the forced unbinding simulations were obtained by adding solvent and ions to fill a 90 × 90 × 130 Å3 volume with a 120 mM KCl electrolyte. After equilibration, the average bulk-like ion concentration away from SSB particles increased to ∼150 mM.
In each simulation, external forces were applied according to the following protocol. Two harmonic springs were used to produce ssDNA unwrapping by pulling the ssDNA ends in opposite directions. One end of each spring was tethered to the CoM of the terminal nucleotide of the DNA. The free ends of the two springs were pulled at a rate of 1 Å/ns along the z-axis of the system, in opposite directions. The springs applied forces only along the z-axis of the system; the force constant was set to 2000 pN/nm. A weaker third spring (kspring = 200 pN/nm) held the CoMs of the two terminal DNA nucleotides within the xy-plane. When a terminal nucleotide moved further than 55 Å away from the center of the SSB along the z-axis the simulation was stopped, the length of the pulling spring was recorded, and the terminal nucleotide was removed from the system along with a potassium counterion. The spring was reattached to the nascent terminal nucleotide and the free end of the spring was moved so that its length and, hence, the tension were preserved. With the free end of the spring held stationary, the system was minimized briefly and then equilibrated for 19 ps in the NPT ensemble while the non-hydrogen atoms of the protein and DNA were harmonically restrained (kspring = 1390 pN/nm). Pulling of the DNA was resumed in the NVT ensemble. During the pulling simulations, the temperature was maintained at 310 K.
The simulations in this section were performed prior to the availability to the CHARMM36 force field or the CUFIX corrections (45,46), and so they employed the widely used CHARMM27 force field with CMAP corrections (41,43,51).
Association of ssDNA with SSB
Models were assembled containing an SSB tetramer surrounded by fifteen dT3 fragments. The 5′ end of each DNA fragment was phosphorylated so that each fragment had a net charge of −3e, where e is the elementary charge of a proton. The initial configuration of each DNA fragment was taken from previously performed simulations of DNA (52). To place a fragment, a set of trial coordinates was generated by randomly rotating and translating the fragment through system. If the fragment was found to have a 4-Å contact with the protein or another previously placed fragment, or if the fragment’s atoms lay within 4 nm of the center of SSB, then the trial position was rejected. This process was repeated until a trial position was accepted for each of the DNA fragments. Here and elsewhere in the study, two groups of atoms are considered to be in contact if a pair of their non-hydrogen atoms is located within a specified distance of one another.
Water was added to the system to form a cube that was 106 Å on each side. Neutralizing counterions were added to half of the systems to a 1000 mM concentration. After equilibration, the concentration of counterions 25 Å away from SSB and DNA atoms was seen to be 40 and 1200 mM, respectively for systems without and with additional ions. After minimization, the non-hydrogen SSB atoms that were resolved in the crystal structure were restrained about their initial coordinates during the production simulation.
The configuration of each system after 340 ns of simulation was used to create a new system with higher ssDNA fragment density by adding ten additional fragments to the system as described above. Counterions were added to neutralize the charge of newly added DNA fragments; the system was minimized and simulated as described above. The bulk-like counterion concentration of the initially 40 mM system rose to 65 mM.
Similar procedures were used to assemble systems for the study of competition between short ssDNA fragments and peptide fragments of the acidic C-terminal tip of SSB. These systems contained 15 dT3 fragments and 8 peptide fragments each having either MDF, DDD or IPF amino acid sequence. The peptide fragments were introduced into the system following the same protocols as for the ssDNA fragments. Upon minimization, the systems were simulated having SSB atoms restrained as described above. The effective ion concentration was 40 or 1200 mM, depending on the system.
Long timescale simulations of SSB65 in solution
The 10-μs-long explicit solvent MD simulations of the SSB65 complex were performed using a structural model that had the C-terminal tails truncated after Arg115. The simulations were performed using the D.E. Shaw Research Anton supercomputer (49). The simulations employed a set of parameters equivalent to those used for the NAMD runs (see the first paragraph of the ‘Materials and Methods’ section), except the Nosé Hoover thermostat (53,54) and the Martyna-Tobias-Klein barostat (37) were employed, and a longer 12-Å cutoff was used for non-bonded interactions. The 50 or 1100 mM simulation systems were assembled in the same way as the 160 mM system described in the second part of the ‘Materials and Methods’ section; the 50 mM system contained only neutralizing K+ ions. A fourth system was prepared for the SSB-GG mutant initially in 120 mM KCl with each SSB monomer truncated after residue 112 and extended to include an eleven-residue amino acid fragment (GG-MDFDDDIPF) comprised of two glycine residues followed by the terminal nine amino acids of the wild-type SSB sequence. The bulk-like ion concentration in the fourth simulation rose to 170 mM during equilibration. NAMD was used to minimize and equilibrate the systems with restraints applied to coordinates resolved in the crystal structure as described in the second part of the ‘Materials and Methods’ section, prior to simulation on Anton.
RESULTS
SSB–bound ssDNA rearranges in solution but is stable in crystallographic geometry
The reptation model of SSB diffusion implies that DNA is mobile on the SSB surface, which is at apparent odds with well-defined placement of DNA nucleotides in the crystal structure. There are, however, substantial differences between the in vivo solution environment and the environment realized during the structure determination process. To determine what effect the environment surrounding an SSB complex has on mobility of its bound DNA, we simulated two systems: one where an SSB–dC70 complex, which we refer hereafter as SSB65, was submerged in 160 mM KCl electrolyte and one where four complexes, each containing an SSB protein and two dC35 fragments, were arranged in the crystallographic geometry in a 100 mM KCl electrolyte, Figure 2A.
Figure 2.

Mobility of ssDNA on SSB surface in solution and crystal structure environments. (A) Simulation unit cells for solution (left) and crystal (right) environments. The simulation unit cell for the solution environment contains one SSB tetramer (white molecular surface) wrapped with dC70 (green vdW spheres). Ions are depicted as orange and yellow spheres and water is depicted as a semi-transparent surface. The unit cell for the crystal environment contains four SSB oligomers (light blue) and eight 35-nt ssDNA fragments (green) submerged in solvent. One crystallographic unit cell is highlighted using green vdW spheres for DNA and molecular surfaces for the SSB proteins. Several copies of the crystallographic unit cells are depicted in the background using a less detailed molecular surface representation. Water and ions are not shown. (B) Root-mean squared deviation (RMSD) of the DNA and protein backbone from their crystallographic coordinates during simulations in the crystal (gray; 100 mM KCl) and solution (blue; 160 mM KCl) environments. For DNA, only the nucleotides resolved in the crystal structure were used for the calculation. For the crystal structure system, the RMSD values were averaged over the four copies of the SSB–ssDNA complex. (C) Root-mean squared fluctuations (RMSF) of the center of mass (CoM) of the backbone of each nucleotide during 500 ns of simulation. The annotation at the top of the figure indicates the nucleotides resolved in the crystal structure. Each resolved nucleotide in the crystal structure had a corresponding nucleotide in the model of SSB65. The tinted background indicates nucleotides interacting with the N-terminal arginine-rich patch (yellow) and the DBGs (orange). Motion of nucleotides through these regions was seen to be extremely rare and very small in magnitude, so ascribing the regions to ranges of nucleotide index is a good approximation.
During 500 ns of simulation in a solution environment, the protein remained stable, except for loops, while the DNA departed somewhat from its initial configuration, see Supplementary Movie S1. Strikingly, the last 10 nts of the DNA’s 3′ end moved ∼2 nm away from their initial configuration to another region on the SSB surface. While this stretch of DNA had not been resolved in the crystal structure, many local rearrangements were observed for the nucleotides that were.
Next, we simulated a complex of SSB with two ssDNA fragments in the crystallographic environment; Supplementary Movie S2 illustrates the resulting MD trajectory. Although some DNA nucleotides were observed to undergo conformational fluctuations, the magnitude of the fluctuations was considerably less than in the solution environment. No large structural transformations were observed, as indicated by the low root-mean squared (RMS) deviation from the initial coordinates, Figure 2B. Furthermore, the RMS fluctuation of almost every nucleotide was lower in the crystal than in solution, Figure 2C. In the solution environment, the RMS fluctuation of the DNA ends was seen to be quite high. By contrast, in the crystal environment, the value of the RMS fluctuation for the DNA ends was almost the same as for interior nucleotides.
We attribute the 2-fold difference between the RMS deviation in solution and crystal environments to interactions with neighboring proteins and DNA. At any given time during the simulation in the crystal environment, half of the nucleotides bound to one SSB had at least one 4-Å contact with an atom from a neighboring SSB protein. In summary, our MD simulations demonstrate that ssDNA binds to SSB complex more stably in the crystallographic environment than in solution. The increased stability in a crystal can be attributed to the large number of contacts between ssDNA and the neighboring SSB molecules. Thus, the crystallographic resolution of ssDNA does not contradict the possibility of ssDNA rearranging its wrapping geometry around SSB in solution.
Unwrapping of ssDNA from SSB stalls at reproducible sites
The energy landscape of SSB–ssDNA interactions was recently probed through single-molecule experiments revealing that ssDNA unwraps from SSB in discrete steps when tension is applied to the ssDNA (11). While force-spectroscopy experiments can probe the kinetic and thermodynamic properties of a macromolecular complex (55), they cannot directly elucidate the rupture pathway. For that reason, we decided to apply external forces to remove ssDNA from the SSB surface. Models of the SSB35 and SSB65 complexes were assembled with a 150 mM KCl electrolyte and equilibrated for several hundred nanoseconds. In subsequent simulations, rupture of SSB35 and SSB65 complexes was achieved by pulling each end of the DNA in opposite directions. The pulling simulations were performed three times for each binding mode using, as initial conditions for the simulations, three different microscopic states attained by the system during equilibration simulations (after about 100, 300 and 500 ns). The pulling protocol is illustrated schematically in Figure 3A and B and is described in detail in the ‘Materials and Methods’ section.
Figure 3.

Forced unbinding of ssDNA from SSB. (A and B) Schematic illustration of the simulation protocol. Each end of a DNA strand is tethered to one end of a virtual harmonic spring. The other end of each spring is pulled away from SSB along the z-axis of the system at a steady rate of 1 Å/ns. (C and D) The index of the first (5′ end) and last (3′ end) nucleotide of the ssDNA fragment bound to SSB during forced unwrapping of SSB35 (C) and SSB65 (D). The nucleotides are numbered in ascending order from 5′ to 3′ ends of ssDNA. Three simulations were performed for each SSB model after approximately 100 (black), 300 (red) and 500 (blue) ns of unbiased equilibration. At any 1 ns interval of the simulation, a nucleotide is considered bound to the SSB if it has at least one atomic 4-Å contact that was also present at the onset of the simulation. (E and F) Total time spent by a terminal nucleotide prior to parting with the SSB surface in the simulations of the SSB35 (E) and SSB65 (F) systems. The blocks of color in the background of panels C–F indicate nucleotides that were closely associated with the DBGs (orange) or with the N-terminal arginine-rich patch (yellow). (G and H) Force applied during removal of ssDNA from SSB. The force applied to the ssDNA, averaged over the two springs, is plotted against simulation time for each unwrapping trajectory. Each data point represents a 5 ns time-average of the tension in the springs sampled at 100 ps intervals. The values of the force averaged over the entire MD trajectory are 75 (black), 61 (red) and 71 (blue) pN for SSB35 and 58 (black), 55 (red) and 39 (blue) pN for SSB65.
Figure 3C and D; Supplementary Movies S3 and 4 illustrate the simulated unwrapping processes. For each simulation, the index of the first and last nucleotide of the ssDNA fragment bound to SSB is plotted versus simulation time. A change of the index indicates detachment (or reattachment) of a nucleotide from (or to) the SSB surface. A time series of the first and last nucleotide index fully characterizes the pathway of forced unwrapping of ssDNA from SSB. As ssDNA was unwrapped from SSB, the number of bound nucleotides reduced from 40 (SSB35) or 70 (SSB65) to ∼5. The unwrapping was asymmetric: more nucleotides were removed from one end of ssDNA than from the other. However, in each of the three simulations of the same system, the nucleotides unwrapped in a unique order. In the case of SSB35, all three trajectories ended with the same stretch of ssDNA bound to the same DBG. For SSB65, however, one of the trajectories ended with a different DBG bound to ssDNA than in the other two trajectories. Supplementary Note 1 in the Supplementary Data provides a complete description of the rupture process.
Unwrapping of ssDNA occurred in a step-wise manner, with several nucleotides detaching from the protein surface at the same time. No force was applied directly to SSB, so when the rupture process stalled at one ssDNA end, it could proceed at the other end. When both ends stalled, tension in the DNA strand would build until one or more nucleotides detached from the SSB at either end of the bound fragment. The rupture process was seen to stall at roughly six distinct locations, Figure 3C–F. Four of these locations are the symmetry-related ‘DNA binding grooves between L and L23’ that include Arg56, Phe60, Arg21, Arg84, Arg86, Lys87, Trp88, Trp40, Arg41 and Trp54. We refer to the two remaining symmetry-related locations, which include residues Arg3, Arg115, Lys73, Tyr70 and Arg72, as ‘N-terminal arginine-rich patches’ because they contain numerous arginines, see Figure 1A. Interestingly, in some pulling trajectories, unwrapping from a DBG would proceed while the other end remained stalled at one of the N-terminal arginine patches.
and L23’ that include Arg56, Phe60, Arg21, Arg84, Arg86, Lys87, Trp88, Trp40, Arg41 and Trp54. We refer to the two remaining symmetry-related locations, which include residues Arg3, Arg115, Lys73, Tyr70 and Arg72, as ‘N-terminal arginine-rich patches’ because they contain numerous arginines, see Figure 1A. Interestingly, in some pulling trajectories, unwrapping from a DBG would proceed while the other end remained stalled at one of the N-terminal arginine patches.
Analysis of the interaction energy between the DNA and each residue in these stall sites indicates that arginine residues play the dominant role in DNA binding, both in terms of total enthalpy and in terms of enthalpy per nucleotide, Supplementary Figure S1. The stronger interaction of ssDNA with arginine than lysine may be because arginine interacts with nucleotides through stacking interactions in addition to electrostatics (56,57). To our knowledge, lysine and arginine residues have not been targets of site-directed mutagenesis experiments with the exception of Arg72, which was shown to have little impact on DNA binding (58). Nevertheless, our finding is consistent with a study showing reduced rate of acetylation of SSB lysines upon ssDNA binding (59) and with a recent coarse-grained simulation study of E. coli SSB interacting with DNA (32), which demonstrated that electrostatic interactions are essential for DNA binding in the crystallographic geometry.
The force applied to the DNA ends during the six unwrapping simulations exhibited large peaks, Figure 3G and H. With rupture occurring via multiple stochastic events, the peaks of the force were not distributed with a discernible pattern or regularity because the precise unwrapping path taken by the ssDNA varied in each simulation. The force may have depended on the orientation of the SSB or on intrinsic differences in the instantaneous conformation of DNA–protein contacts. We were unable to find a direct correlation between changes in the force and the occurrence of unbinding events. The apparent lack of correlation may be due to the noisy nature of the force data. Overall, the average forces applied in the simulations were considerably greater than those applied in single-molecule experiment (29). Given the difference in the time scales of the simulation and experiment, the difference in the rupture force magnitudes is expected (60).
One of the forced unwrapping simulations was continued for an extra 145 ns, stretching ssDNA up to 200% of its force-free contour length. Complete dissociation of SSB from a taut stretch of ssDNA was not observed. In qualitative agreement with this observation, single-molecule studies have shown that complete removal of ssDNA from SSB comes with an 8 kBT free energy penalty (29).
In summary, we found unwrapping of ssDNA to proceed as a sequence of stochastic steps. Unwrapping repeatedly stalls at distinct arginine-rich patches on the protein surface whereas the force required to unwrap ssDNA exhibits large fluctuations. However, we also found the force peak patterns to differ among repeat simulations of the same system and no correlation between the occurrence of the force peaks and the unbinding events.
Spontaneous association of ssDNA with SSB
The process of DNA association with SSB would ideally be studied through direct simulation of full-length ssDNA wrapping an SSB tetramer. However, because the relaxation times of long (35–65 nts) ssDNA polymers are considerably greater than the typical simulation time, such brute force simulations would not provide adequate conformational sampling. Since the interactions of SSB with small ssDNA fragments are microscopically similar to those with polymeric DNA, we instead probed kinetics of ssDNA association with SSB and its dependence on ionic condition using an ensemble of short ssDNA fragments. This approach contrasts with usual methods for studying association between biomolecules which enhance sampling of a ligand that binds to a single region of the protein (61). However, similar approaches to the one employed here were recently used to study the interactions between the amino acid side chains and DNA (62) and to investigate binding of molecular oxygen to a respiratory protein (63).
Spontaneous association of ssDNA with SSB was observed using an ensemble of six systems, each containing one SSB protein, fifteen dT3 fragments (45 nts total) randomly placed in electrolyte solution, see Figure 4A. Two sets of simulations were performed corresponding to the low salt (40 mM Na+, each system contained the minimum number of sodium ions required to neutralize the system) and high salt (1200 mM NaCl) environments. During the 340 ns simulations, many DNA fragments quickly became associated with the SSB, see Figure 4A and Supplementary Movie S5. After about 200 ns from the beginning of the simulation, the association and dissociation rates became roughly equal; the number of bound nucleotides approached a steady state value in the 40 mM system, Figure 4B. At the end of each 340 ns simulation, 10 extra dT3 fragments were added to each system along with neutralizing counterions, bringing the total number of nucleotides in each system to 75. The low salt systems were each simulated for another 250 ns; the 1200 mM electrolyte systems were each simulation for another 450 ns. In the low salt simulation, the concentration of sodium away from solute increased from 40 to 65 mM due to the additional neutralizing counterions.
Figure 4.

Spontaneous association of ssDNA with SSB. (A) Microscopic configuration of one of six simulation systems at the beginning (left) and after 340 ns (right) of equilibration simulation. Each system contains 15 dT3 fragments (green vdW spheres), one SSB protein truncated after amino acid 112 (gray molecular surface), water (semitransparent blue surface) and ions (small spheres). Initially, DNA fragments were placed at random positions and orientations such that no fragment possessed a 4-Å contact with the protein. During the equilibration simulations, the α carbon atoms of amino acids forming β sheets and α helices were harmonically restrained about their initial coordinates (ksp = 1 kcal/mol) while DNA fragments spontaneously bind to and dissociate from the SSB surface. (B) Ion concentration-dependent binding of ssDNA fragments to SSB. Purple and blue lines indicate data for the systems containing 40 and 1200 mM electrolyte, respectively. For each system, six simulations were performed in parallel starting with fifteen dT3 fragments randomly placed around SSB; no DNA fragment was initially in contact with SSB. After 340 ns, another ten dT3 fragments plus neutralizing counterions were added to the simulation systems. This brought the bulk-like counterion concentration up to 65 mM for the 40 mM system. The solid/dashed lines indicate the average (over the six simulation systems) number of DNA nucleotides bound to/unbound from SSB. The gray regions highlight similar steady-state concentrations of free (not bound to SSB) nucleotides in the 40 and 1200 mM NaCl systems containing 45 and 75 ssDNA fragments, respectively. The green regions emphasize the dependence of ssDNA binding on ion concentration. (C) Equilibrium density of DNA nucleotides observed in MD simulations of spontaneous binding of the DNA fragments (top row) and in the simulations of the SSB65 complex in 160 mM KCl (bottom row). The mass density of DNA atoms was obtained using the volmap plugin of VMD with a 1-Å resolution, averaged over all equivalent simulation trajectories (except the first 100 ns), and the symmetry axes of the homotetramer. The left-most column depicts the SSB as a gray molecular surface and the DNA mass density isosurface as a semi-transparent green surface. The surface isovalue was taken to be one tenth of the peak value in each density. The four vertical lines labeled i–iv depict the locations of the DNA mass density tomograms shown to the right. From left to right, the slices move away from the center of the SSB.
Once bound to the SSB, ssDNA fragments could stay bound to the same location for the remainder of the simulation, dissociate from the SSB back to solution or move to another location along the protein surface. The most prevalent nucleotide binding locations were seen to be the DBGs, which bound the DNA almost irreversibly, and the adjacent crevices between the L and L23 loops, see Figure 1A. Nucleotides were also seen to transiently bind across the β-sheet preceding loop L23, toward the seam between the upper and lower dimers. By averaging local densities of DNA nucleotides over the simulation trajectories, the symmetry axes of the protein and the six simulation systems, we obtained three-dimensional maps detailing local concentrations of DNA along the SSB surface; one such map is shown in Figure 4C. For comparison, we show in Figure 4C a nucleotide density map obtained from the simulation of the SSB65 complex in solution environment. The two maps exhibit very similar features, although there are some obvious differences in the local concentration values; the Pearson correlation of the two maps is 0.72. A slightly lower correlation (Pearson coefficient of 0.62) is observed when comparing the DNA density obtained from the simulations of spontaneous binding to that obtained from the simulation of the ssDNA–SSB complex in a crystal environment, see Supplementary Figure S2. For reference, the correlation between the DNA density maps obtained from the simulations of the SSB–ssDNA complexes in solution and crystal environments is 0.83. The correlation between DNA densities in various systems described in this manuscript is specified in Supplementary Figure S3.
and L23 loops, see Figure 1A. Nucleotides were also seen to transiently bind across the β-sheet preceding loop L23, toward the seam between the upper and lower dimers. By averaging local densities of DNA nucleotides over the simulation trajectories, the symmetry axes of the protein and the six simulation systems, we obtained three-dimensional maps detailing local concentrations of DNA along the SSB surface; one such map is shown in Figure 4C. For comparison, we show in Figure 4C a nucleotide density map obtained from the simulation of the SSB65 complex in solution environment. The two maps exhibit very similar features, although there are some obvious differences in the local concentration values; the Pearson correlation of the two maps is 0.72. A slightly lower correlation (Pearson coefficient of 0.62) is observed when comparing the DNA density obtained from the simulations of spontaneous binding to that obtained from the simulation of the ssDNA–SSB complex in a crystal environment, see Supplementary Figure S2. For reference, the correlation between the DNA density maps obtained from the simulations of the SSB–ssDNA complexes in solution and crystal environments is 0.83. The correlation between DNA densities in various systems described in this manuscript is specified in Supplementary Figure S3.
The outcome of the spontaneous binding simulations provide insights into the effect of ion concentration on ssDNA association to SSB. The nucleotide association in the low electrolyte systems is seen to reach dynamic equilibrium considerably faster than in other systems, saturating first at ∼25 nts bound to DNA (45-nt systems) and, upon increasing the nucleotide concentration, again at ∼35 nts per SSB. It is interesting that 35 nts is also the number of DNA polymer nucleotides that SSB will bind under low salt conditions. To provide a fair comparison to experiment, we must estimate the effective concentration of unbound nucleotides around each SSB in measurements with excess ssDNA polymers. We assume that in a typical experiment, the free nucleotide concentration near an SSB molecule is dominated by the two strands emerging from the SSB. Integrating over the end-to-end distance of a worm-like chain model, the concentration of nucleotides around an SSB in a volume matching that of our simulation system was estimated to be ∼50 mM, close to the ∼56 mM steady-state concentration of unbound nucleotides realized in our 65 mM electrolyte system containing 75 nts. Hence, the binding of 35 nucleotides to SSB during the simulation at low salt appears to be consistent with the number of nucleotides that bind SSB in experiments.
In simulations with high salt conditions, ∼55 nucleotides were seen to associate with SSB at steady state. This is similar to but slightly lower than the 65 nts known to bind SSB under high salt conditions. The free nucleotide concentration was only ∼30 mM in the simulations, lower than the effective concentration of nucleotides in a similar volume around an SSB molecule bound to an ssDNA polymer. The regions highlighted in Figure 4B show a fair comparison of the number of bound nucleotides (green background) at different ionic conditions, possible only because the two systems have the same concentration in bulk solution (gray background). We find that, at a concentration of free nucleotides a little below the effective concentration within a long DNA polymer, roughly 25 and 55 nts bind SSB at low and high salt, respectively, consistent with experiments that measure the number of nucleotides that bind SSB (8).
Kinetic analysis of the simulation data yields ion concentration-dependent rates for the nucleotides’ association to and dissociation from SSB. We assumed DNA binding to be described by the following kinetic model: 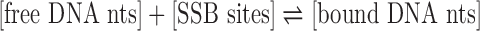 . To reduce the number of fitting parameters to a tractable number, we assumed that an SSB tetramer has independent binding sites available for 65 nts, regardless of the ion concentration. The only remaining parameters were the kinetic rates for DNA nucleotides associating to and dissociating from the protein. Supplementary Note 2 provides a complete description of the model. Optimization of the remaining parameters yielded good fits to the data, see Supplementary Figure S4. Going from low- to high-salt solutions, the binding rate decreased slightly from 2.7 × 108 to 1.2 × 108 s−1M−1 whereas the unbinding rate decreased substantially, from 4.3 × 106 to 2.9 × 105 s−1. The dissociation constant, Kd, computed using these rates drops from 16 to 2.4 mM with increasing ion concentration. The dissociation constants estimated directly from the steady state numbers of bound nucleotides are within ∼10% of the above values. We attribute the decreases in the kinetics to increased screening of electrostatic interactions as well as increased solvent viscosity (64,65).
. To reduce the number of fitting parameters to a tractable number, we assumed that an SSB tetramer has independent binding sites available for 65 nts, regardless of the ion concentration. The only remaining parameters were the kinetic rates for DNA nucleotides associating to and dissociating from the protein. Supplementary Note 2 provides a complete description of the model. Optimization of the remaining parameters yielded good fits to the data, see Supplementary Figure S4. Going from low- to high-salt solutions, the binding rate decreased slightly from 2.7 × 108 to 1.2 × 108 s−1M−1 whereas the unbinding rate decreased substantially, from 4.3 × 106 to 2.9 × 105 s−1. The dissociation constant, Kd, computed using these rates drops from 16 to 2.4 mM with increasing ion concentration. The dissociation constants estimated directly from the steady state numbers of bound nucleotides are within ∼10% of the above values. We attribute the decreases in the kinetics to increased screening of electrostatic interactions as well as increased solvent viscosity (64,65).
Similar studies of ssDNA–SSB binding kinetics have been performed experimentally using longer ssDNA fragments in 20–1000 mM monovalent electrolyte (25,66,67). Using various theoretical and empirical relations, it is possible to obtain semi-quantitative comparisons with the results of these studies. The study baring the greatest similarity to the simulations performed here included measurements of the binding kinetics dT8 and dT16 with SSB in a 220 mM monovalent electrolyte (66). The association rate is expected to be proportional to the diffusion rate, which in turn should be proportional to 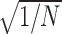 where N is the number of nucleotides in the DNA fragment, according to an ideal Zimm polymer model (68). Using the association rate of dT3 in a high-salt solution as a reference, the simulation data suggest association rates of 7.3 × 107 and 5.2 × 107 s−1M−1 for dT8 and dT16, respectively, in perfect agreement with the corresponding experimental values of 7 × 107 and 5 × 107 s−1M−1.
where N is the number of nucleotides in the DNA fragment, according to an ideal Zimm polymer model (68). Using the association rate of dT3 in a high-salt solution as a reference, the simulation data suggest association rates of 7.3 × 107 and 5.2 × 107 s−1M−1 for dT8 and dT16, respectively, in perfect agreement with the corresponding experimental values of 7 × 107 and 5 × 107 s−1M−1.
As shown below, the simulated dissociation rate of 29,000 s−1 for dT3 in high salt is consistent with the experimental dissociation rates for dT8 and dT16, 1700 and 40 s−1, respectively (66). According to the Arrhenius equation, 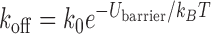 , where Ubarrier is the height of the free energy barrier for unbinding, kBT is the thermal energy, and k0 is an intrinsic unbinding rate. Assuming Ubarrier = uN + u0, where u is the energy per nucleotide, N is the number of nucleotides and u0 is a constant offset, a least squares fit to the simulated and experimental kinetic rates provides u = 0.56 kBT/nt. Using this energy and the simulated rate, we calculate dissociation rates of 1760 and 20 s−1 for dT8 and dT16, respectively. In addition, the energy per nucleotide is in good agreement with the 0.44 kBT/nt value obtained from single-molecule experiment (11).
, where Ubarrier is the height of the free energy barrier for unbinding, kBT is the thermal energy, and k0 is an intrinsic unbinding rate. Assuming Ubarrier = uN + u0, where u is the energy per nucleotide, N is the number of nucleotides and u0 is a constant offset, a least squares fit to the simulated and experimental kinetic rates provides u = 0.56 kBT/nt. Using this energy and the simulated rate, we calculate dissociation rates of 1760 and 20 s−1 for dT8 and dT16, respectively. In addition, the energy per nucleotide is in good agreement with the 0.44 kBT/nt value obtained from single-molecule experiment (11).
The ion dependence of experimentally measured kinetic rates for dT70 interacting with SSB were very similar to the dependence observed in our simulations of the dT3 systems (25,67). Going from 20 to 1000 mM, the experimentally measured association rate dropped by a factor of 3 (25). In our simulations, the rate dropped by a factor of 2 upon changing the effective counterion concentration from 40 to 1200 mM. The experimentally measured dissociation rate decreased by a factor of 5 for the concentration change from 20 to 200 mM (67) and by another factor of 2 from 200 to 1000 mM (25). Hence, increasing the ion concentration from 20 to 1000 mM decreases the unbinding rate of dT70 by a factor of 10 in experiment, which is similar to the 15-fold decrease of the dT3 unbinding rate observed in the simulations of the 40 and 1200 mM systems.
The acidic C-terminal tips of wild-type E. coli SSB may compete with ssDNA for SSB binding sites (69,70). To examine such a possibility, we constructed twelve replica systems, each containing one copy of SSB, 15 dT3 fragments, 40 mM Na+ or 1200 mM NaCl solution and eight 3-residue peptides; the peptides had the amino acid sequence of the acidic C-terminal tip of SSB. We found that, regardless of ion concentration, DNA binding was unhampered by the competition with the tips, see Supplementary Figure S5 and Movie S6. Indeed, SSB affinity for the peptides was seen to be overall quite low, suggesting that non-specific binding of acidic tips, e.g. through electrostatic interactions, does not directly mediate interaction between SSB molecules. Given the experimental observation that acidic tips promote cooperative ssDNA binding (69,70), our simulations suggest that SSB may possess a specific binding site for the acidic tips that our simulations could not identify because of their relatively short duration.
Thus, we have shown that simulation of spontaneous binding can map local affinity of DNA to the SSB surface, revealing features that are in good agreement with the crystallographic studies. Good quantitative agreement between the simulated and crystallographic affinity maps suggest that our simulation method can be applied to other DNA binding proteins to predict and characterize the DNA binding sites. We find the kinetic and equilibrium properties of DNA binding to SSB to depend on electrolyte conditions: ∼35 nts bind SSB under low salt conditions; the number of bound nucleotides increase to ∼55 at high salt concentration. The rate of DNA binding decreases twofold when changing salt conditions from 40 to 1200 mM, whereas the unbinding rate drops by an order of magnitude. Finally, we find acidic tips of the SSB tails to not affect DNA binding to SSB.
Local reptation of ssDNA during microseconds-long simulations of SSB65 in solution
Using the D.E. Shaw Research Anton supercomputer (49), we examined interactions of ssDNA with SSB at the microseconds time scale. In addition to extending our simulation of the SSB65 complex in 160 mM KCl, we also simulated the same SSB65 complex in 50 mM K+ (neutralizing condition) and 1100 mM KCl electrolyte. Each system was simulated for 10 μs, see Supplementary Movies S7–9 in the Supplementary Data. We also performed a fourth simulation of the SSB-GG mutant (70), which had the tips fused directly to the SSB core without a linker, see Supplementary Movie S10. The SSB-GG mutant was simulated in 170 mM KCl. This mutant was selected because it included the C-terminal tip, but the tails were nevertheless short enough to allow good sampling during an MD simulation.
A number of observations are common to all simulation systems. First, the DNA is rather mobile at the surface of SSB, Figure 5A and Supplementary Figure S6, but explores a similar volume of conformational space regardless of the ion concentration or the presence of C-terminal tips. Second, dynamics of ssDNA at the surface of SSB is consistent with diffusion by reptation. An exemplary, but not unique, bulge formation event is highlighted in the panels of Figure 5B. This bulge was seen to move short distances through the nucleotides on the 100-ns timescale, but did not appear to disturb adjacent nucleotides bound to a DBG.
Figure 5.

Motion of DNA bound to SSB in solution during microseconds-long simulations. (A) Extent of motion of DNA backbone. Each row shows the DNA backbone conformation sampled every 96 ns during the simulation. The DNA is depicted as a tube that is colored from green to blue every 10 nt. The top row corresponds to the simulation of the GG-tip mutant and shows the acidic tip conformation using a purple tube representation. The bottom row depicts the simulation trajectory of SSB with C-terminal tails truncated. The protein–DNA complex was surrounded by a 160 mM KCl electrolyte (not depicted). Each image depicts the protein using a molecular surface representation. Each column shows the complex rotated by 90° compared to the previous column. (B) Highlight of the formation of a bulge. After nearly 4 μs of simulation in 160 mM KCl, a prominent bulge was seen to form adjacent to a DBG. The DNA backbone is shown every 300 ns from left to right and top to bottom using a vdW representation with nucleotides colored between dark green and purple every 10 nt. The bulge continued to change its conformation during the remainder of the simulation. Supplementary Movie S13 illustrates the formation of the bulge.
Bulges were quantified by examining those nucleotides with fewer than fifteen 6-Å contacts with the protein, Figure 6A. A number of bulges were seen to form, with most located on or adjacent to a DBG and involving 1–6 nts, Figure 6B. In general, the bulges underwent conformation changes and sometimes dissipated on the microsecond timescale, Figure 6C. Other local conformational changes were observed where nucleotides would change how they contact the SSB surface. Although we observed many local conformational changes, displacement of the DNA along the wrapping pathway was seen to be quite rare on this timescale. Events consistent with diffusion by reptation are depicted in Supplementary Movies S11–13 in the Supplementary Data.
Figure 6.

Quantification of DNA bulges on the SSB surface. (A) DNA bulges on the SSB surface during 10-μs simulations of the SSB complex, as characterized by nucleotides having fifteen or fewer 6-Å contacts between backbone atoms and non-hydrogen SSB atoms. For reference, the nucleotides each had ∼30 such contacts on average. The contact data were sampled for each nucleotide every 2.4 ns and were smoothed using a 1.5-nt by 20-ns wide 2D Gaussian kernel. Motion of the density shown in the plot corresponds to propagation of the bulge through the DNA. (B) Distribution of bulge sizes during 10-μs simulations of the SSB complex. The bulge sizes were determined from the contact data shown in panel A for each microscopic conformation. (C) Distribution of bulge lifetimes during 10-μs simulations of the SSB complex. The lifetime of each bulge was determined from the contact data shown in panel A.
The DNA strand was also observed to undergo several large-scale cooperative rearrangements. In each of the four simulations, a similar change of a DNA wrapping configuration occurred within the first 200 ns: the 6 nts forming the 3′ DNA end migrated along the SSB surface to adopt a new wrapping pathway, see Supplementary Figure S7A and Supplementary Movies S7–10, increasing the distance between the DNA ends from 25 to 31–54 Å depending on the system. Another dramatic conformational change was observed after 6.3 μs of simulation of SSB-GG, where a group of nearly 8 nts was seen to move across the SSB surface orthogonal to the wrapping path before returning to the original location, see Supplementary Movie S14 and Supplementary Figure S7B. One end of these nucleotides remained bound to a DBG and the other end remained bound to one of the N-terminal arginine patches. The observation of concerted motion between arginine-rich sites suggests that DNA may be loosely associated with SSB away from arginine-rich locations. With the exception of the motion of the 3′ DNA end, all of the bulge formations and DNA motions discussed above were observed in nucleotides that were resolved by x-ray crystallography.
Contrary to our initial expectations, the electrolyte conditions and presence of acidic tips had rather modest effects on the manner of ssDNA association with SSB despite a multitude of statistical characterizations, including RMS displacements and fluctuations, the number of nucleotides making 4-Å contacts with the protein, and the number of initial 4-Å atomic contacts between DNA and protein that remained intact, see Supplementary Figures S8 and S9A and B. A small but statistically significant difference was observed in the average RMS fluctuation of the CoMs of resolved nucleotides, which was ∼4 Å for complex in a 50 mM solution and ∼3 Å for SSB in the higher salt solutions. We also found that it was possible to distinguish the systems using pairwise-q, a measure of the similarity of two polymer configurations, see Supplementary Figure S9C and D. The mean of the pairwise-q distributions increased with the ion concentration from ∼0.3 in the absence of added ions to ∼0.4 for 160 and 1100 mM, indicating that the DNA was slightly less dynamic under higher salt conditions. The reduced RMS fluctuations and increased mean pairwise-q values for systems in higher salt solution are both in qualitative agreement with the reduction of the ssDNA dissociation rate observed in our MD simulations of spontaneous binding of ssDNA fragments to SSB (see previous section). In the simulation of the SSB-GG system, the acidic tips were seen to explore a large number of possible configurations, but remained adsorbed to the SSB surface, which contrasts with the results of the simulations of competitive binding of small ssDNA and acidic tips fragments, Supplementary Figure S5. The strong association of the acidic tips to the SSB surface seen in the simulations of the SSB-GG system is caused by the very high local concentration of the tips resulting from their covalent attachment to the SSB core via a short, two-residue tether.
Thus, in our long timescale simulations of the SSB65 complex, we directly observed formation of bulges in the ssDNA conformation that likely represent the microscopic mechanism underlying diffusion of SSB along ssDNA (29,71). The extensive dynamics of ssDNA observed in our simulations suggests that the majority of ssDNA bound to SSB can be easily repositioned. Unexpectedly, we found the concentration of ions to not substantially affect the dynamics of ssDNA bound to SSB, despite seeing considerable effects of ion concentration on rates of ssDNA fragment binding to and unbinding from the SSB surface. We attribute the apparent discrepancy to strong association of the DNA polymer to the high-affinity regions at the SSB surface (e.g. DBGs) that greatly reduce ability of loosely bound DNA nucleotides to dissociate from the SSB surface. We expect that much longer simulation would be required to observe partial spontaneous unraveling of the SSB65 complex at low salt condition.
DISCUSSION
Using several complimentary MD approaches, we have characterized the interactions between ssDNA and SSB at the microscopic level. Stated concisely, our main findings are that ssDNA bound to SSB is highly dynamic, stabilized by arginine-rich patches of the protein surface, stabilized by polymerization, insensitive to competition for binding from C-terminal tips of SSB, and able to diffuse through the propagation of small bulges around the SSB surface. Below we discuss each of these findings.
Our simulations indicate that ssDNA bound to SSB remains highly mobile and can rearrange its wrapping of the SSB protein within microseconds. The strongest evidence supporting this statement comes from the direct observation of substantial DNA motion in all four 10-μs simulations of SSB, Figures 5 and 6; Supplementary Figures S6 and S7, quantified by the average RMS deviation values of 10 Å for DNA fragments resolved in the crystal structure, Supplementary Figure S8A and the low fraction of native 4-Å contacts Supplementary Figure S9B. At the same time, we find spontaneous displacement of DNA nucleotides to be suppressed in the crystal environment, which is consistent with the existence of an atomic structure derived from x-ray crystallography. Further support of the highly dynamic DNA–SSB binding interface is derived from the simulations of spontaneous binding of ssDNA fragments, where the characteristic timescale of a fragment becoming unbound was as low as ∼100 ns, depending on the ion concentration. Taken together, our simulations support a view of SSB where contacts between ssDNA and the protein can readily be traded to realize many possible microscopic conformations.
We also demonstrated that DNA bound to SSB is stabilized by arginine-rich patches on the protein surface. During the simulated process of forced unwrapping of ssDNA from SSB, we observed the unwrapping process to stall at several arginine-rich patches that included the DBGs, Figure 3C–F. Furthermore, rearrangement of DNA bound to the DBGs was minimal during equilibrium simulations of the SSB complex in solution, Figure 2C. Such rearrangement or unbinding of DNA from the DBGs and other arginine-rich regions may represent a rate-limiting step during the process of SSB sliding along ssDNA. Finally, in the simulations of spontaneous binding of ssDNA fragments to SSB, Figure 4C, the highest affinity sites were the DBGs (peak DNA density 50–140 times bulk density), followed by grooves in the SSB surface bordered by Lys3, Arg41, Tyr60, Arg3 and Arg115 (peak DNA density 30-85 times bulk density).
Most of our simulations used models of SSB that had the C-terminal tails truncated due to the difficulty of simulating intrinsically disordered proteins. The acidic C-terminal tips play an important role in the coordination of proteins involved in DNA metabolism (17). Furthermore, it was recently suggested that the C-terminal tips could compete with DNA for binding SSB, which could have implications for their ability to recruit DNA processing proteins. We found, however, that the acidic C-terminal tips of SSB do not effectively compete with ssDNA for SSB binding, Supplementary Figure S5, at least not for unspecific binding. This observation is consistent with the notion that the intrinsically disordered linkers, and not the C-terminal tips, directly mediate SSB–SSB interactions (72). In our long time-scale MD simulation of the SSB-GG mutant the local concentration of the tips was very high, and we observed the acidic tips to sample a diverse range of microscopic conformations while remaining strongly associated with the grooves at the dimer–dimer interface of the SSB.
Finally, we directly observed the microscopic steps that occur when SSB diffuses along a DNA strand in multi-microsecond simulations of SSB65 in electrolytes ranging from 50 mM K+ to 1100 mM KCl. Specifically, we observed the formation and short-ranged diffusion of several small bulges, usually 1-5 nts in size (see Figure 6), consistent with the results of the single-molecule fluorescence experiments that found SSB to move along DNA according to the reptation model of diffusion, with DNA bulges averaging 3-nt in size (28). None of the bulges were observed diffusing through a DBG, suggesting that the motion of bulges through the DNA binding grooves represents a rate-limiting step during the process of diffusion. One implication of this conjecture is that the rate of diffusion of SSB along ssDNA should be faster for SSB35 than it is for SSB65 because a bulge needs to propagate through fewer DBGs.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs T. Ha and Y. Chemla (UIUC) and members of their research groups for their illuminating discussions. Supercomputer time was provided via XSEDE allocation MCA05S028, University of Illinois Blue Waters petascale system, the Argonne Leadership Computing Facility’s Mira via a discretionary allocation and the National Resource for Biomedical Supercomputing at the DESRES supercomputer Anton at the Pittsburgh Supercomputing Center via allocation PSCA00052.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [PHY-1430124]. Funding for open access charge: National Science Foundation [PHY-1430124].
Conflict of interest statement. None declared.
REFERENCES
- 1. Halazonetis T.D., Gorgoulis V.G., Bartek J.. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319:1352–1355. [DOI] [PubMed] [Google Scholar]
- 2. Yang H., Jeffrey P.D., Miller J., Kinnucan E., Sun Y., Thoma N.H., Zheng N., Chen P.-L., Lee W.-H., Pavletich N.P.. BRCA2 function in DNA binding and recombination from a BRCA2–DSS1–ssDNA structure. Science. 2002; 297:1837–1848. [DOI] [PubMed] [Google Scholar]
- 3. Johnson A., O’Donnell M.. Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 2005; 74:283–315. [DOI] [PubMed] [Google Scholar]
- 4. Duderstadt K.E., Reyes-Lamothe R., van Oijen A.M., Sherratt D.J.. Replication-fork dynamics. Cold Spring Harb. Perspect. Biol. 2014; 6:a010157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bochkarev A., Bochkareva E.. From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr. Opin. Struct. Biol. 2004; 14:36–42. [DOI] [PubMed] [Google Scholar]
- 6. Rosche W.A., Jaworski A., Kang S., Kramer S.F., Larson J.E., Geidroc D.P., Wells R.D., Sinden R.R.. Single-stranded DNA-binding protein enhances the stability of CTG triplet repeats in Escherichia coli. J. Bacteriol. 1996; 178:5042–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meyer R.R., Laine P.S.. The single-stranded DNA-binding protein of Escherichia coli. Microbiol. Rev. 1990; 54:342–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lohman T.M., Ferrari M.E.. Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and coperativities. Annu. Rev. Biochem. 1994; 63:527–570. [DOI] [PubMed] [Google Scholar]
- 9. Lohman T.M., Overman L.B.. Two binding modes in Escherichia coli single strand binding protein–single stranded DNA complexes. J. Biol. Chem. 1985; 260:3594–3603. [PubMed] [Google Scholar]
- 10. Bujalowski W., Lohman T.M.. Escherichia coli single-strand binding protein forms multiple, distinct complexes with single-stranded DNA. Biochemistry. 1986; 25:7799–7802. [DOI] [PubMed] [Google Scholar]
- 11. Suksombat S., Khafizov R., Kozlov A.G., Lohman T.M., Chemla Y.R.. Structural dynamics of E. coli single-stranded DNA binding protein reveal DNA wrapping and unwrapping pathways. Elife. 2015; 4:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferrari M.E., Bujalowski W., Lohman T.M.. Co-operative binding of Escherichia coli SSB tetramers to single-stranded DNA in the (SSB)35 binding mode. J. Mol. Biol. 1994; 236:106–123. [DOI] [PubMed] [Google Scholar]
- 13. Kozlov A.G., Jezewska M.J., Bujalowski W., Lohman T.M.. Binding specificity of Escherichia coli single-stranded DNA binding protein for the χ subunit of DNA pol III holoenzyme and PriA helicase. Biochemistry. 2010; 49:3555–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naue N., Fedorov R., Pich A., Manstein D.J., Curth U.. Site-directed mutagenesis of the χ subunit of DNA polymerase III and single-stranded DNA-binding protein of E. coli reveals key residues for their interaction. Nucleic Acids Res. 2011; 39:1398–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Antony E., Weiland E., Yuan Q., Manhart C.M., Nguyen B., Kozlov A.G., McHenry C.S., Lohman T.M.. Multiple C-terminal tails within a single E. coli SSB homotetramer coordinate DNA replication and repair. J. Mol. Biol. 2013; 425:4802–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Umezu K., Kolodner R.D.. Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J. Biol. Chem. 1994; 269:30005–30013. [PubMed] [Google Scholar]
- 17. Shereda R.D., Kozlov A.G., Lohman T.M., Cox M.M., Keck J.L.. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008; 43:289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castellana M., Wilson M.Z., Xu Y., Joshi P., Cristea I.M., Rabinowitz J.D., Gitai Z., Wingreen N.S.. Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat. Biotech. 2014; 32:1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cho W.K., Jayanth N., English B.P., Inoue T., Andrews J.O., Conway W., Grimm J.B., Spille J.H., Lavis L.D., Lionnet T. et al. . RNA Polymerase II cluster dynamics predict mRNA output in living cells. Elife. 2016; 5:1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Japrung D., Bahrami A., Nadzeyka A., Peto L., Bauerdick S., Edel J.B., Albrecht T.. SSB binding to single-stranded DNA probed using solid-state nanopore sensors. J. Phys. Chem. B. 2014; 118:11605–11612. [DOI] [PubMed] [Google Scholar]
- 21. Mahmood M.A.I., Ali W., Adnan A., Iqbal S.M.. 3D structural integrity and interactions of single-stranded protein-binding DNA in a functionalized nanopore. J. Phys. Chem. B. 2014; 118:5799–5806. [DOI] [PubMed] [Google Scholar]
- 22. Raghunathan S., Kozlov A.G., Lohman T.M., Waksman G.. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat. Struct. Mol. Biol. 2000; 7:648–652. [DOI] [PubMed] [Google Scholar]
- 23. Khamis M., Casas-Finet J., Maki A.. Investigation of the role of individual tryptophan residues in the binding of Escherichia coli single-stranded DNA binding protein to single-stranded polynucleotides. J. Biol. Chem. 1987; 262:10938–10945. [PubMed] [Google Scholar]
- 24. Casas-Finet J.R., Khamis M.I., Maki A.H., Chase J.W.. Tryptophan 54 and phenylalanine 60 are involved synergistically in the binding of E. coli SSB protein to single-stranded polynucleotides. FEBS Lett. 1987; 220:347–352. [DOI] [PubMed] [Google Scholar]
- 25. Kozlov A.G., Lohman T.M.. Stopped-flow studies of the kinetics of single-stranded DNA binding and wrapping around the Escherichia coli SSB tetramer. Biochemistry. 2002; 41:6032–6044. [DOI] [PubMed] [Google Scholar]
- 26. Joo C., McKinney S.A., Nakamura M., Rasnik I., Myong S., Ha T.. Real-time observation of RecA filament dynamics with single monomer resolution. Cell. 2006; 126:515–527. [DOI] [PubMed] [Google Scholar]
- 27. Wu C.A., Zechner E.L., Marians K.J.. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. I. Multiple effectors act to modulate Okazaki fragment size. J. Biol. Chem. 1992; 267:4030–4044. [PubMed] [Google Scholar]
- 28. Roy R., Kozlov A.G., Lohman T.M., Ha T.. SSB protein diffusion on single-stranded DNA stimulates RecA filament formation. Nature. 2009; 461:1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou R., Kozlov A.G., Roy R., Zhang J., Korolev S., Lohman T.M., Ha T.. SSB functions as a sliding platform that migrates on DNA via reptation. Cell. 2011; 146:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Römer R., Schomburg U., Krauss G., Maass G.. Escherichia coli single-stranded DNA binding protein is mobile on DNA: proton NMR study of its interaction with oligo- and polynucleotides. Biochemistry. 1984; 23:6132–6137. [DOI] [PubMed] [Google Scholar]
- 31. Kuznetsov S.V., Kozlov A.G., Lohman T.M., Ansari A.. Microsecond dynamics of protein–DNA interactions: direct observation of the wrapping/unwrapping kinetics of single-stranded DNA around the E. coli SSB tetramer. J. Mol. Biol. 2006; 359:55–65. [DOI] [PubMed] [Google Scholar]
- 32. Mishra G., Levy Y.. Molecular determinants of the interactions between proteins and ssDNA. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:5033–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scaltriti E., Polverini E., Grolli S., Eufemi E., Moineau S., Cambillau C., Ramoni R.. The DNA binding mechanism of a SSB protein from Lactococcus lactis Siphophage p2. Biochim. Biophys. Acta. 2013; 1834:1070–1076. [DOI] [PubMed] [Google Scholar]
- 34. Wang C., Nam S.-W., Cotte J.M., Jahnes C.V., Colgan E.G., Bruce R.L., Brink M., Lofaro M.F., Patel J.V., Gignac L.M.. Wafer-scale integration of sacrificial nanofluidic chips for detecting and manipulating single DNA molecules. Nat. Commun. 2017; 8:14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 36. Brünger A.T. X-PLOR, Version 3.1: A System for X-ray Crystallography and NMR The Howard Hughes Medical Institute and Department of Molecular Biophysics and Biochemistry. 1992; New Haven: Yale UniversityPress. [Google Scholar]
- 37. Martyna G.J., Tobias D.J., Klein M.L.. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994; 101:4177–4189. [Google Scholar]
- 38. Feller S.E., Zhang Y.H., Pastor R.W., Brooks B.R.. Constant pressure molecular dynamics simulation—the Langevin Piston Method. J. Chem. Phys. 1995; 103:4613–4621. [Google Scholar]
- 39. Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wells D.B., Bhattacharya S., Carr R., Maffeo C., Ho A., Comer J., Aksimentiev A.. Gracheva ME. Optimization of the molecular dynamics method for simulations of DNA and Ion transport through biological nanopores. Nanopore-based Technology: Single Molecule Characterization and DNA Sequencing. 2012; 870:NY: Humana Press; 165–186. [DOI] [PubMed] [Google Scholar]
- 41. MacKerell A.D. Jr, Bashford D., Bellott M., Dunbrack R.L. Jr, Evanseck J., Field M.J., Fischer S., Gao J., Guo H., Ha S. et al. . All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998; 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 42. Hart K., Foloppe N., Baker C.M., Denning E.J., Nilsson L., MacKerell A.D. Jr. Optimization of the CHARMM additive force field for DNA: improved treatment of the BI/BII conformational equilibrium. J. Chem. Theory Comput. 2012; 8:348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. MacKerel A.D., Feig M., Brooks III C.L.. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004; 25:1400–1415. [DOI] [PubMed] [Google Scholar]
- 44. Beglov D., Roux B.. Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J. Chem. Phys. 1994; 100:9050–9063. [Google Scholar]
- 45. Yoo J., Aksimentiev A.. Improved parametrization of Li+, Na+, K+, and Mg2 + ions for all-atom molecular dynamics simulations of nucleic acid systems. J. Phys. Chem. Lett. 2012; 3:45–50. [Google Scholar]
- 46. Yoo J., Aksimentiev A.. Improved parameterization of amine–carboxylate and amine–phosphate interactions for molecular dynamics simulations using the CHARMM and AMBER force fields. J. Chem. Theory Comput. 2016; 12:430–443. [DOI] [PubMed] [Google Scholar]
- 47. Matsumoto T., Morimoto Y., Shibata N., Kinebuchi T., Shimamoto N., Tsukihara T., Yasuoka N.. Roles of functional loops and the CD-terminal segment of a single-stranded DNA binding protein elucidated by X-ray structure analysis. J. Biochem. 2000; 127:329–335. [DOI] [PubMed] [Google Scholar]
- 48. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graphics. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 49. Shaw D.E., Dror R.O., Salmon J.K., Grossman J.P., MacKenzie K.M., Bank J.A., Young C., Deneroff M.M., Batson B., Bowers K.J. et al. . Millisecond-scale molecular dynamics simulations on Anton. Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis. 2009; Portland: ACM; 39:1–39:11. [Google Scholar]
- 50. Grubmüller H. SOLVATE 1.0 Manual. 1996; http://www.mpibpc.mpg.de/grubmueller/solvate.
- 51. MacKerell A.D. Jr, Banavali N.K.. All-atom empirical force field for nucleic acids: II. application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000; 21:105–120. [Google Scholar]
- 52. Maffeo C., Ngo T.T.M., Ha T., Aksimentiev A.. A coarse-grained model of unstructured single-stranded DNA derived from atomistic simulation and single-molecule experiment. J. Chem. Theory Comput. 2014; 10:2891–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nosé S. A unified formulatoion of the constant temperature molecular dynalmics methods. J. Chem. Phys. 1984; 81:511–519. [Google Scholar]
- 54. Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985; 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 55. Dudko O.K., Hummer G., Szabo A.. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:15755–15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mascotti D.P., Lohman T.M.. Thermodynamics of oligoarginines binding to RNA and DNA. Biochemistry. 1997; 36:7272–7279. [DOI] [PubMed] [Google Scholar]
- 57. Bhattacharya S., Derrington I.M., Pavlenok M., Niederweis M., Gundlach J.H., Aksimentiev A.. Molecular dynamics study of MspA arginine mutants predicts slow DNA translocations and ion current blockades indicative of DNA sequence. ACS Nano. 2012; 6:6960–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Witte G., Fedorov R., Curth U.. Biophysical analysis of Thermus Aquaticus single-stranded DNA binding protein. Biophys. J. 2008; 94:2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen J., Smith D.L., Griep M.A.. The role of the 6 lysines and the terminal amine of Escherichia coli single-strand binding protein in its binding of single-stranded DNA. Protein Sci. 1998; 7:1781–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Walton E.B., Lee S., Van Vliet K.J.. Extending Bell’s model: how force transducer stiffness alters measured unbinding forces and kinetics of molecular complexes. Biophys. J. 2008; 94:2621–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gumbart J.C., Chipot C.. Efficient determination of protein–protein standard binding free energies from first principles. J. Chem. Theory Comput. 2013; 9:3789–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Andrews C.T., Campbell B.A., Elcock A.H.. Direct comparison of amino acid and salt interactions with double-stranded and single-stranded DNA from explicit-solvent molecular dynamics simulations. J. Chem. Theory Comput. 2017; 13:1794–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Husen P., Solov’yov I.A.. Spontaneous binding of molecular oxygen at the Qo-Site of the bc1 complex could stimulate superoxide formation. J. Am. Chem. Soc. 2016; 138:12150–12158. [DOI] [PubMed] [Google Scholar]
- 64. Goldsack D.E., Franchetto R.C.. The viscosity of concentrated electrolyte solutions. I. Concentration dependence at fixed temperature. Can. J. Chem. 1977; 55:1062–1072. [Google Scholar]
- 65. Isono T. Density, viscosity, and electrolytic conductivity of concentrated aqueous electrolyte solutions at several temperatures. Alkaline-earth chlorides, LaCl3, Na2SO4, NaNO3, NaBr, KNO3, KBr, and Cd(NO3)2. J. Chem. Eng. Data. 1984; 29:45–52. [Google Scholar]
- 66. Krauss G., Sindermann H., Schomburg U., Maass G.. Escherichia coli single-strand deoxyribonucleic acid binding protein: stability, specificity, and kinetics of complexes with oligonucleotides and deoxyribonucleic acid. Biochemistry. 1981; 20:5346–5352. [DOI] [PubMed] [Google Scholar]
- 67. Kunzelmann S., Morris C., Chavda A.P., Eccleston J.F., Webb M.R.. Mechanism of interaction between single-stranded DNA binding protein and DNA. Biochemistry. 2010; 49:843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zimm B.H. Dynamics of polymer molecules in dilute solution: viscoelasticity, flow birefringence and dielectric loss. J. Chem. Phys. 1956; 24:269–278. [Google Scholar]
- 69. Kozlov A.G., Cox M.M., Lohman T.M.. Regulation of single-stranded DNA binding by the C termini of Escherichia coli single-stranded DNA-binding (SSB) protein. J. Biol. Chem. 2010; 285:17246–17252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kozlov A.G., Weiland E., Mittal A., Waldman V., Antony E., Fazio N., Pappu R.V., Lohman T.M.. Intrinsically disordered C-terminal Tails of E. coli single-stranded DNA binding protein regulate cooperative binding to single-stranded DNA. J. Mol. Biol. 2015; 427:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee K.S., Marciel A.B., Kozlov A.G., Schroeder C.M., Lohman T.M., Ha T.. Ultrafast redistribution of E. coli SSB along long single-stranded DNA via intersegment transfer. J. Mol. Biol. 2014; 426:2413–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bianco P.R., Lyubchenko Y.L.. SSB and the RecG DNA helicase: an intimate association to rescue a stalled replication fork. Protein Sci. 2017; 26:638–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.