

Abstract
Early recognition of acute kidney injury (AKI) is critical to prevent its associated complications as well as its progression to long term adverse outcomes like chronic kidney disease. A growing body of evidence from both laboratory and clinical studies suggests that inflammation is a key factor contributing to the progression of AKI regardless of the initiating event. Biomarkers of inflammation are therefore of interest in the evaluation of AKI pathogenesis and prognosis. There is evidence that the renin angiotensin aldosterone system is activated in AKI, which leads to an increase in angiotensin II (Ang II) formation within the kidney. Ang II activates pro-inflammatory and pro-fibrotic pathways that likely contribute to the progression of AKI. Angiotensinogen is the parent polypeptide from which angiotensin peptides are formed and its stability in urine makes it a more convenient marker of renin angiotensin system activity than direct measurement of Ang II in urine specimens, which would provide more direct information. The potential utility of urinary angiotensinogen as a biomarker of AKI is discussed in light of emerging data showing a strong predictive value of AKI progression, particularly in the setting of decompensated heart failure. The prognostic significance of urinary angiotensinogen as an AKI biomarker strongly suggests a role for renin–angiotensin system activation in modulating the severity of AKI and its outcomes.
Keywords: acute decompensated heart failure, AKI, angiotensin II, angiotensinogen, renin–angiotensin system
Introduction
Acute kidney injury (AKI) is a global health problem with its increasing incidence [1], association with significant morbidity and mortality [2], and lack of effective therapeutic interventions [3]. Early recognition of AKI is therefore critical to translate into timely intervention to prevent its associated complications like volume overload, hyperkalemia, acidosis and uremia [4], as well as its progression to long-term adverse outcomes like chronic kidney disease (CKD) [5–7] and cardiovascular events [7]. The current diagnostic criteria of AKI are based on oliguria and acute rise in serum creatinine. It usually takes 2–3 days to increase significantly because of the rate-limiting step of creatinine production and release by skeletal muscle [8, 9]. Therefore, interventions administered at the time of diagnosis of AKI using elevated serum creatinine may not be effective, as judged by several clinical trials of promising therapies for AKI in humans [10–12]. The possibility of a more timely and accurate diagnosis of AKI early on is therefore critical to prevent kidney dysfunction and damage [13]. The kinetic glomerular filtration rate (GFR) formula developed by Chen [14] is, in our opinion, a sensitive tool to recognize AKI early and more precisely estimate the decline and recovery of kidney function. Although early recognition of the fall in GFR is critical, some biomarkers may provide insights on AKI progression that the GFR itself cannot provide [15].
In the last few years, we have witnessed an explosion of research directed towards the discovery of novel biomarkers for AKI. Biomarkers examined reflect physiological and pathophysiological processes and have the potential ability to identify either the injured tubular system and/or assist in identifying the high-risk patients at an earlier stage, thereby fostering preventive and protective measures well before clinical AKI is manifest [13, 16]. These include neutrophil gelatinase-associated lipocalin (NGAL) [17], kidney injury molecule-1 (KIM-1) [18], interleukin 18 (IL-18) [19], Cystanin C [20], liver-type fatty acid-binding protein [21], tissue inhibitor of metalloproteinases (TIMP-2), IGF-binding protein 7 (IGFBP7) [22] and angiotensinogen [23] (Table 1). Two markers of G1 cell cycle arrest, TIMP-2 and IGFBP7, have been viewed as a promising biomarker [22]. However, they still need substantial validation to define their clinical role [31, 33]. The widespread clinical use of biomarkers in AKI, while promising, in our opinion is not yet warranted. In large, prospective, multicenter trials of the aforementioned biomarkers have failed to show ‘troponin-like’ diagnostic performance [35, 36], to use the comparison of what is expected to diagnose acute coronary syndrome as commonly done in the clinical setting.
Table 1.
Biomarkers of AKI
| AKI biomarker | Characteristics/functions | AUCs for AKI prediction | Settings (sample collection) | Limitations |
|---|---|---|---|---|
| NGAL | A 25-kDa protein of the family of lipocalins with bacteriostatic function | 0.87 | All hospitalized patients [24] | May be elevated in the settings of sepsis, CKD and UTI [25] |
| 0.72 | Cardiac surgery [26] | The lack of specific cut-off values [27] | ||
| 0.80 | ICU [28] | |||
| KIM-1 | A 38.7-kDa type I transmembrane glycoprotein | 0.85 | Cardiac surgery [18] | May be elevated in the settings of chronic proteinuria and inflammatory diseases [16] |
| 0.77 | ICU and others [18] | High cost and poor availability [27] | ||
| L-FABP | A 14-kDa protein from the large superfamily of lipid binding proteins | 0.81 | Cardiac surgery [21] | Strongly associated with anemia in nondiabetic patients |
| IL-18 | A 24-kDa cytokine from the IL-1 family of cytokines | 0.72 | Cardiac surgery in adults [26] | No certain prediction of AKI in adults [16] |
| 0.75 | ICU [29] | |||
| [TIMP-2]X[IGFBP7] | TIMP-2: a 21-kDa protein, endogenous inhibitor of meralloproteinase activities | 0.80 | ICU [22] | May be elevated in the setting of diabetes |
| IGFBP7: a 29-kDa secreted protein known to bind to and inhibit signaling through IGF-1 receptors | Needs validation to define clinical role [31] | |||
| 0.84 | Cardiac surgery [30] | |||
| uAGT | A 453-amino acid-long protein with 10 N-terminal amino acids | 0.84 | Acute decompensated heart failure [32] | Needs validation in other clinical settings. May be considered as a prognostic biomarker. Data for use as a diagnostic biomarker are limited [33] |
| 0.70 | Cardiac surgery patients [34] | |||
| 0.73 | Other etiologies of AKI [23] |
L-FABP, liver-type fatty acid-binding protein; TIMP-2, tissue inhibitor of metalloproteinase-2; IGFBP7, IGF binding protein 7; ICU, intensive care unit; UTI, urinary tract infection; AUC, area under the curve. Modified from Kashani et al. [33]
The pathogenesis of AKI is not very well understood but a growing body of evidence from both laboratory and clinical studies suggests that inflammation and its associated molecular markers may be a key factor [37]. Activation of the renin-angiotensin–aldosterone system (RAAS) has long been recognized to contribute to chronic renal injury [38]. Angiotensin II (Ang II) has been shown to be capable of increasing the expression of pro-inflammatory and pro-fibrotic cytokines such as transforming growth factor (TGF) and tumor necrosis factor (TNF), thereby indicating the involvement of RAAS activation in AKI as well [39]. Urinary angiotensinogen (uAGT) has been described as a biomarker of renin–angiotensin system (RAS) overactivity in kidney diseases and hypertension. Its potential utility as a biomarker in AKI is also being increasingly recognized [23, 31]. The prognostic significance of uAGT as an AKI biomarker stems from its potential value as an index of RAS activation in modulating the severity of AKI [31]. In this review, we will discuss the potential utility and significance of uAGT as a biomarker by summarizing the studies that investigated the performance of uAGT in prognostication and risk stratification of AKI patients along with highlighting the relevance of RAS in the pathogenesis of AKI.
RAS in AKI: role of Ang II
Recent studies have suggested that the pathogenesis behind AKI and its progression is largely the result of acute inflammation [40, 41] involving complex interactions between inflammatory cytokines and chemokines that are primarily responsible for leukocyte activation and recruitment, leading to endothelial activation and injury associated with increased expression of adhesion molecules, further leukocyte infiltration and vascular congestion [41]. The kidney contains all components of the RAAS system [42, 43] (Figure 1). There is growing evidence that the locally produced intrarenal Ang II, which is the key effector peptide of RAAS, might contribute to the pathogenesis of AKI by increasing the expression of pro-inflammatory and pro-fibrotic cytokines such as TNF and TGF beta, thereby modulating inflammation, proliferation and fibrosis [37, 39]. These actions are to be viewed with its hemodynamic actions, namely, preferential vasoconstriction of efferent arterioles, which is protective in terms of maintenance of GFR, during volume depletion decompensated heart failure and reduced kidney perfusion in general [44]. These actions are primarily exerted via angiotensin type I (AT1) receptors [45].
Fig. 1.

Scheme of main RAS pathway: angiotensinogen is cleaved by rennin to form angiotensin I, which is subsequently converted to Ang II by ACE. Ang II binds to Ang II receptors (AT1-R) and (AT2-R). Ang II can also be cleaved by ACE2 to form the vasodilatory and anti-inflammatory peptide angiotensin (1–7) that binds to its specific receptor (Mas-R) to exert its actions. ACE, angiotensin converting enzyme; ACE2, angiotensin-converting enzyme 2; APA, aminopeptidase A; cGMP, cyclic GMP; PGF2, prostaglandin F2; ROS, reactive oxygen species.
Ischemia is one of the most frequent causes of AKI [46, 47]. The proximal tubule is a major component of renal cortex and its S3 segment in the outer stripe of the outer medulla is most susceptible to ischemia/reperfusion injury [48–50]. Ang II is expressed in the proximal tubule [51] where it also known to stimulate the synthesis of TGF beta [52] and an increase in the level of TGF beta mRNA and protein has been demonstrated early after acute ischemic injury in the rats [53]. In the proximal tubule, Ang II level was found to be increased by ∼3.5-fold on reduction of renal perfusion pressure, which correlated with the levels of whole kidney Ang II in male Sprague Dawley rats [51]. Kontogiannis and Burns [54] demonstrated an increase in intrarenal Ang II levels 24 h following bilateral renal ischemia accompanied by decrease in proximal tubular AT1 mRNA receptor expression, along with decreased cortical and medullary receptor binding [54]. The levels of Ang II and AT1 receptors returned back to the level of sham-operated rats at 72 and 120 h, respectively [54]. These findings are in concordance with those reported by Allred et al. [55] who studied the levels of renal and plasma angiotensin peptides in Sprague Dawley rats after left renal artery occlusion. They observed a significant increase in renal tissue Ang II by 64% in the ischemic kidneys 24 h post-surgery compared with the non-ischemic and sham-operated kidneys. Whereas Ang II receptor density was reduced in both the ischemic as well as contralateral non-ischemic kidney 24 h post-surgery, greater reductions in the ischemic kidney were observed. In contrast, no significant difference was observed between plasma Ang II levels 24 h post-ischemia between ischemic and sham-operated rats [55].
An increase in Ang II in the urine was also reported during the first 24 h post-ischemia. These findings also provide evidence in support of the intrarenal RAS independent of the circulatory RAS during ischemic injury to the kidneys [55]. In another study, intrarenal Ang II was increased in male Wistar rats 4 h after ischemic/reperfusion injury while the plasma Ang II levels remained unchanged. Similar to the previous studies mentioned above, the rise in intrarenal Ang II levels was associated with a significant decrease in AT1 receptor mRNA expression 4 h post-ischemia/reperfusion [56]. The observation that AT1 receptors undergo rapid internalization on binding of Ang II [57, 58] has been proposed as the most probable mechanism behind the subsequent downregulation of AT1 mRNA following the increase in Ang II in ischemic kidneys [54, 55].
Ang II is also responsible for producing aldosterone, the role of which in development and progression of renal injury has been described in both experimental and human models [59, 60]. Beyond its role in renal sodium and electrolyte transport, aldosterone elicits kidney tissue injury independently of blood pressure and renal hemodynamics [61]. Whether the mineralocorticoid antagonist, Spirinolactone, administered before or after AKI protects against development of CKD was examined in a rat model of bilateral renal ischemia [62]. Treatment with spironolactone either before or after ischemia prevented subsequent CKD by avoiding activation of fibrotic and inflammatory pathways, indicating that Spirinolactone may be a promising treatment for the prevention of AKI-induced CKD [62]. Studies in patients in the setting of AKI, to our knowledge, are not available. The use of RAS blockers in a way may provide an indirect effect by preventing the formation of Aldosterone (see below).
Treatment with AT1 receptor antagonists has been shown to hasten the recovery in experimental animal models of ischemic reperfusion injury [54]. Some studies have also demonstrated the efficacy of high doses of ARBs to not only halt the progression of ischemic injury, but also retard the inflammatory processes by blocking TNF, IL-1beta and IL-6 up-regulation-and prevent leucocyte infiltration that ensued 24 h post-ischemia in experimental models of AKI, thereby conferring renoprotection against ischemic renal injury [45]. Some authors have provided data against the renoprotective roles of ARBs in post-ischemic renal injury [63, 64] while others have also shown protective effects of captopril [65] as well as Aliskiren [66] on ischemia/reperfusion injury, which further exemplifies the role of RAAS activation in the pathogenesis of ischemic renal injury. The relevance of intrarenal RAS in patients with AKI is suggested by the study of Cao et al. [67] who examined the status of various RAS components in kidney biopsy-proven acute tubular necrosis (ATN) patients. Intrarenal Ang II was found to be upregulated in the distal tubule along with angiotensinogen in the proximal tubule. This augmentation of intrarenal RAS was also found to be associated with severity of ATN [67]. Moreover, this increase in intrarenal Ang II was correlated with uAGT in these patients. Although the Cao et al. [67] study did not determine the effects of RAAS blockade on uAGT and intrarenal Ang II, the findings of upregulation of intrarenal RAAS in patients with ATN strongly supports the notion that intrarenal RAAS may indeed play a major role in the pathogenesis of AKI.
Angiotensinogen as a biomarker of RAS over-activity in AKI
In transgenic mice, angiotensinogen excess has been shown to lead to activation of RAS, leading to the pathogenesis of progressive renal injury [68–72]. Angiotensinogen locally produced in the proximal tubules [73] has been suggested to be the primary source of uAGT [74, 75]. The locally formed angiotensinogen is primarily responsible for the subsequent formation of Ang II along the nephron [74], and both animal and clinical studies have documented uAGT to be a potential indicator of intrarenal RAS activity [74, 76]. However, plasma angiotensinogen can be filtered, particularly in states of altered glomerular permeability, and provide a major source of uAGT [15]. A strong correlation between intrarenal Ang II and uAGT has been previously described in animal models of CKD [77] and hypertension [78, 79]. Intrarenal angiotensinogen expression was found to be increased in patients with ATN, suggesting the potential involvement of RAS in AKI as well [67].
Alge et al. [23, 34] proposed that uATG could be used as a prognostic indicator of AKI to identify patients with AKI at risk of developing adverse outcomes. They first studied 97 patients who underwent cardiac surgery and developed AKI within 2 days of surgery and classified them into three groups depending upon the stage of AKI [Acute Kidney Injury Network (AKIN) Stage 1, Stage 2 and Stage 3] at the time of sample collection to evaluate uAGT and creatinine ratio (uAGT/CR). It was found that uAGT/CR correlated well with peak serum creatinine in these patients corresponding with the stage of AKI (Figure 2A). To evaluate how the uAGT/CR correlated with the progression of AKI they further sub-analyzed these patients by classifying them as AKI Stage 1 at the time of collection (n = 79) and found a statistically significant increase in uAGT/CR in the patients who subsequently progressed to AKI Stage 3 (n = 10) compared with the patients who remained at Stage 1 (n = 59) (Figure 2B). Thus, uAGT/CR discriminated between patients who experienced worsening of AKI after sample collection and those who did not, in both the whole cohort and in the subset classified as AKI Stage 1 at collection (Figure 3). Additionally, they also demonstrated that those with higher uAGT/CR concentration had a need of renal replacement therapy, longer duration of hospital stay and risk of death. This shows that uAGT also had the ability to predict the patients with increased risk of these adverse outcomes. This study was limited to patients with AKI secondary to cardiac surgery [23]. The ability of uAGT as a prognostic marker of AKI from other causes was examined in a retrospective case control study [34]. Alge et al. followed up to measure the levels of uAGT in intensive care unit patients who developed AKI from diverse etiologies [34]. The stratified analysis showed that patients with AKI secondary to ischemic ATN had the highest median uAGT/CR (260.2 ng/mg) while the patients with pre-renal AKI had the lowest median uAGT/CR (11.3 ng/mg). They demonstrated that increased uAGT levels were associated with increased risk of (i) worsening AKI, (ii) longer hospital duration, (iii) need for renal replacement therapy or (iv) death. Thus, it was concluded that uAGT was a strong independent predictor of these adverse outcomes. However, this study was limited by the small sample size and also failed to show if uAGT could serve as biomarker of early AKI [34].
Fig. 2.

Box and whisker plots showing the distribution of uAnCR (uAGT/creatinine ratio) by group in patients who developed AKI after cardiac surgery. (A) Among patients who had AKI of any AKIN stage at the time of sample collection (n = 97), uAnCR increased in a graded manner with AKI severity (as determined by maximum AKIN stage). (B) uAnCR increased in a graded manner with AKI (urinary Angiotensinogen and creatinine ratio) severity in the subset of patients who were classified as AKIN Stage 1 AKI at the time that their urine samples were collected (n = 79). Box plots show the median (solid line), 25th and 5th percentiles. Error bars represent the 5th and 9th percentiles. AKIN stage groups were compared with the Kruskal–Wallis test (P-value shown in bottom right). The asterisk represents a P < 0.05 compared with AKIN Stage 1 in the post hoc Dunn’s test for pairwise comparison. Figure adapted with permission from Alge et al. [23].
Fig. 3.

Angiotensinogen is associated with worsening of AKI. Receiver operator characteristic (ROC) curves were used to test the ability of uAnCR (uAGT/creatinine ratio) to predict worsening of AKI after sample collection among (A) patients who were any stage AKI at the time of collection (n = 97) and (B) the subset who were classified as AKIN Stage 1 at collection (n = 79). Figure adapted with permission from Alge et al. [23].
ATN is the prototypic cause of AKI and the observation of increased uAGT in patients with ATN was further explored in another study that investigated the role of intrarenal angiotensinogen in the pathogenesis of AKI. Increased expression of intrarenal angiotensinogen was found not only in proximal tubules but also in the distal tubules, which was in contrast to the findings in normal kidney where angiotensinogen is expressed only in the proximal tubule. Increased expression of intrarenal angiotensinogen was found to be positively correlated with increase in intrarenal Ang II and increased uAGT in these patients (Figure 4). Serum angiotensinogen levels were found to be comparable in both the control and ATN groups, suggesting a role of intrarenal RAS in the pathogenesis of ATN independent of circulatory RAS. Furthermore, the increase in urinary angiotensinogen was associated with increase in ATN severity (Figure 5). This correlation prompted the conclusion that uAGT reflects intrarenal RAS status, and could be used as an indicator to evaluate the severity of ATN-induced AKI [67].
Fig. 4.

Expression of intrarenal RAS associated with uATG level in patients with ATN. (A) uAGT at the time of renal biopsy in patients with ATN and in healthy volunteers. (B) Serum angiotensinogen at the time of renal biopsy in patients with ATN and in healthy volunteers. (C) Correlation between intrarenal angiotensinogen expression and uAGT in patients with ATN. (D) Correlation between intrarenal Ang II expression and uAGT in patients with ATN. t-test, *P < 0.05 versus healthy volunteers in (A) and (B). Pearson correlation, *P < 0.05 in (C) and (D). Figure adapted with permission from Cao et al. [67].
Fig. 5.
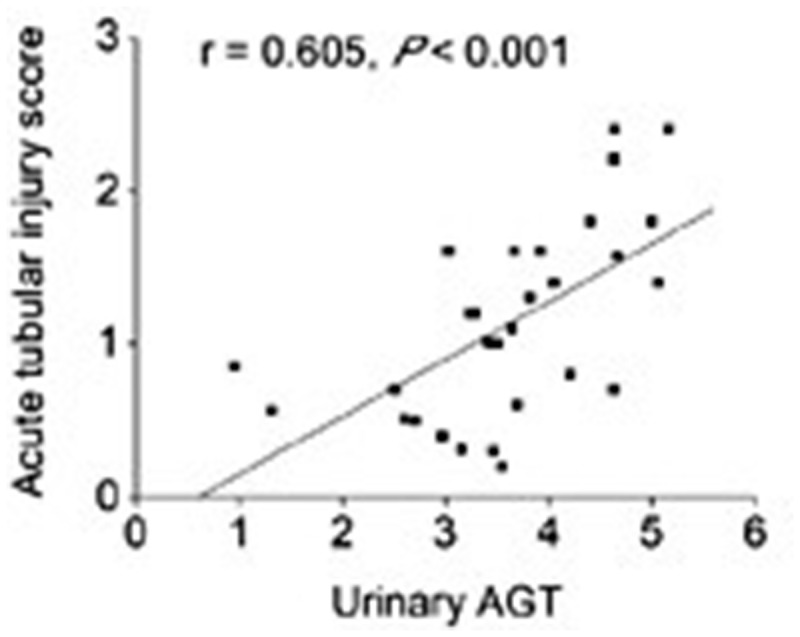
uAGT level associated with ATN severity. Correlation between uAGT and acute tubular injury score in patient with ATN. Figure adapted with permission from Cao et al. [67].
Angiotensinogen as a biomarker of AKI associated with decompensated heart failure and its comparison with other AKI biomarkers
AKI is a frequent complication of acute decompensated heart failure (ADHF) [15]. Yang et al. [32] explored the power of uAGT in predicting the progression of AKI in patients with ADHF by conducting a prospective two-stage study. They examined 317 patients with newly diagnosed ADHF in Stage 1 (test set), while 119 patients with ADHF were included to assess the validity of uAGT as a biomarker in Stage 2 (validation set). The level of uAGT measured had a characteristic peak on the first day of hospitalization and was found to be associated with a 50-fold increased risk of AKI progression, thus indicating it to be a powerful predictor for AKI. They further compared its performance with other biomarkers of AKI [NGAL, urine albumin to creatinine ratio, N-terminal pro-B-type natriuretic peptide (NT-proBNP)] and found it to be superior in predicting AKI not only in patients without preexisting CKD, but also in patients with preexisting CKD. The measured uAGT in patients with preexisting CKD was found to be higher compared with those without preexisting CKD. Furthermore, in a prospective follow-up of these patients after hospital discharge uAGT was found to be an independent predictor for 1-year mortality and re-hospitalization in these patients with AKI associated with ADHF [32]. This was confirmed by the same group in a prospective study involving 732 hospitalized adults with ADHF to examine the potential utility of uAGT in combination with other kidney injury biomarkers (NGAL, IL-18 and KIM-1) in predicting AKI progression [81]. All biomarkers were measured at the time of initial diagnosis. In the adjusted model, patients with the highest tertile of uAGT had a 10.8-fold increase in the progression of AKI compared with the ones with the lowest tertile, while increased uNGAL was associated with 4.7-fold odds of AKI progression, whereas increased urinary IL-18 revealed a 3.6-fold risk. Urinary AGT outperformed not only each one of these three biomarkers, but also the performance of their combination for predicting AKI progression [81] (Figure 6).
Fig. 6.
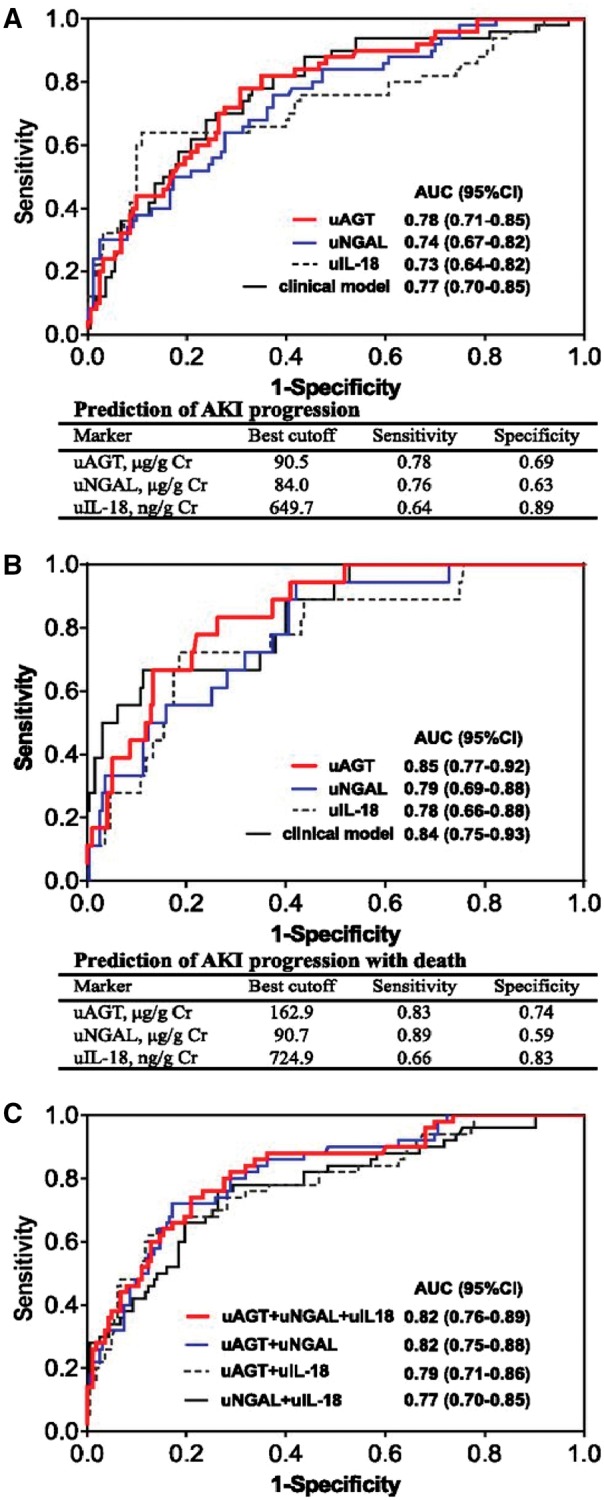
ROC analyses for predicting AKI progression or AKI progression with death. (A) The area under the curve (AUC) of renal injury biomarkers [urinary NGAL (uNGAL), urinary IL-18 (uIL-18)] and clinical model, at the time of AKI diagnosis, for predicting AKI progression. (B) The AUCs of renal injury biomarkers (uAGT, uNGAL, uIL-18) and clinical model, at the time of AKI diagnosis, for predicting AKI progression with subsequent death. (C) The performance of combination of renal injury biomarkers for predicting AKI progression. The clinical risk model includes age, gender, hypertension, diabetes, preadmission eGFR, NT-proBNP, serum albumin, hemoglobin, diuretic dosage before AKI, use of spironolactone before AKI, use of RAS inhibitors before AKI and change of serum creatinine from baseline at the time of AKI diagnosis. Figure adapted with permission from Chen et al. [81].
Possible significance of increased uAGT and therapeutic implications in AKI
While intrarenal RAS activation seems to play a prominent role in modulating the severity of AKI, the possibility of filtered angiotensinogen influencing intrarenal angiotensinogen levels and consequent intrarenal Ang II formation cannot be ruled out [15]. Studies in transgenic mice of podocyte-selective injury have shown that circulation-derived angiotensinogen can activate kidney RAS when the glomerular filtration barrier is altered, indicating the dependency of kidney Ang II generation on filtered angiotensinogen [82, 83]. In the setting of ADHF, activation of systemic RAS likely precedes any additional intrarenal RAS activation as AKI develops [15]. To initiate RAS, an increase in circulating renin is usually needed, the rate-limiting step that is responsible for the cleavage of angiotensinogen and formation of angiotensin I that subsequently leads to circulating Ang II overactivity (Figure 1). An increase in plasma angiotensinogen, by contrast, is not necessary, because this protein is abundant in plasma and therefore not a limiting step in RAS activation. At the local kidney level, however, uAGT is lower than in the circulation; therefore, an increase in kidney angiotensinogen could trigger RAS activation by providing the substrate for downstream formation of angiotensin peptides [15]. Thus, in AKI secondary to ADHF, an increase in uAGT may likely indicate kidney RAS activation, reflecting the role of intra renal RAS in modulating the severity of AKI. Henceforth, the question regarding the bona fide source of uAGT in AKI to be the result of intrarenal formation or increased glomerular passage or impaired tubular reabsorption remains to be answered [15].
Regardless of its origin, an increase in uAGT likely reflects kidney RAS activation, and the finding that increase in intrarenal Ang II correlates strongly with increase in uAGT but not plasma angiotensinogen, further supports this notion [74]. With such a proposition, it could be argued RAS blockers would be beneficial in AKI. There is evidence that Ang II upregulates angiotensinogen synthesis both within the kidney and in the liver [79, 84]. This implies that a positive feedback may perpetuate Ang II formation in states of Ang II overactivity, suggesting a rationale for using RAS blockers in AKI. In experimental models of AKI, intrarenal Ang II increased after renal ischemia perfusion injury while the levels of angiotensin (1–7) decreased [55]. Pretreatment with RAS blockers [angiotensin-converting enzyme inhibitor (ACEi), angiotensin receptor blockers (ARB) or direct renin inhibitors] was shown to alleviate the severity of AKI by reducing inflammation, thereby mitigating the renal ischemic-reperfusion injury [45, 65, 66]. RAS blockers, however, can exacerbate AKI by interfering with Ang II-dependent glomerular efferent arteriole vasoconstriction, which leads to a decrease in GFR during renal under-perfusion. Because of this concern, it is a common (and reasonable) clinical practice to withhold RAS inhibitors during AKI or in anticipation of procedures that may cause AKI such as cardiac surgery.
What is the evidence for this practice? The evidence is actually limited and largely based on studies post-cardiac surgery. Cittanova et al. [85] sought to identify preoperative risk factors responsible for post-operative renal impairment in patients undergoing aortic surgery. Chronic treatment with ACEi was the only factor significantly associated with post-operative GFR decline [85]. Arora et al. [86] more recently examined whether long-term use of ACEi/ARB is associated with an increased incidence of AKI after cardiac surgery. Intra- and post-operative factors that were associated with post-operative AKI were hypotension during surgery, use of vasopressors and post-operative hypotension. Overall, 40.2% of patients who were on chronic ACEi and underwent cardiac surgery developed AKI. Multiple regression logistic model showed an independent and significant association of AKI and preoperative use of ACEi/ARB [79]. While these studies support the practice of withholding RAS blockers in patients at risk for AKI, it should be noted, however, that a reduction in GFR is not necessarily equivalent to more severe renal injury. Recent studies have emphasized the need to distinguish structural AKI that involves renal parenchymal damage from functional AKI that primarily involves decrease in GFR [87]. Therefore, studies to investigate the potential benefits of RAS downregulation in AKI taking into account not only GFR itself as the outcome, but also other important outcomes such as progression to CKD would be desirable. It is unlikely, however, that such studies will ever be done with existing RAS blockers considering their potential detrimental hemodynamic effects on GFR.
Rather than blocking Ang II formation and action, an approach to downregulate RAS overactivity that could be safer would consist in fostering the degradation of Ang II to favor angiotensin (1–7) formation [42]. Studies with recombinant ACE2 administration should be considered as a newer approach to counteract some of the undesirable effects of RAS overactivity in AKI [80]. Administration of recombinant ACE2 can lower circulatory plasma Ang II levels and increase angiotensin (1–7) levels, although the large molecular size of this enzyme does not render it filterable by the kidney under normal conditions [88]. It is nevertheless important to consider newer approaches to counteract RAS overactivity in AKI. The first step is to detect RAS overactivity and in this regard an increase in uAGT provides a convenient tool. Regardless of the pathophysiological significance of increased uAGT in AKI, it serves as a promising biomarker capable of identifying at an early stage high risk patients with AKI that have the propensity to progress to higher stages of AKI and with worse outcomes.
Funding
This work was supported by National Institute of Diabetes and Digestive Kidney Diseases (grant R01DK104785) and a gift to Northwestern University by the Joseph and Bessie Feinberg Foundation.
Conflict of interest statement
None declared.
References
- 1. Ali T, Khan I, Simpson W. et al. Incidence and outcomes in acute kidney injury: a comprehensive population-based study. J Am Soc Nephrol 2007; 18: 1292–1298 [DOI] [PubMed] [Google Scholar]
- 2. Waikar SS, Curhan GC, Wald R. et al. Declining mortality in patients with acute renal failure, 1988 to 2002. J Am Soc Nephrol 2006; 17: 1143–1150 [DOI] [PubMed] [Google Scholar]
- 3. Okusa MD, Molitoris BA, Palevsky PM. et al. Design of clinical trials in acute kidney injury: a report from an NIDDK workshop–prevention trials. Clin J Am Soc Nephrol 2012; 7: 851–855 [DOI] [PubMed] [Google Scholar]
- 4. Doyle JF, Forni LG.. Acute kidney injury: short-term and long-term effects. Crit Care 2016; 20: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishani A, Xue JL, Himmelfarb J. et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 2009; 20: 223–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chawla LS, Eggers PW, Star RA. et al. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 2014; 371: 58–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palant CE, Amdur RL, Chawla LS.. Long-term consequences of acute kidney injury in the perioperative setting. Curr Opin Anaesthesiol 2017; 30: 100–104 [DOI] [PubMed] [Google Scholar]
- 8. Ronco C. Biomarkers for acute kidney injury: is NGAL ready for clinical use? Crit Care 2014; 18: 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murugan R, Kellum JA.. Acute kidney injury: what’s the prognosis? Nat Rev Nephrol 2011; 7: 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Allgren RL, Marbury TC, Rahman SN. et al. Anaritide in acute tubular necrosis. Auriculin anaritide acute renal failure study group. N Engl J Med 1997; 336: 828–834 [DOI] [PubMed] [Google Scholar]
- 11. Hirschberg R, Kopple J, Lipsett P. et al. Multicenter clinical trial of recombinant human insulin-like growth factor I in patients with acute renal failure. Kidney Int 1999; 55: 2423–2432 [DOI] [PubMed] [Google Scholar]
- 12. Tumlin JA, Finkel KW, Murray PT. et al. Fenoldopam mesylate in early acute tubular necrosis: a randomized, double-blind, placebo-controlled clinical trial. Am J Kidney Dis 2005; 46: 26–34 [DOI] [PubMed] [Google Scholar]
- 13. Ronco C. Acute kidney injury: from clinical to molecular diagnosis. Crit Care 2016; 20: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen S. Retooling the creatinine clearance equation to estimate kinetic GFR when the plasma creatinine is changing acutely. J Am Soc Nephrol 2013; 24: 877–888 [DOI] [PubMed] [Google Scholar]
- 15. Wysocki J, Batlle D.. Urinary angiotensinogen: a promising biomarker of AKI progression in acute decompensated heart failure: what does it mean? Clin J Am Soc Nephrol 2016; 11: 1515–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schrezenmeier EV, Barasch J, Budde K. et al. Biomarkers in acute kidney injury - pathophysiological basis and clinical performance. Acta Physiol 2017; 219: 554–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mishra J, Ma Q, Prada A. et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 2003; 14: 2534–2543 [DOI] [PubMed] [Google Scholar]
- 18. Shao X, Tian L, Xu W. et al. Diagnostic value of urinary kidney injury molecule 1 for acute kidney injury: a meta-analysis. PloS One 2014; 9: e84131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parikh CR, Jani A, Melnikov VY. et al. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am J Kidney Dis 2004; 43: 405–414 [DOI] [PubMed] [Google Scholar]
- 20. Herget-Rosenthal S, Marggraf G, Husing J. et al. Early detection of acute renal failure by serum cystatin C. Kidney Int 2004; 66: 1115–1122 [DOI] [PubMed] [Google Scholar]
- 21. Portilla D, Dent C, Sugaya T. et al. Liver fatty acid-binding protein as a biomarker of acute kidney injury after cardiac surgery. Kidney Int 2008; 73: 465–472 [DOI] [PubMed] [Google Scholar]
- 22. Kashani K, Al-Khafaji A, Ardiles T. et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care 2013; 17: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alge JL, Karakala N, Neely BA. et al. Urinary angiotensinogen and risk of severe AKI. Clin J Am Soc Nephrol 2013; 8: 184–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Singer E, Elger A, Elitok S. et al. Urinary neutrophil gelatinase-associated lipocalin distinguishes pre-renal from intrinsic renal failure and predicts outcomes. Kidney Int 2011; 80: 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Devarajan P. Emerging urinary biomarkers in the diagnosis of acute kidney injury. Expert Opin Med Diagn 2008; 2: 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ho J, Tangri N, Komenda P. et al. Urinary, plasma, and serum biomarkers' utility for predicting acute kidney injury associated with cardiac surgery in adults: a meta-analysis. Am J Kidney Dis 2015; 66: 993–1005 [DOI] [PubMed] [Google Scholar]
- 27. Medic B, Rovcanin B, Vujovic KS. et al. Evaluation of novel biomarkers of acute kidney injury: the possibilities and limitations. Curr Med Chem 2016; 23: 1981–1997 [DOI] [PubMed] [Google Scholar]
- 28. de Geus HR, Bakker J, Lesaffre EM. et al. Neutrophil gelatinase-associated lipocalin at ICU admission predicts for acute kidney injury in adult patients. Am J Respir Crit Care Med 2011; 183: 907–914 [DOI] [PubMed] [Google Scholar]
- 29. Doi K, Negishi K, Ishizu T. et al. Evaluation of new acute kidney injury biomarkers in a mixed intensive care unit. Crit Care Med 2011; 39: 2464–2469 [DOI] [PubMed] [Google Scholar]
- 30. Meersch M, Schmidt C, Van Aken H. et al. Urinary TIMP-2 and IGFBP7 as early biomarkers of acute kidney injury and renal recovery following cardiac surgery. PloS One 2014; 9: e93460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alge JL, Arthur JM.. Biomarkers of AKI: a review of mechanistic relevance and potential therapeutic implications. Clin J Am Soc Nephrol 2015; 10: 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang X, Chen C, Tian J. et al. Urinary angiotensinogen level predicts AKI in acute decompensated heart failure: a prospective, two-stage study. J Am Soc Nephrol 2015; 26: 2032–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kashani K, Cheungpasitporn W, Ronco C.. Biomarkers of acute kidney injury: the pathway from discovery to clinical adoption. Clin Chem Lab Med 2017; doi: 10.1515/cclm-2016-0973 [DOI] [PubMed] [Google Scholar]
- 34. Alge JL, Karakala N, Neely BA. et al. Urinary angiotensinogen predicts adverse outcomes among acute kidney injury patients in the intensive care unit. Crit Care 2013; 17: R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parikh CR, Thiessen-Philbrook H, Garg AX. et al. Performance of kidney injury molecule-1 and liver fatty acid-binding protein and combined biomarkers of AKI after cardiac surgery. Clin J Am Soc Nephrol 2013; 8: 1079–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parikh CR, Coca SG, Thiessen-Philbrook H. et al. Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol 2011; 22: 1748–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wen X, Murugan R, Peng Z. et al. Pathophysiology of acute kidney injury: a new perspective. Contrib Nephrol 2010; 165: 39–45 [DOI] [PubMed] [Google Scholar]
- 38. Siragy HM, Carey RM.. Role of the intrarenal Renin-Angiotensin-Aldosterone system in chronic kidney disease. Am J Nephrol 2010; 31: 541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruiz-Ortega M, Ruperez M, Esteban V. et al. Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dial Transplant 2006; 21: 16–20 [DOI] [PubMed] [Google Scholar]
- 40. Bonventre JV, Weinberg JM.. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 2003; 14: 2199–2210 [DOI] [PubMed] [Google Scholar]
- 41. Bonventre JV, Zuk A.. Ischemic acute renal failure: an inflammatory disease? Kidney Int 2004; 66: 480–485 [DOI] [PubMed] [Google Scholar]
- 42. Batlle D, Wysocki J, Soler MJ. et al. Angiotensin-converting enzyme 2: enhancing the degradation of angiotensin II as a potential therapy for diabetic nephropathy. Kidney Int 2012; 81: 520–528 [DOI] [PubMed] [Google Scholar]
- 43. Chappell MC. Nonclassical renin-angiotensin system and renal function. Compr Physiol 2012; 2: 2733–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ichikawi I, Harris RC.. Angiotensin actions in the kidney: renewed insight into the old hormone. Kidney Int 1991; 40: 583–596 [DOI] [PubMed] [Google Scholar]
- 45. Molinas SM, Cortes-Gonzalez C, Gonzalez-Bobadilla Y. et al. Effects of losartan pretreatment in an experimental model of ischemic acute kidney injury. Nephron Exp Nephrol 2009; 112: e10–e19 [DOI] [PubMed] [Google Scholar]
- 46. Bonventre JV, Yang L.. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 2011; 121: 4210–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sharfuddin AA, Molitoris BA.. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 2011; 7: 189–200 [DOI] [PubMed] [Google Scholar]
- 48. Glaumann B, Glaumann H, Trump BF.. Studies of cellular recovery from injury. III. Ultrastructural studies on the recovery of the pars recta of the proximal tubule (P3 segment) of the rat kidney from temporary ischemia. Virchows Archiv B Cell Pathol 1977; 25: 281–308 [PubMed] [Google Scholar]
- 49. Bonventre JV. Mechanisms of ischemic acute renal failure. Kidney Int 1993; 43: 1160–1178 [DOI] [PubMed] [Google Scholar]
- 50. Witzgall R, Brown D, Schwarz C. et al. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest 1994; 93: 2175–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boer WH, Braam B, Fransen R. et al. Effects of reduced renal perfusion pressure and acute volume expansion on proximal tubule and whole kidney angiotensin II content in the rat. Kidney Int 1997; 51: 44–49 [DOI] [PubMed] [Google Scholar]
- 52. Wolf G, Mueller E, Stahl RA. et al. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-beta. J Clin Invest 1993; 92: 1366–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Basile DP, Rovak JM, Martin DR. et al. Increased transforming growth factor-beta 1 expression in regenerating rat renal tubules following ischemic injury. Am J Physiol 1996; 270: F500–F509 [DOI] [PubMed] [Google Scholar]
- 54. Kontogiannis J, Burns KD.. Role of AT1 angiotensin II receptors in renal ischemic injury. Am J Physiol 1998; 274: F79–F90 [DOI] [PubMed] [Google Scholar]
- 55. Allred AJ, Chappell MC, Ferrario CM. et al. Differential actions of renal ischemic injury on the intrarenal angiotensin system. Am J Physiol Renal Physiol 2000; 279: F636–F645 [DOI] [PubMed] [Google Scholar]
- 56. da Silveira KD, Pompermayer Bosco KS, Diniz LR. et al. ACE2-angiotensin-(1-7)-Mas axis in renal ischaemia/reperfusion injury in rats. Clin Sci (Lond) 2010; 119: 385–394 [DOI] [PubMed] [Google Scholar]
- 57. Thomas WG, Thekkumkara TJ, Baker KM.. Molecular mechanisms of angiotensin II (AT1A) receptor endocytosis. Clin Exp Pharmacol Physiol 1996; 3: S74–S80 [PubMed] [Google Scholar]
- 58. Hunyady L. Molecular mechanisms of angiotensin II receptor internalization. J Am Soc Nephrol 1999; 10 (Suppl 11): S47–S56 [PubMed] [Google Scholar]
- 59. Chrysostomou A, Pedagogos E, MacGregor L. et al. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin II receptor blocker. Clin J Am Soc Nephrol 2006; 1: 256–262 [DOI] [PubMed] [Google Scholar]
- 60. Zhou X, Ono H, Ono Y. et al. Aldosterone antagonism ameliorates proteinuria and nephrosclerosis independent of glomerular dynamics in L-NAME/SHR model. Am J Nephrol 2004; 24: 242–249 [DOI] [PubMed] [Google Scholar]
- 61. Rafiq K, Hitomi H, Nakano D. et al. Pathophysiological roles of aldosterone and mineralocorticoid receptor in the kidney. J Pharmacol Sci 2011; 115: 1–7 [DOI] [PubMed] [Google Scholar]
- 62. Barrera-Chimal J, Perez-Villalva R, Rodriguez-Romo R. et al. Spironolactone prevents chronic kidney disease caused by ischemic acute kidney injury. Kidney Int 2013; 83: 93–103 [DOI] [PubMed] [Google Scholar]
- 63. Jerkic M, Miloradovic Z, Jovovic D. et al. Relative roles of endothelin-1 and angiotensin II in experimental post-ischaemic acute renal failure. Nephrol Dial Transplant 2004; 19: 83–94 [DOI] [PubMed] [Google Scholar]
- 64. Kim SJ, Lim YT, Kim BS. et al. Mechanism of reduced GFR in rabbits with ischemic acute renal failure. Ren Fail 2000; 22: 129–141 [DOI] [PubMed] [Google Scholar]
- 65. Efrati S, Berman S, Hamad RA. et al. Effect of captopril treatment on recuperation from ischemia/reperfusion-induced acute renal injury. Nephrol Dial Transplant 2012; 27: 136–145 [DOI] [PubMed] [Google Scholar]
- 66. Wang Z, Liu Y, Han Y. et al. Protective effects of aliskiren on ischemia-reperfusion-induced renal injury in rats. Eur J Pharmacol 2013; 718: 160–166 [DOI] [PubMed] [Google Scholar]
- 67. Cao W, Jin L, Zhou Z. et al. Overexpression of intrarenal Renin-Angiotensin system in human acute tubular necrosis. Kidney Blood Press Res 2016; 41: 746–756 [DOI] [PubMed] [Google Scholar]
- 68. Kobori H, Nishiyama A, Abe Y. et al. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension 2003; 41: 592–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sachetelli S, Liu Q, Zhang SL. et al. RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney Int 2006; 69: 1016–1023 [DOI] [PubMed] [Google Scholar]
- 70. Kobori H, Ozawa Y, Satou R. et al. Kidney-specific enhancement of ANG II stimulates endogenous intrarenal angiotensinogen in gene-targeted mice. Am J Physiol Renal Physiol 2007; 293: F938–F945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lavoie JL, Lake-Bruse KD, Sigmund CD.. Increased blood pressure in transgenic mice expressing both human renin and angiotensinogen in the renal proximal tubule. Am J Physiol Renal Physiol 2004; 286: F965–F971 [DOI] [PubMed] [Google Scholar]
- 72. Liu F, Brezniceanu ML, Wei CC. et al. Overexpression of angiotensinogen increases tubular apoptosis in diabetes. J Am Soc Nephrol 2008; 19: 269–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Darby IA, Congiu M, Fernley RT. et al. Cellular and ultrastructural location of angiotensinogen in rat and sheep kidney. Kidney Int 1994; 46: 1557–1560 [DOI] [PubMed] [Google Scholar]
- 74. Kobori H, Harrison-Bernard LM, Navar LG.. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int 2002; 61: 579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nakano D, Kobori H, Burford JL. et al. Multiphoton imaging of the glomerular permeability of angiotensinogen. J Am Soc Nephrol 2012; 23: 1847–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yamamoto T, Nakagawa T, Suzuki H. et al. Urinary angiotensinogen as a marker of intrarenal angiotensin II activity associated with deterioration of renal function in patients with chronic kidney disease. J Am Soc Nephrol 2007; 18: 1558–1565 [DOI] [PubMed] [Google Scholar]
- 77. Urushihara M, Kondo S, Kagami S. et al. Urinary angiotensinogen accurately reflects intrarenal Renin-Angiotensin system activity. Am J Nephrol 2010; 31: 318–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kobori H, Alper AB Jr, Shenava R. et al. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients. Hypertension 2009; 53: 344–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kobori H, Prieto-Carrasquero MC, Ozawa Y. et al. AT1 receptor mediated augmentation of intrarenal angiotensinogen in angiotensin II-dependent hypertension. Hypertension 2004; 43: 1126–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wysocki J, Ye M, Batlle D.. Delivery of murine recombinant ACE2 in different mouse models of albuminuria [Abstract]. J Am Soc Nephrol 2016: 435A [Google Scholar]
- 81. Chen C, Yang X, Lei Y. et al. Urinary biomarkers at the time of AKI diagnosis as predictors of progression of AKI among patients with acute cardiorenal syndrome. Clin J Am Soc Nephrol 2016; 11: 1536–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matsusaka T, Niimura F, Shimizu A. et al. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol 2012; 23: 1181–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Matsusaka T, Niimura F, Pastan I. et al. Podocyte injury enhances filtration of liver-derived angiotensinogen and renal angiotensin II generation. Kidney Int 2014; 85: 1068–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Herrmann HC, Dzau VJ.. The feedback regulation of angiotensinogen production by components of the renin-angiotensin system. Circ Res 1983; 52: 328–334 [DOI] [PubMed] [Google Scholar]
- 85. Cittanova ML, Zubicki A, Savu C. et al. The chronic inhibition of angiotensin-converting enzyme impairs postoperative renal function. Anesth Analg 2001; 93: 1111–1115 [DOI] [PubMed] [Google Scholar]
- 86. Arora P, Rajagopalam S, Ranjan R. et al. Preoperative use of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers is associated with increased risk for acute kidney injury after cardiovascular surgery. Clin J Am Soc Nephrol 2008; 3: 1266–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Coca SG, Garg AX, Swaminathan M. et al. Preoperative angiotensin-converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dial Transplant 2013; 28: 2787–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wysocki J, Ye M, Khattab AM. et al. Angiotensin-converting enzyme 2 amplification limited to the circulation does not protect mice from development of diabetic nephropathy. Kidney Int 2017; 91: 1336–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]