

Abstract
Background
Glioblastoma represents an archetypal example of a heterogeneous malignancy. To understand the diverse molecular consequences of this complex tumor ecology, we analyzed RNA-seq data generated from commonly identified intratumoral structures in glioblastoma enriched using laser capture microdissection.
Methods
Raw gene-level values of fragments per kilobase of transcript per million reads mapped and the associated clinical data were acquired from the publicly available Ivy Glioblastoma Atlas Project database and analyzed using MetaboAnalyst (v3.0). The database includes gene expression data generated from multiple structural features commonly identified in glioblastoma enriched by laser capture microdissection.
Results
We uncovered a relationship between subtype heterogeneity in glioblastoma and its unique tumor microenvironment, with infiltrating cells harboring a proneural signature while the mesenchymal subtype was enriched in perinecrotic regions. When evaluating the tumors’ transcriptional profiles in the context of their derived structural regions, there was a relatively small amount of intertumoral heterogeneity in glioblastoma, with individual regions from different tumors clustering tightly together. Analyzing the transcriptional profiles in the context of evolutionary progression identified unique cellular programs associated with specific phases of gliomagenesis. Mediators of cell signaling and cell cycle progression appear to be critical events driving proliferation in the tumor core, while in addition to a multiplex strategy for promoting angiogenesis and/or an immune-tolerant environment, transformation to perinecrotic zones involved global metabolic alterations.
Conclusion
These findings suggest that intratumoral heterogeneity in glioblastoma is a conserved, predictable consequence to its complex microenvironment, and combinatorial approaches designed to target these unequivocally present tumor biomes may lead to therapeutic gains.
Keywords: glioblastoma, next-generation sequencing, tumor evolution, tumor heterogeneity
Importance of the study
Intertumoral heterogeneity in glioblastoma has been well described, with transcriptional profiles clustering into specific subtypes. Further, intratumoral heterogeneity in glioblastoma has also been recently recognized, with the identification of multiple molecular subtypes within an individual tumor, although factors contributing to this level of heterogeneity have not been defined. This study evaluates multiple transcriptional profiles within an individual tumor in the context of their geographic location. We uncovered a relationship between subtype heterogeneity in glioblastoma and its unique tumor microenvironment and demonstrated that when evaluating the tumors’ transcriptional profiles in the context of their derived regions, there was a relatively small amount of intertumoral heterogeneity in glioblastoma. Collectively, these findings suggest that intratumoral heterogeneity in glioblastoma is a conserved, predictable consequence to its complex microenvironment, and combinatorial approaches designed to target these unequivocally present tumor biomes may lead to therapeutic gains.
Glioblastoma (GBM) is the most common primary malignant brain tumor in adults.1 Despite aggressive, multimodal treatment regimens consisting of surgical resection, radiation therapy, and chemotherapy, outcomes remain poor, with a median survival of 15 months and few long-term survivors.2 Therefore, considerable effort is being made to better understand the underlying biology of these tumors to help guide the development of novel therapeutic strategies. Accordingly, GBM is one of the first cancer types systematically studied at the genomic and transcriptomic level.3–5 Transcriptional profiling has revealed a landscape of intertumoral heterogeneity in GBM, identifying distinct molecular tumor subtypes that offer the promise of subtype-specific therapeutic strategies.3,5 These include the proneural and mesenchymal subtypes, which have been identified most consistently in GBM. Their transcriptional profiles are mutually exclusive and can be applied to approximately one half of tumors. The proneural subtype is characterized by mutations in isocitrate dehydrogenase 1, frequent alterations in expression of p53 and platelet-derived growth factor receptor alpha polypeptide, and a transcriptional signature typically present in low-grade glioma,6,7 whereas mesenchymal GBM is characterized by mesenchymal gene expression, including CD44 and chitinase 3-like 1 (YKL40) and attributed to more aggressive disease. Other subtypes include classical and neural, which are characterized by epidermal growth factor receptor and the expression of neuronal markers, respectively.5
In addition to well-defined intertumoral heterogeneity in GBM, increasing evidence suggests that individual tumors comprise a diverse mixture of cells and multiple molecular subtypes.8–10 This intratumoral heterogeneity consists of genetically and transcriptionally distinct populations of cells that likely play a contributory role in therapeutic resistance and disease progression. However, since GBM represents an archetypal example of a heterogeneous malignancy, harboring regions of invasion, necrosis, and vascularization,1 this level of intratumoral heterogeneity should not be unexpected. To begin to understand the diverse molecular consequences of this complex tumor ecology, we analyzed RNA-seq data generated from commonly identified intratumoral structures in GBM that were isolated and enriched using laser capture microdissection.11,12 We uncovered a relationship between subtype heterogeneity in GBM and its unique tumor microenvironment and demonstrated that when evaluating the tumors’ transcriptional profiles in the context of their derived structural regions, there was a relatively small amount of intertumoral heterogeneity, suggesting that intratumoral heterogeneity in GBM is a conserved, predictable consequence to its complex microenvironment.
Materials and Methods
For RNA sequencing and analysis, raw gene-level values of fragments per kilobase of transcript per million reads mapped (FPKM) and the associated clinical data were acquired from the publicly available Ivy Glioblastoma Atlas Project database (Ivy GAP; http://glioblastoma.alleninstitute.org).12 The database includes gene expression data for multiple structural features commonly identified in GBM enriched by laser capture microdissection. Multiple cores of the same structure from an individual tumor were averaged. All data analyses were performed using the MetaboAnalyst (v3.0).13 Raw FPKM values were filtered based on the median intensity value, normalized to the median values, and log transformed for further analysis. For GBM subtype classification, gene expression values of 840 transcripts were normalized, as previously described.5 Unsupervised hierarchical clustering was performed using Euclidean distance matrix and Ward’s linkage rule. The 4 subtype classifiers were applied with numeric code annotation representing genes of each subtype with Bonferroni statistical significance (P-value < 0.05). Variations in gene expression patterns between structural features were visualized using principal component analysis (PCA) and partial least squares discriminant analysis. Score plots were generated with 95% confidence regions and models were validated using the permutation testing function (2000 iterations). Permutation testing was performed to establish whether the observed differences between groups were statistically significant. Variable importance in projection (VIP) plots were used to determine which genes are most important to each cohort. This model ranks genes based on their ability to differentiate between groups in a given model. Higher VIP values (x-axis) correlate with higher discriminating capacity. Pathways analysis was completed in MetaboAnalyst (v3.0)13 using the integrated pathway tool and selecting the gene-centric pathway option. Normalized gene expression data from different structural regions were subjected to univariate analysis, and volcano plots were generated. Based on the results of the Mann–Whitney t-test, genes with a P-value < 0.05 and having a log fold change >2 were used for gene enrichment analysis based on Fisher’s exact test.
Results
Analyzed were transcriptional expression profiles obtained from the Ivy GAP database, which included geographically distinct regions within individual patient derived GBM specimens.11,12 Briefly, in this database, laser capture microdissection was used to isolate and enrich GBM cells from commonly identified intratumoral structures using a semi-automated annotation application based on statistical machine learning algorithms performed on ~12000 hematoxylin and eosin (H&E) histologic images.11 The structural regions included: the tumor leading edge (LE; outermost boundary of tumor where the ratio of tumor to normal cells is ~1–3/100), infiltrating tumor (IT; the intermediate zone between the LE and cellular tumor [CT], where the ratio of tumor cells to normal cells is about 10–20/100), and the CT (constitutes the major part of the core tumor, where the ratio of tumor cells to normal cells is ~100/1 to 500/1). Within the CT, regions included: microvascular proliferation (MVP; 2 or more blood vessels sharing a common vessel wall), hyperplastic blood vessels (HBVs; regions of increased density of blood vessels that appear to have thickened walls or endothelial cell proliferation), pseudopalisading cells around necrosis (PAN; tumor cells that aggregate in rows 10–30 nuclei wide at higher density than the surrounding CT to form pseudopalisading cells that appear to point toward a common center of necrosis), and the perinecrotic zone (PNZ; a boundary of tumor cells typically 10–30 nuclei wide along the edge of necrosis that lacks a clear demarcation of PAN). RNA-seq was then performed on enriched cells isolated from these individual regions. A total of 119 regions from 37 individual tumors were evaluated. As an initial investigation, we sought to validate the methods utilized to isolate the above-described structural regions, identifying expected increases in endothelial and hypoxia markers in the vascular (MVP, HBV) and hypoxic (PAN, PNZ) regions, respectively (Supplementary Figure S1). Next, using this RNA-seq dataset, nonvascular structural regions (n = 90) were subtyped as proneural, neural, classical, or mesenchymal using methods described by Verhaak et al5 (Fig. 1A). The specific structural regions of individual tumors utilized in this analysis are provided in Supplementary Table S1 and patient characteristics of tumors utilized in this study are provided in Supplementary Table S2. As an initial investigation, we determined the level of subtype heterogeneity within an individual tumor. In 20 tumors that had 2 regions represented, there was a 60% likelihood of observing multiple molecular subtypes. The likelihood for multiple subtypes increased to 86% if 3 regions were represented (n = 7), and 100% if more than 3 regions were represented (n = 6; Supplementary Table S1). Next, we examined the distribution of subtypes within the described structural regions. As demonstrated in Fig. 1B, all 8 tumors that had tissue available from the LE were subtyped as proneural and when combined with the IT tumors, collectively representing the peripheral, invasive tumor front, were nearly exclusively subtyped as proneural (n = 13/16). Conversely, the mesenchymal subtype was almost exclusively observed in regions of necrosis (PAN and PNZ; n = 27/30). A majority of the central tumors comprised both classical and neural subtypes (n = 34/37). To further validate these findings, we extended studies by evaluating regional expression of genes associated with molecular subtypes in GBM using mRNA in situ hybridization data also included in Ivy GAP.11 Oligodendrocyte transcription factor (Olig2), platelet derived growth factor subunit A (PDGFA), and thrombospondin 1 (THBS1) were used to serve as molecular markers for the proneural, classical, and mesenchymal subtypes, respectively. Region-specific expression of these molecular subtypes in an individual tumor were confirmed, with Olig2 being localized in the LE and IT, PDGFA localized in the CT, and THBS1 localized in PNZ (Supplementary Figure S2). In addition to validating recent investigations identifying intratumoral subtype heterogeneity in GBM,9,10 these findings suggest that this heterogeneity is not random but a direct consequence of predictable regional adaptations within a given tumor microenvironment, with the proneural subtype representing the infiltrating, leading edge of a tumor, the neural and classical subtypes representing the CT, and the mesenchymal signature localized to PNZs.
Fig. 1.

Intratumoral molecular subtype heterogeneity in GBM is directly influenced by the tumor microenvironment. Transcriptional expression profiles were obtained from the Ivy GAP database,11 which used laser capture microdissection to enrich geographically distinct regions within individual patient derived tumors. This RNA-seq dataset, which included 90 structural regions obtained from 37 individual tumors, was molecularly subtyped using gene expression values of 840 transcripts as described by Verhaak et al. (A) Heat map and (B) graphical depiction of molecular subtypes identified in the individual structural regions. Subtypes: proneural (PN), neural (N), classical (CL), mesenchymal (M). Structural regions: leading edge (LE), infiltrating tumor (IT), cellular tumor (CT), perinecrotic zone (PNZ), pseudopalisading cells around necrosis (PAN). Numbers of samples comprising each structural region are included in parentheses.
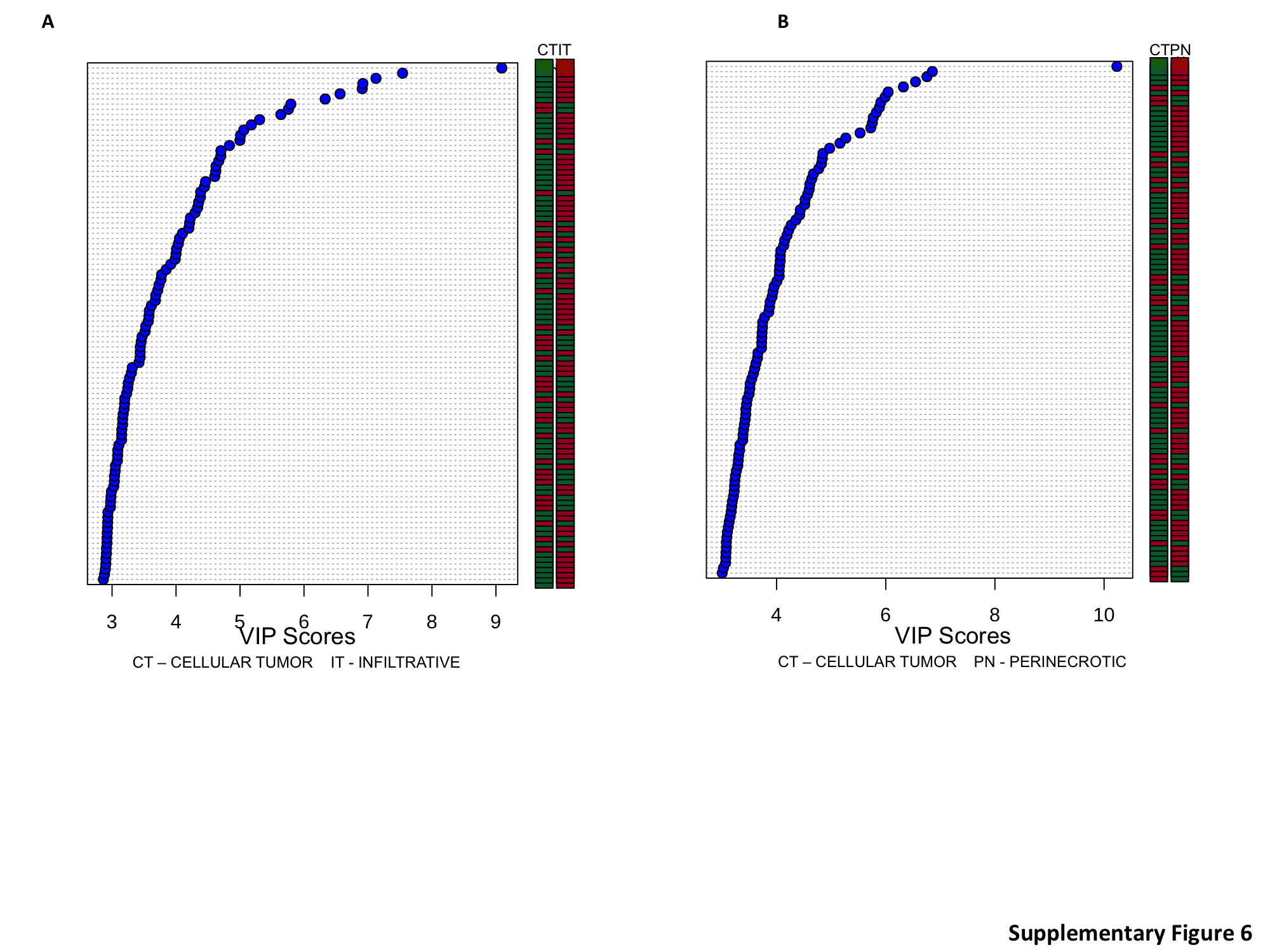
As individual structural regions and/or microenvironments appeared to play a contributory role in influencing molecular subtypes in GBM, we subsequently analyzed the entire RNA-seq dataset generated from the individual regions to better understand the degree of inter- and intratumoral heterogeneity in this malignancy. Based on observed clustering patterns and biologic similarities, we combined the IT with the LE and the PAN with the PNZs, thereby representing the invasive and perinecrotic components of the tumor, respectively, and compared these with the CT. PCA performed on this cohort again demonstrated strong clustering of transcriptional programs based on location (Fig. 2A and Supplementary Figure S3A). Similar to the proneural and mesenchymal molecular subtypes, the invasive cells and the perinecrotic RNA-seq signatures appeared to be mutually exclusive, while the CT shared features with the other regions. What was particularly striking was the apparent lack of intertumor heterogeneity in GBM, as similar regions from different tumors clustered tightly together (P < 5 × 10–4). This suggests that genomic alterations contributing to growth in these tumor niches are highly conserved and strategies developed to target their biologic machinery may have broad activity.
Fig. 2.

PCA of RNA-seq profiles in the context of their structural regions demonstrates limited intertumoral heterogeneity in GBM. (A) PCA was performed using the entire RNA-seq profile of individual tumor regions, including the infiltrating tumor (LE + IT), perinecrotic region (PNZ, PAN), and the central tumor core. Symbols identify tumors with all 3 regions represented. (B, C) VIP analysis was performed to identify the most important genes differentiating individual regions (red: upregulation, green: downregulation). IT: infiltrative tumor; CT: cellular tumor; PN: perinecrotic region.
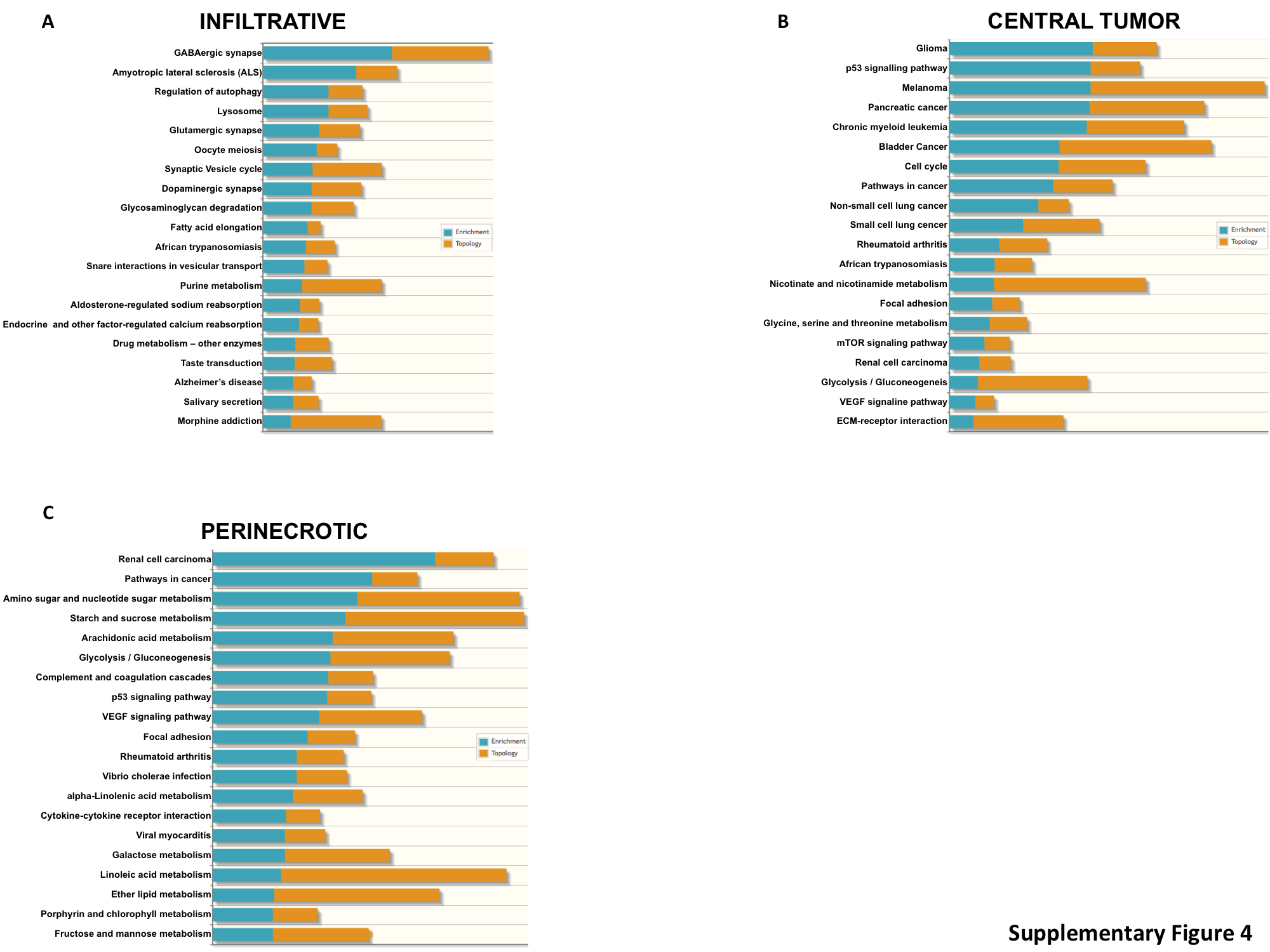
To begin to understand the specific molecular events driving regional growth and their contributory role in gliomagenesis, we analyzed the transcriptional programs in the context of evolutionary progression. In this model, as the proneural signature was exclusively found in infiltrative cells (LE/IT; Fig. 1A, B), and this molecular subtype has previously been ascribed to low-grade glioma,6,7 we used the transcriptional programs of the infiltrative cells to define early events in gliomagenesis. The primary assumption of the model is that these cells would progress to a highly cellular CT, typical of malignant glioma, and then finally advance to regions of necrosis (PAN/PNZ) consistent with GBM. Using this evolutionary model as a framework, we performed pathway analysis on overexpressed genes (P < 0.05, log 2 fold change >2), initially determining unique molecular features harbored by the infiltrative cells compared with the CT. This level of analysis identified numerous pathways associated with neuronal receptor signaling, autophagy, and fatty acid metabolism unique to early stages of gliomagenesis (Supplementary Figure S4A). Genes enriched in these pathways included a number of G-proteins and calmodulin family members (Supplementary Table S3), which have been previously described to play an early role in Olig2-mediated gliomagenesis.14 Based on our model, we went on to determine the molecular events required for these cells to progress to a highly cellular CT. Interestingly, traditional cancer-related signaling pathways emerged as key mediators driving progression, with glioma and p53 signaling ranking highest on the list. Genes enriched in these pathways included insulin-like growth factor receptor, mitogen-activated protein kinase, and mammalian target of rapamycin signaling,15,16 along with mediators of cell cycle progression, including cyclin-dependent kinase 4/6 and WEE1, which have all been previously described to play a role in gliomagenesis,4,6,17 further validating our findings. As the tumor continues to evolve from CT to regions of necrosis, in addition to expected changes in angiogenesis and tumor signaling, we uncovered significant alterations in global metabolic programs associated with this level of progression, with metabolic pathways, which included amino acid and nucleotide metabolism and glycolysis, comprising 10 of the top 20 pathways (Supplementary Figure S4). Subsequently, we performed a VIP analysis to identify the most important genomic features distinguishing these levels of tumor progression. Progression from infiltrative cells to a highly cellular tumor appeared to be driven by a downregulation of a number of genes (Fig. 2B), including the top 2 genes, VSNL1 and NEFL, whose miRNA-driven downregulation has been previously attributed to gliomagenesis,18,19 and SERPINI1, whose loss has been associated with epithelial-mesenchymal transition.20 Key genetic events driving continued progression to PNZs include upregulation of interleukin-8, which has been previously described to correlate with histologic grade and to be highly expressed in regions of PAN 21; N-myc downregulated gene, a marker for hypoxia 22,23; and the mesenchymal cell marker SERPINE1,24 further validating our proposed model and approach (Fig. 2C). In addition to expected increases in vascular endothelial growth factor, we uncovered angiopoietin-like protein 4 (ANGPTL4)25,26 and baculoviral IAP repeat containing 327 as potential mediators of angiogenesis and apoptosis inhibition, respectively, in the perinecrotic niche.
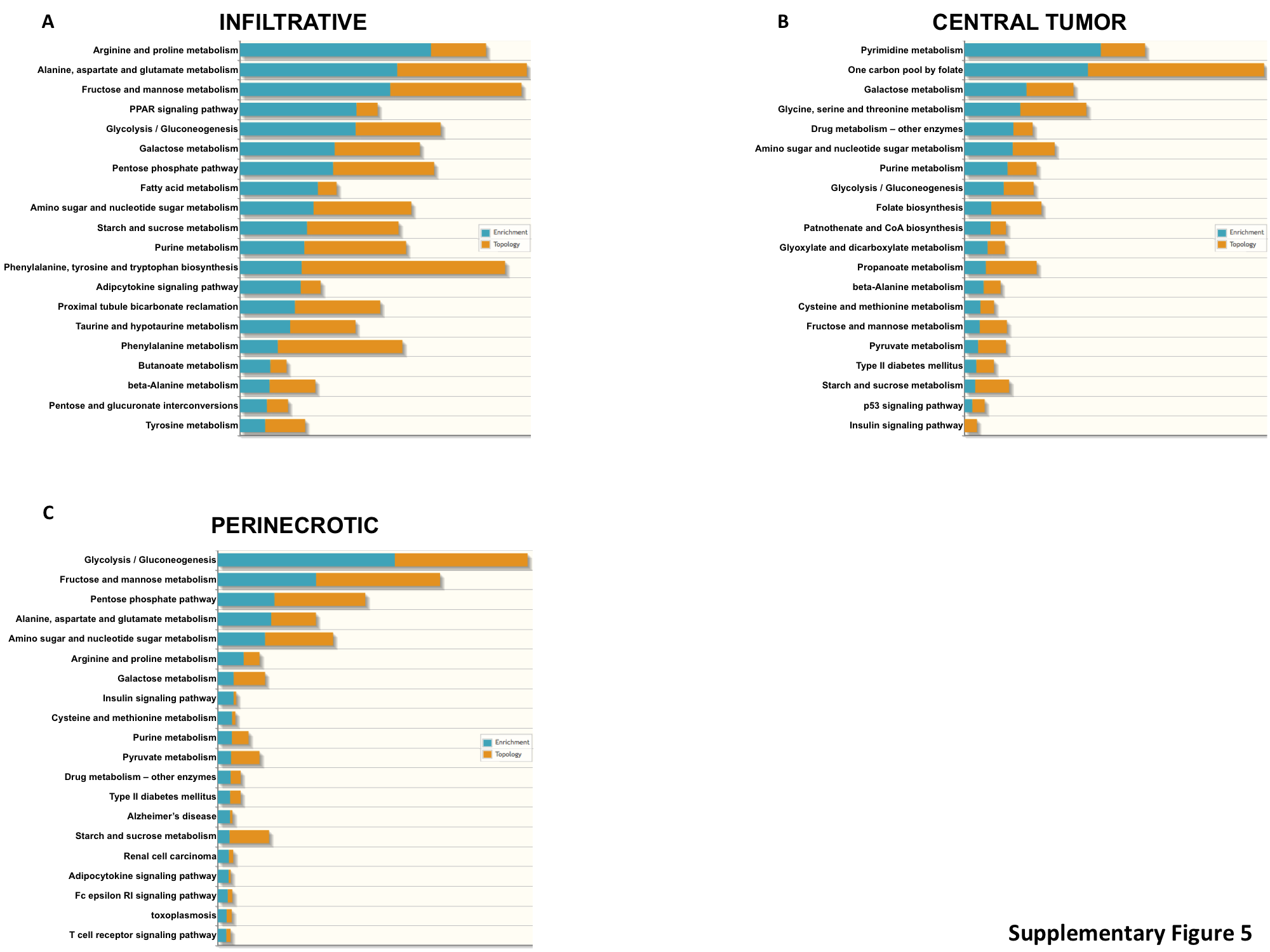
As we identified significant regional alterations of cellular metabolism in GBM, particularly in the progression from the highly cellular CT to PNZs, we went on to evaluate these pathways in further detail. PCA performed using a panel of 255 genes involved in cellular metabolism (Supplementary Table S4) once again demonstrated significant clustering of transcriptional profiles from different tumors based on region, with the PNZ demonstrating the largest degree of separation, and again, mutually exclusive to the infiltrative region (Fig. 3A and Supplementary Figure S3B). Pathway analyses identified amino acid, fatty acid, and pentose phosphate pathway metabolism as unique to the infiltrative cells, while pyrimidine and folate metabolism appeared to play a more contributory role in progression to a CT. Alterations in glycolysis played a dominant role in continued progression to the PNZ (Supplementary Figure S5). VIP analysis suggested that infiltrative cells appeared to rely heavily on glutamine, nucleotide, and fatty acid metabolism, and progression to a highly cellular tumor appeared to consist of a glycolytic switch from hexokinase (HK)1 to HK2, which is consistent with previous reports in GBM28 (Fig. 3B). This glycolytic switch was even more striking when progressing to PNZs, with continued increases in HK2 expression coupled with glucose transporters (solute carrier family 2 members 1 and 3 [SLC2A1/3]), its continued downstream metabolism (phosphofructokinase platelet, lactate dehydrogenase A), and inhibition of pyruvate dehydrogenase kinase isozyme 1 (PDK1) (Fig. 3C).
Fig. 3.

Structural regions in GBM are driven by unique metabolic programs. (A) PCA was performed on individual tumor regions, including the infiltrating tumor (LE + IT), perinecrotic region (PNZ, PAN), and the central tumor core using a 255-gene list associated with metabolism. (B, C) VIP analysis was performed to identify the most important metabolic genes differentiating individual regions (red: upregulation, green: downregulation).
Discussion
Comprehensive genomic-based classifications have provided a framework for developing precision therapy-based treatment strategies in GBM. These investigations have demonstrated clear intertumoral heterogeneity in this malignancy, identifying specific molecular subtypes that may provide insight into unique cellular origins of individual tumors and offer the potential for subtype-specific therapeutic strategies.3,5 However, a number of recent studies have identified significant intratumoral heterogeneity in GBM,9,10 representing a clear obstacle to achieving the intended clinical benefits offered by precision medicine. For example, single cell RNA-seq demonstrated the presence of multiple molecular subtypes in each of 5 primary GBMs,10 and multiple subtypes were observed when tissue was obtained from spatially distinct regions within an individual tumor.9 Therefore, studies designed to better understand the cause and/or consequence of this observed heterogeneity in GBM may guide development of novel therapeutic strategies in this otherwise lethal malignancy.
The variety of cells and structural regions making up the complex ecosystem in GBM is well established using standard H&E staining. This includes an infiltrating, leading edge of a tumor and a central tumor core, which consists of a variety of distinct regions, including cells proliferating within areas of microvasculature and pseudopalisading cells around necrosis.1 In this study, we hypothesized that intratumoral molecular heterogeneity in GBM was not random, but a direct consequence of its complex microenvironment. Therefore, we sought to determine if cells in these different structural regions might be considered as unique biomes harboring predictable phenotypes. To test this, we utilized a unique data repository generated by the Ivy GAP, which used laser capture microdissection to isolate RNA from over 100 structural regions in 37 individual tumors that were subsequently molecularly profiled using RNA-seq.11,12 Mining this database, which allows for an unprecedented window into molecular consequences of the complex tumor microenvironment, we validated significant molecular subtype heterogeneity in GBM. The heterogeneity of transcriptional signatures was particularly evident when evaluated in the context of their derived structural region, as tumors that had more than 2 regions represented in the cohort almost always harbored multiple molecular subtypes. This line of investigation also uncovered a previously undescribed relationship between subtype heterogeneity in GBM and its unique tumor microenvironment, with nearly all of the subtypes generated from the infiltrating, leading edge of a tumor harboring a proneural signature, while the mesenchymal subtype was almost exclusively observed in perinecrotic regions within an individual tumor. These findings are consistent with our current understanding of the different molecular subtypes in GBM and their potential biologic relevance. For example, the mutual exclusivity of these transcriptional subtypes3,5 is recapitulated by their derived structural location, and the mesenchymal signature has been previously associated with regions of necrosis.29–31 These results are among the first to be able to integrate these observations within an individual tumor and reinforce the concept that GBM consists of a very heterogeneous group of cells, and a classification of an individual tumor as a specific subtype based on a single tissue sample is likely a misnomer that does not recognize this malignancy’s underlying complexity.
Although a considerable amount of intratumoral heterogeneity was noted in GBM, a particularly striking observation when evaluating the transcriptional profiles in the context of their derived structural regions was the relatively small amount of intertumoral heterogeneity in this malignancy as a whole. Specifically, we observed significant clustering of transcriptional profiles of specific structural regions from different tumors, suggesting that the observed intratumoral heterogeneity may be a predictable consequence to its given microenvironment. This concept has recently been elegantly described by Lloyd et al, who, using evolutionary game theory, demonstrated that cellular spatial heterogeneity in a tumor is largely governed by regional variations in environmental conditions and went on to biologically validate these findings in breast cancer.32 Using this approach as a framework, we analyzed the transcriptional programs from the different regions in the context of evolutionary progression to provide insight into specific molecular events driving regional growth and malignant transformation. In this model, the individual regions within a tumor were defined as specific stages of tumor evolution, with infiltrative cells, which harbored the proneural subtype typically ascribed to low-grade glioma,6,7,33 serving as a window into early events in gliomagenesis that progressed to a highly cellular CT, and finally to advanced regions of necrosis consistent with GBM. In addition to identifying molecular pathways that appeared to validate this model—including the identification of Olig2-regulated genes in infiltrative cells, which have been previously described to play an early role in gliomagenesis,14 downmodulation of genes previously identified as important suppressors of gliomagenesis in the CT,18,19 and alterations in angiogenesis and hypoxia commonly attributed to GBM21,28,33—this line of investigation offers a unique perspective on the molecular machinery contributing to gliomagenesis, thereby offering insight into novel therapeutic strategies. For example, an overwhelming majority of genes appeared to be downregulated when transforming from infiltrative cells to the CT (Supplementary Figure S6A), suggesting that global epigenetic modifications and/or miRNAs may play a contributory role for this level of transformation. In addition, upregulation of several genes previously described to be important mediators of cell cycle progression in GBM, including cyclin-dependent kinase 4/6 and WEE1, appear to be critical events driving proliferation in the CT.4,6,17 Conversely, an overwhelming majority of genes appeared to be upregulated when transforming to the PNZ (Supplementary Figure S6B), suggesting that additional epigenetic events and/or activation of key transcription factors may be contributing to this later stage of progression. Further, in addition to a multiplex strategy for promoting angiogenesis and/or an immune-tolerant environment, including upregulation of vascular endothelial growth factor A, interleukin-8,21,34 and ANGPTL4,25,26 transformation to PNZs demonstrated global alterations in metabolism. The most dominant pathway was enhanced aerobic glycolysis by switching from HK1 to HK2,28 increased expression of the glucose transport SLC2A3/glucose transporter 3,35,36 and inhibition of pyruvate dehydrogenase through PDK1, which represents a critical node regulating the Warburg effect.37,38 It is important to note that although the approach we utilized provides a framework to understand gliomagenesis, experimental data are not provided demonstrating that these molecular profiles actually provide a survival advantage in a particular microenvironment. Therefore, future studies designed to demonstrate evolutionary selection of a particular phenotype in a given microenvironment are warranted. In addition, it will be important to validate regional expression of these identified genes to more definitely establish their role in the tumor ecology of GBM. An illustration summarizing these findings is provided in Fig. 4.
Fig. 4.

Intratumoral heterogeneity in GBM is a direct consequence of predictable cellular biomes driven by evolutionary progression. A summary of our findings is presented, demonstrating that observed molecular subtypes in GBM are directly influenced by regional changes in the tumor microenvironment. In addition, a summary of key molecular events driving individual cell biomes is provided, including global transcriptional repression and activation of growth factor signaling and cell cycle progression driving proliferation in the central tumor, to global transcriptional activation and increases in angiogenesis and reliance on glycolysis in the perinecrotic region. Therefore, combinatorial strategies designed to target these biomes individually may be important to further clinical advancement in GBM.
Collectively, the significant level of intratumoral heterogeneity observed in GBM suggests that targeting a single molecular pathway would have a low likelihood of clinical success, which has been repeatedly observed in this and other malignancies.39 Conversely, the relative lack of intertumoral heterogeneity supports the intriguing possibility that rather than the “n-of-1” approach of developing unique, personalized therapeutic strategies designed to target a specific genotype or phenotype of an individual tumor, perhaps reverting back to a “one size fits all” multi-agent therapeutic strategy rationally designed to individually target these unique and predictable tumor cell biomes present in all tumors may lead to more durable tumor control and overcome acquired and/or innate therapeutic resistance. Further studies should be designed to develop and utilize preclinical models that accurately recapitulate these unique biomes, comprehensively evaluate their molecular consequences, and then, using this framework, design multitargeted approaches that incorporate innovative dosing regimens based on evolutionary principles to maximize control40–42 and improve tolerability to target the complex, yet predictable, heterogeneity observed in GBM.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This work was supported by the NIH/NINDS (R21NS090087), ACS (RSG-11-029-01), and the Herb & Betty Fisher Seed Research Grant (P.C.).
Conflict of interest statement. None declared.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. [DOI] [PubMed] [Google Scholar]
- 2. Gilbert MR, Wang M, Aldape KD et al. . Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31(32):4085–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phillips HS, Kharbanda S, Chen R et al. . Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. [DOI] [PubMed] [Google Scholar]
- 4. TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verhaak RG, Hoadley KA, Purdom E et al. ; Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guan X, Vengoechea J, Zheng S et al. . Molecular subtypes of glioblastoma are relevant to lower grade glioma. PLoS One. 2014;9(3):e91216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ozawa T, Riester M, Cheng YK et al. . Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26(2):288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meyer M, Reimand J, Lan X et al. . Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci U S A. 2015;112(3):851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parker NR, Hudson AL, Khong P et al. . Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci Rep. 2016;6:22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patel AP, Tirosh I, Trombetta JJ et al. . Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allen Institute for Brain Science. Ivy Glioblastoma Atlas Project 2015. Available from: glioblastoma.alleninstitute.org.
- 12. Puchalski R, Shah N, Miller J et al. . GENO-32. An anatomic transcriptional atlas of glioblastoma. Neuro Oncol. 2015;17(suppl 5):v99. [Google Scholar]
- 13. Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. 2015;43(W1):W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsigelny IF, Kouznetsova VL, Lian N, Kesari S. Molecular mechanisms of OLIG2 transcription factor in brain cancer. Oncotarget. 2016;7(33):53074–53101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Soroceanu L, Kharbanda S, Chen R et al. . Identification of IGF2 signaling through phosphoinositide-3-kinase regulatory subunit 3 as a growth-promoting axis in glioblastoma. Proc Natl Acad Sci U S A. 2007;104(9):3466–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pachow D, Wick W, Gutmann DH, Mawrin C. The mTOR signaling pathway as a treatment target for intracranial neoplasms. Neuro Oncol. 2015;17(2):189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarcar B, Kahali S, Prabhu AH et al. . Targeting radiation-induced G(2) checkpoint activation with the Wee-1 inhibitor MK-1775 in glioblastoma cell lines. Mol Cancer Ther. 2011;10(12):2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barbagallo D, Condorelli A, Ragusa M et al. . Dysregulated miR-671-5p / CDR1-AS / CDR1 / VSNL1 axis is involved in glioblastoma multiforme. Oncotarget. 2016;7(4):4746–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Z, Yang J, Xu G et al. . Targeting miR-381-NEFL axis sensitizes glioblastoma cells to temozolomide by regulating stemness factors and multidrug resistance factors. Oncotarget. 2015;6(5):3147–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsuda Y, Miura K, Yamane J et al. . SERPINI1 regulates epithelial-mesenchymal transition in an orthotopic implantation model of colorectal cancer. Cancer Sci. 2016;107(5):619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005;7(2):122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cangul H. Hypoxia upregulates the expression of the NDRG1 gene leading to its overexpression in various human cancers. BMC Genet. 2004;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Said HM, Polat B, Staab A et al. . Rapid detection of the hypoxia-regulated CA-IX and NDRG1 gene expression in different glioblastoma cells in vitro. Oncol Rep. 2008;20(2):413–419. [PubMed] [Google Scholar]
- 24. Sullivan JP, Nahed BV, Madden MW et al. . Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014;4(11):1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Babapoor-Farrokhran S, Jee K, Puchner B et al. . Angiopoietin-like 4 is a potent angiogenic factor and a novel therapeutic target for patients with proliferative diabetic retinopathy. Proc Natl Acad Sci U S A. 2015;112(23):E3030–E3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu K, Babapoor-Farrokhran S, Rodrigues M et al. . Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget. 2016;7(7):7816–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang D, Berglund A, Kenchappa RS, Forsyth PA, Mulé JJ, Etame AB. BIRC3 is a novel driver of therapeutic resistance in glioblastoma. Sci Rep. 2016;6:21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wolf A, Agnihotri S, Micallef J et al. . Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011;208(2):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Conroy S, Wagemakers M, Walenkamp AM, Kruyt FA, den Dunnen WF. Novel insights into vascularization patterns and angiogenic factors in glioblastoma subclasses. J Neurooncol. 2017;131(1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mathew LK, Skuli N, Mucaj V et al. . miR-218 opposes a critical RTK-HIF pathway in mesenchymal glioblastoma. Proc Natl Acad Sci U S A. 2014;111(1):291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cooper LA, Gutman DA, Chisolm C et al. . The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am J Pathol. 2012;180(5):2108–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lloyd MC, Cunningham JJ, Bui MM, Gillies RJ, Brown JS, Gatenby RA. Darwinian dynamics of intratumoral heterogeneity: not solely random mutations but also variable environmental selection forces. Cancer Res. 2016;76(11):3136–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chinnaiyan P, Kensicki E, Bloom G et al. . The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012;72(22):5878–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eikawa S, Ohue Y, Kitaoka K et al. . Enrichment of Foxp3+ CD4 regulatory T cells in migrated T cells to IL-6- and IL-8-expressing tumors through predominant induction of CXCR1 by IL-6. J Immunol. 2010;185(11):6734–6740. [DOI] [PubMed] [Google Scholar]
- 35. Boado RJ, Black KL, Pardridge WM. Gene expression of GLUT3 and GLUT1 glucose transporters in human brain tumors. Brain Res Mol Brain Res. 1994;27(1):51–57. [DOI] [PubMed] [Google Scholar]
- 36. Nishioka T, Oda Y, Seino Y et al. . Distribution of the glucose transporters in human brain tumors. Cancer Res. 1992;52(14):3972–3979. [PubMed] [Google Scholar]
- 37. Michelakis ED, Sutendra G, Dromparis P et al. . Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2(31):31ra34. [DOI] [PubMed] [Google Scholar]
- 38. Prabhu A, Sarcar B, Miller CR et al. . Ras-mediated modulation of pyruvate dehydrogenase activity regulates mitochondrial reserve capacity and contributes to glioblastoma tumorigenesis. Neuro Oncol. 2015;17(9):1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12(7):487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Enriquez-Navas PM, Kam Y, Das T et al. . Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nichol D, Jeavons P, Fletcher AG et al. . Steering evolution with sequential therapy to prevent the emergence of bacterial antibiotic resistance. PLoS Comput Biol. 2015;11(9):e1004493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Silva AS, Kam Y, Khin ZP, Minton SE, Gillies RJ, Gatenby RA. Evolutionary approaches to prolong progression-free survival in breast cancer. Cancer Res. 2012;72(24):6362–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.