


Abstract
Aprataxin and PNKP-like factor (APLF) is a DNA repair factor containing a forkhead-associated (FHA) domain that supports binding to the phosphorylated FHA domain binding motifs (FBMs) in XRCC1 and XRCC4. We have characterized the interaction of the APLF FHA domain with phosphorylated XRCC1 peptides using crystallographic, NMR, and fluorescence polarization studies. The FHA–FBM interactions exhibit significant pH dependence in the physiological range as a consequence of the atypically high pK values of the phosphoserine and phosphothreonine residues and the preference for a dianionic charge state of FHA-bound pThr. These high pK values are characteristic of the polyanionic peptides typically produced by CK2 phosphorylation. Binding affinity is greatly enhanced by residues flanking the crystallographically-defined recognition motif, apparently as a consequence of non-specific electrostatic interactions, supporting the role of XRCC1 in nuclear cotransport of APLF. The FHA domain-dependent interaction of XRCC1 with APLF joins repair scaffolds that support single-strand break repair and non-homologous end joining (NHEJ). It is suggested that for double-strand DNA breaks that have initially formed a complex with PARP1 and its binding partner XRCC1, this interaction acts as a backup attempt to intercept the more error-prone alternative NHEJ repair pathway by recruiting Ku and associated NHEJ factors.
INTRODUCTION
DNA damage repair generally requires multiple enzymatic steps that are facilitated by formation of multi-protein complexes assembled by scaffold proteins. XRCC1 serves as a primary scaffold for formation of a single-strand break repair (SSBR) complex, but also participates in additional repair pathways, including base excision repair (1,2), nucleotide excision repair (3) and others (4–7). It contains specialized N- and C-terminal domains that interact specifically with DNA polymerase β and DNA Ligase3α, respectively, as well as a casein kinase 2 (CK2)-phosphorylated FHA domain binding motif (FBM) that interacts with at least three alternate binding partners: polynucleotide kinase/phosphatase (PNKP), aprataxin (APTX), and aprataxin and PNKP-like factor (APLF) (8–10). The nature of the interaction between the FBMs and FHA domains has been elucidated in a series of structural and binding studies (11–15); the structural similarities of the corresponding FHA domains result in a complex web of competitive binding partners that can be challenging to unravel (16–18). In the cases of PNKP and APTX, both binding partners can modify the termini at the strand break, making the ends more suitable for subsequent repair by a polymerase or ligase, and hence can contribute to SSBR. Although originally suggested to exhibit enzymatic activity (19), APLF appears to lack a structure that would support such activity (20). Instead, the protein appears to function as another scaffold protein, interacting primarily with XRCC4-Ligase4, Ku and with poly(ADP-ribose) through its C-terminal PAR-binding Zinc finger (PBZ) domains to support non-homologous end joining (NHEJ) (21,22). The advantage of complex formation involving two scaffold proteins, XRCC1 and APLF, that support different repair pathways is at present not completely clear, particularly since as noted above, the formation of the XRCC1-APLF complex competes with formation of the XRCC1-PNKP and XRCC1-APTX complexes whose role in SSBR is better established. Further, it becomes necessary to sort out the factors that determine the relative levels of the competing repair complexes that form under different cellular conditions.
Formation of the FBMs in XRCC1 and XRCC4 relies on constitutive phosphorylation by CK2 (8,9,23), a kinase known to exhibit strong selectivity for sequences rich in anionic residues (24,25). This selectivity implies that most phosphopeptides formed by CK2-dependent phosphorylation contain multiple anionic residues and are often multiply phosphorylated. Such highly anionic peptide sequences will exhibit several general characteristics, including a distribution of apparent pK values that differ from those of their component amino acid residues. Phosphate pK values for the protonation/deprotonation of dianionic/monoanionic free phosphoserine (pSer) and phosphothreonine (pThr) are approximately 5.75 and 5.95, respectively (26–28), increasing to values near 6.1 in short peptides (29). Since the pSer and pThr pK values are above those of Asp (pK ∼ 3.8) and Glu (pK ∼ 4.5) residues (30), phosphate deprotonation with increasing pH will occur last so that pSer and pThr residues in CK2-phosphorylated peptides will in general exhibit elevated pK values that arise from the tendency of the polyanionic peptide to share the last remaining protons with nearby deprotonated Asp and Glu carboxyl groups. This behavior is expected to have several functional consequences, including pH sensitivity that extends further into the normal physiological range and, for binding sites such as those in the FHA domain, an increased mismatch between the protonation states of the free and bound peptide.
Interactions of XRCC1 and XRCC4 binding motifs with the FHA domains of PNKP, APTX and APLF have been characterized structurally (12–15) and quantified primarily with isothermal titration calorimetry (ITC) (12,15). These studies have established the essential role of the pThr residues in XRCC1 (pThr519) and XRCC4 (pThr233) for complex formation, as well as an ∼10-fold affinity enhancement produced by additional phosphorylation of the Ser residue that immediately precedes the phosphorylated Thr. Further affinity enhancements resulting from additional phosphorylations of Ser and Thr residues downstream of the binding motif have also been demonstrated (12,15). However, most ITC studies also indicate that these additional phosphorylations alter binding stoichiometry, and the APLF–XRCC1 interaction appears to be somewhat of an outlier (15). For the APLF–XRCC1 interaction, additional phosphorylation of Ser518 preceding pThr519 increased binding affinity by <4-fold, and additional phosphorylation of downstream Thr523 and Ser525 residues did not further increase binding affinity (15). In addition to the reported ITC studies, a fluorescence study of the interaction of full-length, phosphorylated XRCC1 with the PNKP FHA domain, indicated 1:1 binding stoichiometry corresponding to Kd = 3.5 nM (31).
The studies described here were performed to more completely characterize the factors that are involved in the APLF–XRCC1 interaction in order to further understand its role in DNA repair.
MATERIALS AND METHODS
Protein preparation
The FHA domain coding sequence (174–488 bp) of human APLF (APLF FHA), (NCBI reference sequence: NM_173545), with additional sequences at both ends for Gateway LR reaction, was synthesized and cloned into the pUC57 vector by GenScript. The expression clone was obtained by LR reaction using pDest527 destination vector by following the manufacturer's recommended protocol, resulting in a plasmid that contains a His6-tag and a TEV cleavage site at the N-terminus.
The pDest527-APLF FHA constructs were transformed into Escherichia coli stain Rosetta2 (DE3). Cells were grown in the shaker at 37°C in one tenth of a total culture volume for seed culture overnight, and inoculated into the main Luria-Bertani (LB) media after cell harvest and washing cell pellets with fresh LB media. The main cultures were incubated at 30°C for 1 h in the shaker for temperature equilibration, induced with 0.1 mM isopropyl β-d-1-thiogalactopyranoside, and incubated further for 4 h at the same temperature. Cells were disrupted by sonication and centrifuged at 35 000 x g for 30 min at 4°C. The supernatant was applied to a Ni-NTA column (GE healthcare), eluted with imidazole, and treated with TEV protease to cleave the N-terminal His6-tag which was subsequently removed by an additional passage through the Ni-NTA column. A Superdex 75 prep grade gel-filtration column (GE healthcare) was employed for further purification in 25 mM Tris–HCl pH 7.4, 50 mM NaCl, 1 mM EDTA.
To produce isotope-labeled APLF FHA, cells were grown in M9 medium containing 1 g l−1 of 15NH4Cl and 2 g l−1 of 12C- or 13C-d-glucose (Cambridge Isotope Laboratory), following the previous overexpression and purification procedure. All protein samples were concentrated and stored at −80°C. The concentration of APLF FHA protein was determined using the calculated extinction coefficient value at 280 nm (ε280 = 9970 M−1 cm−1).
Crystallization, data collection and structure determination
Crystallization screening of APLF FHA and APLF FHA/phosphopeptides was carried out using sitting drop vapor diffusion with 250 nl sample mixed with 250 nl screening buffer per each condition, and incubated at 20°C. APLF FHA (9–10 mg/ml; MW = 11.57 kDa; 0.7–0.8 mM) was used for crystallization of uncomplexed and peptide-complexed studies. The latter studies contained a 9-residue XRCC1 diphosphopeptide, XRCC1pSpT-9 (P514YAGpSpTDEN522) or monophosphopeptide, XRCC1EpT-9 (P514YAGEpTDEN522) at a 1:1 peptide:protein ratio. Plate-like crystals from apo and the APLF FHA/ XRCC1EpT-9 complex were obtained using condition 25 of the Index HR2–144 (Hampton Research) screen containing 3.5 M sodium formate pH 7.0, and using condition 16 of the PEGII (Qiagen) screen containing 0.1 M Tris pH 8.5, 30% (w/v) PEG1000, respectively. Rhombic-shaped crystals were grown from the APLF FHA/ XRCC1pSpT-9 complex using condition 83 of the PEGII (Qiagen) screen containing 0.5 M lithium chloride, 0.1 M Tris–HCl pH 8.5, 28% (w/v) PEG6000. No further optimization of conditions was required. Samples were subsequently frozen in liquid nitrogen in the presence of 5–10% ethylene glycol as a cryoprotectant. Data were collected using a 007HF Rigaku X-ray generator equipped with a Saturn92 CCD detector.
Data were processed using HKL2000 (32). Phasing was performed by molecular replacement (MR) with PHASER (33) using apo-PNKP coordinates (PDB accession code: 2BRF) for apo-APLF FHA, which was subsequently used as the search model in MR for the complexes. The structures were refined using PHENIX (34) and iterative model building using Coot (35). The final model structure exhibited favorable geometry with 100% of the residues in the allowed region of the Ramachandran plot. Crystallographic statistics are summarized in Supplementary Table S1. All structure figures were prepared using UCSF Chimera (http://www.cgl.ucsf.edu/chimera).
NMR spectroscopy
All NMR experiments were performed at 35°C on a Varian INOVA 500 MHz spectrometer equipped with a triple-resonance, pulsed-field gradient cryo-probe, with the exception of the 31P experiments, which were collected on an Agilent/Varian DD2 600 MHz spectrometer equipped with a heteronuclear broadband probe. The backbone chemical shift assignments were made from 3D HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO and HN(CA)CO experiments that were obtained using Agilent BioPack pulse sequences (Agilent). The 3D experiments were acquired with non-uniform sampling to speed up the data acquisition (36,37). Since several of the amide resonances were broadened as a result of the intermediate amide proton exchange dynamics at pH 7.4, a low pH buffer, 25 mM sodium citrate, 50 mM NaCl, and 1 mM EDTA, pH 5.5, was used for the amide backbone assignments. For phosphopeptide titration experiments, 2D 1H,15N-HSQC spectra were acquired on a sample containing 0.1 mM U-[15N] APLF-FHA in 25 mM HEPES, 25 mM MES, 150 mM NaCl, 1 mM EDTA, pH 7.4 as a function of phosphopeptide concentration. The interaction of sodium phosphate, sodium sulfate, and sodium ethylsulfate with U-[15N] APLF FHA sample was also studied by monitoring the 1H,15N-HSQC spectrum of the labeled protein as a function of the anion concentration. The volume of peptide or anion added never exceeded more than 10% of the total volume.
1D 31P-NMR experiments were employed to determine the pK values of the phosphate groups in phosphopeptides and to characterize the interaction of the phosphopeptide with the APLF-FHA domain. Experimental parameters included a 31P offset of 2.48 ppm, a sweep width of 20 ppm, an acquisition time of 1 sec, a relaxation delay of 1 sec, and 1024 scans were acquired per experiment. Phosphopeptide concentrations of 0.5 mM were used in all 1D 31P NMR experiments. A two-dimensional selective 1H,31P-HSQC experiment (38) was used to assign the phosphoserine and phosphothreonine resonances. The 2D experiment was acquired with the following 1H and 31P parameters: offsets of ∼4.7 and 2.48 ppm, sweep widths of 12 and 20 ppm and acquisitions times of 256 ms and 13.2 ms. The relaxation delay was 1 s and 256 scans were acquired per increment.
Measurements of the pK values of the free phosphopeptides were performed in 50 mM sodium acetate, 50 mM sodium cacodylate, 1 mM EDTA as a function of pH with 0.5 mM tri-methylphosphate (TMP) added as a chemical shift reference. Small amounts of 0.1 N NaOH or HCl were titrated to vary the pH in the range from 8.5 to 4.0 in 0.2–0.4 pH decrements. pK values were determined by fitting the pH-dependent chemical shift data to a Henderson–Hasselbalch equation:
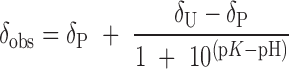 |
where δP and δU correspond to the protonated and unprotonated chemical shifts. The dependence of the phosphopeptide 31P NMR shifts as a function of APLF FHA domain concentration were determined in 25 mM HEPES, 25 mM MES, 150 mM NaCl, 1 mM EDTA, pH 7.4, with 0.25 mM TMP added as a chemical shift reference. The volume of APLF FHA domain added never exceeded more than 10% of the total volume.
Peptide binding assays
Apparent dissociation constants for peptide-protein interactions were determined based on fluorescence polarization measurements of fluorescein-labeled peptides on a POLARstar Omega fluorescence polarization (FP) reader (BMG Labtech) using an excitation wavelength of 485 nm and the emission measured at 520 nm at room temperature. XRCC1 phosphopeptides used in FP studies (Table 1) were labeled with an N-terminal FITC-Gly-Gly fluorophore. Measurements were made as a function of pH in the range from pH 7.4 to 5.5 in FP buffer consisting of 25 mM HEPES, 25 mM MES, 150 mM NaCl, 1 mM EDTA, 2 mM DTT and 0.05% Tween 20. Samples of the APLF FHA domain in FP buffer were prepared at pH 7.4, 7.0, 6.5, 6.0 and 5.5. Samples corresponding to each titration step were put in a 96-well black flat-bottomed plate and the fluorescence emission polarization was determined. The FP results were normalized to a 0–1 scale after curve-fitting for the direct comparison of results from different conditions. Data for the interaction of a phosphopeptide (L) with APLF FHA (M) were fit to a single-site binding equation as described previously (39). All phosphopeptides used were synthesized by either GenScript or Thermo Fisher.
Table 1. K d values of XRCC1 phosphopeptides at pH 7.4.
| Peptide | Sequencea | K d (μM) | Method |
|---|---|---|---|
| XRCC1pSpT-9 | P514YAGpSpTDEN522 | 80 | NMR |
| XRCC1pSpT-18 | D513PYAGpSpTDENTDSEEHQE530 | 3.31 ± 0.15 | FP |
| XRCC1SpT-18 | D513PYAGSpTDENTDSEEHQE530 | >20 | FP |
| XRCC1EpT-18 | D513PYAGEpTDENTDSEEHQE530 | 9.94 ± 4.60 | FP |
| XRCC1pSpTEE-18 | D513PYAGpSpTDENEDEEEHQE530 | 0.30 ± 0.07 | FP |
| XRCC1pSpT-24 | Q507EENGEDPYAGpSpTDENTDSEEHQE530 | 0.16 ± 0.02 | FP |
aPhosphorylated or mutated residues are underlined.
Lineshape simulations
The lineshape simulations utilized a Mathematica algorithm (Wolfram Research, Champaign, IL, USA) based on the two-site exchange formalism given by Rogers and Woodbrey (40). The fraction of bound ligand, pB, is initially calculated using the above relation, the FHA domain and peptide concentrations and an assumed Kd value which is then varied to optimize the agreement with the experimental spectra. The simulation also requires values for the transverse relaxation times of the uncomplexed, T2F, and peptide-complexed, T2B nuclei, the chemical shift difference Δν for the free and bound FHA domain resonances and the lifetime of the protein-peptide complex, τB. For the simulations performed in this study, T2A and T2B were both set at 0.03 s, the Δν value for each exchanging resonance was obtained from the spectra at 0 and maximal peptide concentrations, and the values of Kd and τB optimized to simulate the observed spectral data. Although some variation of T2F and T2B leads to further improvement of the fits, this varied from resonance to resonance and did not significantly influence optimization of the Kd and τB parameters.
RESULTS
Structural basis of the interaction of phosphorylated XRCC1 with the APLF FHA domain
Crystal structures of the apo APLF FHA domain and its complexes with a mono- and di-phosphorylated XRCC1 peptide were determined (Figure 1, and Supplementary Table S1). Apo-APLF FHA adopts the classical FHA β-sandwich topology composed of 10 β-stands forming 2 β-sheets (β1–β2–β6–β7–β9–β10 and β3–β4–β5–β8) facing each other as a core structure, and nine connecting loops (Figure 1A). The domain exhibits high structural similarity with the FHA domains of human PNKP [root mean-square deviation (RMSD) = 1.082 Å using 73 Cα atoms] and human APTX (RMSD = 1.166 Å using 77 Cα atoms) (Figure 1B), which also mediate interactions with CK2-phosphorylated XRCC1 and XRCC4 (13,15,41–43).
Figure 1.

Crystal structures of the APLF FHA domain unliganded and bound to a phosphorylated XRCC1 peptide. (A) Cartoon representation of apo-APLF FHA domain (green) with the 10 β-strands numbered. (B) Stereo view showing an overlay of α-carbon traces of APLF FHA (green) with the FHA domains of PNKP (blue) and APTX (magenta). The APLF FHA structures in complex with (C) XRCC1pSpT-9 diphosphopeptide (protein, light gray; peptide, blue) and (D) XRCC1EpT-9 monophosphopeptide (tan, protein; pink, peptide) are represented. Simulated annealing Fo-Fc omit maps of each phosphopeptide (green mesh) contoured at 3.0 σ for the diphosphopeptide and 2.5 σ for the monophosphopeptide are displayed. The peptide residues are annotated in blue with underlined, italic residue names, and the APLF FHA residues important for binding are annotated in black. Hydrogen-bond interactions are also depicted (red dotted line). (E) The topologies of the β3-β4 and β5-β6 loops in the APLF FHA domain for the apo (green), the XRCC1pSpT-9 diphosphopeptide-complexed (light gray), or the XRCC1EpT-9 -monophosphopeptide-complexed (tan) structures are represented. Hydrogen-bonds that sustain a binding-ready conformation are indicated by black, red or cyan dashed lines, respectively.
The structures of the complexes formed between the APLF FHA domain and a nine-residue XRCC1pSpT-9 peptide, P514YAGpSpTDEN522 or the analog containing a Glu residue at position 518, P514YAGEpTDEN522 (XRCC1EpT-9) were determined. The S518E peptide analog was of interest due to the large effect of Ser518 phosphorylation on binding affinity (vide infra). The structures of the mono- and diphosphorylated peptide complexes with electron density observed for peptide residues 514–521 are shown in Figure 1C and D. As anticipated, structural differences between the uncomplexed and XRCC1pSpT-9 diphosphopeptide-complexed domain (RMSD = 0.326 Å using 99 Cα atoms) and between the uncomplexed and XRCC1EpT-9 monophosphopeptide-complexed domain (RMSD = 0.373 Å using 100 Cα atoms) are quite small. Both peptide complexes contain the same set of peptide–protein interactions, involving the tips of loops β3–β4 and β5–β6, similar to observations in previous studies of FHA domain/phosphopeptide complexes (11,13,15,44,45). The peptide complexes are further buttressed by the hydrophobic interactions of Pro29 with the XRCC1 Tyr515 sidechain in position pT-4 (i.e. four residues N-terminal to the pThr) and hydrogen-bonds of the Asn60 sidechain with pT+1 carbonyl (Asp520-CO), the Lys36 sidechain with the pT-3 carbonyl (Ala516-CO), and the Lys36 carbonyl with the pT+1 amide proton (Asp520-NH). There are multiple direct contacts between pThr519 and the FHA domain, including hydrogen-bonds with Arg27, Ser39, Arg40, and His58 (Figure 1C and D). Alternative conformations of His58 form a hydrogen-bond either with a water molecule or with pThr519, implying less involvement in the direct interaction compared with the other residues. Although Arg27, Ser39 and Arg40 are well conserved among the FHA domains, His58 is not, and corresponds to a Gly residue in PNKP and APTX (10). Alternatively, the pSer518/Glu518 sidechains extend into the solvent and make no direct contacts with APLF FHA domain in our crystal structures.
The phosphorylated XRCC1 peptide complexes with the APLF and PNKP FHA domains are generally in close agreement for residues Pro514 through Asp520, but begin to diverge at Glu521. This appears to result primarily from the interaction of PNKP Asn97 with XRCC1 Asn522 in the PNKP complex (PDB accession code: 2W3O) (12), while we detect no significant interactions for residues near the C-terminus of the bound XRCC1 peptide. However, despite the apparent absence of interactions for residues after Glu521, additional, non-specific electrostatic interactions of residues downstream of the binding motif significantly contribute to binding affinity (vide infra).
Pre-organization of the phosphopeptide binding site
As illustrated in Figures 1C and D, the bound phosphopeptides run along the surface of the protein rather than sitting within a cleft, and are stabilized mainly by interaction with seven residues in the β3–β4 or β5–β6 loops of the APLF FHA domain (Arg27, Pro29, Lys36, Ser39, Arg40, His58, and Asn60). A comparison of the uncomplexed and peptide-complexed structures indicates that multiple hydrogen bonding interactions help to stabilize the positions of critical sidechains of the protein in a binding-ready mode (Figure 1E). Although the Lys36 and Arg40 sidechain positions are not strongly constrained, the sidechains of Arg27 and Ser39 are constrained by hydrogen bonding with the Val38 carbonyl and the Arg41 amide NH, respectively, sustaining the topology of the β3-β4 loop in a binding-ready conformation. The Asn60 sidechain in the β5–β6 loop forms a hydrogen-bond with the Arg37 carbonyl in the β3-β4 loop to maintain the topology between the two loops. The arrangement of the β3–β4 and β5–β6 loops is additionally supported by His42 hydrogen bonding interactions. The position of Pro29 is maintained by a hydrogen-bond between the Gly28 amide NH and the Ile33 carbonyl, and the His58 sidechain is likely constrained by hydrogen bonding with the Asn60 amide NH.
The FHA domain binding motifs exhibit elevated pK values characteristic of CK2-phosphorylated peptides
CK2-dependent phosphorylation targets polyanionic consensus sequences (24), yielding polyanionic peptides. Such molecules typically exhibit a broad distribution of pK values as a result of electrostatic interactions and hydrogen-bond formation, analogous to the effects seen in simple polyanionic molecules such as citrate. Since isolated pSer and pThr have higher pK values than the Asp or Glu residues in the peptide (26,29), they will generally be the last residues to deprotonate as the pH is raised and are expected to exhibit elevated pK values as a consequence of the effect of nearby anionic sidechains that effectively share the last remaining protons. In order to evaluate the pK values for the phosphorylated FBMs of XRCC1 and XRCC4, we performed pH titration studies of the diphosphorylated XRCC1 and XRCC4 peptides using 31P NMR (Figure 2A and B). For peptides containing both pSer and pThr residues, we assigned resonances using a 1H,31P-HSQC experiment in which the resulting spectra exhibit passive coupling interactions with Hα in the case of pSer, or with the β-CH3 protons in the case of pThr (Supplementary Figure S1). As anticipated, the pK values for both phosphorylated residues are well above the values of 6.1 reported for pThr and for short, neutral peptides containing these residues (26–29). Indeed, the pK values for the pThr residues in the XRCC1 and XRCC4 peptides are 6.86 and 7.22, respectively (Figure 2; Supplementary Table S2). The pK values of the preceding pSer residues are also elevated (6.56 for XRCC1, and 6.90 for XRCC4), but are not as high as the values for pThr. The absence of a negative charge in non-phosphorylated Ser results in the reduction of the pK value of adjacent pThr, while there is little effect on the pK value when the pSer is replaced by Glu, consistent with a contribution of the anionic environment to elevation of the pK values (Supplementary Figure S2, and Supplementary Table S2). These high pK values suggest that FHA binding affinity may exhibit a significant pH sensitivity in the physiological range.
Figure 2.

31P-NMR titration data for diphosphorylated XRCC1 and XRCC4 peptides. (A) 1D 31P-NMR spectra of XRCC1pSpT-9 at the pH values indicated are shown (upper panel). 31P chemical shifts for pSer (△) and pThr (▴) were plotted as a function of pH, and fit to a Henderson-Hasselbalch equation as described in Methods (lower panel). (B) 1D 31P NMR spectra of XRCC4pSpT-12 at the pH values indicated for pSer (O) and pThr (•), are shown (upper panel), with pK determinations in the lower panel. Samples contained trimethylphosphate (TMP) as an internal shift standard, and 50 mM sodium acetate, 50 mM sodium cacodylate, and 1 mM EDTA buffer.
Preference of the phosphothreonine binding site for dianionic phosphate
The presence of Arg27, Ser49 and Arg40 and probably His58 residues in the phosphothreonine binding site of the APLF FHA domain create a net positive charge of at least +2, and consequently an expected preference for a dianionic phosphate group. We evaluated this preference based on NMR titration studies using inorganic phosphate performed at pH 5.5, corresponding to monoanionic phosphate and at pH 7.4, corresponding to a dianionic-monoanionic mixed charge state. Over this pH range, the FHA domain amide resonances exhibit only small shift changes in the absence of titrants, consistent with the conclusion that no major conformational change occurs during pH alteration (data not shown). Some amide resonances are broadened and disappear near neutral pH, presumably as a result of conformational and/or proton exchange. Observation of the more strongly shifted amide resonances yielded phosphate Kd values of 14.1 ± 3.7 mM (pH 7.4) and Kd = 92.5 ± 17.8 mM (pH 5.5) (Supplementary Figure S3). The only titrable residue in the phosphate binding site, His58, becomes more positive at lower pH, which would presumably lead to higher binding affinity in contrast with the observed effect. Thus, the higher affinity at pH 7.4 indicates a preference of the APLF FHA domain for a dianionic charge state of the phosphate group. This conclusion was further supported by a comparison between the amide shift changes produced by sodium sulfate and sodium ethyl sulfate, indicating that dianionic sulfate perturbs the same resonances that are sensitive to phosphate, although to a lesser extent, while monoanionic ethylsulfate up to 150 mM fails to produce any significant amide shifts (Supplementary Figure S4). The smaller shifts produced by sulfate may be related to its weaker hydrogen-bonding ability, and particularly its poor tendency to form bidentate interactions (46) that are observed for pThr with Arg40. Taken together, the preferential interaction of dianionic phosphate and sulfate groups with the APLF FHA domain indicates that the dianionic state of pThr in phosphorylated XRCC1 is the preferred binding state.
The nature of the phosphopeptide-FHA domain interaction was further evaluated using 31P NMR. Figure 3 illustrates the titration of XRCC1 phosphorylated at Ser518/Thr519 and XRCC4 phosphorylated at Ser232/Thr233 with the APLF FHA domain at the protein/peptide ratios indicated. In each of the examples shown, both the pSer and pThr resonances shift in the direction of greater deprotonation, however the shift magnitude is much greater for the pThr phosphate (Figure 3A and B). For both peptides, the phosphate resonance of pThr exhibits slow exchange while the pSer exchange is only a bit below the coalescence rate. The large shift of the pThr 31P resonance is consistent with the high selectivity of the site for dianionic phosphate. The relatively small shift of the pSer phosphate indicates that the bound peptide environment is mildly selective for the more highly ionized pSer, presumably as a consequence of proximity to positively charged Lys36 and Arg40 residues.
Figure 3.

31P NMR titration data for diphosphorylated XRCC1 and XRCC4 peptides as a function of the APLF FHA domain concentration. 31P NMR spectra of samples containing (A) 0.5 mM of an 18-residue XRCC1 peptide phosphorylated on Ser518/Thr519 (XRCC1pSpT-18), and (B) 0.5 mM of an 12-residue XRCC4 peptide phosphorylated on Ser232/Thr233 (XRCC4pSpT-12) were titrated with the APLF FHA domain at the ratios indicated. The pSer resonance is indicated by a blue arrow, and the pThr resonance by a red dotted arrow. Titration studies were performed in 25 mM HEPES, 25 mM MES, 150 mM NaCl, 1 mM EDTA, pH 7.4 in the presence of a 0.25 mM TMP chemical shift reference. Resonances arising from impurities in the XRCC1pSpT-18 sample are indicated with an X.
Separation of binding specificity and binding affinity
The phosphorylated FBM is located in an unstructured, polyanionic linker region of XRCC1 that has an isoelectric point (pI) of 4.2, and presumably becomes even more acidic after multiple phosphorylations. In contrast, the pI of the APLF FHA domain is 9.6. Although the crystal structure of the APLF FHA domain complex with the XRCC1 phosphopeptide defines the positions of only seven XRCC1 residues, the charge complementarity suggests the possibility that non-specific interactions may extend well beyond the recognition motif. Contributions of binding motif residues as well as residues flanking the binding site motif were evaluated by studies of a series of peptides, summarized in Table 1. The binding affinity of a nine-residue peptide corresponding to the peptide observed in the crystal structure, XRCC1pSpT-9, was determined by NMR spectroscopy to be extremely weak, ∼80 μM. Based on studies indicating the importance of residues downstream of the binding motif (12,15), further Kd measurements were performed using an 18-residue construct phosphorylated at Ser518 and Thr519 corresponding to residues Asp513-Glu530 (XRCC1pSpT-18). For more accurate determinations of the lower Kd values, these measurements used fluorescence polarization of the peptides labeled with an N-terminal FITC-Gly-Gly fluorophore. The Kd value for XRCC1pSpT-18 is 3.3 μM, weakening to >20 μM if only Thr519 is phosphorylated (XRCC1SpT-18). Replacement of Ser518 with a Glu residue (XRCC1EpT-18) gave an intermediate value of 9.9 μM. In order to evaluate whether the additional phosphorylations at Thr523 and Ser525 could enhance binding affinity due to a non-specific electrostatic interaction with the positively charged FHA domain, these residues were replaced with Glu (XRCC1pSpTEE-18). Consistent with this hypothesis, the Kd of XRCC1pSpTEE-18 was reduced 10-fold to 0.30 μM. Given the significant binding contributions of residues downstream of the binding motif, we also evaluated the binding affinity of a 24-residue construct from Gln507-Glu530 extended with both upstream and downstream residues, and phosphorylated at Ser518 and Thr519 (XRCC1pSpT-24). This construct had the highest affinity measured, Kd = 0.16 μM. Combining the last two results, we can estimate the effects of the Thr523 and Ser525 phosphorylations on the affinity of the longer construct, XRCC1pSpTEE-24, as Kd ∼ 16 nM, approaching the value of 3.5 nM reported by Lu et al. (31) for the interaction of the full length phosphorylated XRCC1 with the FHA domain of PNKP. Thus, despite lacking some of the interactions observed in the complex with the PNKP-FHA domain, the interaction with APLF FHA can be quite significant.
These binding studies demonstrate that even in the absence of additional phosphorylations, there is a significant enhancement of the binding affinity resulting from extension of the recognition motif to include additional anionic residues. Increasing the length of the phosphorylated XRCC1 peptide does not, however, alter the chemical shift perturbation pattern that results from binding of the short, 9-residue binding motif (Figure 4A). Thus, no qualitative change in the peptide-FHA domain interaction is associated with extension of the interacting peptide, consistent with the non-specific nature of these additional interactions.
Figure 4.

Chemical shift perturbations vs. peptide length. (A) Amide chemical shift changes for 0.1 mM U-[15N]APLF FHA domain in the presence of 0.8 mM XRCC1pSpT-9 (blue bars) or XRCC1pSpT-18 (orange bars). (B) 1H,15N-HSQC spectra of U-[15N]APLF FHA domain as a function of XRCC1pSpT-9 (left panel) or XRCC1pSpT-18 (right panel). Peptide concentrations (in μM): 0 (red), 10 (orange), 20 (yellow), 50 (green), 100 (blue), 200 (purple), 400 (cyan) and 800 (pink). Titration studies were performed in a NMR buffer containing 25 mM HEPES, 25 mM MES, 150 mM NaCl, 1 mM EDTA, pH 7.4.
Further insight into this effect is derived from 1H,15N-HSQC spectra of APLF FHA obtained as a function of the concentration of XRCC1 diphosphopeptides of varying length. Results of two titration studies using peptide XRCC1pSpT-9 and peptide XRCC1pSpT-18 are shown in Figure 4B. As is immediately apparent from these spectra, many of the ligand-sensitive resonances in the protein, particularly Gly26, Arg27, Gly28, Leu30, Ile33 and His42 that exhibit fast exchange kinetics in the presence of the shorter XRCC1pSpT-9 peptide, exhibit slow to intermediate exchange behavior with the longer XRCC1pSpT-18 peptide. For the slowly exchanging resonances, these FHA domain amide resonances broaden and disappear, and a second resonance at the end of the titration range narrows and becomes more readily observed as the binding site becomes saturated.
The spectra of APLF FHA domain with the longer peptide XRCC1pSpT-18 also exhibit a rather unusual characteristic: resonances showing the largest Δ1H shift differences exhibit slow exchange characteristics, while resonances shifting primarily in the 15N dimension are in fast exchange. Although for many of these resonances, the Δ15N shifts exceed the Δ1H shifts when measured in ppm, the shifts measured in Hz are larger in the 1H dimension. The 1H spectra for the slowly exchanging resonances were simulated as described in Materials and Methods. An optimal fit for the slowly exchanging peptide resonances was obtained using Kd = 4 μM, τB = 0.9 ms, very similar to the Kd value obtained by fluorescence polarization (Figure 5, and Table 1). The longer, higher affinity phosphopeptide constructs thus also exhibit slower dissociation rate constants (longer bound lifetimes).
Figure 5.

Simulation of amide exchange data. Amide 1H NMR shifts obtained in the study shown in Figure 3B were simulated as described in Materials and Methods. The total shift differences between uncomplexed and fully complexed resonances were taken directly from the spectra. Color coding of the spectra and simulations as a function of peptide concentration (μM) is: 0 (black); 10 (orange); 20 (yellow); 50 (green); 100 (blue); 200 (purple); 400 (cyan); 800 (red).
pH dependence of binding affinity
All of the peptides studied exhibit significant pH-dependent binding affinity, as anticipated from the high pK values and the selectivity of the APLF FHA domain for the dianionic pThr residue. We performed a series of APLF FHA domain titration studies with the diphosphorylated 18-residue XRCC1pSpT-18 peptide containing a fluorescein label at various pH values (Figure 6A). A large change in the Kd value is observed as the pH is decreased; below pH 6.5 the Kd values rise more significantly (Table 2). A more abrupt effect of pH on binding affinity is evident for the longer, higher-affinity 24-residue construct, XRCC1pSpT-24 (Figure 6B). Above pH 6.5, the enhanced binding affinity of the longer peptide tends to more significantly dominate the pH-dependence that is more apparent with the shorter construct; however, in both cases the Kd values rise significantly below pH 6.5. Interestingly, replacement of pSer with Glu in position pT-1, which reduces the binding affinity at pH 7.4 ∼3-fold, also leads to more pH-sensitive binding (Table 2). The binding affinity of XRCC1pSpT-18 phosphopeptide to the APLF FHA domain is reduced by less than 2.5-fold at pH 6.5, and by >8-fold at pH 6.0, whereas that of XRCC1EpT-18 phosphopeptide is reduced by more than 5- and 30-fold at pH 6.5 and 6.0, respectively (Figure 6C, and Table 2). Combined with the results from comparisons of the binding affinity of the pSpT-, SpT- and EpT-XRCC1 phosphopeptide analogs (Figure 6D, and Table 1), these results suggest that pSer is important not only for a non-specific electrostatic interaction, but also for reducing the pH sensitivity of the interaction.
Figure 6.

Apparent Kd values as a function of pH. pH-dependent Kd values were measured by fluorescence polarization (FP) using N-terminal FITC-fluorophore labeled (A) XRCC1pSpT-18, (B) XRCC1pSpT-24 or (C) XRCC1EpT-18 (pH 7.4: •, ─; pH 7.0: □, ···; pH 6.5: *, —; pH 6.0: △, -·-, pH 5.5: ж, -··-). (D) The titration curves of the three phosphopeptides obtained at pH 7.4 are compared. The FP values were normalized to 1.0 to facilitate comparison of the results. Data were fit to a single-site binding equation as described in Materials and Methods. Studies were performed in 25 mM HEPES, 25 mM MES, 150 mM NaCl, 1 mM EDTA, 2 mM DTT, and 0.05% Tween 20.
Table 2. pH-dependent Kd values of XRCC1 diphosphopeptides with varying length.
| K d (μM) | |||
|---|---|---|---|
| pH | XRCC1pSpT-18 | XRCC1pSpT-24 | XRCC1EpT-18 |
| 7.4 | 3.31 ± 0.15 | 0.16 ± 0.02 | 9.94 ± 4.60 |
| 7.0 | 4.67 ± 0.04 | 0.20 ± 0.03 | — |
| 6.5 | 8.23 ± 1.89 | 0.32 ± 0.09 | >50 |
| 6.0 | >25 | 3.65 ± 0.91 | >300 |
| 5.5 | >60 | 6.65 ± 2.35 | — |
DISCUSSION
The studies described here reveal two general features of the phosphopeptide-FHA domain interaction: 1) The binding affinity of the XRCC1-derived FBM peptides for APLF FHA and presumably for the FHA domains of PNKP and APTX as well, exhibit a significant pH dependence. This pH-dependence extends into the physiological range and is probably characteristic of the elevated pK values of pSer and pThr residues in polyanionic peptides phosphorylated by CK2. This behavior contrasts with the pK values of isolated pSer and pThr (26,28), and of pSer or pThr-containing peptides that are not polyanionic (29,47). 2) The binding affinity characterizing the phosphopeptide-FHA domain interaction involves both an FBM corresponding to residues identified in crystal structures, as well as flanking residues that extend well beyond the recognition motif. The additional binding effects appear to involve non-specific electrostatic contributions, since they do not result in specific, identifiable changes in the 1H,15N-HSQC spectra of the FHA–FBM complex, but nevertheless make large contributions to the observed binding affinity. In this context, we note that the PNKP, APTX and APLF FHA domains all have isoelectric points >9.0 that can support such interactions. This behavior is reminiscent of the role that residues preceding the XRCC1 BRCTb domain play in augmenting the interaction with the Ligase3 BRCT domain (48).
The mismatch between the protonation state of the bound (dianionic) and free pThr phosphate is unusual and in general, considered undesirable for biological interactions, which are usually designed to minimize binding-dependent changes in protonation state at physiologically relevant pH values (49,50). Although the pThr519 residue in XRCC1 interacts extensively with the FHA binding site, the affinity enhancement produced by pSer518 appears to depend on an electrostatic contribution, rather than on specific interactions with FHA domain residues. However, the pSer518 residue can exert an additional effect by facilitating deprotonation/protonation of pThr519 that accompanies peptide association and dissociation. For example, a proton that is initially shared by the pSer-pThr phosphate groups in the associating peptide can be fully transferred to the pSer residue as the pThr dianionic phosphate more fully binds to the cationic FHA domain residues, while transfer of the proton from pSer to pThr facilitates dissociation. In this way, the pSer, lacking strong direct interactions with the domain, acts as a portable buffer that facilitates proton-dependent binding and release. A Glu residue at this position whose pK value is ∼4.5, will generally be less effective since it is not a good buffer near pH 6.5. This analysis is also consistent with the greater pH sensitivity of the interaction of APLF FHA with XRCC1EpT-18 compared with that of the XRCC1pSpT-18 phosphopeptide (Figure 6C, and Table 2), although the pK values of the adjacent pThr residue of 6.8 are almost unchanged (Supplementary Table S2). This difference does not arise from changes of the interaction since the structures of the XRCC1pSpT-9 and XRCC1EpT-9 phosphopeptide complexes are essentially identical (RMSD = 0.320 Å using 101 Cα atoms) (Supplementary Figure S5). Analogous roles for buffers are well established in the literature. For example, the activity of carbonic anhydrase, which interconverts HCO3– and CO2, is strongly dependent on buffer concentration to neutralize the protons that are produced or consumed in the reaction (51). Rapid binding kinetics are presumably useful for these interactions, since in any specific repair process, it may be necessary to evaluate several alternate binding partners in order to effect the repair before the XRCC1 repair complex collapses.
Isothermal titration calorimetry (ITC) studies of the binding of phosphorylated XRCC1 and XRCC4 peptides with the FHA domains of PNKP, APTX and APLF have indicated a strong preference for sequential pSpT residues, and further binding enhancement resulted from phosphorylation of XRCC1 Thr523 and Ser525 residues that are downstream of the FBM (12,15). However, the latter effect was not observed for the XRCC1–APLF interaction (15). Nevertheless, replacement of Thr523 and Ser525 with Glu residues in our fluorescence polarization assay led to a further increase in binding affinity, consistent with non-specific electrostatic contributions (Table 1). Additional charge-dependent binding enhancement is supported by studies of the interaction of full-length, tetra-phosphorylated XRCC1 with the PNKP FHA domain, indicating 1:1 binding stoichiometry corresponding to Kd = 3.5 nM (31), compared with Kd = 140 nM for a short, tetraphosphorylated XRCC1 peptide (12). These increases in affinity are consistent with the effects observed in the present study resulting from additional non-specific binding interactions involving residues beyond the FBM.
Although dissociation constants determined using various FBM phosphopeptide constructs indicate greater affinity of XRCC1 for PNKP and APTX than for APLF (12,15,31), the additional contributions of flanking residues make comparisons somewhat more difficult. More important, however, the independent recruitment of APLF by PARP1 is mediated by the interaction of the C-terminal PBZ domains with PAR. Since APLF appears to exhibit significant structural disorder (20), it is likely possible for it to simultaneously bind to both PAR and XRCC1 FBM, potentially biasing the interactome toward formation of this complex.
Cells exposed to hypoxia and low pH have been reported to exhibit a diminished capacity for DNA repair compared with control cells grown under standard culture conditions (52). Significant inhibition of rejoining double-stand DNA breaks (DSBs) also has been observed when irradiated cells are maintained at pH 6.0 (53). Although other effects, including the pH-dependence of polymerase enzymes (54,55) contribute to this behavior, pH-dependent interactions between XRCC1/4 and FHA domain proteins, including PNKP, APTX and APLF, may be an important contributor under some circumstances. Tumor cells, while not universally acidic, often experience hypoxia and low pH (56). Strenuous exercise has been found to lower intracellular pH for periods of time that are generally brief (57), but can become more protracted in cells that have been compromised by various illnesses (58), and consequently could influence DNA repair processes ongoing during this period.
The role of the XRCC1-APLF complex in DNA repair and particularly SSBR has been unclear. As summarized above, APLF competes with two other proteins, PNKP and APTX that are involved in modifying the termini of DNA breaks to facilitate subsequent biochemical steps required to complete the repair. Alternatively, APLF functions as a scaffold supporting NHEJ (21,22,59), and hence may even be viewed as an inhibitor of efficient SSBR. We suggest that the XRCC1-APLF interaction is representative of a group of interactions that constitute competitive DNA repair pathways. As proposed by Wang et al. (60), Ku and PARP1 compete for binding to the termini at DSBs and recruit different sets of repair factors that support either NHEJ or a more highly error-prone alternative NHEJ (alt-NHEJ) repair pathway. In alt-NHEJ, PARP1 recruits its major binding partner XRCC1 by a PAR-dependent binding of the BRCTa domain (61–63), which in turn forms a complex that includes Ligase3α (48,64) and the MRN set of proteins (6,65) that participates in this repair pathway (Figure 7). Studies supporting the involvement of PNKP in alt-NHEJ (66) further suggest that participation of XRCC1 may support recruitment of additional enzymes besides Lig3α. The interaction domains of APLF may function as interception or backup attempts to re-introduce proteins that support the higher fidelity NHEJ pathway. APLF may intercept the alt-NHEJ pathway at two points, binding directly to PARP1-associated PAR using its PBZ domains, and additional binding to the XRCC1 FBM with its FHA domain, the interaction characterized in this study. The role of APLF to intercept XRCC1-dependent repair is further supported by its apparent reliance on XRCC1-mediated nuclear transport (10).
Figure 7.

XRCC1 at the nexus of three different repair pathways. Schematic illustrating NHEJ backup recruitment by APLF. In addition to the standard abbreviations, KuBM—Ku binding motif; FBM—FHA domain binding motif. NHEJ proteins (blue), alt-NHEJ proteins (orange), proteins involved in multiple pathways (gray).
To the extent that the PAR signal is inherently ambiguous, analogous recruitment may also involve other PARP family members. PARP2 appears to function similarly to PARP1 in SSBR (67). PARP3 has been reported to act as a sensor of DSBs, and to recruit APLF (59). Although recognition of DSBs by PARP3 disfavors NHEJ pathways (68), limited recruitment of XRCC1 to the PARylated site may occur. The additional interaction of APLF with XRCC1 could similarly function to intercept the XRCC1 repair complex after initial recruitment by PARP3. The XRCC1 would subsequently be replaced by higher FBM-affinity XRCC4 (15).
Many of the interactions summarized in Figure 7 represent competition among alternative repair pathways, rather than coordination of an efficient repair. As summarized recently (17), the accuracy of damage recognition is limited, leading to a trial and error process in which alternate complexes are formed in an effort to move the repair process forward. The complex web of repair protein interactions is presumably weighted by a combination of damage probability and damage toxicity, with the latter factor an important consideration for the XRCC1–APLF interaction.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession numbers: 5W7W, 5W7X and 5W7Y.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Lori Edwards and Bob Petrovich of the protein expression core facility for optimization of the FHA domain expression, to Dr R. Scott Williams for providing access to his fluorescent plate reader and for helpful discussions about this project, and to Dr Geoff Mueller for many useful discussions and suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health [ZIA-ES050111 to R.E.L., ZIA-ES102645 to L.C.P.]; KRIBB Research Initiative Program (Korean Biomedical Scientist Fellowship Program), Korea Research Institute of Bioscience and Biotechnology, Republic of Korea. Funding for open access charge: NIEHS Intramural Research Program [ZIA-ES050111 to R.E.L., ZIA-ZIA-ES102645 to L.C.P.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dianova I.I., Sleeth K.M., Allinson S.L., Parsons J.L., Breslin C., Caldecott K.W., Dianov G.L.. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004; 32:2550–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cappelli E., Taylor R., Cevasco M., Abbondandolo A., Caldecott K., Frosina G.. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem. 1997; 272:23970–23975. [DOI] [PubMed] [Google Scholar]
- 3. Moser J., Kool H., Giakzidis I., Caldecott K., Mullenders L.H.F., Fousteri M.I.. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell. 2007; 27:311–323. [DOI] [PubMed] [Google Scholar]
- 4. Gabel S.A., DeRose E.F., London R.E.. XRCC1 interaction with the REV1 C-terminal domain suggests a role in post replication repair. DNA Repair (Amst.). 2013; 12:1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogi T., Limsirichaikul S., Overmeer R.M., Volker M., Takenaka K., Cloney R., Nakazawa Y., Niimi A., Miki Y., Jaspers N.G. et al. . Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell. 2010; 37:714–727. [DOI] [PubMed] [Google Scholar]
- 6. Dutta A., Eckelmann B., Adhikari S., Ahmed K.M., Sengupta S., Pandey A., Hegde P.M., Tsai M.S., Tainer J.A., Weinfeld M. et al. . Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2017; 45:2585–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saribasak H., Maul R.W., Cao Z., McClure R.L., Yang W., McNeill D.R., Wilson D.M., Gearhart P.J.. XRCC1 suppresses somatic hypermutation and promotes alternative nonhomologous end joining in Igh genes. J. Exp. Med. 2011; 208:2209–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo H., Chan D.W., Yang T., Rodriguez M., Chen B.P.C., Leng M., Mu J.J., Chen D., Zhou S.Y., Wang Y. et al. . A new XRCC1-Containing complex and its role in cellular survival of methyl methanesulfonate treatment. Mol. Cell. Biol. 2004; 24:8356–8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loizou J.I., El-Khamisy S.F., Zlatanou A., Moore D.J., Chan D.W., Qin J., Sarno S., Meggio F., Pinna L.A., Caldecott K.W.. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004; 117:17–28. [DOI] [PubMed] [Google Scholar]
- 10. Iles N., Rulten S., El-Khamisy S.F., Caldecott K.W.. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell. Biol. 2007; 27:3793–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Durocher D., Taylor I.A., Sarbassova D., Haire L.F., Westcott S.L., Jackson S.P., Smerdon S.J., Yaffe M.B.. The molecular basis of FHA Domain: phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol Cell. 2000; 6:1169–1182. [DOI] [PubMed] [Google Scholar]
- 12. Ali A.A.E., Jukes R.M., Pearl L.H., Oliver A.W.. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic Acids Res. 2009; 37:1701–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bernstein N.K., Williams R.S., Rakovszky M.L., Cui D., Green R., Karimi-Busheri F., Mani R.S., Galicia S., Koch C.A., Cass C.E. et al. . The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol. Cell. 2005; 17:657–670. [DOI] [PubMed] [Google Scholar]
- 14. Williams R.S., Bernstein N., Lee M.S., Rakovszky M.L., Cui D., Green R., Weinfeld M., Glover J.N.. Structural basis for phosphorylation-dependent signaling in the DNA-damage response. Biochem. Cell Biol. 2005; 83:721–727. [DOI] [PubMed] [Google Scholar]
- 15. Cherry A.L., Nott T.J., Kelly G., Rulten S.L., Caldecott K.W., Smerdon S.J.. Versatility in phospho-dependent molecular recognition of the XRCC1 and XRCC4 DNA-damage scaffolds by aprataxin-family FHA domains. DNA Repair (Amst.). 2015; 35:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Becherel O.J., Jakob B., Cherry A.L., Gueven N., Fusser M., Kijas A.W., Peng C., Katyal S., McKinnon P.J., Chen J.J. et al. . CK2 phosphorylation-dependent interaction between aprataxin and MDC1 in the DNA damage response. Nucleic Acids Res. 2010; 38:1489–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. London R.E. The structural basis of XRCC1-mediated DNA repair. DNA Repair (Amst.). 2015; 30:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caldecott K.W. DNA single-strand break repair. Exp. Cell Res. 2014; 329:2–8. [DOI] [PubMed] [Google Scholar]
- 19. Kanno S.I., Kuzuoka H., Sasao S., Hong Z.H., Lan L., Nakajima S., Yasui A.. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J. 2007; 26:2094–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hammel M., Yu Y., Radhakrishnan S.K., Chokshi C., Tsai M.S., Matsumoto Y., Kuzdovich M., Remesh S.G., Fang S., Tomkinson A.E. et al. . An intrinsically disordered APLF Links Ku, DNA-PKcs, and XRCC4-DNA ligase IV in an extended flexible non-homologous end joining complex. J. Biol. Chem. 2016; 291:26987–27006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grundy G.J., Rulten S.L., Zeng Z.H., Arribas-Bosacoma R., Iles N., Manley K., Oliver A., Caldecott K.W.. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J. 2013; 32:112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Macrae C.J., McCulloch R.D., Ylanko J., Durocher D., Koch C.A.. APLF (C2orf13) facilitates nonhomologous end-joining and undergoes ATM-dependent hyperphosphorylation following ionizing radiation. DNA Repair (Amst.). 2008; 7:292–302. [DOI] [PubMed] [Google Scholar]
- 23. Montenarh M. Protein kinase CK2 in DNA damage and repair. Transl Cancer Res. 2016; 5:49–63. [Google Scholar]
- 24. Meggio F., Marin O., Pinna L.A.. Substrate-Specificity of Protein-Kinase Ck2. Cell Mol. Biol. Res. 1994; 40:401–409. [PubMed] [Google Scholar]
- 25. Meggio F., Pinna L.A.. One-thousand-and-one substrates of protein kinase CK2. FASEB J. 2003; 17:349–368. [DOI] [PubMed] [Google Scholar]
- 26. Vogel H.J., Bridger W.A.. P-31 nuclear magnetic-resonance pH titration studies of the phosphoproteins tropomyosin and glycogen phosphorylase-A. Can. J. Biochem. Cell Biol. 1983; 61:363–369. [DOI] [PubMed] [Google Scholar]
- 27. Robitaille P.M.L., Robitaille P.A., Brown G.G., Brown G.G.. An analysis of the pH-dependent chemical-shift behavior of phosphorus-containing metabolites. J. Magn. Reson. 1991; 92:73–84. [Google Scholar]
- 28. Xie Y., Jiang Y., Ben-Amotz D.. Detection of amino acid and peptide phosphate protonation using Raman spectroscopy. Anal Biochem. 2005; 343:223–230. [DOI] [PubMed] [Google Scholar]
- 29. Hoffmann R., Reichert I., Wachs W.O., Zeppezauer M., Kalbitzer H.R.. H-1 and P-31 NMR-spectroscopy of phosphorylated model peptides. Int. J. Pept. Prot. Res. 1994; 44:193–198. [DOI] [PubMed] [Google Scholar]
- 30. Tollinger M., Forman-Kay J.D., Kay L.E.. Measurement of side-chain carboxyl pK(a) values of glutamate and aspartate residues in an unfolded protein by multinuclear NMR spectroscopy. J. Am. Chem. Soc. 2002; 124:5714–5717. [DOI] [PubMed] [Google Scholar]
- 31. Lu M., Mani R.S., Karimi-Busheri F., Fanta M., Wang H., Litchfeld D.W., Weinfeld M.. Independent mechanisms of stimulation of polynucleotide kinase/phosphatase by phosphorylated and non-phosphorylated XRCC1. Nucleic Acids Res. 2010; 38:510–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 33. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. . PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D, Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 36. Kazimierczuk K., Orekhov V.Y.. Accelerated NMR spectroscopy by using compressed sensing. Angew. Chem. Int. Edit. 2011; 50:5556–5559. [DOI] [PubMed] [Google Scholar]
- 37. Orekhov V.Y., Jaravine V.A.. Analysis of non-uniformly sampled spectra with multi-dimensional decomposition. Prog. Nucl. Mag. Res. Sp. 2011; 59:271–292. [DOI] [PubMed] [Google Scholar]
- 38. Wu Z.R., Tjandra N., Bax A.. P-31 chemical shift anisotropy as an aid in determining nucleic acid structure in liquid crystals. J. Am. Chem. Soc. 2001; 123:3617–3618. [DOI] [PubMed] [Google Scholar]
- 39. Kirby T.W., Gassman N.R., Smith C.E., Pedersen L.C., Gabel S.A., Sobhany M., Wilson S.H., London R.E.. Nuclear localization of the DNA repair scaffold XRCC1: uncovering the functional Role of a bipartite NLS. Sci. Rep. 2015; 5:13405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rogers M.T., Woodbrey J.C.. Proton magnetic resonance study of hindered internal rotation in Som substituted N,N-dimethylamides. J. Phys. Chem.-US. 1962; 66:540–546. [Google Scholar]
- 41. Date H., Igarashi S., Sano Y., Takahashi T., Takano H., Tsuji S., Nishizawa M., Onodera O.. The FHA domain of aprataxin interacts with the C-terminal region of XRCC1. Biochem. Biophys. Res. Commun. 2004; 325:1279–1285. [DOI] [PubMed] [Google Scholar]
- 42. Clements P.M., Breslin C., Deeks E.D., Byrd P.J., Ju L., Bieganowski P., Brenner C., Moreira M.C., Taylor A.M., Caldecott K.W.. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst.). 2004; 3:1493–1502. [DOI] [PubMed] [Google Scholar]
- 43. Koch C.A., Agyei R., Galicia S., Metalnikov P., O’Donnell P., Starostine A., Weinfeld M., Durocher D.. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004; 23:3874–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yuan C., Yongkiettrakul S., Byeon I.J., Zhou S., Tsai M.D.. Solution structures of two FHA1-phosphothreonine peptide complexes provide insight into the structural basis of the ligand specificity of FHA1 from yeast Rad53. J. Mol. Biol. 2001; 314:563–575. [DOI] [PubMed] [Google Scholar]
- 45. Pennell S., Westcott S., Ortiz-Lombardia M., Patel D., Li J., Nott T.J., Mohammed D., Buxton R.S., Yaffe M.B., Verma C. et al. . Structural and functional analysis of phosphothreonine-dependent FHA domain interactions. Structure. 2010; 18:1587–1595. [DOI] [PubMed] [Google Scholar]
- 46. Rapp C., Klerman H., Levine E., McClendon C.L.. Hydrogen bond strengths in phosphorylated and sulfated amino acid residues. PLoS One. 2013; 8:e57804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singer A.U., FormanKay J.D.. pH titration studies of an SH2 domain-phosphopeptide complex: Unusual histidine and phosphate pK(a) values. Protein Sci. 1997; 6:1910–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cuneo M.J., Gabel S.A., Krahn J.M., Ricker M.A., London R.E.. The structural basis for partitioning of the XRCC1/DNA ligase III-alpha BRCT-mediated dimer complexes. Nucleic Acids Res. 2011; 39:7816–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Onufriev A.V., Alexov E.. Protonation and pK changes in protein-ligand binding. Q. Rev. Biophys. 2013; 46:181–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petukh M., Stefl S., Alexov E.. The role of protonation states in ligand-receptor recognition and binding. Curr. Pharm. Des. 2013; 19:4182–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Khalifah R.G. Carbon-dioxide hydration activity of carbonic-anhydrase - paradoxical consequences of unusually rapid catalysis. PNAS. 1973; 70:1986–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yuan J.L., Narayanan L., Rockwell S., Glazer P.M.. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res. 2000; 60:4372–4376. [PubMed] [Google Scholar]
- 53. Jayanth V.R., Bayne M.T., Varnes M.E.. Effects of extracellular and intracellular pH on repair of potentially lethal damage, chromosome aberrations and DNA double-strand breaks in irradiated plateau-phase A549 cells. Radiat. Res. 1994; 139:152–162. [PubMed] [Google Scholar]
- 54. Shimazaki N., Yoshida K., Kobayashi T., Toji S., Tamai K., Koiwai O.. Over-expression of human DNA polymerase lambda in E-coli and characterization of the recombinant enzyme. Genes Cells. 2002; 7:639–651. [DOI] [PubMed] [Google Scholar]
- 55. Sucato C.A., Upton T.G., Kashemirov B.A., Osuna J., Oertell K., Beard W.A., Wilson S.H., Florian J., Warshel A., McKenna C.E. et al. . DNA polymerase beta fidelity: Halomethylene-modified leaving groups in pre-steady-state kinetic analysis reveal differences at the chemical transition state. Biochemistry. 2008; 47:870–879. [DOI] [PubMed] [Google Scholar]
- 56. Tannock I.F., Rotin D.. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989; 49:4373–4384. [PubMed] [Google Scholar]
- 57. Taylor D.J., Styles P., Matthews P.M., Arnold D.A., Gadian D.G., Bore P., Radda G.K.. Energetics of human-muscle - exercise-induced ATP depletion. Magnet. Reson. Med. 1986; 3:44–54. [DOI] [PubMed] [Google Scholar]
- 58. Arnold D.L., Bore P.J., Radda G.K., Styles P., Taylor D.J.. Excessive intracellular acidosis of skeletal muscle on exercise in a patient with a post-viral exhaustion/fatigue syndrome. A 31P nuclear magnetic resonance study. Lancet. 1984; 1:1367–1369. [DOI] [PubMed] [Google Scholar]
- 59. Rulten S.L., Fisher A.E., Robert I., Zuma M.C., Rouleau M., Ju L., Poirier G., Reina-San-Martin B., Caldecott K.W.. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell. 2011; 41:33–45. [DOI] [PubMed] [Google Scholar]
- 60. Wang M.L., Wu W.Z., Wu W.Q., Rosidi B., Zhang L.H., Wang H.C., Iliakis G.. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006; 34:6170–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G.. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998; 18:3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Breslin C., Hornyak P., Ridley A., Rulten S.L., Hanzlikova H., Oliver A.W., Caldecott K.W.. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 2015; 43:6934–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim I.K., Stegeman R.A., Brosey C.A., Ellenberger T.. A quantitative assay reveals ligand specificity of the DNA scaffold repair protein XRCC1 and efficient disassembly of complexes of XRCC1 and the poly(ADP-ribose) polymerase 1 by poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 2015; 290:3775–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Caldecott K.W., Mckeown C.K., Tucker J.D., Ljungquist S., Thompson L.H.. An interaction between the mammalian DNA-repair protein Xrcc1 and DNA ligase-III. Mol. Cell. Biol. 1994; 14:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Della-Maria J., Zhou Y., Tsai M.S., Kuhnlein J., Carney J.P., Paull T.T., Tomkinson A.E.. Human Mre11/human Rad50/Nbs1 and DNA ligase III alpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J. Biol. Chem. 2011; 286:33845–33853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Audebert M., Salles B., Weinfeld M., Calsou P.. Involvement of polynucleotide kinase in a poly(ADP-ribose) polymerase-1-dependent DNA double-strand breaks rejoining pathway. J. Mol. Biol. 2006; 356:257–265. [DOI] [PubMed] [Google Scholar]
- 67. Hanzlikova H., Gittens W., Krejcikova K., Zeng Z., Caldecott K.W.. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017; 45:2546–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Beck C., Boehler C., Guirouilh Barbat J., Bonnet M.E., Illuzzi G., Ronde P., Gauthier L.R., Magroun N., Rajendran A., Lopez B.S. et al. . PARP3 affects the relative contribution of homologous recombination and nonhomologous end-joining pathways. Nucleic Acids Res. 2014; 42:5616–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession numbers: 5W7W, 5W7X and 5W7Y.