

Abstract
Herein we report the discovery and SAR of an indole-based protease activated receptor-4 (PAR-4) antagonist scaffold derived from a similarity search of the Vanderbilt HTS collection, leading to MLPCN probe ML354 (VU0099704). Using a novel PAC-1 fluorescent αIIbβ3 activation assay this probe molecule antagonist was found to have an IC50 of 140 nM for PAR-4 with 71-fold selectivity versus PAR-1 (PAR-1IC50 = 10 μM).
Keywords: Protease activated receptor 4, PAR-4 antagonist, ML354
Thrombin, a key enzyme of coagulation, directly activates human platelets through stimulation of the protease activated receptors (PARs).1–3 PAR-4 is a thrombin receptor found on human platelets, along with PAR-1, and represents a potential therapeutic target for the treatment of thrombotic disorders.2–9 Pharmacological inhibition of the PARs, in various animal models, blocks arterial thrombosis, supporting a potential benefit offered by a selective PAR small-molecule antagonist.2,5,6 Recently the PAR-1 antagonist Vorapaxar (Zontivity™) was approved by the FDA as the first in class protease activated receptor antagonist after a phase III trial showed that when used daily with standard of care (e.g., aspirin) it was superior to standard of care alone in reducing the incidence of several combined acute primary and secondary endpoints, including myocardial infarction, stroke, and cardiovascular death. However, in a separate trial Vorapaxar increased intracranial bleeding risk and as a result must not be used in patients with a prior history of stroke or transient ischemic attack (TIA).10 PAR-4 is activated only at high thrombin levels and therefore may not underlie hemostasis. As a result, it is currently unclear whether inhibition of PAR-4 would be a superior target with lower risk of bleeding. In order to understand more fully the potential therapeutic benefit of PAR-4 inhibition, and importantly its safety profile versus PAR-1 antagonism, the development of highly selective PAR-4 antagonists are needed. As part of a collaborative project to address this need we initiated a medicinal chemistry effort within the Molecular Libraries Probe Production Center Network (MLPCN) to develop more potent and selective PAR-4 tool compounds that could progress toward in vivo models of thrombosis.11 Indazole YD-3 is the first non-peptidic, selective antagonist of PAR-4 reported by Lee, et al. in 2002.12 (Fig. 1). Although YD-3 has been successfully used as a tool compound, its high lipophilicity has limited its utility beyond in vitro experiments. A limited optimization effort for YD-3 was reported six years later in 2008,13 however physicochemical properties still remained an issue. Since this report no further modifications around this template have been reported. In 2013, we reported a new indole scaffold,14 affording a potent PAR-4 antagonist 1(PAR-4 IC50 = 66 nM, Fig. 1). Compound 1 validated the indole core as an attractive replacement and encouraged further modifications within this chemotype. Importantly, the indole scaffold avoided reported synthetic challenges associated with separation of N1 and N2 benzyl regioisomers found within the indazole series.13 However recent in vitro DMPK studies in these laboratories indicate microsomal stability and plasma stability are clear issues for both YD-3 and 1, likely due in part to the conserved ester moiety, (Fig. 1).
Figure 1.

Profile of PAR-4 antagonists YD-3 and 1.17
The presence of ethyl benzoate moiety within both YD-3 and 1 led us to conduct a rudimentary structural similarity search around the lead indole. We selected compounds which were somewhat similar to 1 and which were readily available within Vanderbilt’s sample repository. Approximately 160 compounds were selected for single point testing, and Figure 2 illustrates some of the structural diversity which was explored. A handful of ‘actives’ were obtained from this similarity screen and among them, SID 161004645 (VU0099704),15 which contains an aryl nitro group, proved to be robust with a PAR-4 IC50 of 140 nM based upon a PAR-4-AP induced PAC-1 fluorescent αIIbβ3 activation assay.16 Since PAR-1 and PAR-4 are both expressed on human platelets, we determined the selectivity of VU0099704 in the presence of PAR-1-AP and PAR-4-AP agonists. From this assessment we measured a PAR-1 IC50 >10 μM for VU0099704, indicating a roughly 70-fold selectivity for PAR-4 over PAR-1 (Fig. 3). VU0099704 represents an unprecedented PAR-4 antagonist chemotype for the in vitro study of platelet activation and is devoid of the common benzoate ester pharmacophore found within YD-3 and 1. VU0099704 was subsequently declared MLPCN probe compound ML354.11
Figure 2.

Sampling of the diversity explored around the lead compound 1, as obtained from a structural similarity search.
Figure 3.

ML354 (VU0099704) profile and inhibition activity against PAR-4 and PAR-1 using PAC-1.
Due to the presence of the aryl nitro group found within ML354, which can be associated with toxicological challenges,18,19 and its recently measured in vitro DMPK profile (see Fig. 3, high plasma protein binding and metabolic turnover, and poor cytochrome P450 inhibition profile), ML354 remains limited to in vitro experiments using isolated platelets. Thus, in an effort to identify appropriate in vivo PAR-4 antagonist tool compounds we set out to further expand the SAR within ML354 and related hybrid indole subseries in order to identify a viable lead with similar or improved potency and selectivity for PAR-4 while improving DMPK properties. When feasible we pursued an iterative parallel synthesis approach for the chemical optimization of ML354 analogues. Herein we report our efforts detailing the synthesis and SAR resulting from modifications around four major structural regions of ML354 as depicted in Figure 4.
Figure 4.

Optimization strategy for ML354.
Initially in order to assess basic SAR around the nitro indole we surveyed a number of modifications while holding the nitro substituted indole core constant. Key findings from these studies are summarized within Figure 5. Alkylation of the eastern carbinol or replacement with an alkylamino moiety (e.g., VU0478944 and VU0478946) led to active PAR-4 antagonists; however, activity was generally diminished compared to ML354 (~2–30-fold). Interestingly, an attempt to transfer the N-benzyl moiety present in YD-3 and 1 proved unsuccessful in the context of the nitro indole, and led to the inactive compound VU0518413 (Fig. 5).
Figure 5.

PAR-4 activity for select ML354 series nitroindole analogues.
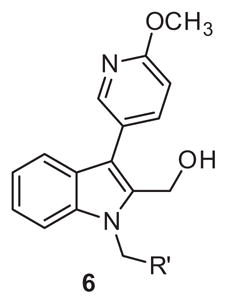
Returning to our strategy outlined in Figure 4 we turned to hybrid molecules targeting our previously identified14 6-methoxypyridin-3-yl indole biaryl ester replacement. In conjunction with this heterocyclic modification we maintained the 2-methyl carbinol present within ML354 as a means to reduce lipophilicity and enhance free fraction, an issue apparent in the previous tool compounds described including ML354 (see Fig. 1 and Fig. 3). The first library 6 was prepared according to Scheme 1, and purified by reverse phase chromatography. Beginning with commercial 1H-indole-2-carboxylate 2, bromination delivered 3 in quantitative yield. Suzuki coupling of 3 with 6-methoxypyridin-3-yl phenylboronic acid provided 4, followed by electrophilic substitution to afford 5. Reduction by lithium borohydride generated hybrid carbinol analogues 6.
Scheme 1.

Reagents and conditions: (a) THF, NBS (1.0 equiv), rt, 2 h, 99%; (b) 6-methoxypyridin-3-yl phenylboronic acid (1.5 equiv), Pd(PPh3)4 (0.1 equiv), Na2CO3 (2.0 equiv), DMF–H2O (4:1), μw, 120 °C, 15 min, 75%; (c) K2CO3 (2 equiv), benzyl bromide (2 equiv, iodomethane applied for 6a), DMF, 45 °C, 16 h, 60–82%; (d) LiBH4, THF, rt, 16 h, 75–90%.
PAR-4 antagonism activity was evaluated via percentage of the max PAC-1 binding assay at a 10 μM single point concentration after PAR-4-AP stimulation. Unfortunately despite the presence of the methyl carbinol, the absence of the nitro functionality proved deleterious for PAR-4 activity (Table 1). No blockade of PAC-1 binding was evident across all N-benzyl analogues (6b–6g PAC-1 >100%) as well as the ML354 N-methyl comparator, 6a.
Table 1.
Structure and activity of analogues 6
| ||
|---|---|---|
| Compd | R′ | % Max PAC-1a |
| 6a |
|
121 |
| 6b |
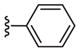
|
109 |
| 6c |
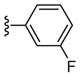
|
126 |
| 6d |
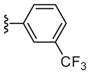
|
133 |
| 6e |
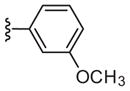
|
134 |
| 6f |
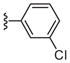
|
135 |
| 6g |
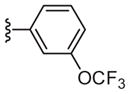
|
135 |
Values indicate the percentage of Max PAC-1 binding after PAR4-AP stimulation of human platelets.16
Given the lack of activity from initial library 6 the next series of library syntheses introduced a 5-methysulfonyl and a 5-nitrile as electron withdrawing functionality to replace the nitro moiety. As shown in Scheme 2 the synthesis of series 11 was accomplished in a manner similar to series 6 in five steps with moderate to excellent yields for each step. Although measurable inhibition of PAC-1 binding was noted at 10 μM for 11a-2 and 11a-3 within the 5-methylsulfonyl subseries bearing 3-substituted benzyl congeners, the PAR-4-AP induced PAC-1 binding response was still significant at >85% for the remaining analogues (Table 2). With the exception of 11b-1, which inhibited PAC-1 binding modestly at 61% and had weak potency with an IC50 >10 μM, the remaining nitrile derivatives were inactive based upon PAC-1 binding. Thus in the context of the 6-methoxypyridin-3-yl biaryl moiety, electron withdrawing groups at the 5-position of the indole had very limited effects on PAR-4 mediated activity.
Scheme 2.

Reagents and conditions: (a) THF, NBS (1.0 equiv), rt, 2 h, 99%; (b) 6-methoxypyridin-3-yl phenylboronic acid (1.5 equiv), Pd(PPh3)4 (0.1 equiv), Na2CO3 (2.0 equiv), DMF–H2O(4:1), μw, 120 °C, 15 min, 70%; (c) K2CO3 (2 equiv), benzyl bromide (2 equiv, iodomethane applied for 11a-1, 11b-1), DMF, 45 °C, 16 h, 60– 90%; (d) LiBH4, THF, rt, 16 h, 55–87%.
Table 2.
Structure and activity of analogues 11
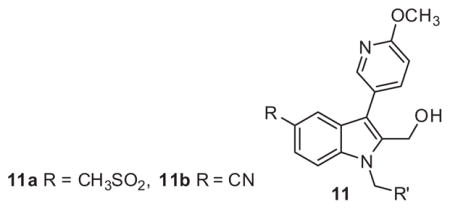
| |||||
|---|---|---|---|---|---|
| Compd | R′ | % Max PAC-1a | Compd | R′ | % Max PAC-1a |
| 11a-1 |
|
96 | 11b-1 |
|
61/>10b |
| 11a-2 |
|
77 | 11b-2 |
|
84 |
| 11a-3 |
|
78 | 11b-3 |
|
83 |
| 11a-4 |
|
85 | 11b-4 |
|
85 |
| 11a-5 |
|
89 | 11b-5 |
|
90 |
| 11a-6 |
|
87 | 11b-6 |
|
114 |
Values indicate the percentage of Max PAC-1 binding after PAR4-AP stimulation of human platelets.
IC50 (μM).16
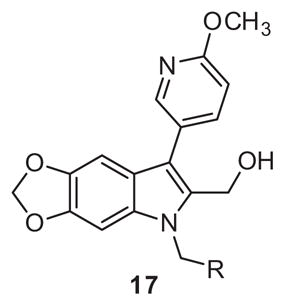
In an attempt to mimic the hydrogen bonding features of the nitro group rather than its polarization of the core phenyl ring, a series of dioxolane derivative 17 were prepared in 5 steps, as depicted in Scheme 3. Starting from 6-bromobenzo[d][1,3]dioxole-5-carbaldehyde 12 and ethyl isocyanoacetate a highly efficient one-pot copper(I) catalyzed condensation/coupling/deformylation process was employed to afford key intermediate indole 13 in 76% yield.20 Subsequent bromination, Suzuki cross-coupling, alkylation, and ester reduction proceeded smoothly as before. Unfortunately, similar to library 11, the third SAR study afforded mostly inactive compounds, with 17b as the most active with a moderate 62% PAC-1 binding response remaining after PAR-4-AP stimulation (Table 3).
Scheme 3.

Reagents and conditions: (a) ethyl 2-isocyanoacetate (1.2 equiv), CuI (0.2 equiv), Cs2CO3 (2 equiv,), DMSO, 80 °C, 16 h, 76%; (b) THF, NBS (1.0 equiv), rt, 2 h, 99%; (c) 6-methoxypyridin-3-yl phenylboronic acid (1.5 equiv), Pd(PPh3)4 (0.1 equiv), Na2CO3 (2.0 equiv), DMF–H2O (4:1), μw, 120 °C, 15 min, 65%; (d) K2CO3 (2 equiv), benzyl bromide (2 equiv, iodomethane applied for 17a), DMF, 45 °C, 16 h, 60–90%; (e) LiBH4, THF, rt, 16 h, 50–80%.
Table 3.
Structure and activity of analogues 17
| ||
|---|---|---|
| Compd | R % | Max PAC-1 bindinga |
| 17a |
|
120 |
| 17b |
|
62 |
| 17c |
|
71 |
| 17d |
|
81 |
| 17e |
|
84 |
| 17f |
|
95 |
Values indicate the percentage of Max PAC-1 binding after PAR4-AP stimulation of human platelets.16
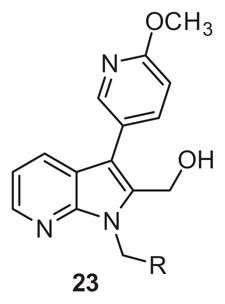
Based on the above data, we switched our nitro replacement strategy towards pyridine incorporation and prepared azaindoles of type 23 (Scheme 4). Pyridine substitution represents a subtle modification of the ring electronics which has proven to be a common and successful nitro isostere replacement in other contexts.21 Prepared in a manner similar to library 17 starting from 2-bromonicotinaldehyde (Scheme 3), the starting indole core 19 was readily prepared in 65% yield using again the cascade annulation chemistry reported by Cai et al.20 Once again, the remaining four steps commenced in moderate to good yields to provide final analogues 23.
Scheme 4.

Reagents and conditions: (a) ethyl 2-isocyanoacetate (1.2 equiv), CuI (0.2 equiv), Cs2CO3 (2 equiv,), DMSO, 80 °C, 16 h, 65%; (b) THF, NBS (1.0 equiv), rt, 2 h, 99%; (c) 6-methoxypyridin-3-yl phenylboronic acid (1.5 equiv), Pd(PPh3)4 (0.1 equiv), Na2CO3 (2.0 equiv), DMF–H2O (4:1), μw, 120 °C, 15 min, 76%; (d) K2CO3 (2 equiv), benzyl bromide (2 equiv, iodomethane applied for 23a), DMF, 45 °C, 16 h, 60–90%; (e) LiBH4, THF, rt, 16 h, 50–85%.
Interestingly, the pyridyl replacement appeared to have a broader impact with several compounds inhibiting PAR-4 mediated PAC-1 binding <80%, in particular the trifluoromethyl derivatives 23c–23d were the most effective at blocking binding of PAC-1, with 23c affording an IC50 of 10 μM in concentration-response studies. No activity was noted in a PAR-1 assay using 23c up to 100 μM. Unfortunately ML354 N-methyl comparator, compound 23a, proved inactive. With the potential exception of 11b-1, across all non-nitro containing subseries, the N-methyl congeners were uniformly inactive (6a, 11a-1, 17a, and 23a). Relative to direct comparators within library 6, the azaindole improved inhibition activity from inactive for 6g to 43% for 23c and similarly for 6d to 23d (Table 4). Although overall these compounds would appear to display modest antagonism, 23c and 23d represent promising starting points devoid of the ester and nitro functionality found within prior PAR-4 tool compounds and indicate that exploration of alternative regioisomeric azaindoles and substituted analogs derived from scaffold 23 are warranted and may provide compounds within the series with potency similar to ML354.
Table 4.
Structure and activity of analogues 23
| |||||
|---|---|---|---|---|---|
| Compd | R | % Max PAC-1a | Compd | R | % Max PAC-1a |
| 23a |
|
88 | 23e |
|
69 |
| 23b |
|
109 | 23f |
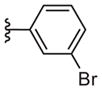
|
76 |
| 23c |
|
43/10b | 23g |
|
81 |
| 23d |
|
49 | 23h |
|
98 |
Values indicate the percentage of Max PAC-1 binding after PAR4-AP stimulation of human platelets.
IC50 (μM).16
Encouraged by these new chemotypes devoid of the ester and nitro functionality we evaluated basic in vitro DMPK properties of 23c and the weakly active phenyl truncated congener 11b-1. In a hepatic microsomal intrinsic clearance assay 11b-1 displayed moderate to high in vitro metabolism with predicted hepatic clearance (CLHEP) = 48 mL min−1 kg−1 in rat and 19 mL min−1 kg−1 in human. Plasma protein binding experiments revealed that 11b-1 was stable in plasma (4 h incubation; 37 °C) and significantly unbound relative to prior PAR-4 tool compounds (1.7% unbound in rat plasma, 2.6% unbound in human plasma). In contrast, compound 23c containing the 3-trifluoromethoxy benzyl moiety maintained high in vitro metabolism based on predicted hepatic clearance (CLHEP = 69 mL min−1 kg−1 in rat and 19 mL min−1 kg−1 in human) and while stable in plasma, 23c was highly protein bound (0.1% unbound in both human and rat plasma). The additional lipophilic character of 23c (cLogP = 3.98 vs 1.88 for 11b-1, calculated using ChemBioDraw 12.0) is likely a contributing factor for the loss of unbound fraction. Future efforts examining subseries 23 actives or regioisomers thereof retaining an N-benzyl moiety may require additional strategies to address metabolism and protein binding should they continue to be challenging in light of the observed profiles described comparing 23c and N-methyl indole 11b-1.
In summary, we have obtained a potent, selective (>71-fold vs PAR-1) PAR-4 antagonist, ML354. The probe compound is low molecular weight and discloses a novel chemotype for PAR-4 antagonist development that can be utilized in vitro to further study PAR-4 activity.22 Based on the profile of ML354 and preliminary efforts described to replace the nitro functionality present we are encouraged by overall improvements made thus far, including plasma stability, free fraction, and potency. Efforts employing alternative azaindole regioisomers, as well as other substituted nitro indole replacements are ongoing. Advances from these studies will be reported in due course.
Acknowledgments
This work was supported by grants from the NIH (U54MH084659, 1R01NS081669), United States and the China Scholarship Council. W. Wen was financially supported by joint-training program (Wu and Lindsley) of China Scholarship Council.
References and notes
- 1.Ruggeri ZM. Platelets Atherothrombosis Nat Med. 2002;8:1227. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 2.Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, Hooper JD. Pharm Ther. 2011;130:248. doi: 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Coughlin S. Nature. 2000;407:258. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 4.Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV, Jr, Tam C, Coughlin SR. Nature. 1998;394:690. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 5.Mao Y, Zhang M, Tuma R, Kunapuli S. J Cereb Blood Flow Metab. 2010;30:1044. doi: 10.1038/jcbfm.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vandendries ER, Hamilton JR, Coughlin SR, Furie B, Furie BC. Proc Natl Acad Sci USA. 2007;104:288. doi: 10.1073/pnas.0610188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cornelissen I, Palmer D, David T, Wilsbacher L, Concengco C, Conley P, Pandey A, Coughlin SR. Proc Natl Acad Sci USA. 2010;107:18605. doi: 10.1073/pnas.1013309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sambrano G, Weiss E, Zheng Y, Huang W, Coughlin S. Nature. 2001;413:74. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- 9.Henrih-Noack P, Riek-Burchardt M, Baldauf K, Reiser G, Reymann K. Brain Res. 2006;1070:232. doi: 10.1016/j.brainres.2005.10.100. [DOI] [PubMed] [Google Scholar]
- 10.See http://www.mercknewsroom.com and http://www.fda.gov for May 2014 Zontivity™ press releases.
- 11.For information on the MLPCN and information on how to request probe compounds, such as ML354, see: http://mli.nih.gov/mli/mlpcn/.
- 12.Lee FY, Lien JC, Huang LJ, Huang TM, Tsai SC, Teng CM, Wu CC, Cheng FC, Kuo SC. J Med Chem. 2001;44:3746. doi: 10.1021/jm010001h. [DOI] [PubMed] [Google Scholar]
- 13.Chen HS, Kuo SC, Teng CM, Lee FY, Want JP, Lee YC, Kuo CW, Huang CC, Wu CC, Huang LJ. Bioorg Med Chem. 2008;16:1262. doi: 10.1016/j.bmc.2007.10.070. [DOI] [PubMed] [Google Scholar]
- 14.Young SE, Duvernay MT, Schulte ML, Lindsley CW, Hamm HE. PLoS ONE. 2013;8:e65528. doi: 10.1371/journal.pone.0065528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VU0099704, (1-methyl-5-nitro-3-phenyl-1H-indol-2-yl)methanol: 1H NMR (400 MHz, CDCl3) δ (ppm): 8.58 (d, J = 2.1 Hz, 1H); 8.17 (dd, J1 = 9.1 Hz, J2 = 2.2 Hz, 1H); 7.55–7.49 (m, 2H); 7.48–7.36 (m, 4H); 4.90 (s, 2H); 3.97 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 142.11; 140.07; 137.73; 133.03; 130.00; 129.14; 127.52; 126.06; 119.46; 118.42; 117.61; 109.37; 55.08; 30.80. HRMS (TOF, ES+) for C16H14N2O3 [M+Na+] calcd mass: 283.1083; found: 283.1081.
- 16.PAC-1 Binding assay: 60 μL of washed platelets (Tyrodes buffer containing 0.1% BSA) at a concentration of 0.15 × 108 platelets/mL were added to 5 mL round bottom polystyrene tubes (BD, Franklin Lakes, NJ). FITC conjugated PAC-1 (BD Biosciences, San Jose, CA) antibody was diluted (to the manufacturers recommended concentration) in Tyrode’s buffer containing 0.1% BSA. 40 μL of diluted antibody was added to the platelets and allowed to incubate for 5 min. Platelets were pre-treated with indicated concentrations of antagonist or DMSO control for 5 min followed by addition of PAR-1-AP (GL Biochem, Shanghai, China) or PAR-4-AP for 10 min. Platelet activity was quenched by the addition ice cold 1.5% paraformaldehyde followed by dilution in 1 × phosphate buffered saline. The final DMSO concentration was 0.5%. Platelets were stored up to 18 h at 4 °C before flow cytometric analysis. Analysis was carried out on a BD FACS Canto II (Franklin Lakes, NJ). Fluorescent intensity was determined for 100,000 events within the platelet gate. Data was collected and analyzed via FACS DiVa software. Flow cytometric data analysis was conducted by the following method. The DMSO-vehicle treated control was subtracted from each data point. 100% response for PAR-4-AP was determined for each individual as the DMSO treated control stimulated with either 200 μM PAR-4-AP, or 20 μM PAR-1-AP. Data was plotted in GraphPad PRISM v.5.0. Dose response curves and subsequent IC50 values were generated using the inhibitory sigmoidal dose response ‘variable slope’ parameter. PAR-4 results were plotted as mean ± SEM.
- 17.After plasma equilibrium binding assay, no drug was detected for either species. Follow-up human plasma stability studies: <1% remaining for both 1 and YD-3 after 4 h incubation. Rat plasma stability studies: 1 and YD-3 ranged from 72% to 89% remaining over 4 h, respectively.
- 18.Smith GF. Prog Med Chem. 2011;50:1. doi: 10.1016/B978-0-12-381290-2.00001-X. [DOI] [PubMed] [Google Scholar]
- 19.Kou-San J. Microbiol Mol Biol Rev. 2012;74:250. doi: 10.1128/MMBR.00006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai Q, Li Z, Wei J, Ha C, Pei C, Ding K. Chem Commun. 2009:7581. doi: 10.1039/b918345k. [DOI] [PubMed] [Google Scholar]
- 21.Meanwell NA. J Med Chem. 2011;54:2529. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 22.In a broad panel selectivity screen at Eurofins Inc. (LeadProfilingScreen™) against 68 GPCRs, ion channels and transporters using 10 μM ML354, moderate binding activity (65–75%) was noted at three targets (5-HT2B, dopamine and norepinephrine transporters); however, functional activity is unknown at these targets and they are not present on human platelets, thus they are not expected to participate in the coagulation cascade.