

Abstract
Mycobacterium tuberculosis synthesizes a thick cell wall comprised of mycolic acids (MA), which are foreign antigens for human T cells. T cell clones from multiple donors were used to determine the fine-specificity of MA recognition by human αβ T cells. Most CD1-presented lipid antigens contain large hydrophilic head groups comprised of carbohydrates or peptides that dominate patterns of T cell specificity. MA diverges from the consensus antigen motif in that it lacks a head group. Using multiple forms of natural and synthetic MA and MA-specific T cells with different T cell receptors, we found that, unlike antigens with larger head groups, lipid length strongly controls T cell responses to MA. In addition, the three forms of MA that naturally occur in M. tuberculosis that differ in modifications on the lipid tail, differ in their potency for activating MA-specific T cell clones. Thus, naturally occurring MA forms should be considered separate, partly cross-reactive antigens. Two of the three forms of MA could be loaded onto human CD1b proteins, creating working CD1b-MA tetramers. The creation of CD1b-MA tetramers represents a new tool for future studies that track the effector functions and kinetics of MA-specific T cells ex vivo.
Keywords: CD1b, mycolic acid, T cells, antigen specificity, lipid antigen, tuberculosis
INTRODUCTION
Mycobacterium tuberculosis has a thick protective cell wall containing two membranes. The key component of the outer membrane is mycolic acid (MA), a lipid defined by an α-branched, β-hydroxy structure. Free MA and mycolyl lipids amount to approximately forty percent of the dry weight of M. tuberculosis [1]. MA is essential for mycobacterial growth in the human host and the target of widely used antibiotics, such as isoniazid. MA, glucose monomycolate (GMM) and glycerol monomycolate all bind to CD1b antigen presenting molecules and are targets of human αβ T cell responses [2–4]. MA was the first known antigen for the CD1 system [2, 5] and it has been used to detect responses in M. tuberculosis-exposed humans [6, 7]. However, nearly all information about the specificity of the human T cell response to MA is derived from three clones, known as DN1, DN.POTT, and GEM18 [2, 8, 9]. The recent validation of CD1b tetramers loaded with GMM prompted our efforts to develop CD1b tetramers loaded with MA. On the molecular level, tetramers loaded with synthetic MA might provide direct evidence for binding to MA-specific T cell receptors (TCRs), avoiding false positive results that can derive from peptide contamination or direct activation of T cells by bacterial products. Further, CD1b-MA tetramers might allow higher throughput isolation of T cell clones and facilitate future study of polyclonal T cells in terms of frequency, functional characteristics, phenotype, and TCR repertoire.
Most CD1-presented antigens are amphipathic glycolipids, phospholipids, sulfolipids or lipopeptides that contain large hydrophilic head groups that dominate the specificity of T cell responses as determined in structure-function studies. That head groups control T cell specificity of CD1b-presented antigens through binding to T cell receptors was recently demonstrated with the first solved trimolecular structure of CD1b-antigen-TCR complex [10]. Here GMM binds within CD1b, such that the glucose moiety protrudes and engages in a large and complex hydrogen bonding network with both the TCR α and β chains. In addition, the TCR contacts the β-hydroxyl group located at the most proximal position of the MA moiety but does not otherwise contact the lipid tail in GMM. This ternary structure involving CD1b, as well as several ternary structures involving CD1d and α-galactosyl ceramide or other antigens [11–13] all point to the carbohydrate unit and the CD1 surface as the determinants of T cell response.
In contrast, MA lacks sugars, peptides, or charged inorganic esters and so might be considered as a ‘headless’ antigen, or more accurately, as having a small head group comprised of a negatively charged carboxylate. A prior study observed loss of T cell response after mutating positive charged residues in the TCR of the MA-specific clone DN1, suggesting TCR recognition of the negatively charged carboxylate moiety [14]. Further, MA with differing naturally occurring distal functional groups on the meromycolate chain known as α-, keto-, or methoxy, influenced recognition by clone DN1 [14]. Whether the distal functional group is buried within CD1b, as predicted by CD1b-GMM structures [10, 15], or folds such that it is somehow directly presented to the TCR at the CD1b surface remains unknown, as no structures of CD1b-MA have been solved.
Determining the role of MA length and distal functional group in CD1b-mediated T cell responses is potentially important in understanding which types of bacteria can be recognized by this system. While most CD1 research focuses on mycobacteria, mycolyl lipids are present among many families belonging to the order of Actinomycetales, including pathogenic and non-pathogenic species. M. tuberculosis and other mycobacteria typically express long chain (C72-86) mycolates with a variety of distal functional groups. Shorter MAs with absent or simple functional groups are synthesized by Rhodococcus spp. (C30-36), Corynebacterium spp. (C30-50), and Nocardia spp. (C40-C66). The role of chain length in control of MA responses is unknown.
To move beyond the few existing, extensively studied MA-specific clones, we sought to develop CD1b tetramers loaded with MA. After synthesizing MA and purifying naturally occurring α-, keto-, and methoxy MA, technical issues in loading extremely hydrophobic long chain mycolates on CD1b were solved and CD1b-MA tetramers were validated using newly derived, MA-specific T cell clones. In sharp contrast to prior studies of glycosylated mycolates [3], we found that MA-specific T cell clones do not recognize short chain MAs and have different preferences for MA forms defined by functional groups. These patterns have implication for the development of lipid vaccines using mixed mycolates or individual molecular species. Further, the data rule in a role for lipid tails in response against MA and contrast with patterns seen for antigens with large head groups, supporting a model of head group positioning on CD1b. Further, the validation of CD1b tetramers creates a new tool for studies of MA responses ex vivo in tuberculosis patients.
RESULTS
Initial attempts to generate CD1b-MA tetramers
Our initial attempts to create MA-loaded CD1b tetramers were based on loading protocols successfully used for GMM and dideoxymycobactin [16, 17]. M. tuberculosis MA (Sigma) (Fig. 1a), which consists of α-, keto-, and methoxy MA forms with an average combined lipid tail length of 80 carbons (referred to as ‘C80 MA mixture’) was used to treat CD1b at pH 4.5 in the presence or absence of the detergent CHAPS. However, as assessed by using the resulting tetramers to stain MA-specific clones DN1 and GEM18, these attempts were unsuccessful. We reasoned that the long chain length and lack of a hydrophilic head group rendered C80 MA to be an extreme hydrophobe that could not be loaded into CD1b using conditions that worked for amphipathic lipids that are more polar than MA. In the past, a key point for loading GMM onto recombinant CD1b in vitro was acidic pH (4.5) [16], which is the pH of the late endosomal compartment where CD1b is loaded in cells, and which functions to release interdomain tethers allowing access to the CD1b cleft [18, 19]. However, we reasoned that low pH also protonates MA, which has a predicted pKa of 4.5. Protonation of MA is expected to decrease further MA solubility in aqueous solutions. Therefore we tried to load MA into CD1b at neutral pH, but this also failed. Prior experiments showed that a less hydrophobic, short chain GMM derived from Rhodococcus equi (C32 GMM) could be successfully loaded onto CD1b [16] and substitute for long chain M. tuberculosis GMM (C80 GMM) in activation assays of the GMM-specific T cell line LDN5 [20]. However, short chain MA from R. equi (C32 MA) could not activate MA-reactive T cell clones even when presented by antigen presenting cells (APC) that could present both C80 and C32 GMM (Fig. 1b). This result documents a roadblock to the use of short chain lipids to overcome the proposed hydrophobicity problem.
Fig. 1. M. tuberculosis MA and R. equi MA have different capacity to stimulate human T cells.
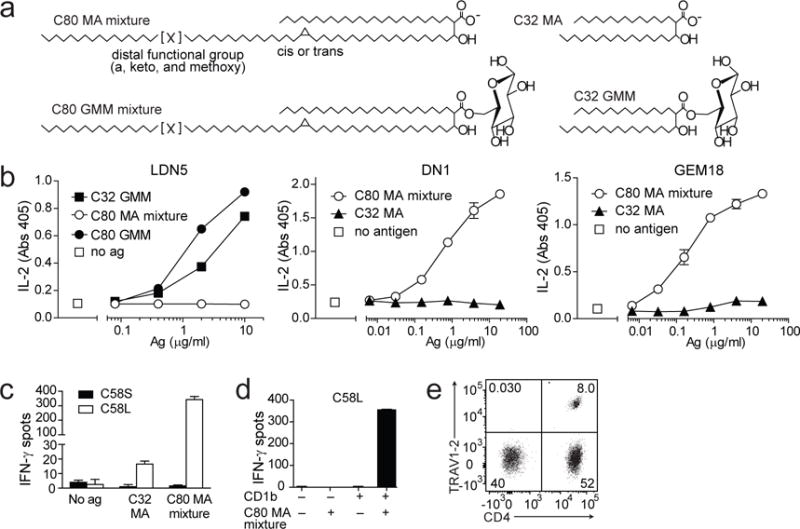
a Simplified chemical structure of C80 MA mixture, the naturally occurring mixture of MA that is synthesized by M. tuberculosis, C80 GMM, C32 MA, which is synthesized by R. equi and C32 GMM [24, 34]. C80 is the average tail length of a reported naturally occurring range of 72-86 carbon atoms, and C32 is the average length of a reported range of 30–36 carbon atoms [24, 34]. b LDN5, DN1, or GEM18 T cells (5×104) were stimulated with monocyte-derived dendritic cells (5×104) and the indicated antigens in duplicate wells. After 20 hours the supernatants were tested for IL-2 content. Cytokine standard curves are shown in Supplemental Fig. 5. Error bars indicate SEM. c The T cell line C58S was generated by polyclonal stimulation of T cells that were isolated based on IFN-γ release after stimulation with C32 MA. The T cell line C58L was obtained after stimulation with C80 MA mixture. Both T cell lines were tested for recognition of C32 MA and C80 MA mixture in an IFN-γ ELISPOT assay with duplicate wells using K562 cells transfected with CD1b as APC. d To determine dependence on CD1b for its activation, C58L was tested in an IFN-γ ELISPOT assay using mock transfected or CD1b-transfected K562 cells as APC. e Flow cytometric analysis of C58L stained with antibodies against CD4+ and TRAV1-2+. In b, one out of three independent experiments using GEM18 or LDN5 is shown. The experiment using DN1 confirming previously published data, was performed once. Experiments shown in c-e were performed two or more times.
Moreover, the marked differences in influence of chain length between the CD1b-MA system and the CD1b-GMM system [20] were striking and unexpected given the similarity of their lipid anchors. In particular, the distal lipid tail of GMM was not required for recognition, whereas similar truncation of the MA tail abolished recognition. Therefore, we decided to study the effect of lipid tail length and composition on MA recognition in more detail.
A panel of human T cell clones recognizing C80 MA
Because the existing MA-specific T cell clones DN1 and GEM18 were both derived based on recognition of APC incubated with M. tuberculosis extract [5, 9], which contains C80 MA mixture, but not C32 MA, we tested whether C32 MA-specific T cells are also present among cryopreserved PBMC. We stimulated PBMC with APC and C32 MA and used an IFN-γ cell surface catch reagent consisting of anti-IFN-γ and anti-CD45 bifunctional antibody and magnetic enrichment to isolate T cells. A T cell line was generated from the isolated T cells by stimulation with anti-CD3 monoclonal antibody (OKT3) and feeder cells. As a control, we performed the procedure in parallel using C80 MA mixture. Stimulation with C32 MA did not lead to the isolation of antigen-specific T cell lines, but stimulation with C80 MA mixture did (Fig. 1c). Activation of this new MA-specific T cell line, named C58L, was dependent on CD1b (Fig. 1d). C58L contained CD4+, CD4−, TRAV1-2+, and TRAV1-2− T cells, demonstrating its polyclonal nature (Fig. 1e). Using the same C80 MA stimulation and IFN-γ catch reagent-based method on another aliquot of cryopreserved PBMC from the same blood donor, we isolated three T cell clones, named clone 11, clone 20, and clone 28. All three newly derived T cell clones showed a strong IFN-γ response to MA that was dependent on CD1b expression (Fig. 2a). These three new clones and the new cell line C58L, along with the recent derivation of GEM18 confirmed and extended the earliest reports of CD1-mediated T cell recognition of MA by clone DN1 [2, 5], suggesting that MA-reactive T cells can be derived from multiple human donors. Further, the growing panel of T cell lines provided new reagents to determine T cell specificity for chain length and other aspects of MA structure.
Fig. 2. Mycolic acid-specific T cell clones and T cell receptors.
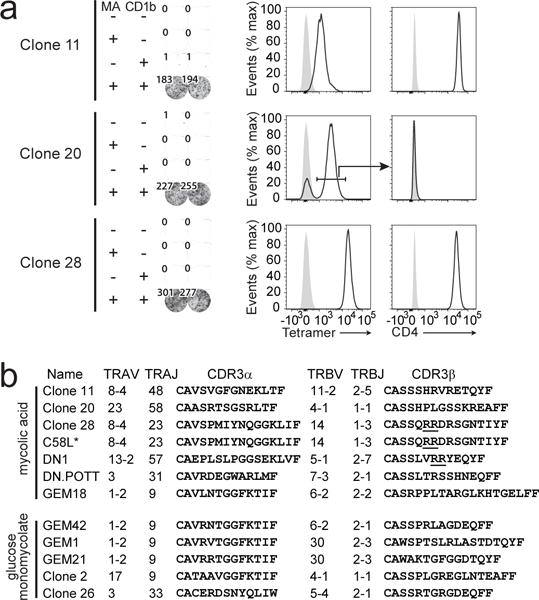
a T cell clones that were generated by limiting dilution and polyclonal stimulation were stained with CD1b-MA tetramer and antibodies against CD3 and CD4. Cells were pre-gated based on forward scatter, side scatter, and CD3. Each clone was tested for recognition of C80 MA mixture in an IFN-γ ELISPOT assay using mock transfected or CD1b-transfected K562 cells as APC. Tetramer+ cells were sorted from clone 20 before the ELISPOT assay. b TCR sequences were determined using a PCR- and Sanger sequencing-based approach. Sequences of DN1, DN.POTT, and GEM18 were previously published [9, 22]. Double arginine motifs are underlined [14]. ELISPOT as shown in a was performed three times and flow cytometry was performed once. Newly derived sequences shown in b were derived from at least two independent PCR reactions. *The C58L-derived TCR sequence was derived from a subpopulation of cells that was sorted based on high staining with CD1b-MA tetramer as shown in Supplemental Fig. 4.
Expression of diverse TCRs
Until recently, CD1b-reactive TCRs were thought to be diverse. However, two different TCR patterns can be distinguished among CD1b specific T cells [9, 21]. GEM TCRs recognize GMM using an invariant TCR α chain comprised of TRAV1-2 and TRAJ9 genes and a biased selection of TCR β chains, favoring TRBV6-2 and TRBV30. LDN5-like cells recognize GMM and use TRAV17 and TRBV4-1-biased TCRs. Having isolated new CD1b and MA-reactive lines and clones, we wanted to compare the TCR sequences of all available MA-specific T cell clones. The TCR sequences of the three new clones were determined, and compared to the previously published MA-specific TCRs of DN1, DN.POTT, and GEM18 [9, 22](Fig. 2b). Although two different TCRs (clone 11 and clone 28) used the TRAV8-4 gene segment, the relatively low number of available MA-specific TCR sequences precludes any conclusion on the question whether MA-specific invariant TCRs exist. Whereas small panels of CD1b and GMM reactive clones were sufficient to discover TCR motifs, we could not identify patterns of shared V or J gene usage or CDR3 length among the available MA-specific TCR sequences.
The TCR of clone 28 uses an arginine doublet in CDR3β. This result supports a prior mutagenesis study showing that this positively charged motif was necessary for MA recognition by clone DN1. Both results predict that that carboxylate group of the mycolic acid moiety could be mediated by positively charged residues in the CDR3 loop [14, 22]. Whereas these arginine residues are predicted to bind the anionic mycolate head group, the other MA-specific TCRs lack this motif and likely have different antigen recognition modes.
Fine specificity for MA variants
Because the first known MA-specific T cell clone, DN1, was reported to recognize some, but not all MA forms that are synthesized by M. tuberculosis [14], and because we found that lipid tail length matters for MA-specific T cells, we next determined the specificity of the new clones for the different forms of MA. We tested whether C80 MA mixture and its three components separated according to their α, keto, and methoxy functional groups, were able to cause cytokine release by DN1, clone 20 and GEM18. The C80 MA mixture potently stimulated all three clones, but the clones showed differing responses based on functional groups, with GEM18 showing preferential activation by α MA with lower responses to methoxy MA and weakest responses to keto MA. Strikingly, clone 20 showed an opposite hierarachy based on functional group (Fig. 3). Lastly, DN1 showed comparable, strong responses to keto and methoxy MA, and a much weaker response to α MA, as previously reported [14]. Because all three forms of MA were recognized by at least one clone, this suggested that each functional group allowed loading into CD1b when presented by an APC. Thus, the differences in potency of the different forms of MA were likely controlled by differing TCR recognition modes of CD1b-MA and these differences were present in distal structures of the lipid tail.
Fig. 3. T cell clones differ in their response against M. tuberculosis MA forms.
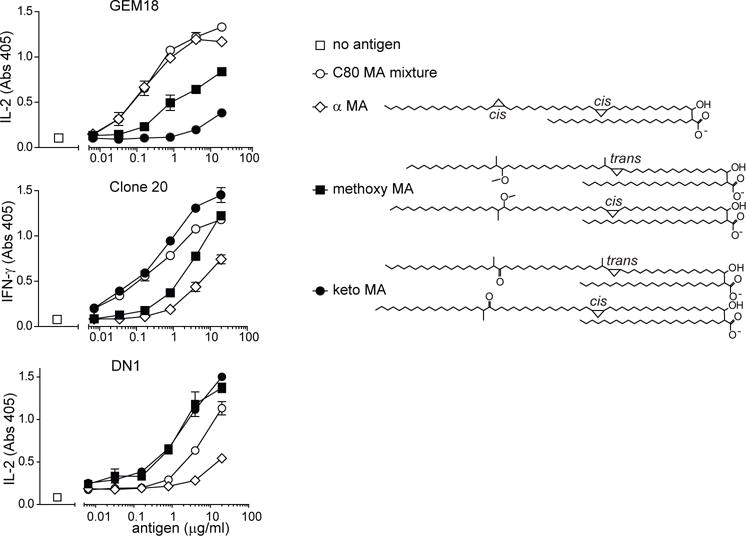
GEM18, clone 20, and DN1 T cells (5×104) were stimulated with allogeneic monocyte-derived dendritic cells (5×104) and antigen in duplicate wells. After 20 hours, supernatants were tested for the presence of IL-2 (GEM18) or IFN-γ (clone 20 and DN1). Structures of α-, keto-, and methoxy MA are drawn as previously reported and represent the average of a naturally occuring range of tail lengths [24, 34]. One out of two independent experiments is shown. Cytokine standard curves are shown in Supplemental Fig. 5. Error bars indicate SEM.
T cell response to synthetic MA
The clone DN1 started the field of CD1 antigen presentation and provided the first example of a human αβ T cell response to a bacterial lipid antigen [2, 5]. Although prior and current experiments were performed with highly purified bacterial MA, contamination with any unrecognized immunostimulant, such as mycobacterial peptides or toll like receptor agonists, could not be formally excluded as the cause of T cell activation. Therefore, we carried out the complete synthesis of long chain MA based on previously published methods [23]. Among the various natural forms of MA present in bacteria, we designed the synthesis to generate a C83 form of methoxy MA with the proximal cyclopropyl in the cis configuration (Fig 4a). Natural methoxy MA is a mixture of C79-C87 in which C83 is the dominant form (Supplemental Fig. 1c), and the proximal cyclopropyl is a mixture of cis and trans, where trans is always associated with an extra methyl group (Fig. 3) [24]. The distal methyl and methoxy groups were synthesized as R, R. Synthetic MA demonstrated the expected mass (m/z 1248.257) of a sodium adduct of C83H164O4 in Ion Trap-Orbitrap Fourier Transform Mass Spectrometry (Fig. 4b and Supplemental Fig. 2) and a diagnostic nuclear magnetic resonance spectrum (Supplemental Fig. 3). Thus, this synthetic MA is chemically identical to a subpopulation of natural methoxy MA purified from M. tuberculosis.
Fig. 4. T cell responses against synthetic MA.
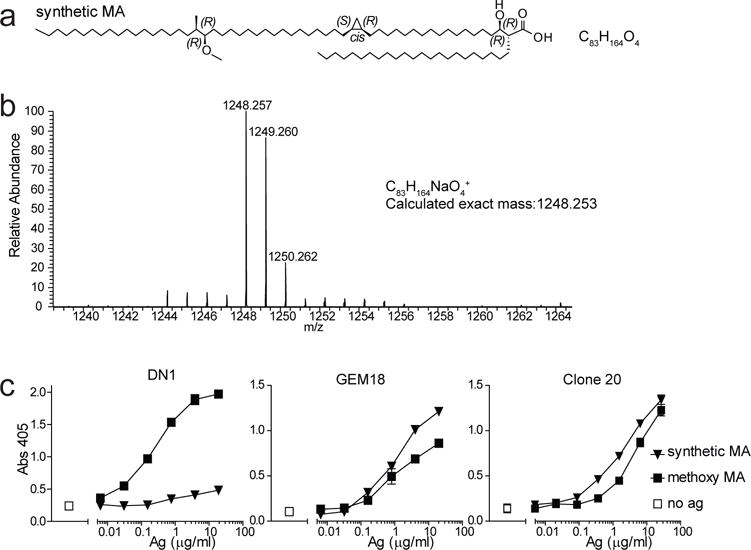
a Chemical structure of synthetic methoxy MA. Unlike natural MAs, this molecule has no tail length variants. b Mass spectrometry and NMR analysis (Supplemental Fig. 3) confirmed the structure shown in a. c DN1, GEM18 and clone 20 T cells (5×104) were stimulated with allogeneic monocyte-derived dendritic cells (5×104) and antigen in duplicate wells. After 20 hours, supernatants were tested for the presence of IL-2 (GEM18) or IFN-γ (clone 20 and DN1). Experiments with DN1 and clone 20 were performed twice, and with GEM18 three times indepenently. Cytokine standard curves are shown in Supplemental Fig. 5.
GEM18 and clone 20 recognized both natural methoxy MA and synthetic methoxy MA with similar dose response curves (Fig. 4c), formally ruling the lipid mycolic acid in as a true antigen for αβ T cells. Unexpectedly, clone DN1 showed only trace response to synthetic methoxy MA but strong responses to natural methoxy MA. Specifically, this result suggests that the configuration of the proximal cyclopropyl or the methyl group with which the trans configuration is associated, is important for methoxy MA recognition by DN1. Overall, these data indicated that MA is the true target of the T cell response but that individual MA-specific T cells with distinct TCRs (Fig. 2b) recognize different forms of MA in different ways (Figs. 3, 4c), with naturally occurring chemical variants of the lipid tail, which are distant from the carboxylate head group, providing strong influence on recognition. This pattern of response is highly distinct from GMM-specific T cell clones, where individual T cell clones show readily recognizable structural motifs in the TCR and respond in highly similar patterns to GMM analogs that vary in the mycolate moiety with regard to length, unstaturations, cyclopropyl groups and oxygenation [3, 10, 20].
Generation of working CD1b-MA Tetramers
The new MA-reactive T cell lines and clones and knowledge of their different fine specificity for the distal structures of MA could now be used to validate new CD1b-MA tetramers. We attempted to load α-, keto-, and methoxy MA separately into human CD1b monomers at pH 4.5 in the presence of the detergent CHAPS. After incubation of CD1b and lipid overnight at 37°C we noticed a precipitate which was most notable with α MA, and least with methoxy MA. Therefore we proceeded with methoxy MA and lowered the molar ratio of MA to CD1b from 40:1 to 15:1, 5:1, and 1.7:1. In addition, we tested several concentrations of CHAPS detergent. The C58L cell line showed clear staining with CD1b-methoxy MA tetramers with a striking pattern demonstrating optimal staining using a molar ratio of 15:1 of MA:CD1b and 0.6% CHAPS (Fig. 5a). The strong role of detergent in promoting staining, further supported the hypothesis that limited loading of extreme hydrophobes onto CD1b is a key issue in generating this new tool. More generally, these data provide evidence for T cell binding to CD1b-MA complexes, indicating that mixed mycobacterial MA provides cognate antigens for an αβ TCR.
Fig. 5. Optimization of CD1b-MA tetramer loading.
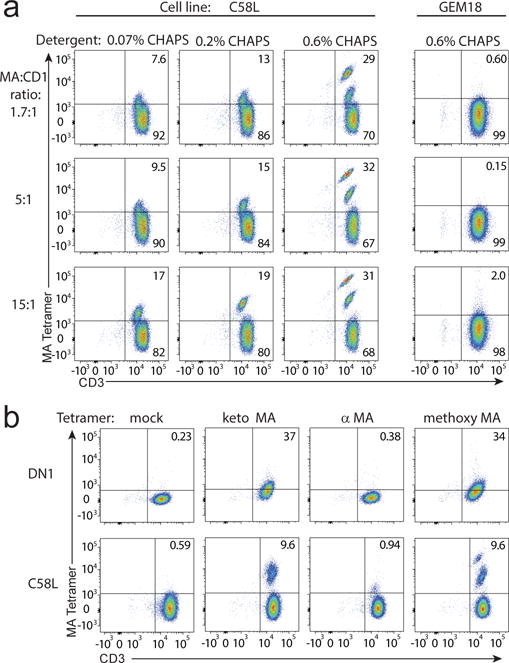
a CD1b monomers were treated with methoxy MA at several molar ratios in buffers containing the indicated percentages of CHAPS detergent, tetramerized using streptavidin-allophycocyanin, and used to stain the indicated T cell line or clone. b Using a 5:1 molar ratio and 0.6% CHAPS, methoxy-, α-, or keto MA were loaded into CD1b tetramers and used to stain the indicated T cell line or clone. Stainings in b and c were performed twice independently. Staining of GEM18 was performed three times independently.
GEM18 responded to methoxy MA with cytokine secretion but did not stain with CD1b-methoxy MA tetramers, which was not unexpected given the relatively low potency of methoxy MA for GEM18 as compared to α MA (Fig. 3). Following the initially inconclusive results, we next tested DN1 for MA-loaded CD1b tetramer staining. Because the different clones required stimulation with different forms of MA for maximum stimulation in vitro, different batches of tetramers were separately treated with each of the MA variants. Keto- and methoxy MA-loaded tetramers weakly stained DN1 whereas α-MA loaded tetramers did not (Fig. 5b). This pattern, carried out with a method that measures the biophysical interaction of CD1b-lipid-TCR, matches the fine specificity pattern observed in activation assays (Fig. 3), providing further evidence for TCR-mediated discrimination of mycolate tails based on a altered physical binding to CD1b-lipid complexes.
We further evaluated the C58L line, which showed two clearly distinct tetramer+ populations when stained with CD1b-methoxy MA (Fig. 5a), for staining with purified MA subclasses (Fig. 5b). As compared with mock loaded tetramers, α-, keto- and methoxy-MA loaded CD1b tetramers all showed staining of C58L, but with markedly differing patterns. The hierarchy of staining of α << keto < methoxy is clearly distinct from the patterns observed for GEM 18 and clone 20 (Fig. 3). After sorting to high purity, cDNA was isolated from each of the two tetramer+ populations that are visible after staining with CD1b-methoxy MA and used for TCR sequencing (Fig. 5b and Supplemental Fig. 4). The population with the highest intensity resulted in a single TCR sequence that was identical to clone 28 at amino acid and nucleotide level (TRAV8-4, TRAJ23, TRB14, TRBJ1-3). Because C58L and clone 28 were independently derived from separate freezes from the same blood draw, we conclude that clone 28 was likely clonally expanded in vivo and mediated extremely high avidity staining with CD1b-MA tetramers. Thus, CD1b-MA tetramers are useful in analysis of human polyclonal T cells and deriving clones therefrom.
DISCUSSION
Building on prior evidence showing that MA isolated from mycobacterial extracts stimulates cytokine production by T cells [2, 5, 14, 22], our results demonstrate that a synthetic MA, which lacks all bacterial molecules has comparable T cell-stimulatory capacity. These experiments prove that the T cell response is not somehow mediated by conventional peptide antigens or bacterial stimulants of TLR-2 or other receptors. Thus, MA is a bona fide, cognate antigen for αβ T cells. Further, tetrameric complexes of MA-loaded CD1b provide a new tool for study of human clonal and polyclonal T cell responses in tuberculosis patients ex vivo.
Previous studies of GMM set the stage for the current structure-function analysis of responses to different forms of MA because these two antigens have the same lipid tail (mycolic acid) but have markedly different head groups (free carboxylate versus glucose) [3, 14, 20, 25]. The strikingly different outcomes of structure-function analyses depending on whether a carbohydrate epitope is present or not, provide insights into the relative roles of the lipid anchor and the hydrophilic head groups of antigens. For GMM, truncation of the mycolic acid from C80 to C32 removes most of the lipid tail as well as functional groups, including unsaturations, cyclopropyl, keto and methoxy groups, but results in only modest effects T cell response or tetramer binding [3, 20, 25].
In contrast, for MA we have been unable to detect any responses to the equivalent truncation leading to C32 MA. Although this initial result might represent insufficient sampling of the T cell repertoire, the hypothesis that T cells distinguish naturally occurring chemical alterations that are distant from the carboxylate does not rest on absence of evidence. Instead, the converse approach in which clones are tested for recognition of MA forms with subtle differences in length or functional groups, shows clear discrimination of natural chemical variance of lipid tails. Fine specificity for lipid tails was observed in all T cells tested in activation assays and in physical interactions with CD1b tetramers. Whereas the low responses of DN1 and clone 20 to α MA might in part result from its extremely low solubility [14], here we found that GEM18 specifically recognizes α MA, suggesting a direct effect of functional group on T cell response to MA and not simply lack of binding to CD1b.
The structural basis of lipid antigen display by CD1b is currently informed by solved binary structures of human CD1b proteins in complex with phosphatidylinositol and other short chain lipids [26, 27], sulfoglycolipid [28] and GMM with C54 [15] or C32 alkyl chains [10]. Most relevant to the current study, the seating of the mycolyl lipid of GMM within the four pockets of the CD1 cleft has been determined experimentally and provides a structural basis for predictions regarding the location of the determinative chemical elements of the mycolyl unit within the groove. The structure-function observations observed here and previously [14] are particularly intriguing since the cyclopropyl group and the keto or methoxy groups are located more than 14 carbon atoms distant from the carboxylate head group. If MA were to be seated similarly to the mycolyl unit of GMM, they would be expected to reside at either end of the T’ tunnel, which is distant from the presumed TCR contact region. Two general models can be considered to explain this surprising outcome.
The previously proposed ‘combinatorial epitope’ model posits that the meromycolate chain folds in an unexpected way so that the α or keto groups are in close proximity to the carboxylate head group and contributes directly to the TCR epitope [14]. Our new data, showing lack of recognition of C32 MA by all available MA-specific T cells, and sensitivity to the distal functional group of C80 MA forms, are consistent with this model. However, taking into account the newly published CD1b-GMM-TCR complex [10], which documents the interactions of the glucose head group with CD1b and the TCR, we propose an alternative ‘epitope stabilization’ model.
Unlike MA, GMM has a large glucose head group. The binary and ternary structures of CD1b-GMM show that the glucose moiety binds the F′ portal region of CD1b, making a hydrogen bond with arginine 79 [15]. This interaction is predicted to stabilize the various GMM analogs with short, medium or long chain tails, as well as analogs with α, keto, or methoxy functional groups, so that the glucose head group lies in nearly the same position in all cases. The role of the head group in stabilizing α-galactosyl ceramide antigens for reproducible interactions with CD1d and NKT TCRs also illustrates the epitope stabilization model [29]. Whereas prior iterations of ‘epitope stabilization’ emphasized antigen interactions with TCRs, our data emphasize head groups stabilizing the antigens within the CD1 groove. In contrast, the lack of a glucose head group in MA might allow the shorter forms of MA to be more mobile or sink inside the CD1b cleft and thereby negatively influence the presentation of the carboxylate group. Similarly, the carboxylate might be more readily subject to any indirect effects of the distantly located functional groups, similar to the known effects of chain length variation on sulfoglycolipid antigens in CD1b [28] or proposed ligand sliding effects documented for ceramide antigens in CD1d [30].
The combinatorial epitope and epitope stabilization models are quite distinct but not mutually exclusive. In both models, tail length and composition may have effects on the conformation of the CD1b protein that influence the docking surface for the TCR. The optimized CD1b loading conditions established here, including the absolute requirement for CHAPS in efficient loading of tetramers, support future strategies to solve binary CD1b-MA crystal structures that are necessary to explain the direct effect of tail length and distal functional group on T cell response to MA and distinguish between two main models.
A second general implication of this study is that the clear MA subclass specificity will inform emerging efforts to develop lipids as vaccine components and efforts to define immunodominant antigens in tuberculosis patients. Prior studies of antigen specific response have typically used mixtures of naturally occurring molecular species present in GMM, glycerol monomycolate or sulfoglycolipid to measure patient responses against CD1b [4, 31, 32]. This approach is straightforward and partly justified by the detailed studies showing nearly equivalent T cell responses to naturally occurring variants of GMM. Thus, GMM occurs in diverse forms but is considered and used as one kind of T cell antigen. Here we found that none of the naturally occurring MA forms of M. tuberculosis is broadly immunodominant or non-stimulatory. Instead, individual T cell clones have specific reactivity patterns to the different M. tuberculosis MA forms. Based on these data, the three forms of MA found in M. tuberculosis should be considered a group of distinct, only partly cross-reactive antigens. The in vivo use of lipid vaccines is moving forward [33] and efforts to determine immunodominant antigens in tuberculosis patients are in progress. These data support a strategy in which individual molecular species, preferably generated through a synthetic approach, should be separately tested, rather than using naturally occurring lipid mixtures from M. tuberculosis. A further practical implication of the fact that each TCR preferentially recognizes one form of MA over other forms is that the tetramers used for the identification of MA-specific T cells in clinical samples should include both the keto MA and the methoxy MA tetramer.
Finally, our results suggest that unlike the strikingly conserved TCRs expressed by CD1d-reactive NKT cells and CD1b-reactive GEM T cells, MA-reactive T cell clones identified to date express diverse TCRs. However, the number of clones that was studied was small, and included clones with different reactivity patters to the different MA forms. Whether CD1b-MA-specific TCR patterns exist that are broadly present among humans is a question that can now be addressed using keto MA or methoxy MA tetramers to isolate large numbers of T cells from many blood donors. Also, the effector functions and kinetics of MA-specific T cells ex vivo can now be studied in cohorts of tuberculosis patients using CD1b-MA tetramers.
MATERIALS AND METHODS
Mycolic acid
A natural mixture of M. tuberculosis MA with chain lengths of 72-86 carbon atoms with mixed α-, keto- and methoxy functional groups (referred to as C80 MA mixture) was obtained from Sigma (M4537). MA with chain lengths of 30–36 carbon atoms (referred to as C32 MA) was isolated from Rhodococcus equi as previously described [20, 22]. We isolated pure αMA (C76-C82), keto MA (C82-C89), and methoxy MA (C79-C87) from M. tuberculosis MA methyl esters by thin layer chromatography, followed by saponification as described previously [14]. Lipids were subjected to thin layer chromatography to measure contaminants and mass spectrometry to determine chain length (Supplemental Fig. 1). Synthetic methoxy mycolic acid was synthesized as previously published [23] with stereochemistry as shown in Fig. 2b as determined by mass spectrometry (Supplemental Fig. 2) and NMR (Supplemental Fig. 3).
Flow cytometry
MA was dried in a glass tube to remove organic solvents and sonicated in a water bath sonicator for 2 hours at 40 °C in 100 μl of a 50 mM citrate buffer pH 4.5 with 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) (Sigma). While keeping the tube at 37 °C, 20 μg of CD1b monomer was added and incubated overnight at 37°C. The next day, the solution was neutralized to pH 7.4 by adding 1M Tris buffer pH 8.5. Tetramers were formed by adding streptavidin-phycoerythrin or streptavidin-allophycocyanin (Life Technologies). For flow cytometry, cells were stained with tetramer (1:100) for 10 min at room temperature. Subsequently, antibodies were added and incubated for 10 min at room temperature, followed by 10 min on ice. The following antibodies were used: CD3-Fitc SK7 (BD Biosciences), TRAV1-2-PE (Vα7.2) (clone 3C10, Biolegend), CD4-BV421 (Biolegend), CD8-PE (eBioscience), TRBV6-2-Fitc (Vβ13.2) (Beckman Coulter). Analysis was performed on a LSRFortessa (BD Biosciences) equipped with 5 lasers. For all analyses, an initial gate was set based on forward scatter and side scatter to exclude debris and doublets. For TCR sequencing, tetramer-positive populations were sorted on a FACSAria Cell Sorter (BD Biosciences) equipped with 3 lasers.
Cell culture and assays
Blood was donated at Massachusetts General Hospital blood, as approved by the institutional review boards of the Lemuel Shattuck Hospital and Partners Healthcare. Biotinylated CD1b monomers were obtained from the NIH tetramer core facility. The polyclonal T cell line named C58L and T cells clones named 11, 20, and 28 were derived after stimulation of peripheral blood mononuclear cells with autologous monocyte-derived dendritic cells and C80 MA mixture for 5 hours, followed by magnetic sorting of IFN-γ producing cells using the IFN-γ secretion assay and cell enrichment kit PE (Miltenyi). For cloning, cells were plated at 1 cell/well in round-bottom 96-well plates containing 2 × 105 irradiated allogeneic PBMCs, 4 × 104 irradiated Epstein Barr Virus-transformed B cells, 30 ng/ml anti-CD3 Ab OKT3, and 1 ng/ml IL-2, which was added on day 2 of the culture. Clones were validated by CD1b-MA tetramer staining and in the case of clone 20, a contaminating population of tetramer-negative cells was removed by cell sorting. Established T cell lines and clones were grown under the same conditions in 25 ml cultures. ELISPOT was performed using Mabtech antibodies according to the manufacturer’s instructions. Cytokine ELISAs were performed with antibodies from eBioscience (human IL-2), Thermo scientific (human IFN-γ), or BD Pharmingen (murine IL-2).
TCR sequencing
RNA was isolated from T cell clones or sorted tetramer-positive populations with an RNeasy kit (Qiagen), followed by cDNA synthesis with a QuantiTect reverse transcription kit (Qiagen). V segment usage was determined by PCR using primer set IPS000029 and IPS000030 as described in the ImMunoGeneTics web site (http://www.imgt.org) in combination with TCRα C region reverse primer GTGGTAGCAGCTTTCACCTCCTTGG and TCRβ C region reverse primer GGTGGCAGACAGGACCCCTTGC. Taq polymerase was used in the supplied buffer (Denville Scientific) under the following cycling conditions: an initial denaturation of 5 min at 95°C, followed by 35 cycles of 1 min at 94°C, 1 min at 59°C, and 1 min at 72°C, followed by a final elongation step of 7 min at 72°C. Sanger sequencing was performed on PCR products.
Supplementary Material
Supplemental Fig. 1 Quality control of purified and commercially available MA
Supplemental Fig. 2 Mass spectrometry of synthetic MA
Supplemental Fig. 3 NMR of synthetic MA.
Supplemental Fig. 4 Sorting strategy for TCR sequencing from C58L
Supplemental Fig. 5 Cytokine standard curves
Acknowledgments
Biotinylated CD1b monomer was obtained through the NIH Tetramer Core Facility. We thank Michael Brenner for providing the DN1 clone and Jamie Rossjohn and Stephanie Gras for advice.
This work was supported by the National Institute of Allergy and Infectious Diseases (Grants AI049313, and AI111224 to D.B.M) and the Netherlands Organization for Scientific Research (NWO Top-punt grant 718.016.001 to A.J.M.)
Nonstandard abbreviations
- MA
mycolic acid
- GMM
glucose monomycolate
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio] -1-propanesulfonate
References
- 1.Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 2.Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature. 1994;372:691–694. doi: 10.1038/372691a0. [DOI] [PubMed] [Google Scholar]
- 3.Moody DB, Reinhold BB, Guy MR, Beckman EM, Frederique DE, Furlong ST, Ye S, Reinhold VN, Sieling PA, Modlin RL, Besra GS, Porcelli SA. Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science. 1997;278:283–286. doi: 10.1126/science.278.5336.283. [DOI] [PubMed] [Google Scholar]
- 4.Layre E, Collmann A, Bastian M, Mariotti S, Czaplicki J, Prandi J, Mori L, Stenger S, De Libero G, Puzo G, Gilleron M. Mycolic acids constitute a scaffold for mycobacterial lipid antigens stimulating CD1-restricted T cells. Chem Biol. 2009;16:82–92. doi: 10.1016/j.chembiol.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Porcelli S, Morita CT, Brenner MB. CD1b restricts the response of human CD4-8- T lymphocytes to a microbial antigen. Nature. 1992;360:593–597. doi: 10.1038/360593a0. [DOI] [PubMed] [Google Scholar]
- 6.Montamat-Sicotte DJ, Millington KA, Willcox CR, Hingley-Wilson S, Hackforth S, Innes J, Kon OM, Lammas DA, Minnikin DE, Besra GS, Willcox BE, Lalvani A. A mycolic acid-specific CD1-restricted T cell population contributes to acute and memory immune responses in human tuberculosis infection. J Clin Invest. 2011;121:2493–2503. doi: 10.1172/JCI46216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seshadri C, Lin L, Scriba TJ, Peterson G, Freidrich D, Frahm N, DeRosa SC, Moody DB, Prandi J, Gilleron M, Mahomed H, Jiang W, Finak G, Hanekom WA, Gottardo R, McElrath MJ, Hawn TR. T Cell Responses against Mycobacterial Lipids and Proteins Are Poorly Correlated in South African Adolescents. Journal of Immunology. 2015;195:4595–4603. doi: 10.4049/jimmunol.1501285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stenger S, Mazzaccaro RJ, Uyemura K, Cho S, Barnes PF, Rosat JP, Sette A, Brenner MB, Porcelli SA, Bloom BR, Modlin RL. Differential effects of cytolytic T cell subsets on intracellular infection. Science. 1997;276:1684–1687. doi: 10.1126/science.276.5319.1684. [DOI] [PubMed] [Google Scholar]
- 9.Van Rhijn I, Kasmar A, de Jong A, Gras S, Bhati M, Doorenspleet ME, de Vries N, Godfrey DI, Altman JD, de Jager W, Rossjohn J, Moody DB. A conserved human T cell population targets mycobacterial antigens presented by CD1b. Nature Immunology. 2013;14:706–713. doi: 10.1038/ni.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gras S, Van Rhijn I, Shahine A, Cheng TY, Bhati M, Tan LL, Halim H, Tuttle KD, Gapin L, Le Nours J, Moody DB, Rossjohn J. T cell receptor recognition of CD1b presenting a mycobacterial glycolipid. Nature communications. 2016;7:13257. doi: 10.1038/ncomms13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, Besra GS, Bharadwaj M, Godfrey DI, McCluskey J, Rossjohn J. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature. 2007;448:44–49. doi: 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]
- 12.Patel O, Pellicci DG, Gras S, Sandoval-Romero ML, Uldrich AP, Mallevaey T, Clarke AJ, Le Nours J, Theodossis A, Cardell SL, Gapin L, Godfrey DI, Rossjohn J. Recognition of CD1d-sulfatide mediated by a type II natural killer T cell antigen receptor. Nat Immunol. 2012;13:857–863. doi: 10.1038/ni.2372. [DOI] [PubMed] [Google Scholar]
- 13.Luoma AM, Castro CD, Mayassi T, Bembinster LA, Bai L, Picard D, Anderson B, Scharf L, Kung JE, Sibener LV, Savage PB, Jabri B, Bendelac A, Adams EJ. Crystal structure of Vdelta1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human gammadelta T cells. Immunity. 2013;39:1032–1042. doi: 10.1016/j.immuni.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grant EP, Beckman EM, Behar SM, Degano M, Frederique D, Besra GS, Wilson IA, Porcelli SA, Furlong ST, Brenner MB. Fine specificity of TCR complementarity-determining region residues and lipid antigen hydrophilic moieties in the recognition of a CD1-lipid complex. Journal of Immunology. 2002;168:3933–3940. doi: 10.4049/jimmunol.168.8.3933. [DOI] [PubMed] [Google Scholar]
- 15.Batuwangala T, Shepherd D, Gadola SD, Gibson KJ, Zaccai NR, Fersht AR, Besra GS, Cerundolo V, Jones EY. The crystal structure of human CD1b with a bound bacterial glycolipid. J Immunol. 2004;172:2382–2388. doi: 10.4049/jimmunol.172.4.2382. [DOI] [PubMed] [Google Scholar]
- 16.Kasmar AG, van Rhijn I, Cheng TY, Turner M, Seshadri C, Schiefner A, Kalathur RC, Annand JW, de Jong A, Shires J, Leon L, Brenner M, Wilson IA, Altman JD, Moody DB. CD1b tetramers bind {alpha}{beta} T cell receptors to identify a mycobacterial glycolipid-reactive T cell repertoire in humans. J Exp Med. 2011;208:1741–1747. doi: 10.1084/jem.20110665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasmar A, Van Rhijn I, Magalhaes KG, Young D, Cheng TY, Turner M, Schiefner A, Kalathur RC, Wilson A, Bhati M, Gras S, Rossjohn J, Shires J, Jakobsen S, Altman JD, Moody DB. CD1a tetramers and dextramers identify human lipopeptide-specific T cells ex vivo. J Immunol. 2013;191:4499–4503. doi: 10.4049/jimmunol.1301660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng TY, Relloso M, Van Rhijn I, Young DC, Besra GS, Briken V, Zajonc DM, Wilson IA, Porcelli S, Moody DB. Role of lipid trimming and CD1 groove size in cellular antigen presentation. Embo J. 2006;25:2989–2999. doi: 10.1038/sj.emboj.7601185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Relloso M, Cheng TY, Im JS, Parisini E, Roura-Mir C, DeBono C, Zajonc DM, Murga LF, Ondrechen MJ, Wilson IA, Porcelli SA, Moody DB. pH-dependent interdomain tethers of CD1b regulate its antigen capture. Immunity. 2008;28:774–786. doi: 10.1016/j.immuni.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moody DB, Briken V, Cheng TY, Roura-Mir C, Guy MR, Geho DH, Tykocinski ML, Besra GS, Porcelli SA. Lipid length controls antigen entry into endosomal and nonendosomal pathways for CD1b presentation. Nat Immunol. 2002;3:435–442. doi: 10.1038/ni780. [DOI] [PubMed] [Google Scholar]
- 21.Van Rhijn I, Gherardin NA, Kasmar A, de Jager W, Pellicci DG, Kostenko L, Tan LL, Bhati M, Gras S, Godfrey DI, Rossjohn J, Moody DB. TCR Bias and Affinity Define Two Compartments of the CD1b-Glycolipid-Specific T Cell Repertoire. Journal of Immunology. 2014;193:5338–5344. doi: 10.4049/jimmunol.1400158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant EP, Degano M, Rosat JP, Stenger S, Modlin RL, Wilson IA, Porcelli SA, Brenner MB. Molecular recognition of lipid antigens by T cell receptors. J Exp Med. 1999;189:195–205. doi: 10.1084/jem.189.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al Dulayymi JR, Baird MS, Roberts E, Deysel M, Verschoor J. The first syntheses of single enantiomers of the major methoxymycolic acid of Mycobacterium tuberculosis. Tetrahedron. 2007;63:2471–2592. [Google Scholar]
- 24.Barry CE, III, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Mycolic acids: structure, biosynthesis and physiological functions. Prog Lipid Res. 1998;37:143–179. doi: 10.1016/s0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- 25.Moody DB, Guy MR, Grant E, Cheng TY, Brenner MB, Besra GS, Porcelli SA. CD1b-mediated T cell recognition of a glycolipid antigen generated from mycobacterial lipid and host carbohydrate during infection. J Exp Med. 2000;192:965–976. doi: 10.1084/jem.192.7.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gadola SD, Zaccai NR, Harlos K, Shepherd D, Castro-Palomino JC, Ritter G, Schmidt RR, Jones EY, Cerundolo V. Structure of human CD1b with bound ligands at 2.3 A, a maze for alkyl chains. Nat Immunol. 2002;3:721–726. doi: 10.1038/ni821. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Alles LF, Versluis K, Maveyraud L, Vallina AT, Sansano S, Bello NF, Gober HJ, Guillet V, de la Salle H, Puzo G, Mori L, Heck AJ, De Libero G, Mourey L. Endogenous phosphatidylcholine and a long spacer ligand stabilize the lipid-binding groove of CD1b. Embo J. 2006;25:3684–3692. doi: 10.1038/sj.emboj.7601244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Alles LF, Collmann A, Versluis C, Lindner B, Guiard J, Maveyraud L, Huc E, Im JS, Sansano S, Brando T, Julien S, Prandi J, Gilleron M, Porcelli SA, de la Salle H, Heck AJ, Mori L, Puzo G, Mourey L, De Libero G. Structural reorganization of the antigen-binding groove of human CD1b for presentation of mycobacterial sulfoglycolipids. Proc Natl Acad Sci U S A. 2011;108:17755–17760. doi: 10.1073/pnas.1110118108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zajonc DM, Cantu C, 3rd, Mattner J, Zhou D, Savage PB, Bendelac A, Wilson IA, Teyton L. Structure and function of a potent agonist for the semi-invariant natural killer T cell receptor. Nat Immunol. 2005;6:810–818. doi: 10.1038/ni1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy C, Shepherd D, Fleire S, Stronge VS, Koch M, Illarionov PA, Bossi G, Salio M, Denkberg G, Reddington F, Tarlton A, Reddy BG, Schmidt RR, Reiter Y, Griffiths GM, van der Merwe PA, Besra GS, Jones EY, Batista FD, Cerundolo V. The length of lipids bound to human CD1d molecules modulates the affinity of NKT cell TCR and the threshold of NKT cell activation. J Exp Med. 2007;204:1131–1144. doi: 10.1084/jem.20062342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilleron M, Stenger S, Mazorra Z, Wittke F, Mariotti S, Bohmer G, Prandi J, Mori L, Puzo G, De Libero G. Diacylated Sulfoglycolipids Are Novel Mycobacterial Antigens Stimulating CD1-restricted T Cells during Infection with Mycobacterium tuberculosis. J Exp Med. 2004;199:649–659. doi: 10.1084/jem.20031097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulrichs T, Moody DB, Grant E, Kaufmann SH, Porcelli SA. T-cell responses to CD1-presented lipid antigens in humans with Mycobacterium tuberculosis infection. Infect Immun. 2003;71:3076–3087. doi: 10.1128/IAI.71.6.3076-3087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larrouy-Maumus G, Layre E, Clark S, Prandi J, Rayner E, Lepore M, de Libero G, Williams A, Puzo G, Gilleron M. Protective efficacy of a lipid antigen vaccine in a guinea pig model of tuberculosis. Vaccine. 2017;35:1395–1402. doi: 10.1016/j.vaccine.2017.01.079. [DOI] [PubMed] [Google Scholar]
- 34.Marrakchi H, Laneelle MA, Daffe M. Mycolic acids: structures, biosynthesis, and beyond. Chemistry and Biology. 2014;21:67–85. doi: 10.1016/j.chembiol.2013.11.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1 Quality control of purified and commercially available MA
Supplemental Fig. 2 Mass spectrometry of synthetic MA
Supplemental Fig. 3 NMR of synthetic MA.
Supplemental Fig. 4 Sorting strategy for TCR sequencing from C58L
Supplemental Fig. 5 Cytokine standard curves